

Abstract
Telomeres are repetitive DNA sequences that protect the ends of linear chromosomes, and they are maintained by a ribonucleoprotein complex called telomerase. Variants in genes encoding for telomerase components have been associated with a spectrum of disease in the lung, skin, bone marrow, and liver. Mutations in the telomerase reverse transcriptase and telomerase RNA component genes have been observed at a higher prevalence in patients with liver disease compared with the general population; however, the presence of variants in other components of the telomerase complex and their impact on clinical outcomes has not been explored. We evaluated 86 patients with end‐stage liver disease for variants in an expanded panel of eight genes, and found that 17 patients (20%) had likely deleterious variants by in silico analysis. Seven unique likely deleterious variants were identified in the regulator of telomere elongation helicase 1 (RTEL1) gene that encodes for a DNA helicase important in telomere maintenance and genomic stability. In gene burden association analysis of their clinical data, the presence of any RTEL1 variant was associated with a 29% lower baseline white blood cell count (95% confidence interval [CI], ‐7% to ‐46%; P Value = 0.01) compared with patients without RTEL1 variants, and the presence of any exonic missense RTEL1 variant was associated with a 42% lower baseline platelet count (95% CI, ‐5% to ‐65%: P Value = 0.03). The presence of any telomerase variant was associated with an increased number of readmissions within 1 year after transplantation demonstrated by an incident rate ratio (IRR) of 3.15 (95% CI, 1.22 to 8.57). No association with survival was observed. Conclusion: Among patients who underwent liver transplantation, the presence of any exonic missense variant was associated with a longer postoperative length of stay with an IRR of 2.16 (95% CI, 1.31 to 3.68).
Abbreviations
- ANNOVAR
Annotate Variation
- CI
confidence interval
- DKC
dyskerin pseudouridine synthase
- ETOH
alcohol
- ExAC
Exome Aggregation Consortium
- FATHMM
Functional Analysis Through Hidden Markov Models
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- IRR
incident rate ratio
- LRT
likelihood ratio test
- PolyPhen 2
polymorphism phenotyping 2
- RTEL1
regulator of telomere elongation helicase 1
- SIFT
Sorting Intolerant From Tolerant
- TERC
telomerase RNA component
- TERT
telomerase reverse transcriptase
- TINF2
telomeric repeat binding factor 1–interacting nuclear factor 2
- WBC
white blood cell
- WRAP53
tryptophan–aspartic acid repeat containing antisense to tumor protein p53
Telomeres are repeating hexanucleotide DNA sequences at the ends of linear chromosomes that protect against successive shortening of genomic DNA during replication, thereby preventing errant DNA repair activities and ultimately cell senescence. Telomere length is maintained by a ribonucleoprotein enzyme called telomerase and other protective telomere‐binding proteins. Multiple single gene–inactivating variants directly affecting telomere length or maintenance have been identified. These mutations have been associated with an early aging phenotype and a spectrum of often overlapping diseases, including bone marrow failure, blood and solid tumor malignancies, pulmonary fibrosis, liver cirrhosis, hair graying, and skin pigmentation.1 As the role of each component of the telomerase complex becomes clearer, new gene candidates become available to explore their relationship to shortened telomeres and organ dysfunction.2
Liver cirrhosis leads to varying degrees of pancytopenia through multiple known mechanisms, including splenic sequestration, decreased thrombopoietin production, nutritional deficiencies, and gastrointestinal bleeding. The potential contribution of telomerase complex gene variants to cytopenias could have significant implications for these patients after liver transplantation, as they take myelosuppressive medications to prevent transplant rejection and infection. Patients with telomerase complex mutations undergoing lung transplantation have increased risk of myelodysplasia and/or bone marrow failure, and shortened telomere length has been associated with worse survival and shorter time to lung allograft dysfunction, although the effect of telomere length on liver transplant outcomes is not known.3, 4 Already, specific variants in the telomerase reverse transcriptase (TERT) gene have been observed at a higher frequency in patients with cirrhosis compared with healthy controls, affecting 2.7% of 521 patients with cirrhosis examined in one large study.5 In another series of 134 patients with cirrhosis, 7% were found to have a missense TERT variant.6 However, these studies did not report clinical characteristics, including cytopenias that may be out of proportion to a patient's degree of liver dysfunction and have been associated with increased morbidity and mortality in patients with cirrhosis.7
The frequency of variants in other genes encoding components of the telomerase machinery and other related proteins outside of TERT and telomerase RNA component (TERC) have not been characterized in patients with cirrhosis. Variants in genes that encode for a DNA helicase (regulator of telomere elongation helicase 1 [RTEL1]), the telomerase complex scaffolding (dyskerin pseudouridine synthase [DKC], NOP10, NHP2), the shelterin complex important for recruiting telomerase to the ends of telomeres (telomeric repeat binding factor 1–interacting nuclear factor 2 [TINF2]), and a telomerase trafficking protein (WD40‐encoding RNA antisense to p53 [WRAP53]) have all been associated with shorter telomere length and bone marrow failure syndromes. Variants in these genes could contribute to the cytopenias seen in patients with cirrhosis and effect posttransplantation outcomes. Thus, similar to other recent studies investigating genetic variants and their association to clinical outcomes, we describe the prevalence of genetic variants in TERT, TERC, RTEL1, DKC, NOP10, NHP2, TINF2, and WRAP53 in a cohort of patients with end‐stage liver disease awaiting liver transplantation, and we assess the association of these variants with their clinical phenotypes and posttransplant outcomes.8, 9
Materials and Methods
The study proposal was deemed consistent with international Good Clinical Practice standards and approved by the University of Southern California Institutional Review Board. Patients with liver disease were identified prospectively and signed informed consent to allow collection of personal health information from the electronic medical record and to donate blood and biopsy samples, if available, as part of a tissue biorepository of patients diagnosed with cirrhosis. For the present study, 86 blood samples were randomly selected from the tissue repository based on the following inclusion criteria: 1) age greater than 18 years of age, 2) diagnosis of end‐stage liver disease secondary to any cause with or without a diagnosis of hepatocellular carcinoma, and 3) sufficient blood sample available for variant analysis.
Three milliliters of whole blood from each patient was sent to Fulgent Diagnostics Laboratory (Temple City, CA). Each sample was tested for the presence of telomere complex variants in the following genes: TERT, TERC, RTEL1, DKC, NOP10, NHP2, TINF2, and WRAP53. Genomic DNA was first isolated from peripheral blood mononuclear cells, then enriched for the coding exons of targeted genes using hybrid capture technology. Prepared DNA libraries were then sequenced using next‐generation sequencing technology. Following alignment, variants were detected in regions of at least 10× coverage.
Clinical data including demographic data, medical and family history, results of diagnostic liver biopsies, and posttransplant outcomes were retrospectively abstracted from the electronic medical records for each patient. Self‐reported ethnicity was abstracted from the medical record, and patient age was calculated from date of blood sample donation. The cause of cirrhosis was determined by the treating physician and abstracted from physician documentation. The underlying cause of cirrhosis was broadly grouped into three categories for statistical analysis: alcohol (ETOH), hepatitis B virus (HBV) or hepatitis C virus (HCV), and other. The presence of hepatocellular carcinoma was recorded from the medical record and defined by pathological specimen or imaging consistent with hepatocellular carcinoma.
For patients who did not undergo liver transplantation, the lowest reported values for each blood cell count were recorded because we hypothesized that any insult commonly experienced by patients with cirrhosis would have a more significant effect on the cell counts in telomere‐mutated patients. We recorded the highest reported values for creatinine, international normalized ratio, and total bilirubin to capture the greatest severity of underlying liver disease or comorbidity. For patients undergoing liver transplantation, we recorded the lowest pretransplant blood counts up to 1 year before transplantation. Posttransplantation laboratory values were reported as averages obtained at 3 months after transplant, although these data were unavailable for patients who had not yet presented for or had died before the initial 3‐month posttransplant evaluation. For patients who underwent transplant, the pretransplant laboratory Model for End‐Stage Liver Disease (MELD) was calculated from the laboratory values taken immediately before transplant.10 For patients who underwent transplant, the posttransplant length of stay and the number of readmissions within 1 year were recorded. Posttransplant survival days were abstracted from the United Network for Organ Sharing UNet follow‐up database.
Variant Annotation and Significance
Genotyped telomere variants were functionally annotated using Annotate Variation (ANNOVAR) software to better categorize variants for gene burden testing.11 Within ANNOVAR, the Homo sapiens hg19 genome assembly (hg19/GRCh37) was selected as the reference genome, and the Reference Sequence (RefSeq) database was used for gene definition.12, 13 Dichotomized predictions for significance of nonsynonymous variants were based on Sorting Intolerant From Tolerant (SIFT), polymorphism phenotyping 2 (PolyPhen 2) HDIV, PolyPhen 2 Hvar, likelihood ratio test (LRT), MutationTaster, Functional Analysis Through Hidden Markov Models (FATHMM), MetaSVM, and MetaLR scores. A variant was considered to be “likely deleterious” if any of the ANNOVAR prediction categories for individual scores were “deleterious,” “probably damaging,” “possibly damaging,” “disease causing automatic,” or “disease causing.” The remaining variants were considered to be “likely tolerated/neutral.” For exonic missense variants, DNA change, protein change, Combined Annotation Dependent Depletion (CADD), and Exome Aggregation Consortium (ExAC) frequency were also extracted from ANNOVAR results. Finally, variants were categorized into nonexclusive groups according to predicted significance and variant type: any, exonic missense, likely deleterious, any RTEL1, exonic missense RTEL1, and likely deleterious RTEL1.
Associations Between Variants and Clinical Values
Gene burden association tests were performed using regression models of categories of variants and various clinical values. Among all patients, we evaluated associations between baseline laboratory values and the presence of variants using natural log–transformed linear regression models. Thus, the exponentiated regression coefficients for laboratory values should be interpreted as the changes in the ratio of the expected geometric means of the original outcome variable. For presentation in outcome tables, results represent the percent change in estimated geometric mean. Follow‐up data on postoperative clinical outcomes (laboratory values, length of stay, number of readmissions within 1 year, and mortality) were available for transplant patients. Using a gene burden test, associations between the presence of variants and pretransplant/posttransplant change in laboratory values were analyzed using natural log–transformed linear regression models; associations with postoperative length of stay and readmissions were analyzed using negative binomial regression models; and associations with mortality were analyzed using Cox proportional hazard regression models.14 All models were adjusted for age at sample, ethnicity/race (Hispanic/non‐Hispanic white/other), diagnosis (ETOH/HBV or HCV/other), and sex (female/male). A conservative Bonferroni adjustment was applied to each group of tests (all variant categories for each outcome) to account for multiple comparisons.
Results
Eighty‐six patients were identified as meeting the inclusion criteria for the study; their demographic information is reported in Table 1. Our cohort was largely Hispanic (44%), with Caucasians (37%) and Asians (9%) constituting the majority of the remaining patients. The mean age of the cohort was 54.9 years, and the patients all had advanced cirrhosis at the time of study participation and sample collection. Cirrhosis due to viral infection accounted for almost half (47%) of the etiologic diagnoses, whereas ETOH was identified as the cause of cirrhosis in 23%. Forty‐three (50%) of the patients underwent liver transplantation.
Table 1.
Patient Characteristics
| N (%) | |||
|---|---|---|---|
| Ethnicity | African American | 2 (2) | |
| Asian | 8 (9) | ||
| Hispanic | 38 (44) | ||
| Middle Eastern | 3 (3) | ||
| Native American | 1 (1) | ||
| Pacific Islander | 1 (1) | ||
| Unknown | 1 (1) | ||
| White | 32 (37) | ||
| Sex | F | 36 (42) | |
| M | 50 (58) | ||
| Diagnosis | ETOH | 20 (23) | |
| HBV/HCV | 40 (47) | ||
| Other | 26 (30) | ||
| Transplant | No transplant | 43 (50) | |
| Transplant | 43 (50) | ||
| Mean ± SD | Median (IQR) | ||
| Age at sample date | 54.9 ± 10.4 | 56 (14.8) | |
| Native Pre‐tx MELD | 25.3 ± 12.7 | 22 (25.5) | |
| Baseline laboratory values | WBC count (×109/L) | 4.3 ± 4.5 | 3.6 (2.2) |
| Absolute neutrophil count (×103/mm3) | 2.9 ± 3.9 | 2.1 (1.8) | |
| Hemoglobin (g/dL) | 9.7 ± 3 | 8.7 (5.3) | |
| Platelets (×109/L) | 68 ± 55 | 50 (63) | |
| Creatinine (mg/dL) | 1.4 ± 1.4 | 0.9 (0.9) | |
| Total bilirubin (mg/dL) | 7 ± 10.6 | 2.4 (5.2) | |
| International normalized ratio | 1.6 ± 0.7 | 1.3 (0.7) | |
Abbreviations: F, female; IQR, interquartile range; M, male; Pre‐tx, pretransplant.
Germline variants in at least one of the eight telomerase complex genes were detected in 68 (79%) patients, with 18 (21%) of the patients harboring no detectable variants (Table 2). A complete description of the variants considered likely deleterious or damaging and likely tolerated or neutral is presented in Table 3. Thirteen variants considered likely deleterious or damaging occurred in 17 patients, and seven of these were in the RTEL1 gene. These variants are displayed by gene locus in Figure 1. There were 3 unrelated patients with the same variant in TINF2 (c.359A>G) and an additional patient with a TINF2 variant (c.721C>T); variants in this gene have not been previously reported outside of patients with dyskeratosis congenita. An additional four variants in RTEL1 were considered likely tolerated or neutral. There were no variants detected in the TERC gene. An additional 122 gene variants were considered to be of unknown significance.
Table 2.
Variant Summary
| N (% variants) | ||
|---|---|---|
| Gene | DKC1 | 9 (6) |
| NHP2 | 10 (7) | |
| NOP10 | 4 (3) | |
| RTEL1 | 42 (28) | |
| TERT | 63 (42) | |
| TINF2 | 11 (7) | |
| WRAP53 | 11 (7) | |
| Variant significance | Likely deleterious/damaging | 18 (12) |
| Likely tolerated/neutral | 10 (7) | |
| Unknown significance | 122 (81) | |
| Variant type | Exonic: missense SNV | 28 (19) |
| Exonic: nonframeshift substitution | 1 (1) | |
| Exonic: synonymous SNV | 79 (53) | |
| Intronic | 23 (15) | |
| UTR3/UTR5 | 19 (13) | |
| Number of variants per patient | 0 | 18 (21) |
| 1 | 21 (24) | |
| 2 | 24 (28) | |
| 3 | 14 (16) | |
| 4 | 6 (7) | |
| 5 | 3 (3) | |
Abbreviations: SNV, single‐nucleotide variant; UTR, untranslated region.
Table 3.
Characteristics of Exonic Missense Variants
| Variant Significance | Gene | N (% Patients) | DNA Change | Protein Change | CADD | ExAC Freq | Highest ExAC Freq Within Any Ethnic Group | Variant Significance Predictions* | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT | PolyPhen 2 HDIV | PolyPhen 2 Hvar | LRT | MutationTaster | FATHMM | MetaSVM | MetaLR | ||||||||
| Likely deleterious/damaging | TINF2 | 3 (3) | c.359A>G | p.Gln120Arg | 9.017 | 0.0033 | 0.033 | T | P | B | N | D | T | T | T |
| TERT | 2 (2) | c.3184G>A | p.Ala1062Thr | 2.954 | 0.0134 | 0.021 | T | B | B | N | A | D | T | T | |
| TERT | 2 (2) | c.835G>A | p.Ala279Thr | 8.235 | 0.0361 | 0.121 | T | B | B | N | A | D | T | T | |
| NHP2 | 2 (2) | c.302G>A | p.Arg101Gln | 22.3 | 0.0046 | 0.007 | T | B | B | N | D | T | T | T | |
| TINF2 | 1 (1) | c.721C>T | p.Pro241Ser | 23.4 | 0.0021 | 0.024 | D | D | D | N | D | D | D | D | |
| RTEL1 | 1 (1) | c.860C>T | p.Thr287Ile | 12.07 | 0.0002 | 0.002 | T | B | B | N | N | D | T | T | |
| RTEL1 | 1 (1) | c.1727G>A | p.Arg576His | 6.641 | 0.0018 | 0.021 | T | B | B | N | N | D | T | T | |
| RTEL1 | 1 (1) | c.2915C>T | p.Thr972Ile | 18.68 | 0.0001 | 0.001 | D | B | B | D | D | T | T | T | |
| RTEL1 | 1 (1) | c.3056A>G | p.Gln1019Arg | 0.276 | 0.0013 | 0.013 | T | B | B | N | N | D | T | T | |
| RTEL1 | 1 (1) | c.3101C>A | p.Pro1034His | 16.95 | 0.0163 | 0.047 | T | D | P | N | N | D | T | T | |
| RTEL1 | 1 (1) | c.3392C>G | p.Thr1131Arg | 24.4 | 8.52E‐06 | 1.561e‐05 | D | D | P | N | N | D | T | D | |
| RTEL1 | 1 (1) | c.3692C>T | p.Thr1231Met | 0.033 | 0.0016 | 0.019 | T | B | B | U | N | D | T | T | |
| TERT | 1 (1) | c.1936C>T | p.Arg646Cys | 14.42 | 4.12E‐05 | 9.625e‐05 | T | B | B | N | N | D | T | D | |
| Likely tolerated/neutral | WRAP53 | 4 (5) | c.407C>G | p.Pro136Arg | 0.524 | 0.0034 | 0.045 | T | B | B | N | N | T | T | T |
| RTEL1 | 2 (2) | c.2546G>A | p.Gly849Asp | 10.29 | 0.013 | 0.081 | T | B | B | N | N | T | T | T | |
| WRAP53 | 1 (1) | c.1565C>G | p.Ala522Gly | 0.005 | 0.3123 | 0.822 | T | B | B | N | P | T | T | T | |
| RTEL1 | 1 (1) | c.2476C>T | p.Pro826Ser | 0.002 | NR | NR | T | B | B | N | N | T | T | T | |
| RTEL1 | 1 (1) | c.2898G>C | p.Glu966Asp | 3.801 | 0.0017 | 0.019 | T | B | B | N | N | T | T | T | |
| RTEL1 | 1 (1) | c.2987C>A | p.Pro996His | 8.765 | 0.0001 | 0.002 | T | B | B | N | N | T | T | T | |
A mutation was considered to be “likely deleterious” and are indicated in bold if any of the following prediction categories were “deleterious,” “probably damaging,” “possibly damaging,” “disease causing automatic,” or “disease causing.” SIFT, FATHMM, MetaSVM, MetaLR: D = deleterious, T = tolerated. PolyPhen 2 HDIV, PolyPhen 2 HVar: D = probably damaging, P = possibly damaging, B = benign. LRT: D = deleterious, N = neutral, U = unknown. MutationTaster: A = disease causing automatic, D = disease causing, N = polymorphism, P = polymorphism automatic.
Abbreviations: Freq, frequency; NR, not reported.
Figure 1.
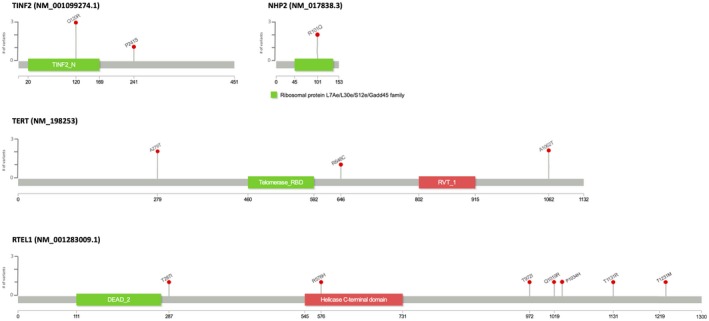
Likely deleterious or damaging telomerase gene variants detected in 86 patients with cirrhosis awaiting liver transplantation, displayed by gene locus.
Given the prevalence of RTEL1 variants in our cohort, we examined the association between any, likely deleterious, and exonic variants in RTEL1 with blood counts and posttransplant outcomes. The presence of any RTEL1 variant was associated with a 29% lower baseline white blood cell (WBC) count (Table 4; 95% confidence interval [CI], ‐7%, to ‐46%; P Value = 0.01), and the presence of any exonic missense RTEL1 variant was associated with a 42% lower baseline platelet count (Table 4; 95% CI, ‐5% to ‐65%; P Value = 0.03). The presence of any telomere variant was associated with a greater increase in platelet count 3 months after transplantation (Table 5). The presence of any exonic missense variant was associated with longer length of stay (Table 6; incident rate ratio [IRR], 2.16; 95% CI, 1.31 to 3.68; P Value = 0.003), and presence of any variant was associated with increased number of readmissions at 1 year after transplant (Table 6; IRR, 3.15; 95% CI, 1.22 to 8.57; P Value = 0.02). However, the presence of telomere variants did not appear to impact overall survival (Table 6). Only the association between postoperative length of stay and the presence of any exonic missense variant survived a conservative Bonferroni correction for multiple comparisons.
Table 4.
Associations Between Presence of Telomere Variants and Baseline Laboratory Values
| Laboratory Test | Variant Type | N (% All Patients) | Estimate* | 95% CI | P Value† | |
|---|---|---|---|---|---|---|
| In Model | With ≥1 Variant | |||||
| WBC count | Any | 86 (100) | 68 (79) | ‒0.17 | ‒0.40, 0.16 | 0.28 |
| Exonic missense | 86 (100) | 24 (28) | 0.06 | ‒0.22, 0.44 | 0.72 | |
| Likely deleterious | 86 (100) | 17 (20) | 0.22 | ‒0.12, 0.70 | 0.24 | |
| Any RTEL1 | 86 (100) | 30 (35) | ‒0.29 | ‒0.46, ‒0.07 | 0.01 | |
| Exonic missense RTEL1 | 86 (100) | 11 (13) | ‒0.30 | ‒0.54, 0.04 | 0.08 | |
| Likely deleterious RTEL1 | 86 (100) | 7 (8) | ‒0.15 | ‒0.49, 0.42 | 0.53 | |
| Absolute neutrophil count | Any | 86 (100) | 68 (79) | ‒0.24 | ‒0.49, 0.12 | 0.17 |
| Exonic missense | 86 (100) | 24 (28) | 0.11 | ‒0.23, 0.60 | 0.56 | |
| Likely deleterious | 86 (100) | 17 (20) | 0.36 | ‒0.08, 1.00 | 0.12 | |
| Any RTEL1 | 86 (100) | 30 (35) | ‒0.26 | ‒0.47, 0.03 | 0.07 | |
| Exonic missense RTEL1 | 86 (100) | 11 (13) | ‒0.30 | ‒0.56, 0.14 | 0.15 | |
| Likely deleterious RTEL1 | 86 (100) | 7 (8) | ‒0.03 | ‒0.47, 0.77 | 0.93 | |
| Hemoglobin | Any | 86 (100) | 68 (79) | ‒0.02 | ‒0.16, 0.14 | 0.79 |
| Exonic missense | 86 (100) | 24 (28) | ‒0.10 | ‒0.22, 0.03 | 0.12 | |
| Likely deleterious | 86 (100) | 17 (20) | ‒0.01 | ‒0.15, 0.15 | 0.89 | |
| Any RTEL1 | 86 (100) | 30 (35) | ‒0.12 | ‒0.22, 0.00 | 0.05 | |
| Exonic missense RTEL1 | 86 (100) | 11 (13) | ‒0.13 | ‒0.27, 0.05 | 0.15 | |
| Likely deleterious RTEL1 | 86 (100) | 7 (8) | 0.05 | ‒0.17, 0.32 | 0.67 | |
| Platelets | Any | 86 (100) | 68 (79) | ‒0.18 | ‒0.46, 0.24 | 0.34 |
| Exonic missense | 86 (100) | 24 (28) | ‒0.03 | ‒0.33, 0.43 | 0.89 | |
| Likely deleterious | 86 (100) | 17 (20) | 0.04 | ‒0.31, 0.57 | 0.84 | |
| Any RTEL1 | 86 (100) | 30 (35) | ‒0.22 | ‒0.45, 0.10 | 0.16 | |
| Exonic missense RTEL1 | 86 (100) | 11 (13) | ‒0.42 | ‒0.65, ‒0.05 | 0.03 | |
| Likely deleterious RTEL1 | 86 (100) | 7 (8) | ‒0.29 | ‒0.62, 0.32 | 0.28 | |
| Creatinine | Any | 86 (100) | 68 (79) | ‒0.02 | ‒0.32, 0.41 | 0.90 |
| Exonic missense | 86 (100) | 24 (28) | 0.22 | ‒0.12, 0.71 | 0.23 | |
| Likely deleterious | 86 (100) | 17 (20) | 0.08 | ‒0.25, 0.56 | 0.66 | |
| Any RTEL1 | 86 (100) | 30 (35) | 0.15 | ‒0.16, 0.56 | 0.38 | |
| Exonic missense RTEL1 | 86 (100) | 11 (13) | 0.24 | ‒0.21, 0.94 | 0.34 | |
| Likely deleterious RTEL1 | 86 (100) | 7 (8) | 0.14 | ‒0.34, 0.99 | 0.63 | |
| Total bilirubin | Any | 86 (100) | 68 (79) | ‒0.33 | ‒0.64, 0.26 | 0.21 |
| Exonic missense | 86 (100) | 24 (28) | 0.08 | ‒0.40, 0.94 | 0.79 | |
| Likely deleterious | 86 (100) | 17 (20) | ‒0.04 | ‒0.49, 0.81 | 0.89 | |
| Any RTEL1 | 86 (100) | 30 (35) | ‒0.36 | ‒0.62, 0.09 | 0.10 | |
| Exonic missense RTEL1 | 86 (100) | 11 (13) | 0.32 | ‒0.40, 1.87 | 0.48 | |
| Likely deleterious RTEL1 | 86 (100) | 7 (8) | ‒0.19 | ‒0.69, 1.13 | 0.67 | |
| International normalized ratio | Any | 82 (95) | 64 (74) | ‒0.12 | ‒0.27, 0.05 | 0.16 |
| Exonic missense | 82 (95) | 22 (26) | ‒0.07 | ‒0.22, 0.11 | 0.44 | |
| Likely deleterious | 82 (95) | 15 (17) | ‒0.09 | ‒0.25, 0.10 | 0.33 | |
| Any RTEL1 | 82 (95) | 28 (33) | ‒0.15 | ‒0.27, 0.00 | 0.05 | |
| Exonic missense RTEL1 | 82 (95) | 10 (12) | 0.02 | ‒0.20, 0.29 | 0.90 | |
| Likely deleterious RTEL1 | 82 (95) | 6 (7) | ‒0.01 | ‒0.27, 0.34 | 0.95 | |
All models are adjusted for age at sample, ethnicity/race (Hispanic/non‐Hispanic white/other), diagnosis (ETOH/HBV or HCV/other), and sex (female/male).
The corresponding regression coefficient was exponentiated, and the presented estimate should be interpreted as the percent change in the expected geometric mean of the laboratory value when at least one mutation is present.
Bolded associations had a P value <0.05, but were not statistically significant after Bonferroni correction for multiple comparisons.
Table 5.
Associations Between Presence of Telomere Gene Variants and Pretransplant/Posttransplant Change in Laboratory Values
| Laboratory Test | Variant Type | N (% Transplant Patients) | Estimate* | 95% CI | P Value† | |
|---|---|---|---|---|---|---|
| In Model | With ≥1 Variant | |||||
| WBC count | Any | 39 (91) | 31 (72) | ‒0.02 | ‒0.31, 0.39 | 0.91 |
| Exonic missense | 39 (91) | 8 (19) | 0.12 | ‒0.22, 0.61 | 0.52 | |
| Likely deleterious | 39 (91) | 5 (12) | 0.05 | ‒0.29, 0.56 | 0.80 | |
| Any RTEL1 | 39 (91) | 11 (26) | 0.22 | ‒0.11, 0.67 | 0.22 | |
| Exonic missense RTEL1 | 39 (91) | 5 (12) | ‒0.06 | ‒0.40, 0.49 | 0.80 | |
| Likely deleterious RTEL1 | 39 (91) | 3 (7) | ||||
| Absolute neutrophil count | Any | 37 (86) | 30 (70) | 0.13 | ‒0.25, 0.70 | 0.56 |
| Exonic missense | 37 (86) | 8 (19) | 0.25 | ‒0.12, 0.79 | 0.21 | |
| Likely deleterious | 37 (86) | 5 (12) | 0.11 | ‒0.26, 0.66 | 0.61 | |
| Any RTEL1 | 37 (86) | 10 (23) | 0.18 | ‒0.16, 0.65 | 0.32 | |
| Exonic missense RTEL1 | 37 (86) | 5 (12) | 0.08 | ‒0.32, 0.71 | 0.74 | |
| Likely deleterious RTEL1 | 37 (86) | 3 (7) | ||||
| Hemoglobin | Any | 39 (91) | 31 (72) | 0.04 | ‒0.07, 0.17 | 0.44 |
| Exonic missense | 39 (91) | 8 (19) | 0.00 | ‒0.11, 0.13 | 0.96 | |
| Likely deleterious | 39 (91) | 5 (12) | ‒0.05 | ‒0.17, 0.08 | 0.40 | |
| Any RTEL1 | 39 (91) | 11 (26) | ‒0.01 | ‒0.11, 0.10 | 0.89 | |
| Exonic missense RTEL1 | 39 (91) | 5 (12) | ‒0.02 | ‒0.15, 0.14 | 0.82 | |
| Likely deleterious RTEL1 | 39 (91) | 3 (7) | ||||
| Platelets | Any | 38 (88) | 30 (70) | 0.68 | 0.09, 1.57 | 0.02 |
| Exonic missense | 38 (88) | 7 (16) | 0.27 | ‒0.24, 1.12 | 0.34 | |
| Likely deleterious | 38 (88) | 4 (9) | ||||
| Any RTEL1 | 38 (88) | 11 (26) | 0.19 | ‒0.19, 0.75 | 0.37 | |
| Exonic missense RTEL1 | 38 (88) | 5 (12) | ‒0.02 | ‒0.46, 0.80 | 0.95 | |
| Likely deleterious RTEL1 | 38 (88) | 3 (7) | ||||
All models are adjusted for age at sample, ethnicity/race (Hispanic/non‐Hispanic white/other), diagnosis (ETOH/HBV or HCV/other), and sex (female/male). Results from models in which fewer than 5 patients have mutations are not shown.
Differences in pretransplant (baseline) and posttransplant (3 months) laboratory values were measured. The corresponding regression coefficient was exponentiated, and the presented estimate should be interpreted as the percent change in the expected geometric mean of pretransplant/posttransplant difference when at least one mutation is present.
Bolded associations had a P value <0.05, but were not statistically significant after Bonferroni correction for multiple comparisons.
Table 6.
Associations Between Presence of Telomere Variants and Posttransplantation Outcomes
| N (% Transplant Patients) | Postoperative Length Of Stay | |||||
|---|---|---|---|---|---|---|
| Variant Type | In Model | With ≥1 Variant | IRR* | 95% CI | P Value | |
| Any | 42 (98) | 34 (79) | 1.60 | 0.89, 2.79 | 0.10 | |
| Exonic missense | 42 (98) | 8 (19) | 2.16 | 1.31, 3.68 | 0.003 | |
| Likely deleterious | 42 (98) | 5 (12) | 2.05 | 1.17, 3.83 | 0.02 | |
| Any RTEL1 | 42 (98) | 14 (33) | 1.07 | 0.67, 1.71 | 0.79 | |
| Exonic missense RTEL1 | 42 (98) | 5 (12) | 1.51 | 0.77, 3.15 | 0.24 | |
| Likely deleterious RTEL1 | 42 (98) | 3 (7) | — | — | — | |
| N (% Transplant Patients) | Posttransplant Readmissions | |||||
| Variant Type | In Model | With ≥1 Variant | IRR* | 95% CI | P Value‡ | |
| Any | 42 (98) | 34 (79) | 3.15 | 1.22, 8.57 | 0.02 | |
| Exonic missense | 42 (98) | 8 (19) | 2.03 | 0.84, 5.29 | 0.08 | |
| Likely deleterious | 42 (98) | 5 (12) | 1.80 | 0.74, 4.77 | 0.21 | |
| Any RTEL1 | 42 (98) | 14 (33) | 1.56 | 0.78, 3.2 | 0.19 | |
| Exonic missense RTEL1 | 42 (98) | 5 (12) | 1.30 | 0.45, 4.04 | 0.61 | |
| Likely deleterious RTEL1 | 42 (98) | 3 (7) | — | — | — | |
| N (% Transplant Patients) | Posttransplant Mortality | |||||
| Variant Type | In Model | With ≥1 Variant | HR‡ | 95% CI | P Value | |
| Any | 39 (91) | 31 (72) | 1.00 | 0.16, 6.16 | >0.99 | |
| Exonic missense | 39 (91) | 7 (16) | 5.15 | 0.84, 31.44 | 0.08 | |
| Likely deleterious | 39 (91) | 5 (12) | 6.15 | 0.84, 44.89 | 0.07 | |
| Any RTEL1 | 39 (91) | 12 (28) | 0.22 | 0.02, 2.06 | 0.19 | |
| Exonic missense RTEL1 | 39 (91) | 4 (9) | — | — | — | |
| Likely deleterious RTEL1 | 39 (91) | 3 (7) | — | — | — | |
All models are adjusted for age at sample, ethnicity/race (Hispanic/non‐Hispanic white/other), diagnosis (ETOH/HBV or HCV/other), and sex (female/male). Results from a model in which fewer than 5 patients have mutations are not shown.
For post‐operative length of stay, the incidence rate ratio (IRR) represents the ratio of LOS when at least one mutation is present vs. when no mutations are present. IRR >1 suggests a risk factor for longer LOS, IRR <1 is protective. For posttransplant readmissions, IRR represents the ratio of readmissions within a year when at least one mutation is present vs. when no mutations are present. IRR >1 suggests a risk factor for more readmissions, IRR <1 is protective.
The HR represents the ratio of mortality rate when at least one mutation is present versus when no mutations are present. An HR >1 suggests a risk factor for mortality; an HR < 1 is protective.
Note: Bolded associations had a P value <0.05, but only the association between exonic missense variants and postoperative stay remained statistically significant after Bonferroni correction for multiple comparisons.
Abbreviation: HR, hazard ratio.
The 3 patients sharing the same TINF2 variant (c.359A>G) were younger than the mean age of the cohort (ages 41, 24, and 46) and had normal to elevated WBC and subnormal platelet counts (34, 81, and 129 × 109/L respectively), but this subgroup was too small to apply statistical comparison and none received transplant (Table 7).
Table 7.
Characteristics of Patients With at least One Likely Deleterious Variant
| Age (Years) | Sex | DNA Change(s) | Allelic Frequency (%) | Gene | Protein Change(s) | Baseline Laboratory Values | Postoperative Outcomes Among Transplant Patients | Telomere Length* | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| WBC (×109/L) | Hgb (g/dL) | Platelet (×109/L) | Length of Stay (Days) | Readmissions Within 1 Year | Mortality | ||||||||
| No transplant | 41 | M | c.3392C>G | 52.1 | RTEL1 | p.Thr1131Arg | 10.1 | 14.3 | 196 | — | — | — | 6,772 |
| 76 | M | c.302G>A | 50.9 | NHP2 | p.Arg101Gln | 6.2 | 9.6 | 115 | — | — | — | N/A | |
| 66 | M | c.302G>A | 49.2 | NHP2 | p.Arg101Gln | 5.3 | 12.5 | 115 | — | — | — | N/A | |
| 55 | F | c.721C>T | 48.7 | TINF2 | p.Pro241Ser | 4.2 | 6.9 | 26 | — | — | — | N/A | |
| 41 | M | c.359A>G | 49.6 | TINF2 | p.Gln120Arg | 5.35 | 7 | 34 | — | — | — | 2,880 | |
| 56 | M | c.3692C>T | 47.7 | RTEL1 | p.Thr1231Met | 3.5 | 14.4 | 45 | — | — | — | 2,565 | |
| 47 | F | c.835G>A | 47.8 | TERT | p.Ala279Thr | 5.4 | 14.5 | 126 | — | — | — | N/A | |
| 60 | M | c.860C>T | 43.2 | RTEL1 | p.Thr287Ile | 3.4 | 8.9 | 12 | — | — | — | 2,353 | |
| 24 | F | c.359A>G | 50.9 | TINF2 | p.Gln120Arg | 41.1 | 6.5 | 81 | — | — | — | N/A | |
| 49 | M | c.1936C>T | 43.2 | TERT | p.Arg646Cys | 2.4 | 7.4 | 92 | — | — | — | 6,211 | |
| 59 | F | c.3184G>A | 49.7 | TERT | p.Ala1062Thr | 1.8 | 7.4 | 25 | — | — | — | N/A | |
| c.3101C>A | 48.9 | RTEL1 | p.Pro1034His | ||||||||||
| 46 | F | c.359A>G | 49.8 | TINF2 | p.Gln120Arg | 4.4 | 7.3 | 129 | — | — | — | N/A | |
| Transplant | 57 | F | c.1727G>A | 47.2 | RTEL1 | p.Arg576His | 3.6 | 10.1 | 151 | 21 | 8 | Alive | N/A |
| 56 | M | c.835G>A | 45.9 | TERT | p.Ala279Thr | 1.2 | 8.9 | 21 | 175 | 3 | Died | N/A | |
| 59 | M | c.3184G>A | 49.7 | TERT | p.Ala1062Thr | 4.2 | 12.1 | 32 | 13 | 7 | Died | N/A | |
| 52 | F | c.2915C>T | 48.3 | RTEL1 | p.Thr972Ile | 1.12 | 6.3 | 10 | 10 | 6 | Alive | 2,842 | |
| 47 | F | c.3056A>G | 49.7 | RTEL1 | p.Gln1019Arg | 4.41 | 8.1 | 35 | 12 | 0 | Alive | 2,521 | |
Relative copy number of the telomere repeat normalized to one single‐copy gene (36B4).
Abbreviations: F, female; Hgb, hemoglobin; M, male; N/A, not available.
Discussion
Using in silico analysis, we identified likely deleterious germline telomere variants in 20% of an unselected population of patients with cirrhosis of various causes. Allelic frequencies of these variants were nearly 50%, suggesting that all variants detected were heterozygous. Strikingly, 8.1% of these patients harbored likely deleterious RTEL1 variants and 4.7% harbored TINF2 variants. We hypothesized that patients harboring telomerase variants would have significantly lower baseline WBC or platelet counts; a relationship is suggested for those with any RTEL1 variant and any RTEL1 exonic missense variant, respectively. However, the association did not retain significance when corrected for multiple comparisons. Interestingly, all 4 patients with variants in TINF2 had subnormal platelet counts (Table 7), and the 3 with c.359A>G were significantly younger than the average age of the cohort, which may support a genetic component to their disease. Finally, the presence of any telomere variant was associated with longer length of stay and increased readmissions.
Our study sequenced eight of the genes known to be involved in telomerase function and maintenance and described a high prevalence of germline RTEL1 variants in a series of patients with a primary diagnosis of cirrhosis. RTEL1 plays a central role in telomere maintenance and genome stability. Telomeres can exist in a protective T‐loop lariat configuration when the 3′ single‐stranded end of telomeric DNA invades an upstream segment of telomere DNA. Mammalian RTEL1 disassembles T‐loops during S phase to allow for telomere replication and also resolves guanine‐quadraplex secondary DNA structures that could cause stalling of replication forks and result in loss of telomere DNA during replication.15, 16, 17, 18 Variants in RTEL1 have been associated with a spectrum of disease, including pulmonary fibrosis, lung cancer, glioma, and bone marrow failure syndromes such as dyskeratosis congenita and its more severe form, Hoyeraal‐Hreidarsson syndrome.19, 20, 21, 22, 23, 24 Notably, among 35 patients with RTEL1 variants in one series from an international bone marrow failure registry, 4 patients had cirrhosis.25 In another series of 27 patients with telomere lengths below the first percentile, 9 had cirrhosis and 1 had an RTEL1 variant.26
One limitation of our study is that we did not have sufficient blood samples to measure telomere length in all patients in this series. We report the lengths obtained in patients with at least one likely deleterious variant in Table 7. Although shorter telomere length has been implicated in disease and is considered supportive evidence of pathogenicity, pathogenicity may not be dependent on telomere length alone, as the components of telomere machinery may have other important functions in DNA repair.22, 27, 28 Recent studies describe pathogenic heterozygous RTEL1 variants in a cohort of patients with aplastic anemia at the National Institutes of Health regardless of telomere length, suggesting that variants in RTEL1 may result in disease due to defects in DNA repair and genome stability rather than telomere length maintenance.25, 28 Nonetheless, we identified 122 gene variants of unknown clinical significance in this cohort, and the corresponding telomere lengths may have provided some insight as to their potential pathogenicity.
The study population consisted of an unselected cohort of patients with cirrhosis of different causes who agreed to donate tissue samples and demographic data to a tissue bank. Although cytopenias result from a variety of mechanisms in patients with chronic liver disease, adjustment for variables related to these mechanisms is not necessary for testing genetic associations, as a germline variation will be independent unless involved in the causal process. For controlling for potential confounding due to population structure, we were limited to adjusting for self‐identified ethnicity, although there exists individual genetic admixture within Hispanic individuals, who composed the majority ethnic group in this cohort.
Our study suggests that rare germline telomere variants may be more prevalent in patients with cirrhosis than previously believed, and they could be associated with lower baseline platelet and WBC counts and a longer posttransplant length of stay. The limited number of likely deleterious variants within each gene limits our power to assess their individual impact on the variables studied. Further investigations into the relationship between germline telomere variants and the development of cirrhosis and into the possibility that cytopenias serve as a surrogate marker of telomere dysfunction in liver disease are warranted. In light of recent reports that that androgen therapy can lengthen telomeres in patients with inherited syndromes of shortened telomeres, our findings also offer a potential therapeutic target to optimize clinical parameters before transplantation and improve posttransplant outcomes.26
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Blackburn EH, Epel ES, Lin J. Human telomere biology: a contributory and interactive factor in aging, disease risks, and protection. Science 2015;350:1193‐1198. [DOI] [PubMed] [Google Scholar]
- 2. Dressen A, Abbas AR, Cabanski C, Reeder J, Ramalingam TR, Neighbors M, et al. Analysis of protein‐altering variants in telomerase genes and their association with MUC5B common variant status in patients with idiopathic pulmonary fibrosis: a candidate gene sequencing study. Lancet Respir Med 2018;6:603‐614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Borie R, Kannengiesser C, Hirschi S, Le Pavec J, Mal H, Bergot E, et al. Severe hematologic complications after lung transplantation in patients with telomerase complex mutations. J Heart Lung Transplant 2015;34:538‐546. [DOI] [PubMed] [Google Scholar]
- 4. Newton CA, Kozlitina J, Lines JR, Kaza V, Torres F, Garcia CK. Telomere length in patients with pulmonary fibrosis associated with chronic lung allograft dysfunction and post‐lung transplantation survival. J Heart Lung Transplant 2017;36:845‐853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hartmann D, Srivastava U, Thaler M, Kleinhans KN, N'Kontchou G, Scheffold A, et al. Telomerase gene mutations are associated with cirrhosis formation. Hepatology 2011;53:1608‐1617. [DOI] [PubMed] [Google Scholar]
- 6. Calado RT, Brudno J, Mehta P, Kovacs JJ, Wu C, Zago MA, et al. Constitutional telomerase mutations are genetic risk factors for cirrhosis. Hepatology 2011;53:1600‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Qamar AA, Grace ND, Groszmann RJ, Garcia‐Tsao G, Bosch J, Burroughs AK, et al. Incidence, prevalence, and clinical significance of abnormal hematologic indices in compensated cirrhosis. Clin Gastroenterol Hepatol 2009;7:689‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, et al. Inherited DNA‐repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016;375:443‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mijuskovic M, Saunders EJ, Leongamornlert DA, Wakerell S, Whitmore I, Dadaev T, et al. Rare germline variants in DNA repair genes and the angiogenesis pathway predispose prostate cancer patients to develop metastatic disease. Br J Cancer 2018;119:96‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, et al. A model to predict survival in patients with end‐stage liver disease. Hepatology 2001;33:464‐470. [DOI] [PubMed] [Google Scholar]
- 11. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high‐throughput sequencing data. Nucleic Acids Res 2010;38:e164 10.1093/nar/gkq603. https://academic.oup.com/nar [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schneider V, Church D: In: Beck J, Benson D, Coleman J, Hoeppner M, Johnson M, Maglott D, et al., eds. The NCBI Handbook. 2nd edn Bethesda, MD: National Center of Biotechnology Information; 2013. [Google Scholar]
- 13. O'Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 2016;44:D733‐D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bansal V, Libiger O, Torkamani A, Schork NJ. Statistical analysis strategies for association studies involving rare variants. Nat Rev Genet 2010;11:773‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Frizzell A, Nguyen JH, Petalcorin MI, Turner KD, Boulton SJ, Freudenreich CH, et al. RTEL1 inhibits trinucleotide repeat expansions and fragility. Cell Rep 2014;6:827‐835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Vannier JB, Pavicic‐Kaltenbrunner V, Petalcorin MI, Ding H, Boulton SJ. RTEL1 dismantles T loops and counteracts telomeric G4‐DNA to maintain telomere integrity. Cell 2012;149:795‐806. [DOI] [PubMed] [Google Scholar]
- 17. Sarek G, Vannier JB, Panier S, Petrini JH, Boulton SJ. TRF2 recruits RTEL1 to telomeres in S phase to promote t‐loop unwinding. Mol Cell 2015;57:622‐635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Uringa EJ, Lisaingo K, Pickett HA, Brind'Amour J, Rohde JH, Zelensky A, et al. RTEL1 contributes to DNA replication and repair and telomere maintenance. Mol Biol Cell 2012;23:2782‐2792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kannengiesser C, Borie R, Ménard C, Réocreux M, Nitschké P, Gazal S, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J 2015;46:474‐485. [DOI] [PubMed] [Google Scholar]
- 20. McKay JD, Hung RJ, Han Y, Zong X, Carreras‐Torres R, Christiani DC, et al. Large‐scale association analysis identifies new lung cancer susceptibility loci and heterogeneity in genetic susceptibility across histological subtypes. Nat Genet 2017;49:1126‐1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Vannier JB, Sarek G, Boulton SJ. RTEL1: functions of a disease‐associated helicase. Trends Cell Biol 2014;24:416‐425. [DOI] [PubMed] [Google Scholar]
- 22. Deng Z, Glousker G, Molczan A, Fox AJ, Lamm N, Dheekollu J, et al. Inherited mutations in the helicase RTEL1 cause telomere dysfunction and Hoyeraal‐Hreidarsson syndrome. Proc Natl Acad Sci U S A 2013;110:E3408‐E3416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Le Guen T, Jullien L, Touzot F, Schertzer M, Gaillard L, Perderiset M, et al. Human RTEL1 deficiency causes Hoyeraal‐Hreidarsson syndrome with short telomeres and genome instability. Hum Mol Genet 2013;22:3239‐3249. [DOI] [PubMed] [Google Scholar]
- 24. Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet 2013;92:448‐453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cardoso SR, Ellison ACM, Walne AJ, Cassiman D, Raghavan M, Kishore B, et al. Myelodysplasia and liver disease extend the spectrum of RTEL1 related telomeropathies. Haematologica 2017;102:e293‐e296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Townsley DM, Dumitriu B, Liu D, Biancotto A, Weinstein B, Chen C, et al. Danazol treatment for telomere diseases. N Engl J Med 2016;374:1922‐1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gutierrez‐Rodrigues F, Kajigaya S, Xingmin Feng X, Fernandez Ibanez MdP, Desierto MJ, Keyvanfar K, et al. Heterozygous RTEL1 variants in patients with bone marrow failure associated with telomere dysfunction in the absence of telomere shortening. Blood 2016;128:1044 http://www.bloodjournal.org [Google Scholar]
- 28. Marsh J, Gutierrez‐Rodrigues F, Cooper J, Jiang J, Gandhi S, Kajigaya S, et al. Heterozygous RTEL1 variants in bone marrow failure and myeloid neoplasms. Blood Adv 2018;2:36‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]