

Abstract
Background
Recent genetic progress has allowed for the molecular diagnosis of Parkinson's disease. However, genetic causes of PD vary widely in different ethnicities. Mutational frequencies and clinical phenotypes of genes associated with PD in Asian populations are largely unknown. The objective of this study was to identify the mutational frequencies and clinical spectrums of multiple PD‐causative genes in a Taiwanese PD cohort.
Methods
A total of 571 participants including 324 patients with early‐onset parkinsonism (onset age, <50 years) and 247 parkinsonism pedigrees were recruited at a tertiary referral center in Taiwan from 2002 to 2017. Genetic causes were identified by an integrated approach including gene dosage analysis, a targeted next‐generation sequencing panel containing 40 known PD‐causative genes, repeat‐primed polymerase chain reaction, and whole‐exome sequencing analysis.
Results
Thirty of the 324 patients with early‐onset parkinsonism (9.3%) were found to carry mutations in Parkin, PINK1, or PLA2G6 or had increased trinucleotide repeats in SCA8. Twenty‐nine of 109 probands with autosomal‐recessive inheritance of parkinsonism (26.6%) were found to carry mutations in Parkin, PINK1, GBA, or HTRA2. The genetic causes for the 138 probands with an autosomal‐dominant inheritance pattern of parkinsonism were more heterogeneous. Seventeen probands (12.3%) carried pathogenic mutations in LRRK2, VPS35, MAPT, GBA, DNAJC13, C9orf72, SCA3, or SCA17. A novel missense mutation in the UQCRC1 gene was found in a family with autosomal‐dominant inheritance parkinsonism via whole‐exome sequencing analysis.
Conclusions
Our findings provide a better understanding of the genetic architecture of PD in eastern Asia and broaden the clinical spectrum of PD‐causing mutations. © 2019 The Authors. Movement Disorders published by Wiley Periodicals, Inc. on behalf of International Parkinson and Movement Disorder Society.
Keywords: early onset, genetics, next‐generation sequencing, Parkinson's disease, parkinsonism
Parkinson's disease (PD) is a progressive neurodegenerative disorder caused by a combination of genetic and environmental risk factors.1 Approximately 5%‐10% of patients have monogenic forms of the disease with either an autosomal‐dominant (AD) or autosomal‐recessive (AR) pattern of inheritance.2 Mutations in a number of genes implicated in both familial and sporadic PD have been identified.3 Identification of these PD‐causative genes provides biological insights into underlying disease processes.3 Neuromodification therapy by targeting these pathways provides an avenue for future drug discovery to mitigate disease progression.4
Evidence has shown that genetic causes can vary depending on the geographic and ethnic backgrounds of the studied populations.5 For example, the major genetic cause for AD inheritance of parkinsonism is a mutation in leucine‐rich repeat kinase 2 (LRRK2)6, 7 with the LRRK2 p.G2019S mutation having the highest frequency among North African Arab‐Berbers and Ashkenazi Jews.8, 9 However, the LRRK2 p.G2019S mutation is rare in Asian patients.10 As mutations in LRRK2 result in hyperactivation of LRRK2 kinase activity, LRRK2 inhibitors have entered clinical trials that offers the prospect of elaborating disease‐modifying treatments for PD.11 These observations indicate a pressing need to expand the knowledge of ethnically appropriate genetics in diverse populations.
We have previously described the clinical features of Taiwanese patients with early‐onset parkinsonism.12 Here we take an integrative approach, including gene dosage analysis, a targeted next‐generation sequencing (NGS) panel, repeat‐primed polymerase chain reaction (PCR), and whole‐exome sequencing (WES) to elucidate genetic causes and the relationships between genotypes and clinical phenotypes in patients with early‐onset parkinsonism and familial parkinsonism in a Taiwanese population.
Materials and Methods
Subjects
A total of 571 participants including 324 patients with early‐onset sporadic parkinsonism (onset age, <50 years) and 247 probands with familial parkinsonism (at least 1 of the family members in 3 generations affected with parkinsonism) were recruited from the Centre for Parkinson and Movement Disorders at a tertiary referral center in Taiwan from 2002 to 2017. Among the 247 probands with familial parkinsonism, 57 probands had an age at onset younger than 50 years. Of all participants, 522 patients fulfilled the United Kingdom PD Society Brain Bank diagnostic criteria of PD,13 and 49 patients also presented with mixed neurodegenerative features, including cognitive decline (n = 18), ataxia (n = 28), and motor neuron disorders (n = 3). All participants received regular evaluations of motor and cognitive functions. Motor symptom severity was evaluated using the Unified Parkinson's Disease Rating Scale (UPDRS) motor subscale14 and Hoehn‐and‐Yahr staging.15 Cognition was evaluated with the Mini‐Mental State Examination,16 and some patients received complete neuropsychological tests.17 All participants provided informed consent, and the institutional ethics review board of National Taiwan University Hospital approved this study.
Of the 247 probands with familial parkinsonism, 138 were compatible with an AD inheritance pattern, and 109 were compatible with AR inheritance or had at least 1 other affected first‐ and/or second‐degree relative with parkinsonism. Of the 324 patients with early‐onset parkinsonism, 72 had previously been reported to screen for Parkin, PINK1, and DJ‐1 mutations.12 In the current study, we enrolled additional patients with early‐onset parkinsonism and applied an integrated genetic approach.
Genetic Analysis
The flowchart of the genetic analysis is presented in Figure 1.
Figure 1.
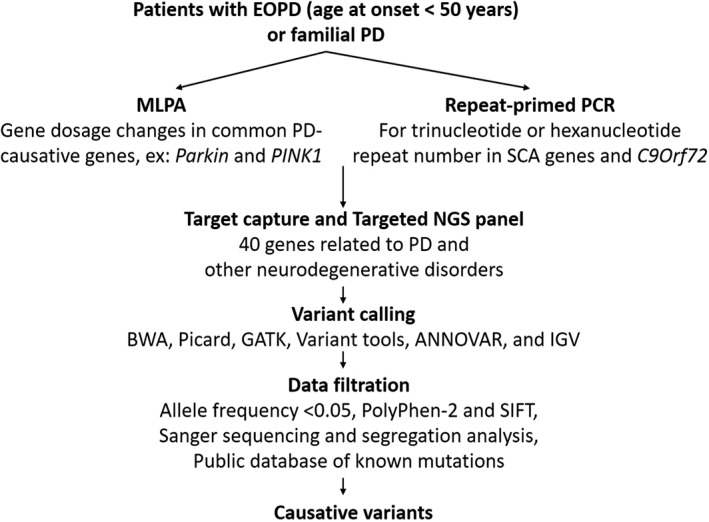
Pipeline for the identification of causative variants in patients with early‐onset parkinsonism or familial parkinsonism.
Multiplex Ligation‐Dependent Probe Amplification
DNA was extracted from venous blood using standard protocols.12 Large deletions or duplications of common PD‐causative genes including SNCA, Parkin, PINK1, DJ‐1, ATP13A2, PLA2G6, FBXO7, DNAJC6, and LRRK2 were detected using the salsa multiplex ligation‐dependent probe amplification (MLPA) kit P051‐c1/P52‐c1 (MRC‐Holland, Amsterdam, The Netherlands). Patents with deletions or duplications then received Sanger sequencing of the target gene to identify missense mutations on the other allele in a compound heterozygous state, and relative quantification of implicated exons was performed to confirm a homozygous deletion state.
Repeat‐Primed PCR
Repeat‐primed PCR was carried out as previously described to screen for GGGGCC hexanucleotide expansions in C9orf72 18 and CAG, CTG, and ATTCT repeats in SCA1, SCA2, SCA3, SCA6, SCA8, and SCA17.19, 20
Targeted NGS Panel
A custom‐designed NGS panel, including 40 genes associated with parkinsonism (Supplementary Table 1), was performed as previously described.21 Figure 1 depicts the criteria for identifying causative variants in the affected families, including target enrichment, variant calling, and data filtering. The details are described in the Supplementary Methods.
Whole‐Exome Sequencing
Affected individuals from 1 multiplex family with AD‐inheritance parkinsonism and without mutations in known PD genes received WES analyses using the Ion Torrent Next‐Generation Sequencing Exon v2 kit and platform (Life Technologies) as previously described.22
Generation and Quantitative Analysis of Neurites of UQCRC1 Knock‐In SH‐SY5Y Cells
We generated mutant UQCRC1 knock‐in (KI) SH‐SY5Y cell lines with clustered, regularly interspaced short palindromic repeats‐associated nuclease 9 (CRISPR‐Cas9) technology, as described in the Supplementary Methods.23 Neurite length for each genotype of SH‐SY5Y cells was quantified manually with Image J software (National Institutes of Health, Bethesda, MD), which is described in the Supplementary Methods.24
Measurement of Mitochondrial Respiratory Chain Activity
We measured mitochondrial respiratory chain activity in wild‐type and mutant UQCRC1 SH‐SY5Y cells with a Seahorse XFe24 extracellular flux analyzer (Seahorse Bioscience, North Billerica, MA), as previously described.25
Results
Genetic Analyses
The mean age at onset of patients with early‐onset parkinsonism was 41.6 ± 6.4 years, and 50.1% were men, whereas the mean age at onset of probands with familial parkinsonism was 54.4 ± 13.7 years, and 53.3% were men. Using target gene capture sequencing, we covered 656 exons in 40 genes representing a total coding region of 158,073 bp. The average coverage was 143‐fold, with 92.3% of sequences having coverage greater than 30‐fold and 89.1% greater than 50‐fold.
Parkin was the most prevalent mutated gene in 324 patients with early‐onset parkinsonism. Of the 14 Parkin mutation carriers (4.3% of patients with early‐onset parkinsonism), 4 had compound heterozygous mutations, and 10 had single heterozygous mutations. Exon deletions were the most common type of mutations, especially exon 5 and between exons 2 and 3 (Fig. 2A). Six patients had missense mutations: 2 had p. R396G, 1 had p.Y267H, and 3 had p.C441R mutations (Fig. 2A).26, 27 We also identified 7 patients with heterozygous PINK1 mutations (2.2% of patients with early‐onset parkinsonism), including 5 missense mutations (2 with p.M341I,28 1 with p.P209A, 1 with p.V350 L, 1 with p.L557P), and 2 heterozygous exon 8 deletions; see Figure 2B. The novel PINK1 p.V350 L and p.L557P mutations were not found in the 1517‐exome database from Taiwan Biobank and have not been described in public databases. In addition, 2 patients from consanguineous families had a common PLA2G6 homozygous mutation, p.D331Y (0.6% of early‐onset parkinsonism; Supplementary Fig. 2A), one of which was previously reported.29 One patient had increased expansion of CTA/CTG trinucleotide repeats in SCA8 (86 repeats). Several novel heterozygous mutations were identified, including 2 with FBXO7 substitutions, p.I87T and p.D328R,30 1 with p.A551V in SYNJ1, and another is p.G394 V in the DNAJC13 gene. An additional 2 patients had heterozygous GBA p.L444P mutations.31
Figure 2.
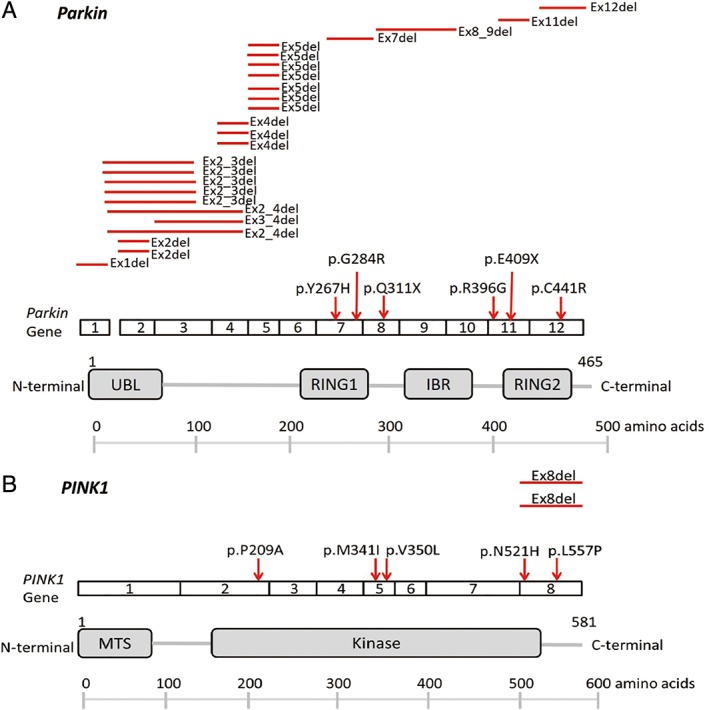
Schematic depiction of (A) Parkin and (B) PINK1 structures and associated disease‐linked mutations, including missense mutations and exonic deletions. [Color figure can be viewed at wileyonlinelibrary.com]
Of 109 probands with AR inheritance of parkinsonism, Parkin was the most prevalent mutated gene. Of the 16 probands carrying Parkin mutations (14.7%), 11 had compound heterozygous or homozygous mutations, and 5 had single heterozygous mutations. All biallelic Parkin mutations were found to segregate with disease status within the affected families (Supplementary Fig. 1). Exon deletions were the most common mutation in the Parkin gene and were found in 11 probands. Three of the 4 p.G284R substitution carriers and all the p.C441R mutation carriers had a compound heterozygous mutation with Parkin exon deletions (Fig. 2A). We also identified 3 probands carrying heterozygous PINK1 mutations, 1 with a p.N521H mutation, 1 with an exon 8 deletion, and another with a p.V350 L mutation. However, the affected family member associated with the proband carrying the heterozygous PINK1 exon 8 deletion was deceased; therefore, we were unable to check the segregation of this substitution in this family. We also identified a proband having a heterozygous HTRA2 p.P143A mutation.32 Nine probands (8.3%) had heterozygous GBA p.L444P mutations.31
Seventeen of the 138 probands with an AD inheritance pattern of parkinsonism (12.3%) were found to carry pathogenic mutations in known parkinsonism‐causative genes. We did not find a LRRK2 p.G2019S mutation, which is the most prevalent mutation in Western populations, but we identified 4 probands with other LRRK2 mutations (2.9%): 1 had a p.I1371V mutation,33, 34 2 had p.R1441H mutations,35, 36 and another had a p.I2012T mutation (Fig. 3A).21 These variants are all known pathogenic mutations and segregated with the parkinsonism phenotype within the families. On the LRRK2 protein, the p.I1371V and p.R1441H substitutions are both on the Ras of complex protein (ROC) domain, and p.I2012T is on the kinase domain of the LRRK2 protein (Fig. 3B). We also identified 2 probands (1.4%) carrying VPS35 mutations, 1 with the previously reported p.D620N37, 38 and another with the p.S679P mutation (Supplementary Fig. 2C). The VPS35 p.D620N variant segregated with the parkinsonism phenotype within the family. However, the affected mother of the proband carrying the heterozygous VPS35 p.S679P was deceased; therefore, we were unable to check the segregation of this substitution in this family. The potentially novel VPS35 p.S679P mutation was not found in the Taiwan Biobank database. Although there are 2 heterozygotes of East Asian descent described in the Genome Aggregation Database (gnomAD; http://gnomad.broadinstitute.org/gnomAD), we speculate this rare variant may cause the disease within the family. Two probands had previously reported MAPT mutations: 1 carried p.R5H,39 and another one had a p.R5C mutation.37, 39 Two novel heterozygous mutations were identified: 1 was p.R1382H in the DNAJC13 gene, and the other was p.R457Q in the GIGYF2 gene. The DNAJC13 p.R1382H was also identified in the affected father of the proband but was absent in the nonaffected sibling. The affected family member of the proband having GIGYF2 p.R457Q was deceased; therefore, we were unable to check the segregation within the family. Given that the finding of GIGYF2 variants causing PD has not been replicated yet, the pathogenicity of GIGYF2 p.R457Q needs further studies to be confirmed. Another 2 patients (1.4%) had heterozygous GBA p.L444P mutations.31 Four probands (2.9% of families) had increased numbers of trinucleotide repeats in genes associated with spinocerebellar ataxia (SCA). Three probands had SCA3 mutations (64, 64, and 68 repeats), and 1 had an increased repeat number in SCA17 (46 repeats). Finally, 1 proband had an increased hexanucleotide repeat expansion in the C9orf72 gene (52 repeats; Supplementary Fig. 3A,B). The abnormal repeat expansions of C9orf72 cosegregated with parkinsonism or other neurodegenerative phenotypes, such as frontotemporal dementia and motor neuron disorder, within the family (Supplementary Fig. 3A).
Figure 3.
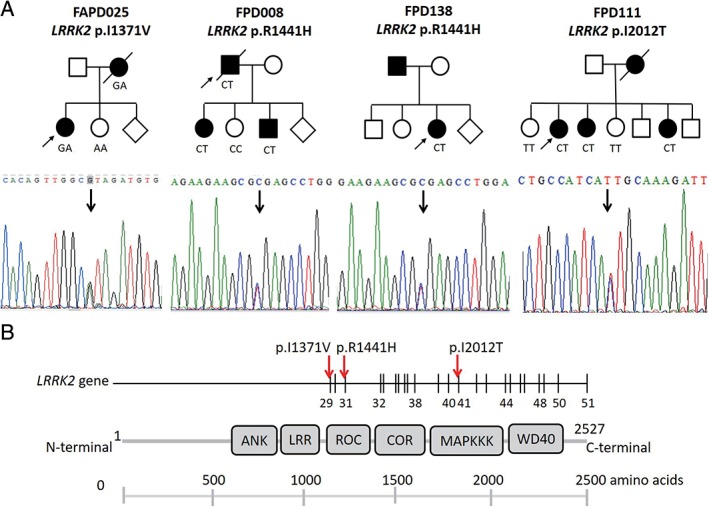
LRRK2 mutations identified in the current study. (A) Pedigrees of the 4 probands carrying LRRK2 mutations. Affected family members are represented with black circles (female) or squares (male). Arrows indicate the proband. Sanger chromatogram sequences are shown in the lower panel of each family pedigree. (B) Schematic depiction of LRRK2 structure and associated disease‐linked missense mutation. [Color figure can be viewed at wileyonlinelibrary.com]
A novel gene, mitochondrial ubiquinol‐cytochrome c reductase core protein I (UQCRC1, MIM 191328), was identified by WES in a family with autosomal‐dominant late‐onset parkinsonism and polyneuropathy. WES performed on 3 affected members of the family (Fig. 4A) with a coverage of 100× read depth showed 217 nonsynonymous variants with minor allele frequency ≤ 0.0001 in the gnomAD. Further comparative analyses identified 3 rare heterozygous variants shared in all exomes that resulted in coding substitutions: UQCRC1 (c.941A > C, p.Y314S), CLCN6 (c.1663G > A, p.D555N), and PTPRG (c.3141C > T, p.P808S). All variants that differed from the consensus sequence were annotated using the ANNOVAR package. The frequency of the variants in the population were also annotated using the gnomAD exome data set. All 3 variants were predicted to be potentially pathogenic based on PolyPhen‐2 and SIFT programs. Only the UQCRC1 p.Y314S cosegregated with disease within the family and was not present in the Taiwan Biobank database (n = 1517 exomes) or 1000 control subjects (Fig. 4A,B). In addition, the gnomAD (n = 123,136 exome and 15,496 whole‐genome sequences) showed UQCRC1 p.Y314S is a novel variant, although CLCN6 p.D555N and PTPRG p.P808S were found at appreciable allele frequencies, 2.4 × 10‐5 and 4.1 × 10‐5, respectively. Therefore, the UQCRC1 p.Y314S was selected as the candidate in this index family. The UQCRC1 p.Y314S substitution was conserved across species (Fig. 4C). UQCRC1 is a subunit of mitochondrial respiratory chain complex III40 and is highly expressed in brain tissue (https://www.proteinatlas.org/ENSG00000010256-UQCRC1/tissue). We therefore selected it as the disease candidate gene. To characterize the impact of the UQCRC1 p.Y314S substitution on human dopaminergic neurons, we established mutant UQCRC1 knock‐in (KI) SH‐SY5Y cell lines with CRISPR‐Cas9 technology. Neurite morphology analysis showed that cells that expressed the UQCRC1 p.Y314S substitution exhibited shortened neurites compared with wild‐type (WT) neurons (SH‐SY5Y control, 80.1 ± 4.9 μm; UQCRC1 WT, 79.2 ± 5.4 μm; UQCRC1 p.Y314S, 45.81 ± 4.3 μm; Fig. 4D; statistics are shown in Fig. 4E; 1‐way analysis of variance [ANOVA], P = 0.008 for UQCRC1 WT vs UQCRC1 p.Y314S; the number of cells assessed in each genotype was 56, 51, and 48 individually, respectively). These results demonstrated that UQCRC1 proteins with the p.Y314S variation impaired neurite morphogenesis and may cause neurite degeneration. As UQCRC1 is a subunit of mitochondrial respiratory complex III protein, we further measured the individual respiratory chain activity in wild‐type neurons and UQCRC1 p.Y314S KI neurons. Treatment with individual mitochondrial respiratory complex inhibitors demonstrated that UQCRC1 p.Y314S expression did not influence on mitochondrial complex I, II, or IV activity, but significantly reduced complex III activity (WT vs UQCRC1 p.Y314S, 5.87% ± 0.23% vs 3.92% ± 0.41%; 1‐way ANOVA, P = 0.01; Fig. 4F). This suggests that UQCRC1 p.Y314S disturbs mitochondrial respiratory chain complex III activity in neurons. Further studies including more families and in vivo functional assays are needed to support the pathogenicity of UQCRC1 in PD.
Figure 4.
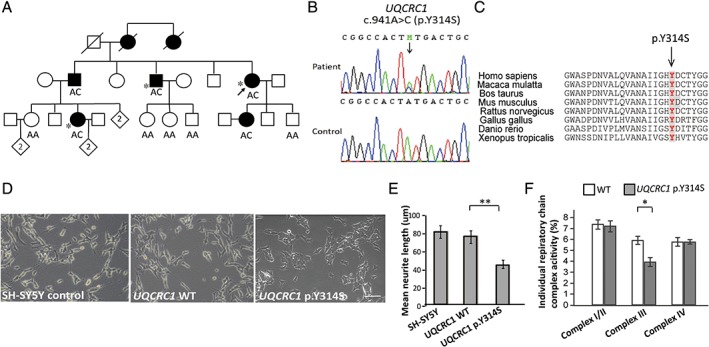
UQCRC1 mutation in a family with multiplex PD and an autosomal‐dominant inheritance pattern. (A) Pedigree of the family with the UQCRC1 c.941A > C (p.Y314S) mutation. Open symbol, unaffected; filled symbol, affected; symbol with a diagonal line, deceased; diamond, total number of children, unknown sex; arrow, proband. *Patients whose whole exomes were sequenced. (B) Sanger sequencing traces confirming the c.941A > C (p.Y314S) variant (RefSeq NM_003365.2). (C) Alignment of multiple UQCRC1 orthologs shows conservation of the p.Tyr314 residue across species. (D) Light microscopic images show SH‐SY5Y neurite morphology (Nikon Eclipse, 80i; 10× magnification) with different genotypes. (E) Quantitative analyses of mean total neurite lengths for the neurons with individual genotypes; n = 25 for each genotype. Data represent the mean ± SEM. **P < 0.01. (F) Percentages of oxygen consumption rate attributable to the activities of complexes I‐IV. In each experiment, data were collected and averaged from 4 separate wells for each individual cell line. Each cell line was assayed in at least 3 independent experiments, and means were calculated. Error bars are standard errors of the mean. P = 0.01, 1‐way ANOVA for UQCRC1 WT versus UQCRC1 p.Y314S for complex III activity. *P < 0.05. [Color figure can be viewed at wileyonlinelibrary.com]
Clinical Phenotypes
A detailed description of all observed major phenotypic features of patients with mutations is summarized in Table 1.
Table 1.
Clinical characteristics of patients with known PD mutations
| Parkin | PINK1 | PLA2G6 | LRRK2 | VPS35 | MAPT | GBA | C9orf72 | SCA | |
|---|---|---|---|---|---|---|---|---|---|
| Homozygous/Compound Heterozygous (n = 15) | Heterozygous (n = 10) | Homozygous (n = 2) | Heterozygous (n = 9 From 4 Families) | Heterozygous (n = 3 From 2 Families) | Heterozygous (n = 4 From 2 Families) | Heterozygous (n = 13) | Heterozygous (n = 2 From 1 Family) | (n = 5) | |
| AAO (mean ± SD) | 28.6 ± 10.5 | 37.7 ± 5.9 | 28.0 ± 2.8 | 58.8 ± 2.9 | 44.5 ± 3.5 | 52.5 ± 10.6 | 52.3 ± 12.9 | 56. 5 ± 2.1 | 41. 8 ± 4.2 |
| Male | 53.3% | 70.0% | 0% | 22.2% | 33.3% | 50% | 38.5% | 100% | 80% |
| Initial symptoms | |||||||||
| Tremor | 46.3 % | 60.0% | 0% | 33.3% | 33.3% | 25.0% | 53.8% | 0% | 0% |
| Disease progression | Slow | Slow | Fast | Variable | Slow | Variable | Variable | Fast | Fast |
| Associate symptoms | Foot dystonia Anxiety Depression |
NA | Anxiety Depression Psychosis |
FTD Anxiety Psychosis |
NA | FTD Depression |
Dementia | Dementia MND |
Ataxia Dementia Polyneuropathy |
AAO, age at onset of symptoms; SD, standard deviation; NA, not applicable; FTD, frontotemporal dementia; MND, motor neuron disorder.
The fast disease progression is defined as a mean annual rate of progression of more than 5 points on the motor subscore of the Unified Parkinson's Disease Rating Scale (UPDRS part III).
Patients Presenting With Levodopa‐Responsive Parkinsonism Without Atypical Features
All patients with mutations in PINK1, FBXO7, SYNJ1, DNAJC13, and GIGYF2 presented with early‐onset levodopa‐responsive parkinsonism. No one reported foot dystonia as an early manifestation. The with‐SCA8 mutation presented with left leg slowness and rigidity at age 38 years. He had good responses to levodopa but complicated by peak‐dose dyskinesia after 4 years of treatment. The clinical phenotypes among the 4 families with LRRK2 mutations were heterogeneous. The mean age of onset for levodopa‐responsive parkinsonism patients with LRRK2 mutations was 58.8 ± 2.9 years.
Patients Presenting With Levodopa‐Responsive Parkinsonism With Dystonia
Foot dystonia is a common and sometimes preceding symptom among patients carrying homozygous or compound heterozygous Parkin mutations (40%; Table 1). The mean age of onset for patients with homozygous or compound heterozygous Parkin mutations was 28.6 ± 10.5 years.
Patients Presenting With Levodopa‐Responsive Parkinsonism and Psychiatric Features
Anxiety and depression are common symptoms for patients with mutations in Parkin and PLA2G6 genes (Table 1).41 Brain MRI scans of patients having homozygous PLA2G6 p.D331Y mutations did not reveal abnormal iron depositions in the basal ganglia (Supplementary Fig. 2B). Of LRRK2 mutation carriers, 1 proband with an LRRK2 p.R1441H mutation had prominent psychotic symptoms 6 years after the onset of motor symptoms under the levodopa dosage of 600 mg/day.
Patients Presenting With Levodopa‐Unresponsive Parkinsonism and FTD
Late‐onset rapid progressive parkinsonism with poor levodopa responses was observed in the proband with an LRRK2 p.1371 V mutation, 2 probands with MAPT mutations, and 1 proband with a hexanucleotide repeat expansion in C9orf72.
Patients Presenting With Motor Neuron Disorders and Parkinsonism With Poor Levodopa Responses
The proband with a hexanucleotide repeat expansion in C9orf72 had progressive leg weakness with fasciculations at the age of 58, and amyotrophic lateral sclerosis was diagnosed. The patient then developed a left‐sided parkinsonism feature and cognition impairment with a neuropsychological test demonstrating behavior variant of frontotemporal dementia (FTD). Brain MRI showed asymmetrical atrophy over the right frontotemporal lobe, and Tc‐99 m TRODAT images revealed decreased uptake over the right putamen (Supplementary Fig. 3C). His brother also had a C9orf72 mutation and developed FTD at age 55 years, followed by rapidly progressive parkinsonism, and was bedridden within 5 years.
Patients Presenting With Parkinsonism and Cerebellar Ataxia
Mild ataxic gait with asymmetrical onset of akinetic‐rigidity was observed in 3 patients with increased numbers of trinucleotide repeats in SCA3 and in 1 patient with a mutation in SCA17.42 The mean age of onset was 42.3 ± 4.0 years.
An Autosomal‐Dominant‐Inheritance PD Family With a Novel UQCRC1 Mutation
The novel UQCRC1 mutation p.Y314S was detected in 5 affected members of the index family (Fig. 4A). All affected UQCRC1 heterozygotic carriers presented with asymmetrical onset tremor‐predominant levodopa‐responsive parkinsonism, and the mean age of onset was 54.2 ± 8.7 years. In addition to parkinsonian features, UQCRC1 p.Y314S heterozygotes also had intrinsic muscle atrophy in the hands and toes, and nerve conduction studies revealed axonal type sensorimotor polyneuropathy.
Discussion
We identified the genetic causes for early‐onset and familial parkinsonism in a large Taiwanese cohort using an integrated genetic approach and established the relationships between genotypes and clinical phenotypes. Our results showed that 9.3% of patients with early‐onset parkinsonism have mutations in Parkin and PINK1, along with fewer than 1% of patients with mutations in PLA2G6, FBXO7, SYNJ1, SCA8, and DNAJC13 genes. Of the AR inheritance families, 26.6% had mutations in Parkin, PINK1, GBA, or HTRA2. Of the AD inheritance families, 12.3% had mutations in LRRK2, VPS35, MAPT, GBA, DNAJC13, C9orf72, SCA3, or SCA17. A potential novel mutation in the UQCRC1 gene was identified in a family with AD inheritance of parkinsonism. Our study represents the most extensive survey of the genetic etiology of early‐onset parkinsonism and within families with parkinsonism in eastern Asia to date and indicates a different mutational spectrum for PD than that seen in white populations.
Parkin was the most commonly mutated gene in patients with early‐onset parkinsonism or probands with AR inheritance of parkinsonism. Exon deletions were the most prevalent mutation types. Concordantly, most of the deleted exons fell into the genomic region between exons 2 and 5, further confirming this region as a mutational hot spot across populations.43 Although biallelic Parkin mutations only account for 1.3% of patients with early‐onset parkinsonism with an onset age younger than 50 and the mutation frequency increases to 4.3% by including heterozygous variants, the biallelic Parkin mutations account for 10.1% of patients with an onset age younger than 40 and 44.4% of patients with an onset before the age of 20 in our population. Familial cases with an AR inheritance pattern were more likely to have biallelic mutations in Parkin than patients with sporadic early‐onset parkinsonism, with a proportion of 10.1% versus 1.3% in our cohort. The Parkin mutation rate seen in our early‐onset parkinsonism cases was comparable with that seen in Norwegian patients, with 2.8% of patients with early‐onset PD (age at onset, <45 years) have biallelic Parkin mutations, and by including heterozygous variants, the mutation frequency increases to 4.6%44 in Korean patients with a mutation frequency of 5% and in Japanese patients with a frequency of 11%.45 However, our Parkin mutation rate is lower than that in some Western populations. A study from a European consortium showed that Parkin mutations account for 18% of early‐onset parkinsonism with onset before age 45 and 50% of families with AR inheritance of parkinsonism.46 The reasons that contribute to the differences in mutation frequency may come from fewer consanguineous marriages in our society because of legal regulations or ethnic differences.
PINK1 is the second most common gene causing early‐onset parkinsonism.47 Familial cases with an AR inheritance pattern were more likely to have PINK1 mutations than patients with sporadic early‐onset parkinsonism, with a proportion of 2.8% versus 2.2% in our cohort. Our findings are consistent with a previous study performed in a Taiwanese population reporting that the frequency of PINK1 mutations was 2% in patients with early‐onset parkinsonism48 and with the results from a Japanese study with a mutation frequency of 1.05% in patients with early‐onset parkinsonism.49 However, the identified mutations were in a heterozygous state in our population. Although evidence has suggested that heterozygous mutations in recessive genes such as PINK1 are indeed susceptibility factors for disease development,50, 51 we suggest that additional modifier genes in combination with environmental exposures may contribute to disease susceptibility in our population. We also identified a homozygous PLA2G6 p.D331Y mutation in 2 patients with early‐onset parkinsonism and parental consanguinity. This mutation was previously reported in a Chinese family with AR inheritance of early‐onset parkinsonism.29 Our findings suggest that PLA2G6 mutations should be considered for patients with early‐onset parkinsonism in our ethnicity.
The genetic causes of AD‐inheritance parkinsonism pedigrees are heterogeneous and include LRRK2, VPS35, MAPT, GBA, DNAJC13, C9orf72, SCA3, SCA8, and SCA17, and the phenotypic spectrums were also variable, ranging from levodopa‐responsive parkinsonism to rapid‐progressive parkinsonism mixed with other neurodegenerative symptoms, including FTD, motor neuron disorders, and ataxia. Our findings support previous observations that patients with increased trinucleotide repeats in SCA genes, especially SCA 2, 3, 8, and 17, may present with parkinsonism, especially in Asia.52, 53, 54 We suggest that screening of abnormal trinucleotide expansions in SCA‐related genes should be considered in patients with parkinsonism in our population.
Our results also identified 4 families with LRRK2 mutations. Although the most common LRRK2 mutation in Western populations is p.G2019S on the LRRK2 kinase domain, we identified p.I1371V and p.R144H mutations, which are on the ROC domain of LRRK2,55 and p.I2012T mutations, which are on the kinase domain of LRRK2, in our families with PD.56, 57, 58 Our results reinforce the observations that LRRK2 mutations have ethnic differences. Current LRRK2 GTP‐binding inhibitors such as FX214959 and kinase inhibitors such as LRRK2‐IN‐160 have been studied in cellular or animal models carrying common mutations in Western populations and may also have therapeutic potential for Asian LRRK2 mutation carriers. We also identified a family presenting with left hemiparkinsonism mixed with motor neuron disorder and FTD, with an increased number of hexanucleotide repeats in the C9orf72 gene. Although mutations in C9orf72 are rarely reported in Asian patients,18, 61, 62 C9orf72 repeat expansions should be considered for patients with parkinsonism that have overt symptoms of FTD or motor neuron disorders in our population. Finally, we identified a mutation in the potential novel parkinsonism‐causing gene UQCRC1, which encodes a mitochondria respiratory complex III protein, in a family with AD‐inheritance parkinsonism. Further studies including more families and functional assays are needed to support the pathogenicity of UQCRC1 in PD.
Although we established a comprehensive genetic screening in patients with early‐onset or familial parkinsonism, there are some limitations in the current study. First, MLPA may occasionally fail to detect variations in the gene dosage, and MLPA is also unable to detect balanced rearrangements. Second, we identified a potential novel mutation, UQCRC1 p.Y314S, which may contribute to familial parkinsonism in our ethnicity. However, because of the rarity of the affected family, future functional analyses using animal models and enrollment of more families having UQCRC1 mutations are needed to prove the pathogenicity of UQCRC1 mutations in the disease pathogenesis. In conclusion, this study presents a systemic genetic analysis of a large Taiwanese cohort of patients to elucidate the genetic architecture of early‐onset and familial parkinsonism in our population. Our results have the potential to facilitate accurate molecular diagnosis and future tailored treatments for susceptible individuals carrying genetic mutations.
Authors’ Roles
Study concept and design: CH Lin and RM Wu. Acquisition of data: CH Lin, PL Chen, CH Tai, HI Lin, CS Chen, ML Chen and RM Wu. Analysis and interpretation of data: CH Lin, PL Chen. Drafting of the manuscript: CH Lin. Critical revision of the manuscript for important intellectual content: RM Wu. Statistical analysis: CH Lin. Obtained funding: CH Lin and RM Wu. Study supervision: RM Wu.
Financial disclosures of all authors (for the preceding 12 months)
nothing to report.
Supporting information
Appendix S1. Supporting Information.
SUPPLEMENTARY FIG. 1 Pedigrees of familial PD patients having biallelic Parkin mutations identified in the current study. Affected family members are represented with black circles (female) or squares (male). Diamonds indicate unknown sex. Arrows indicate the proband.
SUPPLEMENTARY FIG. 2 PLA2G6 and VPS35 mutations identified in the current study. (A) Pedigrees of the 2 patients with early‐onset parkinsonism carrying the PLA2G6 p.D331Y mutation. (B) Brain MRI image of 1 proband having the PLA2G6 p.D331Y mutation. (C) Pedigrees of the 2 probands carrying VPS35 mutations. Affected family members are represented with black circles (female) or squares (male). Arrows indicate the proband. Sanger chromatogram sequences are shown on the lower panel of each family pedigree.
SUPPLEMENTARY FIG. 3 The family carrying increased hexanucleotide (G4C2) repeats in the C9orf72 gene identified in the current study. (A) Pedigrees of the probands carrying C9orf72 mutations. Affected family members are represented with black circles (female) or squares (male) and had variable clinical presentations. The arrow indicates the proband. Wt/m, heterozygous mutation carriers; wt/wt, noncarriers. (B) The electropherograms of the polymerase chain reaction (PCR) products of repeat‐primed PCR reactions investigating the hexanucleotide repeat expansion in C9orf72. Repeat expansions produce a characteristic sawtooth pattern with a 6‐base pair periodicity. The repeat numbers are shown for the proband (upper, 10 of 52 repeats) and age‐matched control (lower, 2 of 7 repeats). (C) Brain MRI (left) and Tc99mTRODAT‐SPECT (right) images of the proband. Arrows indicate asymmetrical atrophy over the right temporal lobe in brain MRI and decreased uptake of Tc99mTRODAT in right putamen.
Supplementary Table 1 The 40 candidate genes involved in PD and related neurodegenerative disorders that were used for targeted NGS in the study.
Acknowledgments
The authors are grateful to the patients who participated in this study. We are very grateful to Dr. Matthew J. Farrer and Dr. Zbigniew K. Wszolek, who helped us to initiate the genetic study in young‐onset and familial parkinsonism in the Taiwanese population. We also thank the staff of the Second Core Lab, Department of Medical Research, National Taiwan University Hospital, for technical support during the study.
Relevant conflicts of interests/financial disclosures: No author reports any conflicts of interest.
Funding agencies: This work was supported by grants from National Taiwan University Hospital (NTUH 107C101‐82), National Taiwan University (106R8805C1), the Ministry of Science and Technology (MOST 106‐2314‐B‐002‐072‐MY3), and the T.H. Wu Foundation.
References
- 1. Kalia LV, Lang AE. Parkinson's disease. Lancet 2015;386:896‐912. [DOI] [PubMed] [Google Scholar]
- 2. Hardy J, Lewis P, Revesz T, Lees A, Paisan‐Ruiz C. The genetics of Parkinson's syndromes: a critical review. Curr Opin Genet Dev 2009;19:254‐265. [DOI] [PubMed] [Google Scholar]
- 3. Singleton A, Hardy J. The evolution of genetics: Alzheimer's and Parkinson's diseases. Neuron 2016;90:1154‐1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lang AE, Espay AJ. Disease Modification in Parkinson's Disease: Current Approaches, Challenges, and Future Considerations. Mov Disord 2018;33:660‐677. [DOI] [PubMed] [Google Scholar]
- 5. Hardy J, Cai H, Cookson MR, Gwinn‐Hardy K, Singleton A. Genetics of Parkinson's disease and parkinsonism. Ann Neurol 2006;60:389‐398. [DOI] [PubMed] [Google Scholar]
- 6. Di Fonzo A, Rohe CF, Ferreira J, et al. A frequent LRRK2 gene mutation associated with autosomal dominant Parkinson's disease. Lancet 2005;365:412‐415. [DOI] [PubMed] [Google Scholar]
- 7. Kachergus J, Mata IF, Hulihan M, et al. Identification of a novel LRRK2 mutation linked to autosomal dominant parkinsonism: evidence of a common founder across European populations. Am J Hum Genet 2005;76:672‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lesage S, Ibanez P, Lohmann E, et al. G2019S LRRK2 mutation in French and North African families with Parkinson's disease. Ann Neurol 2005;58:784‐787. [DOI] [PubMed] [Google Scholar]
- 9. Ozelius LJ, Senthil G, Saunders‐Pullman R, et al. LRRK2 G2019S as a cause of Parkinson's disease in Ashkenazi Jews. N Engl J Med 2006;354:424‐425. [DOI] [PubMed] [Google Scholar]
- 10. Tan EK, Shen H, Tan LC, et al. The G2019S LRRK2 mutation is uncommon in an Asian cohort of Parkinson's disease patients. Neurosci Lett 2005;384:327‐329. [DOI] [PubMed] [Google Scholar]
- 11. Alessi DR, Sammler E. LRRK2 kinase in Parkinson's disease. Science. 2018;360:36‐37. [DOI] [PubMed] [Google Scholar]
- 12. Wu RM, Bounds R, Lincoln S, et al. Parkin mutations and early‐onset parkinsonism in a Taiwanese cohort. Arch Neurol 2005;62:82‐87. [DOI] [PubMed] [Google Scholar]
- 13. Hughes AJ, Daniel SE, Kilford L, Lees AJ. Accuracy of clinical diagnosis of idiopathic Parkinson's disease: a clinico‐pathological study of 100 cases. J Neurol Neurosurg Psychiatry 1992;55:181‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goetz CG, Tilley BC, Shaftman SR, et al. Movement Disorder Society‐sponsored revision of the Unified Parkinson's Disease Rating Scale (MDS‐UPDRS): scale presentation and clinimetric testing results. Mov Disord 2008;23:2129‐2170. [DOI] [PubMed] [Google Scholar]
- 15. Hoehn MM, Yahr MD. Parkinsonism: onset, progression and mortality. Neurology 1967;17:427‐442. [DOI] [PubMed] [Google Scholar]
- 16. Folstein MF, Folstein SE, McHugh PR. “Mini‐mental state.” A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189‐198. [DOI] [PubMed] [Google Scholar]
- 17. Yu RL, Wu RM, Tai CH, Lin CH, Cheng TW, Hua MS. Neuropsychological profile in patients with early stage of Parkinson's disease in Taiwan. Parkinsonism Relat Disord 2012;18:1067‐1072. [DOI] [PubMed] [Google Scholar]
- 18. Lin CH, Chen TF, Chiu MJ, Lin HI, Wu RM. Lack of c9orf72 repeat expansion in Taiwanese patients with mixed neurodegenerative disorders. Front Neurol 2014;5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Orr HT, Chung MY, Banfi S, et al. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat Genet 1993;4:221‐226. [DOI] [PubMed] [Google Scholar]
- 20. Lin CH, Hwu WL, Chiang SC, Tai CH, Wu RM. Lack of mutations in spinocerebellar ataxia type 2 and 3 genes in a Taiwanese (ethnic Chinese) cohort of familial and early‐onset parkinsonism. Am J Med Genet B Neuropsychiatr Genet 2007;144B:434‐438. [DOI] [PubMed] [Google Scholar]
- 21. Fan TS, Wu RM, Chen PL, et al. Clinical heterogeneity of LRRK2 p.I2012T mutation. Parkinsonism Relat Disord 2016;33:36‐43. [DOI] [PubMed] [Google Scholar]
- 22. Bentley SR, Bortnick S, Guella I, et al. Pipeline to gene discovery ‐ Analysing familial Parkinsonism in the Queensland Parkinson's Project. Parkinsonism Relat Disord 2018;49:34‐41. [DOI] [PubMed] [Google Scholar]
- 23. Ran FA, Hsu PD, Wright J, et al. Genome engineering using the CRISPR‐Cas9 system. Nat Protoc 2013;8:2281‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin CH, Lin HI, Chen ML, et al. Lovastatin protects neurite degeneration in LRRK2‐G2019S parkinsonism through activating the Akt/Nrf pathway and inhibiting GSK3β activity. Hum Mol Genet 2016;25:1965‐1978. [DOI] [PubMed] [Google Scholar]
- 25. Salabei JK, Gibb AA, Hill BG. Comprehensive measurement of respiratory activity in permeabilized cells using extracellular flux analysis. Nat Protoc 2014;9:421‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C. Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 2010;31:763‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Funayama M, Li Y, Tsoi TH, et al. Familial Parkinsonism with digenic parkin and PINK1 mutations. Mov Disord 2008;23:1461‐1465. [DOI] [PubMed] [Google Scholar]
- 28. Lee MJ, Mata IF, Lin CH, et al. Genotype‐phenotype correlates in Taiwanese patients with early‐onset recessive Parkinsonism. Mov Disord 2009;24:104‐108. [DOI] [PubMed] [Google Scholar]
- 29. Shi CH, Tang BS, Wang L, et al. PLA2G6 gene mutation in autosomal recessive early‐onset parkinsonism in a Chinese cohort. Neurology 2011;77:75‐81. [DOI] [PubMed] [Google Scholar]
- 30. Lin CH, Chen ML, Lai TT, Tai CH, Wu RM. Mutational analysis of FBXO7 gene in Parkinson's disease in a Taiwanese population. Neurobiol Aging 2013;34:1713 e1711‐1714. [DOI] [PubMed] [Google Scholar]
- 31. Aharon‐Peretz J, Rosenbaum H, Gershoni‐Baruch R. Mutations in the glucocerebrosidase gene and Parkinson's disease in Ashkenazi Jews. N Engl J Med 2004;351:1972‐1977. [DOI] [PubMed] [Google Scholar]
- 32. Lin CH, Chen ML, Chen GS, Tai CH, Wu RM. Novel variant Pro143Ala in HTRA2 contributes to Parkinson's disease by inducing hyperphosphorylation of HTRA2 protein in mitochondria. Hum Genet 2011;130:817‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seki N, Takahashi Y, Tomiyama H, et al. Comprehensive mutational analysis of LRRK2 reveals variants supporting association with autosomal dominant Parkinson's disease. J Hum Genet 2011;56:671‐675. [DOI] [PubMed] [Google Scholar]
- 34. Jankovic MZ, Kresojevic ND, Dobricic VS, et al. Identification of novel variants in LRRK2 gene in patients with Parkinson's disease in Serbian population. J Neurol Sci 2015;353:59‐62. [DOI] [PubMed] [Google Scholar]
- 35. Ross OA, Soto‐Ortolaza AI, Heckman MG, et al. Association of LRRK2 exonic variants with susceptibility to Parkinson's disease: a case‐control study. Lancet Neurol 2011;10:898‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin CH, Tzen KY, Yu CY, Tai CH, Farrer MJ, Wu RM. LRRK2 mutation in familial Parkinson's disease in a Taiwanese population: clinical, PET, and functional studies. J Biomed Sci 2008;15:661‐667. [DOI] [PubMed] [Google Scholar]
- 37. Vilarino‐Guell C, Wider C, Ross OA, et al. VPS35 mutations in Parkinson disease. Am J Hum Genet 2011;89:162‐167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chen YF, Chang YY, Lan MY, Chen PL, Lin CH. Identification of VPS35 p.D620N mutation‐related Parkinson's disease in a Taiwanese family with successful bilateral subthalamic nucleus deep brain stimulation: a case report and literature review. BMC Neurol 2017;17:191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hayashi S, Toyoshima Y, Hasegawa M, et al. Late‐onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol 2002;51:525‐530. [DOI] [PubMed] [Google Scholar]
- 40. Hoffman GG, Lee S, Christiano AM, et al. Complete coding sequence, intron/exon organization, and chromosomal location of the gene for the core I protein of human ubiquinol‐cytochrome c reductase. J Biol Chem 1993;268:21113‐21119. [PubMed] [Google Scholar]
- 41. Lu CS, Lai SC, Wu RM, et al. PLA2G6 mutations in PARK14‐linked young‐onset parkinsonism and sporadic Parkinson's disease. Am J Med Genet B Neuropsychiatr Genet 2012;159B:183‐191. [DOI] [PubMed] [Google Scholar]
- 42. Lin IS, Wu RM, Lee‐Chen GJ, Shan DE, Gwinn‐Hardy K. The SCA17 phenotype can include features of MSA‐C, PSP and cognitive impairment. Parkinsonism Relat Disord 2007;13:246‐249. [DOI] [PubMed] [Google Scholar]
- 43. Kilarski LL, Pearson JP, Newsway V, et al. Systematic review and UK‐based study of PARK2 (parkin), PINK1, PARK7 (DJ‐1) and LRRK2 in early‐onset Parkinson's disease. Mov Disord 2012;27:1522‐1529. [DOI] [PubMed] [Google Scholar]
- 44. Gustavsson EK, Trinh J, McKenzie M, et al. Genetic Identification in Early Onset Parkinsonism among Norwegian Patients. Mov Disord Clinical Practice 2017; doi: 10.1002/mdc3.12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hattori N, Kitada T, Matsumine H, et al. Molecular genetic analysis of a novel Parkin gene in Japanese families with autosomal recessive juvenile parkinsonism: evidence for variable homozygous deletions in the Parkin gene in affected individuals. Ann Neurol 1998;44:935‐941. [DOI] [PubMed] [Google Scholar]
- 46. Lucking CB, Durr A, Bonifati V, et al. Association between early‐onset Parkinson's disease and mutations in the parkin gene. N Engl J Med 2000;342:1560‐1567. [DOI] [PubMed] [Google Scholar]
- 47. Bonifati V, Rohe CF, Breedveld GJ, et al. Early‐onset parkinsonism associated with PINK1 mutations: frequency, genotypes, and phenotypes. Neurology 2005;65:87‐95. [DOI] [PubMed] [Google Scholar]
- 48. Weng YH, Chou YH, Wu WS, et al. PINK1 mutation in Taiwanese early‐onset parkinsonism: clinical, genetic, and dopamine transporter studies. J Neurol 2007;254:1347‐1355. [DOI] [PubMed] [Google Scholar]
- 49. Kumazawa R, Tomiyama H, Li Y, et al. Mutation analysis of the PINK1 gene in 391 patients with Parkinson disease. Arch Neurol 2008;65:802‐808. [DOI] [PubMed] [Google Scholar]
- 50. Puschmann A, Fiesel FC, Caulfield TR, et al. Heterozygous PINK1 p.G411S increases risk of Parkinson's disease via a dominant‐negative mechanism. Brain 2017;140:98‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Klein C, Westenberger A. Genetics of Parkinson's disease. Cold Spring Harb Perspect Med 2012;2:a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Park H, Kim HJ, Jeon BS. Parkinsonism in spinocerebellar ataxia. Biomed Res Int 2015;2015:125273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wu YR, Lin HY, Chen CM, et al. Genetic testing in spinocerebellar ataxia in Taiwan: expansions of trinucleotide repeats in SCA8 and SCA17 are associated with typical Parkinson's disease. Clin Genet 2004;65:209‐214. [DOI] [PubMed] [Google Scholar]
- 54. Kim JS, Son TO, Youn J, Ki CS, Cho JW. Non‐ataxic phenotypes of SCA8 mimicking amyotrophic lateral sclerosis and Parkinson disease. J Clin Neurol 2013;9:274‐279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. West AB, Moore DJ, Choi C, et al. Parkinson's disease‐associated mutations in LRRK2 link enhanced GTP‐binding and kinase activities to neuronal toxicity. Hum Mol Genet 2007;16:223‐232. [DOI] [PubMed] [Google Scholar]
- 56. Hatano T, Funayama M, Kubo SI, et al. Identification of a Japanese family with LRRK2 p.R1441G‐related Parkinson's disease. Neurobiol Aging 2014;35:2656.e17‐2656.e23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wu YR, Chang KH, Chang WT, et al. Genetic variants ofLRRK2 in Taiwanese Parkinson's disease. PLoS One. 2013;8:e82001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Tomiyama H, Li Y, Funayama M, et al. Clinicogenetic study of mutations in LRRK2 exon 41 in Parkinson's disease patients from 18 countries. Mov Disord 2006;21:1102‐1108. [DOI] [PubMed] [Google Scholar]
- 59. Li T, He X, Thomas JM, et al. A novel GTP‐binding inhibitor, FX2149, attenuates LRRK2 toxicity in Parkinson's disease models. PLoS One 2015;10:e0122461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deng X, Dzamko N, Prescott A, et al. Characterization of a selective inhibitor of the Parkinson's disease kinase LRRK2. Nat Chem Biol 2011;7:203‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Chen X, Chen Y, Wei Q, et al. C9ORF72 repeat expansions in Chinese patients with Parkinson's disease and multiple system atrophy. J Neural Transm (Vienna) 2016;123:1341‐1345. [DOI] [PubMed] [Google Scholar]
- 62. Jiao B, Guo JF, Wang YQ, et al. C9orf72 mutation is rare in Alzheimer's disease, Parkinson's disease, and essential tremor in China. Front Cell Neurosci 2013;7:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
SUPPLEMENTARY FIG. 1 Pedigrees of familial PD patients having biallelic Parkin mutations identified in the current study. Affected family members are represented with black circles (female) or squares (male). Diamonds indicate unknown sex. Arrows indicate the proband.
SUPPLEMENTARY FIG. 2 PLA2G6 and VPS35 mutations identified in the current study. (A) Pedigrees of the 2 patients with early‐onset parkinsonism carrying the PLA2G6 p.D331Y mutation. (B) Brain MRI image of 1 proband having the PLA2G6 p.D331Y mutation. (C) Pedigrees of the 2 probands carrying VPS35 mutations. Affected family members are represented with black circles (female) or squares (male). Arrows indicate the proband. Sanger chromatogram sequences are shown on the lower panel of each family pedigree.
SUPPLEMENTARY FIG. 3 The family carrying increased hexanucleotide (G4C2) repeats in the C9orf72 gene identified in the current study. (A) Pedigrees of the probands carrying C9orf72 mutations. Affected family members are represented with black circles (female) or squares (male) and had variable clinical presentations. The arrow indicates the proband. Wt/m, heterozygous mutation carriers; wt/wt, noncarriers. (B) The electropherograms of the polymerase chain reaction (PCR) products of repeat‐primed PCR reactions investigating the hexanucleotide repeat expansion in C9orf72. Repeat expansions produce a characteristic sawtooth pattern with a 6‐base pair periodicity. The repeat numbers are shown for the proband (upper, 10 of 52 repeats) and age‐matched control (lower, 2 of 7 repeats). (C) Brain MRI (left) and Tc99mTRODAT‐SPECT (right) images of the proband. Arrows indicate asymmetrical atrophy over the right temporal lobe in brain MRI and decreased uptake of Tc99mTRODAT in right putamen.
Supplementary Table 1 The 40 candidate genes involved in PD and related neurodegenerative disorders that were used for targeted NGS in the study.