

Abstract
Perfluorooctanoic acid is a ligand for peroxisome proliferator-activated receptor (PPARα). Ammonium perfluorooctanoate (APFO) at 0.1 and 0.3 mg/kg doses activated mouse PPARα, but not human PPARα. This study aimed to clarify whether milligram-order APFO can activate human PPARα, and the receptor is involved in APFO-induced chronic hepatic damage. Male Sv/129 wild-type (mPPARα), Pparα-null, and humanized PPARα (hPPARα) mice (8 weeks old) were divided into three groups. The first was treated with water and the other two with 1.0 and 5.0 mg/kg APFO for 6 weeks, orally, respectively. Both doses activated mouse and human PPARα to a similar or lower degree in the latter. APFO dose dependently increased hepatic triglyceride levels in Pparα-null and hPPARα mice, but conversely decreased those in mPPARα ones. APFO-induced hepatic damage differed markedly among the three genotyped groups: single-cell necrosis was observed in all genotyped mice; inflammatory cells and macrovesicular steatosis only in Pparα-null mice; and microvesicular steatosis and hydropic degenerations in hPPARα and Pparα-null mice. The molecular mechanism underlying these differences may be attributable to those of gene expressions involved in lipid homeostasis (PPARα, β- and ω-oxidation enzymes, and diacylglycerol acyl-transferases) and uncoupling protein 2. Thus, milligram-order APFO activated both mouse and human PPARα in a different manner, which may reflect histopathologically different types of hepatic damage.
Keywords: Hepatic damage, Human, Mouse, Perfluorooctanoic acid, Peroxisome proliferator-activated receptor
Introduction
Perfluorooctanoic acid (PFOA) (CAS, 335-67-1), an organofluoro compound, is used in industrial surfactants, emulsifiers, and numerous consumer products (Butenhoff et al. 2006). Because the biological half-life in humans is reported as 3.5–4.4 years (Olsen et al. 2007), PFOA will probably be added to the list of Persistent Organic Pollutants in the near future (World Wildlife Fund 2005).
A variety of toxicities associated with PFOA exposure have been investigated and revealed by many studies using ammonium perfluorooctanoate (APFO). Since PFOA is hardly excreted from the body (Kennedy et al. 2004) and is accumulated mostly in the liver (Lau et al. 2007), many studies have focused on the risk of hepatic damage, such as peroxisome proliferation (Nakamura et al. 2009), hepato-cyte necrosis (Butenhoff et al. 2002), hepatocellular adenoma (Biegel et al. 2001), and hepatobiliary injury (Minata et al. 2010). Recently, though Minata et al. (2010) showed that APFO caused cholestasis, this finding was seen mainly in peroxisome proliferator-activated receptor α (Pparα)-null mice, not mPPARα mice. Wolf et al. (2008a) reported that lipid droplets were characteristically observed in the livers of Pparα-null mice, but not in mPPARα mice. Thus, these results suggest that PPARα may play an important role in the pathogenesis of APFO-induced hepatosteatosis and cholestasis. However, its precise mechanism has not yet been fully understood.
Epidemiologically, PFOA may influence lipid metabolism: Olsen et al. (2003a) reported a positive association between PFOA and serum total cholesterol (TC) and triglycerides (TG). In contrast, no such association was reported in another factory (Olsen et al. 2003b). Therefore, it is very important to clarify whether PFOA influences the lipid metabolism using experimental animals.
Since PFOA is an agonist for PPARα (Ikeda et al. 1985), its activation enhances the activities of peroxisomal and mitochondrial β-oxidation enzymes for fatty acids and inhibits the secretion of very low-density lipoproteins and cholesterol from the liver, as well as reducing total cholesterol and TG in serum and the accumulation of lipids in the liver (Berthiaume and Wallace 2002). However, the functional activation is thought to differ among species. Additionally, constitutive expression of PPARα is quite different between mice or rats and humans, whose expression is thought to be 1/10 of the experimental animals (Palmer et al. 1998). PPARα-humanized (PPARαTet-OFF) mice that expressed human PPARα only in the liver of Pparα-null background mice have been established (Cheung et al. 2004). This mouse model has been recognized as a useful tool in determining the human PPARα function. As for the effects of Wy-14,643 on hepatic peroxisomal and mitochondrial β-oxidation enzymes, there were few differences in the inductions between mPPARα and hPPARα mice. Ramdhan et al. (2010) reported that one of the trichloroethylene metabolites, trichloroacetic acid, activated not only mouse PPARα but also human PPARα, though the exposure concentration of trichloroethylene was 1,000 and 2,000 ppm, respectively. However, the fact that expressions of human PPARα mRNA and protein are higher in hPPARα mice than in those of mPPARα (Nakamura et al. 2009) may suggest a weaker function of human PPARα compared with mouse PPARα. Indeed, although microgram-order APFO was unable to activate human PPARα, it did activate mouse PPARα (Nakamura et al. 2009). Therefore, it is very important to clarify whether APFO that is higher than that in a previous study (Nakamura et al. 2009) can activate human PPARα and also to determine how the species difference in the function is involved in PFOA-induced hepatic damage when we extrapolate from animal to human data.
Additionally, PFOA is also found to be an agonist for PPARγ (Vanden Heuvel et al. 2006), which has anti-inflammatory power (Jiang et al. 1998), even though contrary opinions have been reported (Chawla et al. 2001). This receptor is also accepted as a master transcriptional regulator of lipid and glucose metabolism (Spiegelman 1998).
In this study, we compared the effects of relatively high dosages of APFO (0, 1.0, and 5.0 mg/kg) on the PPARα and the target gene expressions as well as the involvement of this receptor in hepatic damage using wild-type, Pparα-null, and hPPARα mice. The molecular mechanisms were also clarified by analyzing the mRNA and protein expressions of related genes. Although relatively low-dose APFO could not activate human PPARα (Nakamura et al. 2009), higher doses clearly activated the receptor. Our results also suggest that the species difference in the function may determine the characteristic features of hepatic damage caused by PFOA.
Materials and methods
Experimental animals
This study was conducted according to the Guidelines for Animal Experiments of the Nagoya University Animal Center. Three genotyped male and female mice, i.e., wild-type (mPPARα), Pparα-null (Lee et al. 1995), and hPPARαTet-OFF (hPPARα) (Cheung et al. 2004) mice with an Sv/129 genetic background were bred and reared as described elsewhere (Nakamura et al. 2009). All mice were housed in a temperature- and light-controlled environment (25°C, 12 h light/dark cycle) and maintained on stock rodent chow (Nippon Clea, Tokyo, Japan) and tap water ad libitum. At 8 weeks old, the offspring male mice (n = 8–10) of each strain were assigned to the following treatment groups: treated with distilled water daily for 6 weeks by gavage (control group); treated with 1.0 or 5.0 mg/kg APFO (Tokyo Kasei Kagaku, Tokyo, Japan), respectively, for 6 weeks by gavages (Table 1). Since 0.1 and 0.3 mg/kg APFO activated mouse PPARα, but not human PPARα (Nakamura et al. 2009), about tenfold doses were selected in this experiment. Since we planned to investigate not only PPARα-directed hepatic damage but also reproductive toxicity of PFOA, we selected six-week exposure to the chemicals. The results of reproductive toxicity will be reported elsewhere. Macroscopically, there was no abnormal sign in all animals throughout the treatments. On the day following the last dose (18–20 h later), all mice were killed by decapitation, and the blood and livers were removed. A part of each liver was fixed by 10% buffered formalin. The remaining liver samples were snap frozen in liquid nitrogen and stored at —80°C until used. Plasma was collected after centrifuging blood at 3,500g for 10 min and stored at —80°C until used. The numbers of samples except for the histopathological analyses used were indicated in the Tables and Figure legends, and all measurements were performed in duplicate or triplicate.
Table 1.
Body and liver weight, plasma ALT activity, plasma and hepatic TG and TC levels in mmPPARα, Pparα-null, and hPPARα mice treated with APFO
| APFO |
|||
|---|---|---|---|
| Control | 1.0 mg/kg | 5.0 mg/kg | |
| Body weight (g) at the start of APFO treatments | |||
| mPPARα | 21.0 ± 0.9 (8) | 20.3 ± 2.2 (8) | 20.7 ± 1.6 (9) |
| Pparα-null | 21.1 ± 2.5 (9) | 21.1 ± 2.8 (9) | 21.8 ± 1.5 (9) |
| hPPARα | 20.3 ± 1.9 (9) | 19.4 ± 1.0 (9) | 20.3 ± 2.5 (10) |
| Body weight (g) after APFO treatments | |||
| mPPARα | 24.8 ± 1.0 (8) | 24.7 ± 2.2 (8) | 23.5 ± 1.7 (9) |
| Pparα-null | 24.2 ± 2.4 (9) | 23.6 ± 2.8 (9) | 25.6 ± 2.5 (9) |
| hPPARα | 23.5 ± 1.3 (9) | 22.3 ± 1.7 (9) | 23.5 ± 2.1 (10) |
| Liver weight (g) | |||
| mPPARα | 1.05 ± 0.06 (8) | 1.98 ± 0.22 (8)* | 2.72 ± 0.29 (9)*,# |
| Pparα-null | 1.10 ± 0.10 (9) | 1.57 ± 0.29 (9)* | 3.35 ± 0.48 (9)*,# |
| hPPARα | 1.03 ± 0.08 (9) | 1.48 ± 0.14 (9)* | 2.49 ± 0.35 (10)*,# |
| Liver/body ratio (%) | |||
| mPPARα | 4.23 ± 0.23 (8) | 8.02 ± 0.62 (8)* | 11.54 ± 0.71 (9)*,# |
| Pparα-null | 4.56 ± 0.38 (9) | 6.68 ± 1.27 (9)* | 13.06 ± 1.16 (9)*,# |
| hPPARα | 4.37 ± 0.30 (9) | 6.67 ± 0.42 (9)* | 10.56 ± 0.91 (10)*,# |
| Plasma ALT (IU/L) | |||
| mPPARα | 6.7 ± 0.9 (8) | 7.6 ± 2.6 (8) | 15.6 ± 8.6 (9)*,# |
| Pparα-null | 7.2 ± 1.6 (9) | 8.6 ± 2.5 (9) | 9.3 ± 1.8 (9)* |
| hPPARα | 6.5 ± 1.3 (9) | 8.1 ± 2.7 (9) | 8.9 ± 1.5 (10)* |
| Plasma TG (mg/dl) | |||
| mPPARα | 156.4 ± 43.6 (8) | 107.3 ± 27.4 (8)* | 77.6 ± 19.0 (9)* |
| Pparα-null | 168.7 ± 80.6 (9) | 183.2 ± 67.2 (9) | 164.8 ± 57.7 (9) |
| hPPARα | 195.9 ± 69.2 (9) | 209.5 ± 58.7 (9) | 142.6 ± 36.9 (9)# |
| Hepatic TG (mg/g) | |||
| mPPARα | 13.3 ± 1.5 (8) | 23.4 ± 5.5 (8)* | 11.8 ± 3.8 (9)# |
| Pparα-null | 19.6 ± 11.0 (9) | 58.7 ± 38.6 (9)* | 106.3 ± 34.9 (9)*,# |
| hPPARα | 17.4 ± 4.4 (9) | 34.4 ± 15.7 (9)* | 51.7 ± 11.9 (10)*,# |
| Plasma TC (mg/dl) | |||
| mPPARα | 66.9 ± 22.3 (8) | 63.8 ± 17.6 (8) | 39.4 ± 11.9 (9)*,# |
| Pparα-null | 109.5 ± 27.7 (9)† | 84.5 ± 16.3 (9) | 57.8 ± 21.5 (9)*,# |
| hPPARα | 106.6 ± 13.3 (9)† | 99.2 ± 27.2 (9) | 83.5 ± 25.4 (10) |
| Hepatic TC (mg/g) | |||
| mPPARα | 3.7 ± 0.8 (8) | 3.6 ± 0.8 (9) | 5.3 ± 1.3 (9) |
| Pparα-null | 4.5 ± 0.6 (9) | 6.8 ± 2.5 (9) | 10.4 ± 4.4 (9)* |
| hPPARα | 5.4 ± 0.8 (9) | 6.7 ± 1.5 (9) | 6.1 ± 1.0 (10) |
Data represent mean ± SD. Data in parentheses are the numbers of mice
Significantly different from respective control (0 mg/kg) group (P < 0.05)
Significantly different from respective low-dose (1.0 mg/kg) group (P < 0.05)
Significantly different from control of mPPARα mice (P < 0.05)
Analysis of protein concentrations
Each tissue was homogenized with a threefold volume of 10 mM phosphate buffer (pH 7.4) containing 0.25 M sucrose. Protein concentrations of the homogenate and nuclear fraction samples were measured using a Protein Assay Kit (Bio-Rad, Tokyo, Japan).
Western blotting
A nuclear fraction was extracted from a part of the frozen liver using a CelLytic™ NuCLEAR™ Extraction Kit (SIGMA, Tokyo, Japan). The nuclear fractions (NFκB p65, p50, p52, and PPARα) and liver homogenates (other proteins) for electrophoresis were subjected to 10 or 12.5% polyacrylamide gel electrophoresis and transferred to nitrocellulose membranes. Immunostaining was conducted as described elsewhere (Nakajima et al. 2000).
Real-time quantitative PCR
Total RNA was isolated from the liver using the RNeasy Mini Kit (QIAGEN, Tokyo, Japan). Real-time quantitative PCR analysis was performed as described elsewhere (Nakamura et al. 2009; Ramdhan et al. 2010).
Lipid concentrations in plasma and liver
Lipid from livers was extracted using the method of Folch et al. (1957). TG and TC in the liver and plasma were measured using TG-IE and T-Cho E kits (Wako, Osaka, Japan), respectively.
Histopathological analysis
Small blocks of liver tissues from each mouse (five animals randomly selected from each group) fixed in 10% neutral buffered formalin were embedded in paraffin and sliced into 4-μm sections. Tissue sections of the livers were stained with hematoxylin and eosin (H & E) and examined under a BZ-8000 (Keyence, Osaka, Japan) light microscope. Severities of steatosis, lobular inflammation, and hepatocyte degeneration were scored by a pathologist in a blinded fashion referring to the methods of Brunt et al. (1999) and Ramdhan et al. (2010) with the following minor modifications: (1) grade of steatosis: 0, none (0–5% of parenchymal involvement by steatosis); 1, mild (5–33% of parenchymal involvement by steatosis); 2, moderate (33–66%); 3, severe (>66%); (2) grade of lobular inflammation: 0, none; 1, mild; 2, moderate; 3, severe; (3) single-cell necrosis and hepatocyte hydropic degeneration: 0, absent; 1, present; 2, frequent.
Alanine aminotransferase measurements
Plasma ALT activities were measured using a Transaminase C II Test kit purchased from Wako (Osaka, Japan).
Statistical analysis
The Steel-Dwass method in case of pathological scoring and Tukey-Kramer HSD post hoc test in the other cases were conducted to compare the effects of treatment among each treated group of each genotyped mouse, and also among the control groups of mPPARα, Pparα-null, and hPPARα mice. Values of P < 0.05 were considered to indicate statistical significance.
Results
Body and liver weight
No significant differences were observed in body weight before and after APFO treatments among the control groups of mPPARα, Pparα-null, and hPPARα mice (Table 1). APFO treatments did not induce an increase in body weight in any genotyped mice, whereas they did increase the liver weight and the ratio of liver per body in all genotyped mice in a dose-dependent manner. In the 1.0 mg/kg treatment group, the ratio increases were most prominent in mPPARα mice (1.9-fold)), while in the 5.0 mg/kg dose group, they were most prominent in Pparα-null mice (2.9-fold).
Histopathological evaluation
Apparent macrovesicular and microvesicular steatosis were not observed in the liver of control Pparα-null mice (Fig. 1b, Table 2). APFO induced macrovesicular steatosis only in the liver of Pparα-null mice in dose-dependent fashion, while it induced microvesicular steatosis in both Pparα-null as well as hPPARα mice; the degree of these fat accumulations was not influenced by APFO dosages (Fig. 1, Table 2). High-dose APFO (5.0 mg/kg) induced lobular inflammation only in Pparα-null mice. Although high-dose APFO induced single-cell necrosis in all geno-typed mice, the severity appeared to be greater in mPPARα mice than in Pparα-null and hPPARα mouse lines. Interestingly, high-dose APFO characteristically induced hydropic degeneration of hepatocytes in Pparα-null (arrows in Fig. 1k) and hPPARα mice (Fig. 1i), but not in mPPARα mice. Hypertrophied hepatocytes with prominent eosinophilic cytoplasm, which sometimes appear in the livers of mice treated with potent PPARα activators, were detected in APFO-treated mPPARα (Fig. 1g, arrowheads in Fig. 1j) and hPPARα mice (Fig. 1i). We might note that hepatocholangiole proliferation was only fleetingly observed in Pparα-null mice exposed to 5.0 mg/kg PFOA, but obvious cholestasis, evidenced by intracellular bile droplets and a bile plug, was not observed in the livers of any genotyped mice.
Fig. 1.
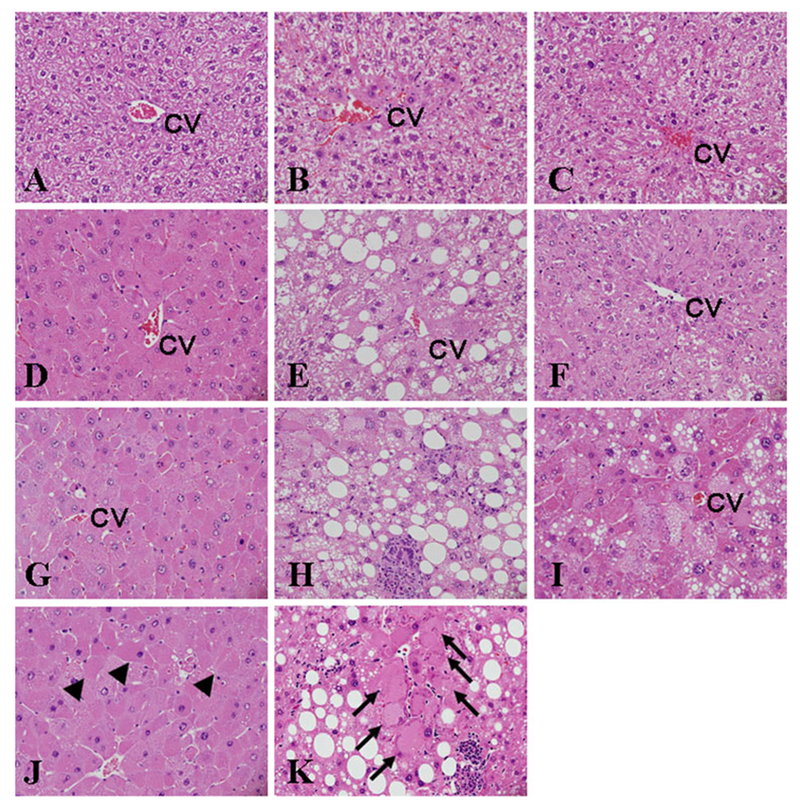
Liver histopathological findings after APFO treatment. Liver section photomicrograph taken from mPPARα (a), Pparα-null (b) and hPPPARα (c) mice treated with 0 mg/kg of APFO. Liver from mPPARα (d), Pparα-null (e) and hPPARα (f) mice treated with 1.0 mg/kg of APFO. Liver from mPPARα (g), Pparα-null (h), hPPARα (i) mice treated with 5.0 mg/kg of APFO. PV, portal vein; CV, central vein; original magnification ×400. Arrowheads (j) and arrows (k) indicate hypertrophied and hydropic hepatocytes with eosinophilic cytoplasm in mPPARα and Pparα-null mice treated with 5.0 mg/kg APFO, respectively
Table 2.
Changes in livers after APFO treatment
| Genotype | mPPARα |
Pparα-null |
hPPARα |
||||||
|---|---|---|---|---|---|---|---|---|---|
| Dose | Control | 1.0 mg/kg APFO | 5.0 mg/kg APFO | Control | 1.0 mg/kg APFO | 5.0 mg/kg APFO | Control | 1.0 mg/kg APFO | 5.0 mg/kg APFO |
| Macrovesicular steatosis | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 1.2 ± 0.8* | 2.0 ± 0.7* | 0.2 ± 0.4 | 0.2 ± 0.4 | 0 ± 0 |
| Microvesicular steatosis | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.6 ± 0.5 | 0.6 ± 0.5 | 0 ± 0 | 0.8 ± 0.4* | 0.8 ± 0.4* |
| Lobular inflammatory cells | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 2.0 ± 0*,# | 0 ± 0 | 0.2 ± 0.4 | 0.2 ± 0.4 |
| Single-cell necrosis | 0 ± 0 | 0 ± 0 | 0.8 ± 0.4 | 0 ± 0 | 0 ± 0 | 0.2 ± 0.4 | 0 ± 0 | 0 ± 0 | 0.4 ± 0.5 |
| Hydropic degeneration | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 2.0 ± 0*,# | 0 ± 0 | 0.2 ± 0.4 | 2.0 ± 0*,# |
Steel–Dwass method was conducted to compare the effects of APFO treatment among each treated group of each genotyped mouse
Data represent mean ± SD for 5 mice per group
Significantly different from respective control (0 mg/kg) group (P < 0.05)
Significantly different from respective low-dose (1.0 mg/kg) group (P < 0.05)
Plasma ALT levels
High-dose APFO increased plasma ALT activities in all genotyped mouse lines, though the elevations were very small, and no differences were noted in these increases among three genotyped mice (mPPARα mice, 2.3-fold vs control; Pparα-null, 1.3-fold; hPPARα mice, 1.4-fold) (Table 1).
Plasma and hepatic TG and TC levels
APFO treatment dose dependently decreased plasma TG levels in mPPARα mice (0.70-fold at 1.0 mg/kg and 0.50-fold at 5.0 mg/kg), while not influencing the levels in Pparα-null and hPPARα mice. In contrast, APFO did not influence hepatic TG levels in mPPARα mice, though 1.0 mg APFO slightly increased those levels. APFO treatments significantly increased hepatic TG levels in Pparα-null mice (3.2-fold and 5.3-fold at 1.0 and 5.0 mg/kg, respectively), but only slightly increased them in hPPARα mice at 1.0 and 5.0 mg/kg (2.0-fold and 3.0-fold, respectively). Consistent with histopathological findings, hepatic TG contents in the control Ppara-null mice were similar to those in the control mPpara mice and hPPARa mice.
High-dose APFO (5.0 mg/kg) treatment dose dependently decreased plasma TC levels in mPPARα and Pparα-null mice, but not in hPPARα mice. In contrast, PFOA increased hepatic TC levels in Pparα-null mice, but not in mPPARα and hPPARα mice.
Analysis of mRNA levels
Because APFO influenced plasma and hepatic TG levels differently in three genotyped mice, we investigated hepatic β-oxidation enzymes that are PPARα-target genes and are involved in fatty acid metabolism. PPARα mRNA expression was significantly greater in hPPARα mice than in mPPARα mice, results similar to those of Nakamura et al. (2009) (Fig. 2). Constitutive expressions of CYP4A10, PT, and VLCAD mRNA were significantly lower in Pparα-null mice than in mPPARα and hPPARα mice, while those of PH were lower only in hPPARα mice.
Fig. 2.
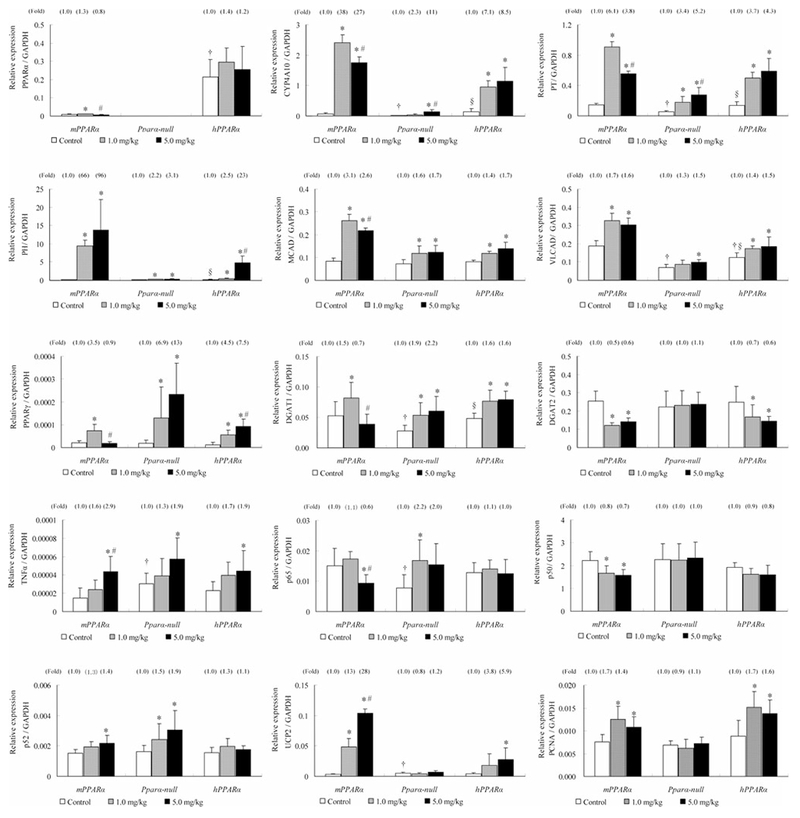
mRNA expressions of PPARα and the related genes in livers from mPPARα, Pparα-null and hPPARα mice treated with APFO. Values represent means ± SD for 8–10 mice per group. *Significantly different from respective control (0 mg/kg) group (P < 0.05). #Significantly different from respective low-dose (1.0 mg/kg) group (P < 0.05). †Significantly different from control of mPPARα (P < 0.05). §Significantly different from control of Pparα-null mice (P < 0.05)
Although APFO treatment did not influence the PPARα-mRNA levels in either mPPARα or hPPARα mice, increased expressions of hepatic PPARα-target genes, CYP4A10, PT, PH, MCAD, VLCAD, and proliferation cell nuclear antigen (PCNA) were observed in both mouse lines. In general, their increases were the same (VLCAD, MCAD, and PCNA) or roughly twofold greater (PT and PH) in the former than in the latter, while CYP4A10-mRNA was around fivefold higher in mPPARα mice compared with that in hPPARα mice. Next, we measured the effects of APFO treatment on triglyceride-synthesizing enzymes (Yen et al. 2008). APFO was found to increase DGAT1-mRNA levels in Pparα-null and hPPARα mice (1.7- to 2.2-fold), but at a 5.0 mg/kg dose alone it decreased the mRNA in mPPARα (0.7-fold). In contrast, APFO slightly reduced DGAT2-mRNA levels in mPPARα and hPPARα mice (~ 0.6-fold). Because DGAT1 and 2 are PPARγ target genes (Ranganathan et al. 2006; Festuccia et al. 2009), the expression was also investigated: both APFO dosages increased PPARγ mRNA levels in Pparα-null mice in a dose-dependent fashion (7- to 13-fold), and only slightly in hPPARα mice (2- to 3-fold); in mPPARα mice, they increased only at a 1.0 mg/kg dose (3.5-fold).
Since obvious inflammatory cell infiltration was seen in Pparα-null mice, but not in mPPARα and hPPARα mice, we measured NFκB subunits: APFO treatments slightly induced p65-mRNA levels only in Pparα-null mice (2.2-fold), but even decreased them in mPPARα (0.6-fold); in p50-mRNA, PFOA significantly decreased at 1.0 and 5.0 mg/kg dosages in mPPARα mice (0.7- to 0.8-fold). As for p52-mRNA, the low- and high-dose APFO significantly raised the levels in Pparα-null (1.5-fold and 1.9-fold) and the high dose in mPPARα mice (1.4-fold), although none of them increased the mRNA levels in hPPARα mice. We also measured the pro-inflammatory cytokine TNFα-mRNA (Moriya et al. 2009) and a mitochondrial antioxidant UCP2-mRNA (Nègre-Salvayre et al. 1997). High-dose PFOA significantly increased TNFα-mRNA in mPPARα, Pparα-null, and hPPARα mice (2.9-fold, 1.9-fold, and 1.9-fold, respectively). An obvious difference was observed in the increase in UCP2-mRNA levels; the increase was dose dependent, with the high dose increasing by 28-fold in mPPARα mice, while it raised the levels fivefold in hPPARα mice. No increase in mRNA was observed in Pparα-null mice.
Western blot analysis
In the control group, PPARα expression was greater in hPPARα mice than in mPPARα mice and surprisingly decreased APFO treatments only in the latter (Fig. 3). The expressions of PT and PH were significantly lower in Pparα-null mice than in mPPARα and hPPARα mice, while that of VLCAD was significantly higher in hPPARα mice than in mPPARα and Pparα-null mice. The levels of PT, PH, and VLCAD in mPPARα mice appeared to reach the maximum at low-dose APFO and did not increase further at the high dose, while in hPPARα mice, the increase tended to elevate in a dose-dependent fashion. APFO elevated MCAD protein only in mPPARα mice at the high dose. The protein expression of the cell proliferation marker, PCNA, was higher in all APFO-treated mPPARα and hPPARα mice compared with respective controls.
Fig. 3.
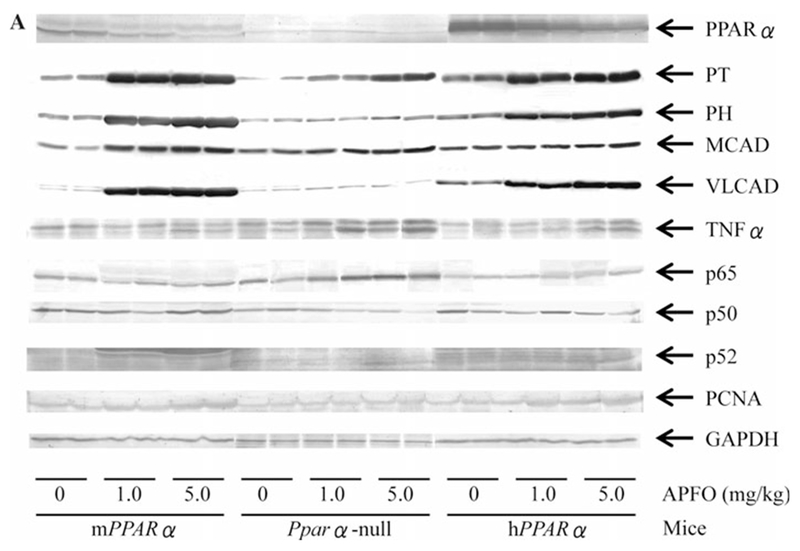
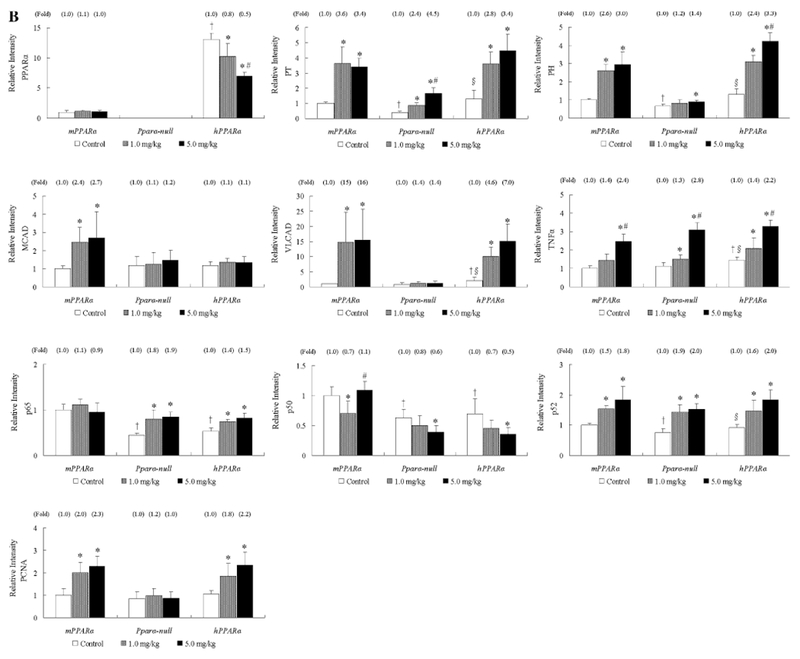
Protein expressions of PPARα and the related genes in livers from mPPARα, Pparα-null and hPPARα mice treated with APFO. a Representative Western blot analyses. As an internal standard, GAPDH was stained. b Each band was quantified by densitometric analysis. Histogram presents means ± SD for 8–10 mice per group, and the mean from each control group in mPPARα mice was assigned a value of 1.0. *Significantly different from respective control (0 mg/kg) group (P < 0.05). #Significantly different from respective low-dose (1.0 mg/kg) group (P < 0.05). †Significantly different from control of mPPARα (P < 0.05). §Significantly different from control of Pparα-null mice (P < 0.05)
The levels of p65 and p52 protein in the control group were lower in Pparα-null and hPPARα mice than in mPPARα mice, while TNFα levels were higher in hPPARα mice than in mPPARα and Pparα-null mice. APFO treatments increased the protein of p65 only in Pparα-null (1.8- to 1.9-fold) and hPPARα mice (1.4- to 1.5-fold), but did not increase p50 levels. In contrast, APFO increased p52 levels in all genotyped mice at both dosages (1.5- to 2.0-fold). High-dose APFO significantly increased TNFα levels in all genotyped mice, while increases were also observed when compared with the low-dose treatment.
Discussion
We reported that microgram-order APFO could not activate human PPARα but did activate mouse PPARα (Nakamura et al. 2009). The current experiment has clearly shown that milligram-order APFO (1.0–5.0 mg/kg) activated both PPARα to a similar or even smaller extent in human PPARα. Therefore, the PPARα function of humans might be weak compared with that of mice, due to the higher expression of human PPARα compared with that of mouse PPARα in the mouse livers used in this experiment. In that connection, Takacs and Abbott (2007) measured the lowest effective concentrations of PFOA at which mouse and human PPARα were activated in an in vitro system, showing them to be 10 and 30 μM, respectively. Wolf et al. (2008b) also reported that the activation of mouse PPARα by APFO was generally higher compared with that of human PPARα. Thus, there were clear-cut species differences in PPARα activation between mice and humans, similar to those by trichloroethylene exposure (Ramdhan et al. 2010).
The first point for discussion in the present study is that APFO-induced hepatic damage was quite different histopathologically among mPPARα, Pparα-null, and hPPARα mice, unlike such differences in plasma ALT activities. Hypertrophic hepatocytes with eosinophilic cytoplasm were observed in mPPARα and hPPARα mice, suggesting the presence of PPARα activation. This finding was consistent with the results that typical PPARα target genes (e.g., CYP4A10, PH, PCNA, and UCP-2) were induced in both mouse groups. In contrast, inflammatory cell infiltrations were frequently observed in Pparα-null mice, which were also supported by the inflammation signaling analysis referring to p65 mRNA and protein expressions. It is of interest to note that APFO decreased p65 and p50 expressions in mPPARα mice, which may have resulted from the strong activation of PPARα in the mouse line. Mouse PPARα induced by APFO treatment may completely inhibit the import of p50 and p65 directly into the nucleus or via IκBα and thereby the inhibiting inflammation, since this receptor was shown to possess such a function (Moriya et al. 2009). However, the function of human PPARα may be weak compared with that of mice and thus could not fully inhibit the inflammatory signaling in hPPARα mice as it does in mPPARα mice. The increase in p52 mRNA and protein were not correlated with inflammatory cell infiltrations and may be related instead to the increase in plasma ALT activity in all genotyped mice.
Our second point involves the histopathological differences in steatosis among three genotyped mice after APFO treatments: microvesicular steatosis was seen in Pparα-null and hPPARα mice, whereas macrovesicular steatosis was observed only in Pparα-null mice. These findings were not replicated in mPPARα mice. In line with these results, APFO treatments also increased hepatic TG and TC levels in Pparα-null and hPPARα mice, though the increase in TC was not significant in the latter mouse line. In mPPARα mice, 5.0 mg/kg dose decreased the TG level, which may be related to the elevation of β-oxidation enzymes. APFO treatments induced fatty acid β-oxidation enzymes more in mPPARα mice than in hPPARα mice, but very few in Pparα-null mice. In addition, APFO increased the expressions of DGAT1 in Pparα-null and hPPARα mice more prominently compared with those in mPPARα mice. As for DGAT2, the exposure decreased the expressions in mPPARα and hPPARα mice, but did not influence those in Pparα-null mice. Taken together, less or lower induction of fatty acid β-oxidation enzymes and higher induction of DGAT1 by APFO may, in part, reflect the increase of macrovesicular and/or microvesicular steatosis in Pparα-null and hPPARα mice. However, we were unable to explain why macrovesicular steatosis was found only in Pparα-null mice. Further study is warranted to determine whether or not a PPARγ or increase in cholesterol is involved in this regard in the liver of Pparα-null mice.
A question may arise as to why DGAT1 was induced in the liver of Pparα-null and hPPARα mice, but not in mPPARα mice, though increased in the 1.0 mg/kg dose group. DGAT1 is expressed in organs that produce large amounts of TG, such as liver, small intestine, and adipose tissue (Cases et al. 1998), which may be regulated by PPARγ (Ranganathan et al. 2006; Festuccia et al. 2009). The mRNA levels of PPARγ were greatly increased in Pparα-null and only slightly in hPPARα and mPPARα mice, but not in mPPARα mice at 5.0 mg/kg dose. Thus, the activation of PPARγ may be related to the increased DGAT1 in all genotyped mice. PFOA treatment also induces mitochondrial biogenesis at the transcriptional level with a preferential stimulation of mitochondrial DNA transcription, which occurs by way of the activation of a PPARγ coactivator-1α pathway (Walters et al. 2009). Indeed, PFOA is also reported to act as an agonist for PPARγ (Vanden Heuvel et al. 2006). However, the question still remains why PPARγ could not be activated by APFO treatment in mPPARα mice. Since the activation of PPARα by APFO was weak in hPPARα mice and showed no activation at all in Ppara-null, this may result in increasing the return PPARγ.
Our third concern in this study is why APFO treatments increased pro-inflammatory cytokine TNFα in all geno-typed mice. TNFα is thought to be an index of Kupffer cell activation (Yoshida et al. 2001), which may be related to the increase in necrotic (Morgan et al. 2008) or inflammatory cells (Dasarathy 2008). APFO treatment increased necrotic cells in the livers of mPPARα and hPPARα mice, and inflammatory cell infiltrations in the livers of Pparα-null mice, both of which may be related to the increase in TNFα levels; these phenomena are also related to the rise in ALT activity.
Finally, in addition to macro/microvesicular steatosis, hydropic degeneration was characteristically observed in hPPARα and Pparα-null mice. Minata et al. (2010) recently reported that PFOA in the liver was easily excreted into the bile duct in mPPARα mice, but that the excretion in Pparα-null mice was less than half that in mPPARα mice. Therefore, PFOA is thought to be much more accumulated in Ppara-null, and perhaps less so in hPPARα mice, compared with that in mPPARα mice. PFOA accumulated in hepatocytes poses a potential risk for mitochondrial dysfunction: APFO treatment results in an increase in the production of oxidative stress (Panaretakis et al. 2001) and in an enhancement of mitochondrial inner membrane permeability, which may disturb the mitochondrial inter-membranous electrochemical gradient, thus reducing the ATP production rate (Starkov and Wallace 2002). A powerful induction of the UCP2 expression following APFO treatment found only in mPPARα mice may serve as protection against mitochondrial damage, because UCP2 is known to be a target gene of PPARα and can inhibit oxidative stress production in mitochondria (Nègre-Salvayre et al. 1997; Kizaki et al. 2002). Very low levels of UCP2 induction in hPPARα mice and non-induction in Pparα-null mice may not be enough to protect mitochondria from PFOA-induced oxidative stress. Furthermore, cellular stress induced by an excessive PFOA accumulation may also damage the endoplasmic reticulum, cytoskeleton, and microtubules, thus impairing excretion of proteins, lipids, and bile acids from hepatocytes. We consider that these abnormalities may eventually lead to the appearance of hydropic degeneration of hepatocytes, preferentially in Ppara-null and hPPARα mice.
In this study, steatosis was not observed in the control Ppara-null mice. This is consistent with the previous report that 24-week-old control Sv/129 Ppara-null mice demonstrated neither apparent steatosis nor increases in hepatic TG contents compared with control wild-type mice (Tanaka et al. 2010). Okiyama et al. (2009) showed the presence of macrovesicular steatosis in the control group of Ppara-null mice, but they have used fat-rich liquid diet as a control. Furthermore, Rosen et al. (2010) reported the presence of significant macrovesicular steatosis in the control Ppara-null mice; such a discrepancy may be derived from the marked difference in mouse age (6- to 9-month-old mice in Rosen’s study vs. 14-week-old mice in this study). It is plausible that steatosis may become obvious with age especially in Pparα-null mice, since mitochondrial β-oxidation ability is constitutively lower in Pparα-null mice than in wild-type mice (Aoyama et al. 1998).
In conclusion, PPARα function may be very important in protecting against hepatic damage caused by PFOA, a result similar to the findings reported by many laboratories (Minata et al. 2010; Wolf et al. 2008b). In this regard, the human PPARα function may be weak compared with that of mice. In addition, the expression of the receptor in human liver was 1/10 lower than that in mice or rats (Palmer et al. 1998). Therefore, hepatic damage may be induced if humans are exposed to high doses of PFOA. In such cases, histopathological findings may resemble those of Ppara-null mice more than mPPARα mice.
Acknowledgments
We wish to thank to Mr. Toshiki Nakamura for his kind assistance with the animal experiments. This study was supported in part by Grants-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (B. 14370121, 17390169).
Abbreviations
- ALT
Alanine aminotransferase
- APFO
Ammonium perfluorooctanoate
- CV
Central vein
- CYP4A10
Cytochrome P450 4A10
- DGAT1
Diacylglycerol acyltransferase 1
- DGAT2
Diacylglycerol acyltransferase 2
- H & E
Hematoxylin and eosin
- hPPARα mice
Humanized PPARα mice
- MCAD
Medium chain acyl-CoA dehydrogenase
- NFκB
Nuclear factor kappa B
- PCNA
Proliferation cell nuclear antigen
- PH
Peroxisomal bifunctional protein
- PPARα
Peroxisome proliferator-activated receptor α
- PPARγ
Peroxisome proliferator-activated receptor γ
- PT
Peroxisomal thiolase
- TC
Total cholesterol
- TG
Triglycerides
- TNFα
Tumor necrosis factor α
- UCP2
Uncoupling protein 2
- VLCAD
Very long chain acyl-CoA dehydrogenase
Footnotes
Conflict of interest The authors declare that there are no conflicts of interest.
Contributor Information
Tomohiko Nakagawa, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan.
Doni Hikmat Ramdhan, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan; Department of Occupational Health and Safety, Faculty of Public Health, University of Indonesia, Depok 16424, Jawa Barat, Indonesia.
Naoki Tanaka, Department of Metabolic Regulation, Shinshu University Graduate School of Medicine, Matsumoto 390-8621, Japan.
Hisao Naito, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan.
Hazuki Tamada, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan.
Yuki Ito, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan; Department of Occupational and Environmental Health, Nagoya City University Graduate School of Medical Sciences, Nagoya 467-8601, Japan.
Yufei Li, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan.
Yumi Hayashi, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan.
Nozomi Yamagishi, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan.
Yukie Yanagiba, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan; National Institute of Occupational Safety and Health, Kawasaki 214-8585, Japan.
Toshifumi Aoyama, Department of Metabolic Regulation, Shinshu University Graduate School of Medicine, Matsumoto 390-8621, Japan.
Frank J. Gonzalez, Laboratory of Metabolism, National Cancer Institute, NIH, Bethesda, MD 20892, USA
Tamie Nakajima, Department of Occupational and Environmental Health, Nagoya University Graduate School of Medicine, 65 Tsurumai-cho, Showa-ku, Nagoya, Aichi 466-8550, Japan.
References
- Aoyama T, Peters JM, Iritani N, Nakajima T, Furihata K, Hashimoto T, Gonzalez FJ (1998) Altered constitutive expression of fatty acid-metabolizing enzymes in mice lacking the peroxisome proliferator-activated receptor α (PPARα). J Biol Chem 273:5678–5684 [DOI] [PubMed] [Google Scholar]
- Berthiaume J, Wallace KB (2002) Perfluorooctanoate, perflourooc-tanesulfonate, and N-ethyl perfluorooctanesulfonamido ethanol: peroxisome proliferation and mitochondrial biogenesis. Toxicol Lett 129:23–32 [DOI] [PubMed] [Google Scholar]
- Biegel LB, Hurtt ME, Frame SR, O’Connor JC, Cook JC (2001) Mechanisms of extrahepatic tumor induction by peroxisome proliferators in male CD rats. Toxicol Sci 60:44–55 [DOI] [PubMed] [Google Scholar]
- Brunt EM, Janney CG, Di Bisceglie AM, Neuschwander-Tetri BA, Bacon BR (1999) Nonalcoholic steatohepatitis: a proposal for grading and staging the histological lesions. Am J Gastroenterol 94:2467–2474 [DOI] [PubMed] [Google Scholar]
- Butenhoff J, Costa G, Elcombe C, Farrar D, Hansen K, Iwai H, Jung R, Kennedy G Jr, Lieder P, Olsen G, Thomford P (2002) Toxicity of ammonium perfluorooctanoate in male cynomolgus monkeys after oral dosing for 6 months. Toxicol Sci 69:244–257 [DOI] [PubMed] [Google Scholar]
- Butenhoff JL, Olsen GW, Pfahles-Hutchens A (2006) The applicability of biomonitoring data for perfluorooctanesulfonate to the environmental public health continuum. Environ Health Perspect 114:1776–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cases S, Smith SJ, Zheng YW, Myers HM, Lear SR, Sande E, Novak S, Collins C, Welch CB, Lusis AJ, Erickson SK, Farese RV Jr (1998) Identification of a gene encoding an acyl CoA: diacylglycerol acyltransferase, a key enzyme in triacylglycerol synthesis. Proc Natl Acad Sci USA 95:13018–13023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM (2001) PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med 7:48–52 [DOI] [PubMed] [Google Scholar]
- Cheung C, Akiyama TE, Ward JM, Nicol CJ, Feigenbaum L, Vinson C, Gonzalez FJ (2004) Diminished hepatocellular proliferation in mice humanized for the nuclear receptor peroxisome proliferator-activated receptor alpha. Cancer Res 64:3849–3854 [DOI] [PubMed] [Google Scholar]
- Dasarathy S (2008) Inflammation and liver. J Parenter Enteral Nutr 32:660–666 [DOI] [PubMed] [Google Scholar]
- Festuccia WT, Blanchard PG, Turcotte V, Laplante M, Sariahmetoglu M, Brindley DN, Richard D, Deshaies Y (2009) The PPARgamma agonist rosiglitazone enhances rat brown adipose tissue lipogenesis from glucose without altering glucose uptake. Am J Physiol Regul Integr Comp Physiol 296:R1327–R1335 [DOI] [PubMed] [Google Scholar]
- Folch J, Lees M, Sloane Stanley GH (1957) A simple method for the isolation and purification of total lipids from animal lipids. J Biol Chem 226:497–509 [PubMed] [Google Scholar]
- Ikeda T, Aiba K, Fukuda K, Tanaka M (1985) The induction of peroxisome proliferation in rat liver by perfluorinated fatty acids, metabolically inert derivatives of fatty acids. J Biochem 98:475–482 [DOI] [PubMed] [Google Scholar]
- Jiang C, Ting AT, Seed B (1998) PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 391:82–86 [DOI] [PubMed] [Google Scholar]
- Kennedy GL Jr, Butenhoff JL, Olsen GW, O’Connor JC, Seacat AM, Perkins RG, Biegel LB, Murphy SR, Farrar DG (2004) The toxicology of perfluorooctanoate. Crit Rev Toxicol 34:351–384 [DOI] [PubMed] [Google Scholar]
- Kizaki T, Suzuki K, Hitomi Y, Taniguchi N, Saitoh D, Watanabe K, Onoe K, Day NK, Good RA, Ohno H (2002) Uncoupling protein 2 plays an important role in nitric oxide production of lipopolysaccharide-stimulated macrophages. Proc Natl Acad Sci USA 99:9392–9397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau C, Anitole K, Hodes C, Lai D, Pfahles-Hutchens A, Seed J (2007) Perfluoroalkyl acids: a review of monitoring and toxicological findings. Toxicol Sci 99:366–394 [DOI] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ (1995) Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol 15:3012–3022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minata M, Harada KH, Karrman A, Hitomi T, Hirosawa M, Murata M, Gonzalez FJ, Koizumi A (2010) Role of peroxisome proliferator-activated receptor-α in hepatobiliary injury induced by ammonium perfluorooctanoate in mouse liver. Ind Health 48:96–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan MJ, Kim YS, Liu ZG (2008) TNFα and reactive oxygen species in necrotic cell death. Cell Res 18:343–349 [DOI] [PubMed] [Google Scholar]
- Moriya T, Naito H, Ito Y, Nakajima T (2009) “Hypothesis of seven balances’’: molecular mechanisms behind alcoholic liver diseases and association with PPARalpha. J Occup Health 51:391–403 [DOI] [PubMed] [Google Scholar]
- Nakajima T, Kamijo Y, Usuda N, Liang Y, Fukushima Y, Kametan K, Gonzalez FJ, Aoyama T (2000) Sex-dependent regulation of hepatic peroxisome proliferation in mice by trichloroethylene via peroxisome proliferator-activated receptor alpha (PPARalpha). Carcinogenesis 21:677–682 [DOI] [PubMed] [Google Scholar]
- Nakamura T, Ito Y, Yanagiba Y, Ramdhan DH, Kono Y, Naito H, Hayashi Y, Li Y, Aoyama T, Gonzalez FJ, Nakajima T (2009) Microgram-order ammonium perfluorooctanoate may activate mouse peroxisome proliferator-activated receptor alpha, but not human PPARalpha. Toxicology 265:27–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nègre-Salvayre A, Hirtz C, Carrera G, Cazenave R, Troly M, Salvayre R, Pénicaud L, Casteilla L (1997) A role for uncoupling protein-2 as a regulator of mitochondrial hydrogen peroxide generation. FASEB J 11:809–815 [PubMed] [Google Scholar]
- Okiyama W, Tanaka N, Nakajima T, Tanaka E, Kiyosawa K, Gonzalez FJ, Aoyama T (2009) Polyenephosphatidylcholine prevents alcoholic liver disease in PPARalpha-null mice through attenuation of increases in oxidative stress. J Hepatol 50:1236–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen GW, Burris JM, Burlew MM, Mandel JH (2003a) Epidemiologic assessment of worker serum perfluorooctanesulfonate (PFOS) and perfluorooctanoate (PFOA) concentrations and medical surveillance examinations. J Occup Environ Med 45:260–270 [DOI] [PubMed] [Google Scholar]
- Olsen GW, Butenhoff JL, Mandel JH (2003b) Assessment of lipid, hepatic and thyroid function in relation to an occupational biologic limit value for perfluorooctanoate 3 M Company, St. Paul. USEPA docket AR-226–1351. US Environmental Protection Agency, Washington [Google Scholar]
- Olsen GW, Burris JM, Ehresman DJ, Froehlich JW, Seacat AM, Butenhoff JL, Zobel LR (2007) Half-life of serum elimination of perfluorooctanesulfonate, perfluorohexanesulfonate, and perfluorooctanoate in retired fluorochemical production workers. Environ Health Perspect 115:1298–1305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer CN, Hsu MH, Griffin KJ, Raucy JL, Johnson EF (1998) Peroxisome proliferator activated receptor-alpha expression in human liver. Mol Pharmacol 53:14–22 [PubMed] [Google Scholar]
- Panaretakis T, Shabalina IG, Grandér D, Shoshan MC, De Pierre JW (2001) Reactive oxygen species and mitochondria mediate the induction of apoptosis in human hepatoma HepG2 cells by the rodent peroxisome proliferator and hepatocarcinogen, perfluorooctanoic acid. Toxicol Appl Pharmacol 173:56–64 [DOI] [PubMed] [Google Scholar]
- Ramdhan DH, Kamijima M, Wang D, Ito Y, Naito H, Yanagiba Y, Hayashi Y, Tanaka N, Aoyama T, Gonzalez FJ, Nakajima T (2010) Differential response to trichloroethylene-induced hepatosteatosis in wild-type and PPARalpha-humanized mice. Environ Health Perspect 118:1557–1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan G, Unal R, Pokrovskaya I, Yao-Borengasser A, Phanavanh B, Lecka-Czernik B, Rasouli N, Kern PA (2006) The lipogenic enzymes DGAT1, FAS, and LPL in adipose tissue: effects of obesity, insulin resistance, and TZD treatment. J Lipid Res 47:2444–2450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosen MB, Schmid JR, Corton JC, Zehr RD, Das KP, Abbott BD, Lau C (2010) Gene expression profiling in wild-type and PPARα-null mice exposed to perfluorooctane sulfonate reveals PPARα-independent effects. PPAR Res 2010 pii: 794739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM (1998) PPARgamma in monocytes: less pain, any gain? Cell 93:153–155 [DOI] [PubMed] [Google Scholar]
- Starkov AA, Wallace KB (2002) Structural determinants of fluoro-chemical-induced mitochondrial dysfunction. Toxicol Sci 66:244–252 [DOI] [PubMed] [Google Scholar]
- Takacs ML, Abbott BD (2007) Activation of mouse and human peroxisome proliferator-activated receptors (alpha, beta/delta, gamma) by perfluorooctanoic acid and perfluorooctane sulfonate. Toxicol Sci 95:108–117 [DOI] [PubMed] [Google Scholar]
- Tanaka N, Zhang X, Sugiyama E, Kono H, Horiuchi A, Nakajima T, Kanbe H, Tanaka E, Gonzalez FJ, Aoyama T (2010) Eicosa-pentaenoic acid improves hepatic steatosis independent of PPARα activation through inhibition of SREBP-1 maturation in mice. Biochem Pharmacol 80:1601–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanden Heuvel JP, Thompson JT, Frame SR, Gillies PJ (2006) Differential activation of nuclear receptors by perfluorinated fatty acid analogs and natural fatty acids: a comparison of human, mouse, and rat peroxisome proliferator-activated receptor-alpha, -beta, and -gamma, liver X receptor-beta, and retinoid X receptor-alpha. Toxicol Sci 92:476–489 [DOI] [PubMed] [Google Scholar]
- Walters MW, Bjork JA, Wallace KB (2009) Perfluorooctanoic acid stimulated mitochondrial biogenesis and gene transcription in rats. Toxicology 264:10–15 [DOI] [PubMed] [Google Scholar]
- Wolf T, Moore BD, Abbott MB, Rosen KP, Das RD, Zehr AB, Lindstrom AB, Strynar MJ, Lau C (2008a) Comparative hepatic effects of perfluorooctanoic acid and WY 14, 643 in PPAR-alpha knockout and wild-type mice. Toxicol Pathol 36:632–639 [DOI] [PubMed] [Google Scholar]
- Wolf C, Takacs M, Schmid J, Lau C, Abbott B (2008b) Activation of mouse and human peroxisome proliferator-activated receptor alpha by perfluoroalkyl acids of different functional groups and chain lengths. Toxicol Sci 106:162–171 [DOI] [PubMed] [Google Scholar]
- World Wildlife Fund (2005) Stockholm convention: ‘‘new POPs’’ screening additional POPs candidates. http://assets.panda.org/downloads/newpopsfinal.pdf
- Yen CL, Stone SJ, Koliwad S, Harris C, Farese RV Jr (2008) Thematic review series: glycerolipids. DGAT enzymes and triacylglycerol biosynthesis. J Lipid Res 49:2283–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Takeda M, Tsutsumi T, Nagata S, Yoshida F, Maita K, Harada T, Ueno Y (2001) Tumor necrosis factor-α expression and Kupffer cell activation in hepatotoxicity caused by micro-cystin-LR in mice. J Toxicol Pathol 14:259–265 [Google Scholar]