

Abstract
Aims/hypothesis
Diabetes in pregnancy is linked to development of obesity in the offspring, but the mechanisms are not fully understood. Gestational diabetes mellitus (GDM) occurs when beta cells are unable to compensate for the normal insulin resistance of late pregnancy. In this study, we used a murine model of beta cell dysfunction to examine the effects of maternal GDM on phenotype in male offspring with and without an inherited predisposition for beta cell dysfunction.
Methods
Beta cell-specific aryl-hydrocarbon receptor nuclear translocator-null (βArnt) mice develop GDM from beta cell dysfunction. βArnt and control female mice were used to induce GDM and non-diabetic pregnancies, respectively.
Results
Offspring from GDM pregnancies became spontaneously obese on a normal-chow diet. They were heavier than offspring from non-diabetic pregnancies, with increased body fat. Respiratory exchange ratio (RER) was higher, indicating decreased capacity to switch to lipid oxidation. Metabolic rate in GDM offspring was decreased prior to onset of obesity. The phenotype was more pronounced in βArnt GDM offspring than in GDM offspring of control genotype, demonstrating an interaction between genotype and pregnancy exposure. βArnt GDM offspring had increased hypothalamic neuropeptide Y (Npy) and decreased pro-opiomelanocortin (Pomc) expression. Weight, body fat, insulin sensitivity and RER in all mice, and hypothalamic Npy in βArnt mice were significantly correlated with AUC of maternal late pregnancy glucose tolerance tests (p<0.01), but not with litter size, maternal weight, triacylglycerol or pre-pregnancy glycaemia.
Conclusions/interpretation
In βArnt mice, exposure to GDM and inheritance of genetic beta cell dysfunction had additive effects on male offspring obesity; severity of the offspring phenotype correlated with maternal glycaemia.
Keywords: ARNT, Beta cell, Energy expenditure, Gestational diabetes, Obesity, Pregnancy, Programming
Introduction
Offspring of women with diabetes in pregnancy are at increased risk of obesity. This is true of type 1 diabetes, type 2 diabetes and gestational diabetes mellitus (GDM) [1–6]. In the diabetes-prone North American Pima Indians, children born after the onset of maternal type 2 diabetes had BMI 2.6 kg/m2 greater than siblings born before maternal hyper-glycaemia and were more likely to develop diabetes [7]. The mechanisms underlying this obesity are not fully elucidated. It has proven difficult to separate the effects of genetic risk and exposure. Improving our understanding of these mechanisms could help to break the cycle of diabetes and obesity begetting diabetes and obesity, an important contributor to the exponential rise in these conditions worldwide [1, 4, 6, 7].
Women become insulin-resistant in the later stages of normal pregnancy. GDM develops when beta cell function cannot compensate for the degree of insulin resistance [8, 9]. Animal models can help determine the contributions of genes, postnatal and early life to offspring phenotypes. Previous studies have investigated the effects of maternal hyperglycaemia using Goto–Kakizaki (GK) rats and leptin receptor heterozygous knockout (db−/+) mice [10]. Transfer of GK embryos to normal mothers did not restore offspring glucose tolerance, whereas exposure of normal embryos to GK mothers caused glucose intolerance [11]. Db/+ mothers, which have greater fat pad weights than their wild-type controls, produce db−/+ offspring with a more pronounced phenotype than db−/+ offspring born to wild-type mothers, showing that exposure to mild maternal obesity worsens offspring phenotype [10]. However, db−/+ mice have normal glucose tolerance, so cannot serve as a model of GDM. Models of maternal overfeeding [12] can produce GDM and are very relevant to the obesity epidemic, but it is not possible to separate contributions of maternal diet, obesity and glucose tolerance.
Genetic beta cell dysfunction such as that caused by the MODY syndromes causes early-onset glucose intolerance, sometimes diagnosed during pregnancy [13]. Polymorphisms in genes that regulate beta cell function are also associated with GDM [14–16]. As many women with GDM will have a genetic predisposition to diabetes, which may or may not be passed on to their offspring, a model that separates the effects of hyperglycaemia, intergenerational effects and inherited genetic risk would be useful. In addition, it has long been debated which nutrients or defects are responsible for offspring effects [17], and a model with deficits only in insulin-secreting cells would help to answer this question.
The beta-cell-specific aryl-hydrocarbon receptor nuclear translocator-null (βArnt) mouse is a model of beta cell dysfunction with normal fasting glucose and mildly impaired glucose tolerance [18]. We previously reported a 90% reduction in ARNT mRNA in islets from patients with type 2 diabetes [18]. Of the five patients with type 2 diabetes in that study, three were women with a known history of GDM. Because the beta cell defect in βArnt mice is inherited in an autosomal dominant manner, it allows examination of the effects of maternal glucose intolerance in offspring with and without inherited beta cell dysfunction. This mimics the human situation, where some children inherit their parents’ susceptibility genes and some do not. The defect is specific to insulin-expressing cells, so allows examination of the effects of abnormalities isolated to this population of cells.
Using this mouse model, the aims of this study were: (1) to examine the mechanisms underlying development of obesity in offspring of GDM mothers; and (2) to examine the interactions between GDM exposure and inherited genetic beta cell defects in offspring risk. Exposure to GDM resulted in spontaneous obesity on normal-chow diet, with a more severe phenotype in βArnt than in floxed-control offspring. GDM offspring had increased body fat and respiratory exchange ratio (RER), and decreased energy expenditure. Maternal glycaemia in pregnancy correlated significantly with offspring weight, insulin insensitivity and RER, and in βArnt mice with hypothalamic neuropeptides.
Methods
Mice βArnt mice were generated as previously reported [19] and bred at Australian BioResources (Mossvale, NSW, Australia). All mice were homozygous Arnt-floxed. The beta cell defect is inherited with one Cre-recombinase allele driven by the rat insulin promoter (RIP). RIP-Cre (+) mice delete Arnt in insulin-expressing cells (flox/flox, RIP-Cre(+) mice=βArnt) and RIP-Cre(−) mice do not (flox/flox, RIP-Cre(−) mice = floxed-controls). In our colony, RIP-Cre transgenic mice have unaltered glucose tolerance vs wild-type or Arnt-flox/flox controls (p>0.6 for all time points, data not shown (dns)). Studies were approved by the Garvan Animal Ethics Committee.
Figure 1a outlines the breeding plan and Fig. 1b, c the timelines for offspring experiments. To induce GDM pregnancies, 12- to 16-week-old βArnt female mice were mated with floxed-control males. Floxed-control females were mated with βArnt males to produce non-diabetic pregnancy (NDP). Day of conception was determined by confirmation of vaginal plugging the following morning. Glucose tolerance test (GTT) was performed at gestational day 16.5 as described below. All mice had free access to chow (energy from fat 8%, from protein 21%, from carbohydrate 71%, 10.6 kJ/g; Gordon’s Specialty Stock Feeds, Yanderra, NSW, Australia). Two separate adult offspring cohorts of six βArnt and six floxed-control females were set up for each cohort to ensure feasibility, starting ~8 weeks apart and the results are combined. After offspring obesity was found in the cohorts indicated in Fig. 1b, a third cohort (Fig. 1c) was started and killed at 6 weeks. The aim for this cohort was to look for changes occurring prior to obesity development, which might therefore be causal rather than secondary to obesity.
Fig. 1.
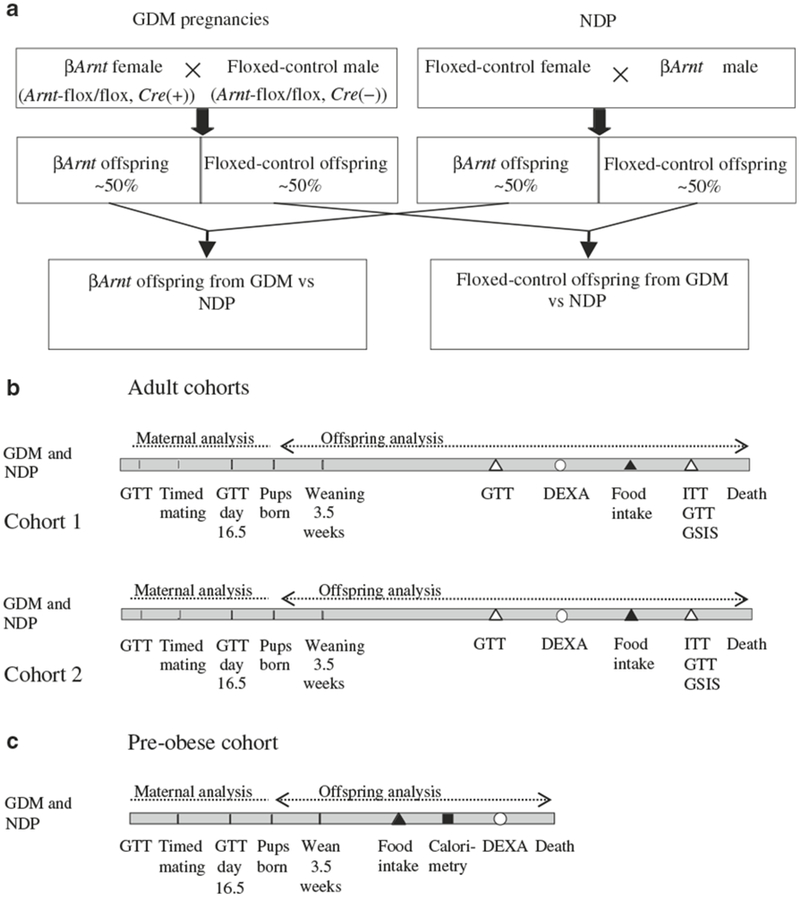
Experimental plans. a Breeding plan for βArnt (from GDM pregnancies) and floxed-control (from control pregnancies) females. b Timeline for testing of mothers and their offspring (adult cohorts). All male offspring were weighed at weaning and every 7 to 10 days thereafter. Glucose metabolic testing is denoted by white triangles: GTT was performed at 8 and 16 weeks, ITT at 15 weeks and GSIS test at 17 weeks of age. DEXA scanning (white circles) was performed at 12 weeks. Food intake (black triangles) was measured at 15 weeks. Physiological tests were spaced in order to minimise mouse stress and allow recovery between studies, with 1 week between ITT, GTT and GSIS at 15, 16 and 17 weeks of age. Offspring were studied in two separate cohorts, about 8 weeks apart, and results were pooled. Results were similar in the two cohorts. Mice were killed at least 3 weeks after the 17 week GSIS. c Timeline for testing of early male offspring phenotype (6 week pre-obese offspring cohort). Offspring were derived from 15 GDM pregnancies and ten non-diabetic pregnancies. Food intake (black triangle) was measured at 5.5 weeks. Half of the mice were killed at 6 weeks of age for collection of plasma and brains for in situ hybridisation. The rest underwent indirect calorimetry (black square) and DEXA scanning (white circle) before killing
Offspring
Litters with fewer than five pups were excluded from analysis, with three litters being excluded each time. Large litters were reduced to seven pups on day 1.5. Pups were weighed at 1.5 days of age and returned to their mothers. Weaning was at 25 days, after which male offspring were weighed every 7–10 days until being killed.
Quantification of food intake
Following acclimatisation to single-housing in cages with absorbent towels rather than usual bedding, food consumption and faecal output were measured daily for 3 consecutive days.
Indirect calorimetry
Calorimetry was performed for 24 h after 24 h acclimatisation (Oxymax Respirometer 0246-002 M; Columbus Instruments, Columbus, OH, USA), as described previously [20]. Free access to food and water was provided. Ambulatory activity was measured using an activity meter (Opto-M3; Columbus Instruments) [20]. RER was calculated as oxygen consumption (O2)/carbon dioxide production (CO2)[21]. Energy expenditure (kJ heat produced) was calculated as calorific value (CV) × O2, where CV=15.966+5.156×RER. O2 and CO2 are expressed corrected for lean body weight, which was measured by dual energy X-ray absorptiometry (DEXA). DEXA was performed within 48 h of calorimetry using a Lunar PIXImus2 (GE Healthcare, Port Washington, NY, USA) as previously reported [20].
Glucose and insulin tolerance tests
Tests were performed after overnight 16 h fasts using a glucometer (Accucheck; Roche, Sydney, NSW, Australia). GTTs employed 2 g/kg of i.p. glucose as previously reported [18]. Glucose-stimulated insulin secretion (GSIS) was measured following administration of 3 g/kg i.p. glucose [18, 22]. Insulin tolerance tests (ITT) used 0.5 units/kg insulin i.p. as previously reported [23].
Tissue and blood collection
Animals were killed after a 16 h fast. Blood was collected. Brains were frozen on dry ice and stored at −70°C. Epigonadal fat was collected and snap frozen in liquid nitrogen.
Assays
Serum insulin and leptin were assayed by ELISA (Crystal Chem, Downers Grove, IL, USA) as per manufacturer’s instructions. Fasting triacylglycerol was assayed using a colorimetric assay (Roche).
Gene expression
Tissue was homogenised in lysis buffer (RLT; Qiagen, Valencia, CA, USA). RNA was extracted as previously described [18]. Real-time PCR was performed using SYBR Green (Applied Biosystems, Foster City, CA, USA) as previously described [18] with a real-time PCR system (ABI 7900HT; Applied Biosystems) (primer sequences available on request).
Brain cryosection and in situ hybridization
After Tissue-Tek (Qiagen) embedding, 20 μm coronal sections of the brain covering the arcuate nucleus were cut. Three sections per mouse (three sections apart) were mounted per slide and stored at −20°C. One slide per mouse was used for each neuropeptide for in situ hybridisation, which was performed as previously described [24]. NIH 1.65 software (http://rsbweb.nih.gov/ij/, accessed 1 December 2009) was used to calculate mean signal density, and slides were dipped in emulsion and counterstained with haematoxylin [24].
Statistical analysis
Two-way ANOVA was used to assess interaction between pregnancy type and offspring genotype. Where interaction was detected, βArnt and control offspring were analysed separately. Characteristics were compared using unpaired t tests unless specified. Bivariate (Spearman) correlations were used. Means ± SEM are shown. Statistical analysis was performed using SPSS 14.0 (SPSS, Chicago, IL, USA). A p value of p<0.05 was considered statistically significant.
Results
βArnt mothers had poorer glucose tolerance in pregnancy
The breeding plan is shown in Fig. 1a. βArnt dams had normal fasting glucose and mildly worse glucose tolerance pre-pregnancy, consistent with our previous report [18]. βArnt dams had normal fertility and litter sizes (6.7±0.5 vs 6.3±0.4 pups per litter in floxed-controls) and remained well during pregnancy. Maternal weight in late pregnancy did not differ significantly (31.9±0.8 vs 30.8±1.1 g, p>0.4).
Fasting glucose did not differ, but glucose levels after glucose loading (GTT) deteriorated significantly at all time points in pregnancy in βArnt mice (p<0.05, t test). In floxed-control mothers, glucose tolerance deteriorated, but not to the same extent. Thus pregnant βArnt females had higher glucose levels than pregnant floxed-control females at all time points except fasting (Fig. 2a). Figure 2b shows total AUCs for GTT (AUCGTT) (p<0.01 for pregnant βArnt vs pregnant floxed-controls).
Fig. 2.
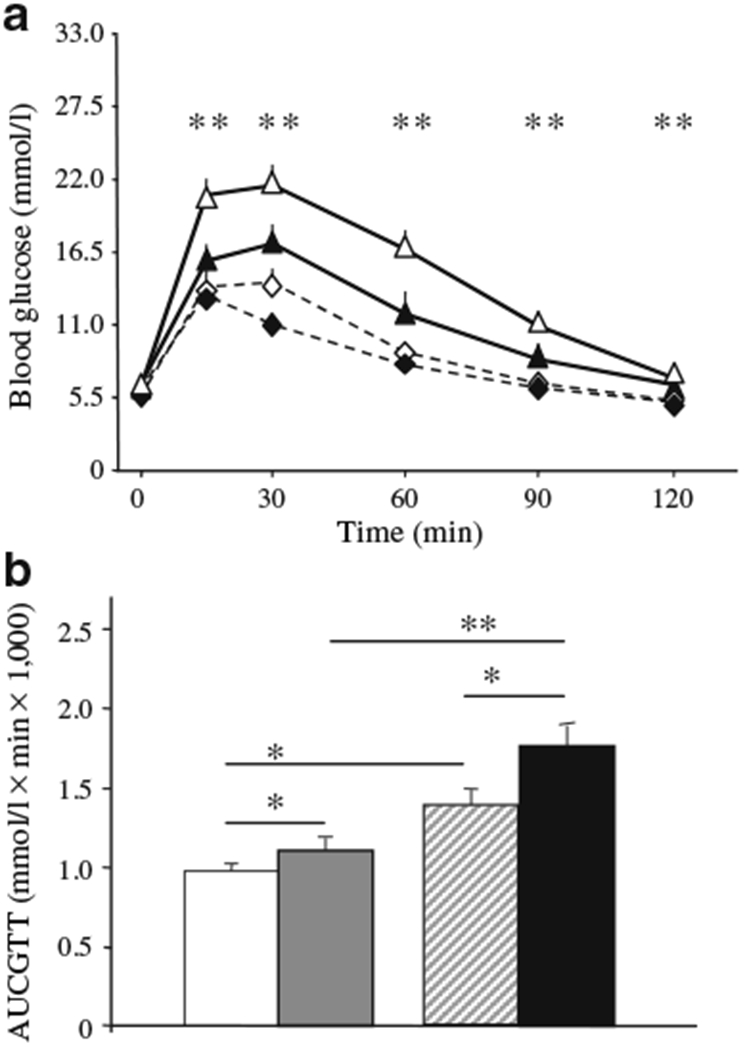
βArnt mice are a model of maternal glucose intolerance in pregnancy. a Pre-gravid and late gestational GTT for βArnt (n=19) and floxed-control (n=19) females. Data presented as means±SEM; **p<0.01 for pregnant βArnt vs pregnant floxed-control. Black diamonds, pre-gravid floxed-control; white diamonds, pregnant floxed-control; black triangles, pre-gravid βArnt; white triangles, pregnant βArnt. b Total AUCGTT. Data presented as means±SEM; *p<0.05 and **p<0.01 for the comparisons shown. White bars, pre-gravid floxed-control; grey bars, pregnant floxed-control; hatched bars, pre-gravid βArnt; black bars, pregnant βArnt
We have previously reported that insulin secretion is impaired in non-gravid βArnt females [18]. Pregnant βArnt females had similar fasting insulin (14.5±3.3 vs 12.4±2.2 pmol/l, p=NS), but during pregnancy first-phase insulin secretion was reduced (increase of 140±57% vs 653±222% in floxed-controls, p<0.05). This is similar to the responses outside of pregnancy. βArnt dams had no differences in circulating fasting triacylglycerol levels (23.2±2.0 vs 29.4±4.0 mmol/l, p=0.17) and there was no correlation between maternal AUCGTT and fasting triacylglycerol.
Maternal GDM resulted in increased offspring weight and adiposity
Similar to human offspring of women with GDM, pups born to GDM mothers were significantly heavier at 1.5 days of age (1.66±0.02 vs 1.57±0.03 g, p=0.02). Pups were not genotyped at this age due to risk of maternal rejection. Instead, genotyping was done at weaning and pups were then placed on a normal-chow diet. Genotyping showed that NDP βArnt offspring were ~10% lighter than GDM ßArnt mice (12.7±0.5 vs 11.6±0.3 g, p=0.034). Floxed-controls were not different (12.3±0.4 vs 12.4±0.2 g). Offspring of GDM gained more weight than NDP offspring (p<0.0001 by ANOVA for repeated measures) resulting in increased body weight from 8 weeks of age (p<0.001 by ANOVA for repeated measures) (Fig. 3a). Interestingly, the increased weight was more pronounced in βArnt GDM offspring than in floxed-control GDM offspring (Fig. 3a) (p<0.01 by ANOVA for repeated measures), showing an interaction between genotype and in utero exposure. There was no difference in weight at any time point between βArnt NDP offspring and floxed-control NDP offspring. Maternal gestational glucose tolerance (AUCGTT) and offspring weight were correlated, with worse maternal glucose tolerance associated with higher offspring weight (Fig. 3b). Results are shown for 15-week-old offspring (p<0.0002), but this correlation was also consistently significant (p<0.05) from 13 through to 19 weeks of age.
Fig. 3.
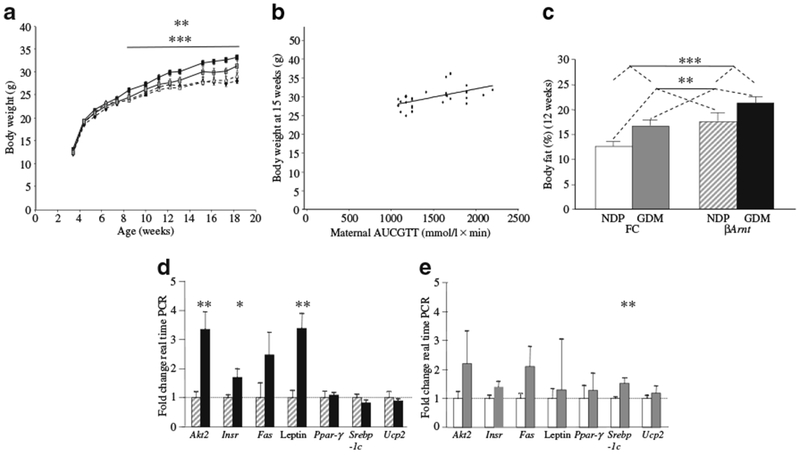
Offspring weight and adiposity were increased in GDM offspring, in proportion to maternal glucose tolerance. a Body weight of floxed-control and βArnt offspring (n=19 floxed-control GDM [grey squares]; n=15 floxed-control NDP [black diamonds]; n=14 βArnt GDM [black rectangles]; n=22 βArnt NDP [white triangles]). **p<0.01 for βArnt GDM offspring vs floxed-control GDM offspring; ***p<0.001 for GDM vs NDP offspring. b The relationship between βArnt offspring body weight at 15 weeks of age and maternal glucose tolerance (total late gestational maternal AUCGTT). r2=0.301, p<0.0005 (Spearman correlation). c Body fat percentage was increased in floxed-control (FC) GDM offspring and FC βArnt offspring at 12 weeks (n=12 FC GDM, n=15 FC NDP, n=9 βArnt GDM, n=15 βArnt NDP). **p<0.01 for GDM vs NDP offspring, ***p<0.001 for βArnt vs FC offspring (ANOVA). d Epigonadal fat gene expression in βArnt GDM (black bars) offspring expressed as percentage of gene expression in NDP βArnt (hatched bars) offspring (n=6 per group); *p<0.05, **p<0.01. e Epigonadal fat gene expression in FC GDM (grey bars) offspring expressed as percentage of gene expression in NDP FC (white bars) offspring (n=6 per group); **p<0.01. Results are corrected for expression of the housekeeping gene Tbp. Data presented as means ± SEM
At 6 weeks of age, 2 weeks before overall weight began to differ in GDM vs NDP offspring, body fat was lower in NDP floxed-controls than in the other three groups (13±1% vs 15±1% in control GDM, 15±1% in βArnt NDP and 15±1% in βArnt GDM offspring, p=0.02). Maternal gestational glucose tolerance (AUCGTT) and offspring body fat at 6 weeks of age were correlated (r2=0.21, p=0.02, Spearman bivariate correlation). Fasting serum leptin levels at 6 weeks were consistent with body fat composition: leptin was similar in βArnt GDM (1.38±0.32 μg/l), βArnt NDP (1.47±0.64 μg/l) and floxed-control GDM (1.25±0.41 μg/l), but was significantly lower in floxed-control NDP (0.37±0.16 μg/l, p<0.01).
Figure 3c shows that by 12 weeks, body fat was 27% greater (95% CI 1.09–1.48-fold) in GDM vs NDP offspring (p=0.002). βArnt offspring had 33% greater body fat than floxed-controls (95% CI 1.14–1.55-fold) (p<0.001, two-way ANOVA). Lean mass contribution was non-significantly lower in GDM than NDP offspring.
Altered gene expression in epigonadal fat
As GDM offspring had greater body fat, we studied the expression of genes involved in lipid accretion and metabolism. At 6 weeks, pre-obesity, there were no significant differences (dns). In βArnt GDM adults, after the onset of obesity, mRNA expression of the following was increased in epigonadal fat: insulin receptor (Insr) (1.70±0.28-fold, p=0.02); Akt2 (3.37±0.56-fold, p<0.01) and leptin (3.39±0.50-fold, p<0.01) (Fig. 3d). In GDM floxed-controls, Srebp-1c (also known as Srebf1) mRNA was increased (1.52±0.18-fold, p=0.01) and there were trends towards an increase in Fas (2.10±0.70-fold, p=0.07) and Insr (1.41±0.17-fold, p=0.08) (Fig. 3e).
Food intake was not increased in GDM offspring
The increased adiposity of GDM offspring was not explained by increased food consumption. Food intake was not increased in GDM offspring and was slightly reduced in floxed-control GDM offspring, perhaps accounting for their less severe obesity phenotype (Fig. 4a). The faeces:food weight ratio was reduced in 15-week-old GDM vs NDP offspring by 0.048 g/g (95% CI 0.018–0.079 g/g) (p=0.003), with the greatest differences observed in βArnt GDM vs βArnt NDP offspring (Fig. 4b). This could be consistent with greater absorption of nutrients or fluid in GDM offspring. Interestingly, faecal output inversely correlated with maternal AUCGTT, suggesting that absorptive capacity may be influenced by in utero exposure (p<0.01, dns).
Fig. 4.
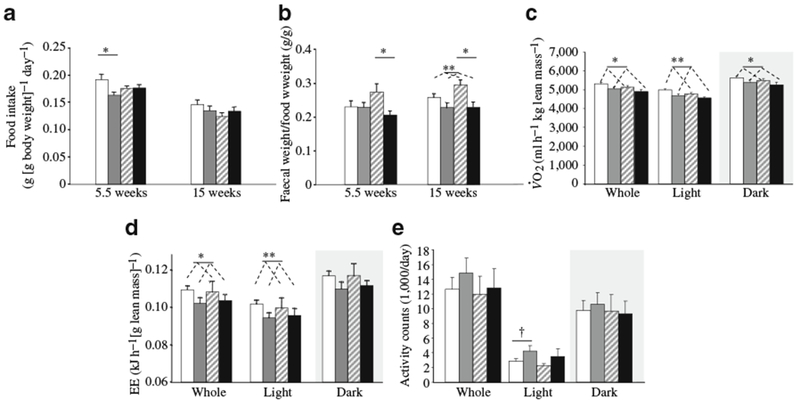
Offspring food intake and metabolic phenotype. a Food intake was not increased in GDM offspring, *p<0.05 (n=10 floxed-control [FC] GDM, n=8 FC NDP, n=10 βArnt GDM, n=6 βArnt NDP at 5.5 weeks; n=10 FC GDM, n=8 FC NDP, n=9 βArnt GDM, n=5 βArnt NDP at 15 weeks). b Faecal weight (wt): food intake ratio, animal numbers as above (a); *p<0.05 (t test) for βArnt GDM vs βArnt NDP, **p<0.01 for GDM vs NDP offspring (ANOVA). c O2 was lower in GDM offspring at 6 weeks of age (n=11 FC GDM, n=15 FC NDP, n=8 βArnt GDM, n=8 βArnt NDP); *p<0.05, **p<0.01 for GDM vs NDP (ANOVA). d Energy expenditure (EE) at 6 weeks of age; *p<0.05, **p<0.01 for GDM vs NDP (ANOVA). e Daily ambulatory activity did not differ; †p=0.08. Data represent means±SEM. White bars, FC NDP; grey bars, FC GDM; hatched bars, βArnt NDP; black bars, βArnt GDM
GDM offspring had decreased metabolic rate, but not decreased locomotion
GDM was associated with significantly decreased O2 at 6 weeks (Fig. 4c). This was associated with significantly reduced calculated energy expenditure for the whole day, and for the light hours during which mice usually rest (Fig. 4d). Ambulatory activity was non-significantly higher in GDM vs NDP offspring, so lower activity did not account for the decrease in energy expenditure (Fig. 4e).
Respiratory exchange ratio was increased in GDM offspring
Increased RER indicates preferential carbohydrate over lipid oxidation, and thus a propensity for lipid storage and increased adiposity [25]. An RER of 1.0 represents 100% carbohydrate oxidation, 0.7 represents 100% lipid oxidation [21]. RER did not differ between βArnt and floxed-control offspring. However, GDM exposure was associated with increased RER during light hours (1.03±0.01 vs 0.99±0.01, p=0.046) (Fig. 5a, b), with failure of the usual daytime switch to lipid oxidation. The fall in RER during light hours was significant, as expected, in NDP offspring (p=0.001, paired t test), but not in GDM offspring (p=0.608). There was a positive correlation between maternal gestational AUCGTT and offspring light hour RER. Thus higher maternal glucose correlated with lack of dip towards lipid oxidation (r2 = 0.23, p<0.01) (Fig. 5c).
Fig. 5.
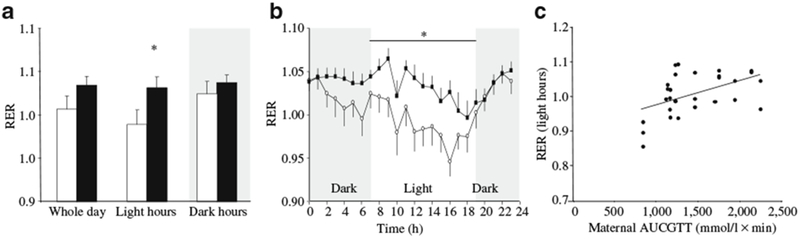
RER was elevated in GDM offspring. a Increased light hours RER in offspring from GDM (black bars) compared with those from NDP (white bars) (n=11 floxed-control [FC] GDM, n=15 FC NDP, n=8 βArnt GDM, n=8 βArnt NDP). b Normal dip in light hours RER in GDM offspring did not occur; *p<0.05 (ANOVA). Data presented as means±SEM. c Correlation between offspring light hours RER and total late gestational maternal AUCGTT. r2=0.23, p<0.01 (Spearman bivariate correlation)
Insulin sensitivity was decreased and insulin secretion pattern impaired in GDM offspring
Consistent with our previous report, βArnt offspring had worse glucose tolerance than floxed-control offspring (p<0.01 at 8 and 16 weeks, two-way ANOVA, 16 week GTT) (Fig. 6a). There was no significant deterioration in glucose tolerance in GDM vs NDP βArnt offspring or in GDM vs NDP floxed-control offspring. Figure 6b shows fasting insulin concentrations in the four offspring groups at 17 weeks. Fasting insulin was significantly greater in GDM vs NDP offspring and also greater in βArnt vs floxed-control offspring. This was consistent with increased weight in GDM offspring.
Fig. 6.
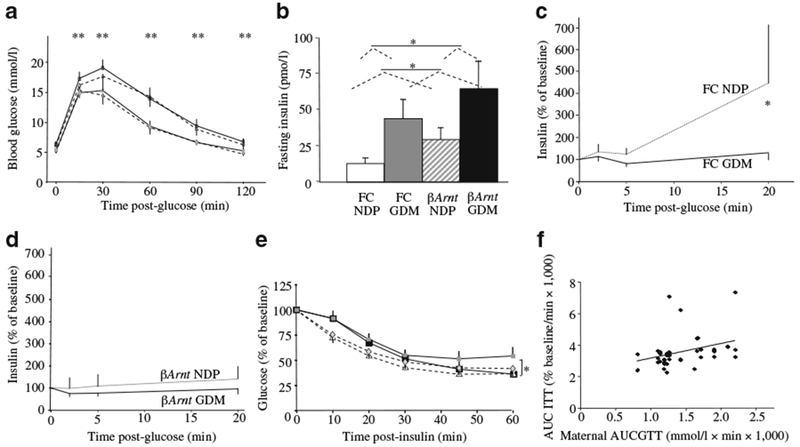
Glucose metabolism in offspring. a GTT at 16 weeks did not differ between GDM and NDP offspring; **p<0.01 for βArnt vs floxed-control (FC) offspring (ANOVA) (n=10 FC GDM, n=16 FC NDP, n=8 βArnt GDM, n=12 βArnt NDP). b Fasting serum insulin at 17 weeks; *p<0.05 (ANOVA) for GDM vs NDP offspring and for βArnt vs FC offspring. c GSIS was impaired in FC GDM (continuous line) vs FC NDP (dotted line) at 17 weeks; *p<0.05 (t test). d GSIS was impaired in all βArnt offspring (dotted line, βArnt NDP; continuous line, βArnt GDM). e ITT at 15 weeks; *p<0.05 for GDM vs NDP offspring (ANOVA). f Correlation between maternal AUCGTT and offspring AUCITT; r2=0.30, p<0.001 (Spearman bivariate correlation). Data represent means±SEM. Key (a, e): grey diamonds, FC NDP; grey squares, FC GDM; white triangles βArnt NDP; black squares, βArnt GDM
Glucose-stimulated insulin secretion was abnormal in floxed-controls from GDM vs those from NDP, with impaired second-phase insulin release at 17 weeks (Fig. 6c) (130±28 vs 447±363% of baseline, p=0.03, t test), although the residual secretion was sufficient to control glucose tolerance. In βArnt mice, GSIS was impaired, consistent with our previous report (Fig. 6d), but there was no further deterioration in GDM βArnt offspring. Consistent with fasting insulin values, GDM offspring at 15 weeks had mildly decreased insulin sensitivity (Fig. 6e). As shown in Fig. 6f, insulin resistance AUC (AUCITT) correlated with maternal glycaemia AUCGTT (higher maternal AUCGTT, higher offspring AUCITT, r2=0.30, p<0.001).
Altered arcuate neuropeptide expression in βArnt GDM offspring
We measured expression of Npy and Pomc mRNA in the arcuate nucleus in 6-week-old offspring using in situ hybridisation. Signal intensity was expressed as a percentage of floxed-control NDP offspring.
There was no difference between floxed-control and βArnt mice from NDP with respect to Npy or Pomc expression. However, in βArnt, but not in floxed-control mice, GDM caused a significant increase in Npy expression and a significant decrease in Pomc expression (Fig. 7a, b).
Fig. 7.
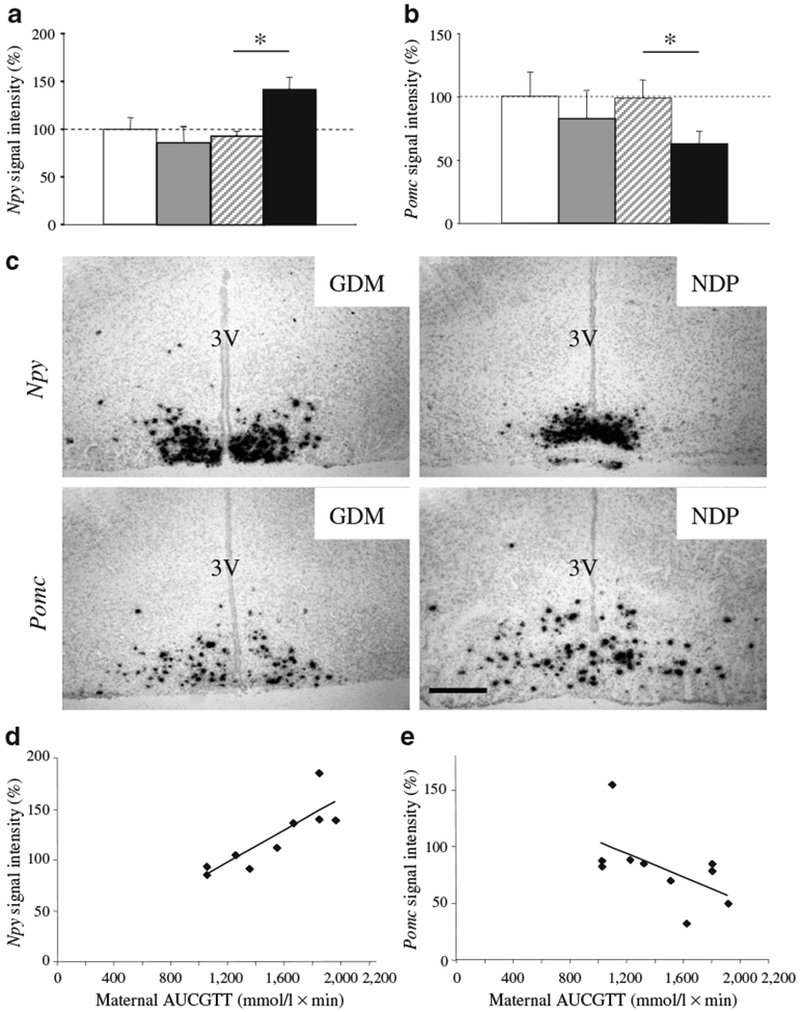
Hypothalamic gene expression in offspring. Arcuate hypothalamic expression of (a) Npy and (b) Pomc by in-situ hybridisation. Signal intensity is expressed as a percentage of NDP floxed-control offspring; n=5 per group; *p<0.05 (t test). Bars represent mean±SEM. White bars, floxed-control NDP; grey bars, floxed-control GDM; hatched bars, βArnt NDP; black bars, βArnt GDM. c Representative images of Npy and Pomc in situ hybridisation in βArnt offspring from GDM vs NDP. 3 V, third ventricle. Scale bar, 25 μm. d Correlation between total maternal AUCGTT and hypothalamic Npy expression in βArnt offspring; r2=0.78, p=0.002 (Spearman bivariate correlation). e Correlation between maternal AUCGTT and Pomc expression in βArnt offspring; r2=0.40, p=0.049
Representative images are shown in Fig. 7c. In βArnt mice, maternal gestational glucose tolerance correlated positively with Npy expression (r2=0.78, p=0.002) (Fig. 7d) and negatively with Pomc expression (r2=0.40, p=0.049).
Discussion
In this mouse model of GDM secondary to beta cell dysfunction, we demonstrated an interaction between GDM exposure and offspring genetic susceptibility to beta cell dysfunction in causing male offspring obesity on a normal-chow diet. Table 1 summarises the phenotypic differences in offspring from GDM vs NDP. While both GDM offspring groups developed obesity, βArnt offspring were more severely affected, such that mice inheriting a predisposition to glucose intolerance were more susceptible to obesogenic effects of GDM than control-genotype mice.
Table 1.
The effects of GDM and offspring genotype on offspring phenotype
| Variable |
Offspring per mother and pregnancy type |
|||
|---|---|---|---|---|
| Floxed-control (NDP) |
βArnt (GDM pregnancy) |
|||
| Floxed-control NDP | βArnt NDP | Floxed-control GDM | βArnt GDM | |
| Body weight | Normala | Increaseda | Normala | Further increaseda |
| Body fat (%) | Normala | Increaseda | Increaseda | Further increaseda |
| Fasting insulin | Normal | Increased | Normal | Increased |
| Food intake | Normal | Normal or decreased | Normal | Normal |
| Energy expenditure | Normal | Decreased | Normal | Decreased |
| Locomotion | Normal | Normal | Normal | Normal |
| RER | Normala | Increaseda | Normala | Increaseda |
| O2 | Normal | Decreased | Normal | Decreased |
| Arcuate nucleus | ||||
| Npy | Normal | Normal | Normala | Increased a |
| Pomc | Normal | Normal | Normala | Decreased a |
Correlated with maternal AUCGTT
Aspects of this model are relevant to human GDM. Thus the glucose intolerance is secondary to impaired beta cell function, with mice having worsening glycaemia in late gestation with preserved normal fasting glucose. Beta cell gene polymorphisms have been linked to type 2 diabetes and GDM in humans; moreover, the gene we studied was decreased in islets from three women with a known history of GDM [18]. Another model that has examined interactions between maternal diabetes and offspring diabetes is the GK rat. GK rats constitute a model of type 2 diabetes secondary to reduced beta cell mass and function. Transfer of GK embryos to Wistar dams did not improve offspring glycaemia, showing that exposure to one generation of normal glucose in utero did not change the GK phenotype [11]. That study did not examine GK rats with no ancestral exposure to GDM in prior generations.
Human studies also demonstrate genotype×pregnancy interactions for offspring phenotype [26, 27]. People with HNF1A MODY mutations were diagnosed with diabetes earlier when their mothers had had diabetes prior to rather, than after their birth [26]. A potential synergism between maternal glucose intolerance and offspring genes is also likely to be important in the normal wider population, where offspring exposed to GDM will often inherit diabetes susceptibility genes.
Exactly how GDM and diabetes in pregnancy increase the risk of offspring obesity is not well understood. In this study, male offspring developed spontaneous obesity on normal chow. A separate cohort of offspring was studied prior to development of the obese phenotype, demonstrating decreased metabolic rate and, in βArnt mice, changes in hypothalamic Npy and Pomc expression prior to obesity. Obesity was preceded by reduced energy expenditure, rather than increased food consumption or decreased locomotion, suggesting an intrinsic metabolic defect. This may be a central defect, as Npy expression was increased and Pomc expression decreased in the arcuate nucleus of βArnt GDM offspring, despite similar body weight, per cent body fat and leptin levels. Floxed-control GDM offspring had similar neuropeptide expression compared with their NDP counterparts. The overall findings suggest central dysregulation.
These data are consistent with previous studies suggesting that central regulation of body weight can be programmed in early life. Thus an inverse correlation between fat mass and leptin receptor expression was observed, with loss of the direct correlation between fat mass and cocaine- and amphetamine-regulated transcript (CART) levels in 30-day-old offspring of overfed ewes [28]. Obese male offspring of rats with streptozotocin-induced diabetes in pregnancy have increased neuropeptide Y (NPY) neuron numbers [29]. Pro-opiomelanocortin- and CART-secreting neurons were not examined in that study.
Respiratory exchange ratio has not been previously reported in offspring of diabetic and GDM pregnancies. In humans, increased RER or respiratory quotient is associated with obesity and decreased lipid oxidation capacity predicts weight gain [30, 31]. Thomas et al. showed that obese individuals could not increase the oxidation of lipids as dietary fat intake increased [32]. In normal mice, RER decreases during the daytime when animals eat less and switch from carbohydrate to lipid oxidation. GDM offspring did not demonstrate a reduction in light hours RER, thus sustaining a preference for carbohydrate over lipid oxidation over the entire 24 h period. These changes may partly explain their increased adiposity and would be consistent with increased expression of lipogenic and insulin-signalling genes in epigonadal fat (favouring fat accretion). Preferential fuel oxidation is also centrally regulated [33]. The lack of normal circadian pattern in the GDM offspring was interesting. Evidence suggests that the adult sleep–wake cycle can be programmed by maternal stress [34]. This work suggests the possibility that in utero GDM exposure might affect programming of the adult circadian metabolic clock and future studies in this area will be of interest.
GDM offspring had decreased faecal output:food intake ratio, suggesting increased capacity for nutrient or fluid absorption. Recent human and animal studies linking gut nutrient absorption capacity with obesity suggest that this finding may contribute to an obese phenotype [35, 36]. Centrally administered NPY inhibits gastric emptying and intestinal transit [37]. Thus increased Npy expression may play a role in the decreased faeces:food weight ratio of GDM offspring. Faeces:food ratio is an approximate measure only, and further studies of the effects of GDM exposure on gastrointestinal function could produce interesting results.
An obvious limitation of this study is that offspring were not cross-fostered. Thus this model did not separate the effects of the intrauterine environment from the early-postnatal environment. GDM mothers may have altered milk composition or subtle differences in the care of their offspring, which (if present) could impact on adult offspring phenotype. Future studies determining whether the effects are diminished by cross-fostering to normal mothers would be of great interest. The offspring underwent multiple metabolic tests (ITT, GTT and GSIS); however, the tests were applied to all groups and mice gained weight at expected trajectories.
In summary, this model of GDM resulted in spontaneous offspring obesity associated with decreased energy expenditure prior to the onset of increased body weight, without increased energy consumption or decreased locomotion. Obesity was preceded by hypothalamic dysregulation.
Human trials demonstrate that treating GDM reduces perinatal complication rates [38], with a linear relationship between maternal glycaemia and short-term offspring outcomes including birthweight [39]. We demonstrated a direct correlation between severity of maternal glucose intolerance and several measures of adult offspring phenotype, including adult offspring weight, body fat, insulin sensitivity and RER, as well as, in βArnt offspring, hypothalamic Npy and Pomc expression. There was no correlation between offspring phenotype and any other examined variables, including maternal weight, plasma triacylglycerol or pre-pregnancy glycaemia, suggesting that glucose was indeed the factor ‘programming’ offspring phenotype.
These data suggest that the dose–response relationship between maternal glycaemia and short-term outcomes in offspring may also exist for longer term outcomes such as adult obesity. Taken together, these novel findings further our understanding of the pathogenesis of obesity in offspring exposed to GDM and provide evidence supporting tight control of maternal hyperglycaemia in humans with a view to avoiding long-term adverse outcomes in their offspring.
Acknowledgements
The authors would like to acknowledge K. Byth (Westmead Hospital) for her assistance with statistics, and A. Dwyer (Concord Hospital, Sydney, NSW, Australia), C. Nolan (Australian National University, Canberra, ACT, Australia) and D. Chisholm (Garvan Institute) for helpful comments on the manuscript. S. M. Lau received salary support from the National Heart Foundation. J. E. Gunton received an NHMRC/DART fellowship and a L’Oreal Australian Women in Science Fellowship. P. A. Baldock, H. Herzog and A. Sainsbury received NHMRC fellowships. The work was partially supported by funds from DART, Endocrine Society of Australia and the Australasian Society for Diabetes in Pregnancy.
Abbreviations
- AUCGTT
AUC for GTT
- AUCITT
AUC for ITT
- βArnt
Beta cell-specific aryl hydrocarbon receptor nuclear translocator null mice
- db−/+
Leptin receptor heterozygous null mice
- DEXA
Dual energy X-ray absorptiometry
- dns
Data not shown
- GDM
Gestational diabetes mellitus
- GK
Goto–Kakizaki
- GSIS
Glucose-stimulated insulin secretion
- GTT
Glucose tolerance test
- ITT
Insulin tolerance test
- NDP
Non-diabetic pregnancy
- NPY
Neuropeptide Y
- RER
Respiratory exchange ratio
- RIP
Rat insulin promoter
- CO2
Carbon dioxide production
- O2
Oxygen consumption
Footnotes
Duality of interest The authors declare that there is no duality of interest associated with this manuscript.
Contributor Information
S. M. Lau, Diabetes and Transcription Factors Group, Garvan Institute of Medical Research, 384 Victoria St, Darlinghurst, NSW, Australia; Department of Diabetes and Endocrinology, Westmead Hospital, Sydney, NSW, Australia; St Vincent’s Clinical School, University of New South Wales, Sydney, NSW, Australia; Faculty of Medicine, University of Sydney, Sydney, NSW, Australia
S. Lin, Neuroscience Program, Garvan Institute of Medical Research, Sydney, NSW, Australia
R. A. Stokes, Diabetes and Transcription Factors Group, Garvan Institute of Medical Research, 384 Victoria St, Darlinghurst, NSW, Australia
K. Cheng, Diabetes and Transcription Factors Group, Garvan Institute of Medical Research, 384 Victoria St, Darlinghurst, NSW, Australia
P. A. Baldock, Bone and Mineral Research Program, Garvan Institute of Medical Research, Sydney, NSW, Australia
R. F. Enriquez, Bone and Mineral Research Program, Garvan Institute of Medical Research, Sydney, NSW, Australia
M. McLean, Centre for Diabetes and Endocrinology Research, Westmead Hospital, Sydney, NSW, Australia
N. W. Cheung, Centre for Diabetes and Endocrinology Research, Westmead Hospital, Sydney, NSW, Australia
A. Sainsbury, Neuroscience Program, Garvan Institute of Medical Research, Sydney, NSW, Australia; School of Medical Sciences, University of New South Wales, Sydney, NSW, Australia
F. J. Gonzalez, Laboratory of Metabolism, National Institutes of Health, Bethesda, MD, USA
H. Herzog, Neuroscience Program, Garvan Institute of Medical Research, Sydney, NSW, Australia
J. E. Gunton, Diabetes and Transcription Factors Group, Garvan Institute of Medical Research, 384 Victoria St, Darlinghurst, NSW, Australia; Department of Diabetes and Endocrinology, Westmead Hospital, Sydney, NSW, Australia; St Vincent’s Clinical School, University of New South Wales, Sydney, NSW, Australia; Faculty of Medicine, University of Sydney, Sydney, NSW, Australia
References
- 1.Plagemann A, Harder T, Kohlhoff R, Rohde W, Dorner G (1997) Overweight and obesity in infants of mothers with long-term insulin-dependent diabetes or gestational diabetes. Int J Obes Relat Metab Disord 21:451–456 [DOI] [PubMed] [Google Scholar]
- 2.Plagemann A, Harder T, Kohlhoff R, Rohde W, Dorner G (1997) Glucose tolerance and insulin secretion in children of mothers with pregestational IDDM or gestational diabetes. Diabetologia 40:1094–1100 [DOI] [PubMed] [Google Scholar]
- 3.Silverman BL, Metzger BE, Cho NH, Loeb CA (1995) Impaired glucose tolerance in adolescent offspring of diabetic mothers. Relationship to fetal hyperinsulinism. Diab Care 18:611–617 [DOI] [PubMed] [Google Scholar]
- 4.Silverman BL, Rizzo TA, Cho NH, Metzger BE (1998) Long-term effects of the intrauterine environment. The Northwestern University Diabetes in Pregnancy Center. Diab Care 21(Suppl 2):B142–B149 [PubMed] [Google Scholar]
- 5.Pettitt DJ, Knowler WC, Bennett PH, Aleck KA, Baird HR (1987) Obesity in offspring of diabetic Pima Indian women despite normal birth weight. Diab Care 10:76–80 [DOI] [PubMed] [Google Scholar]
- 6.Silverman BL, Rizzo T, Green OC et al. (1991) Long-term prospective evaluation of offspring of diabetic mothers. Diabetes 40(Suppl 2):121–125 [DOI] [PubMed] [Google Scholar]
- 7.Dabelea D, Hanson RL, Lindsay RS et al. (2000) Intrauterine exposure to diabetes conveys risks for type 2 diabetes and obesity: a study of discordant sibships. Diabetes 49:2208–2211 [DOI] [PubMed] [Google Scholar]
- 8.Homko C, Sivan E, Chen X, Reece EA, Boden G (2001) Insulin secretion during and after pregnancy in patients with gestational diabetes mellitus. J Clin Endocrinol Metab 86:568–573 [DOI] [PubMed] [Google Scholar]
- 9.Xiang AH, Peters RK, Trigo E, Kjos SL, Lee WP, Buchanan TA (1999) Multiple metabolic defects during late pregnancy in women at high risk for type 2 diabetes. Diabetes 48:848–854 [DOI] [PubMed] [Google Scholar]
- 10.Lambin S, van Bree R, Caluwaerts S, Vercruysse L, Vergote I, Verhaeghe J (2007) Adipose tissue in offspring of Leprdb/+ mice: early-life environment vs genotype. Am J Physiol Endocrinol Metab 292:E262–E271 [DOI] [PubMed] [Google Scholar]
- 11.Gill-Randall R, Adams D, Ollerton RL, Lewis M, Alcolado JC (2004) Type 2 diabetes mellitus—genes or intrauterine environment? An embryo transfer paradigm in rats. Diabetologia 47:1354–1359 [DOI] [PubMed] [Google Scholar]
- 12.Holemans K, Caluwaerts S, Poston L, Van Assche FA (2004) Diet-induced obesity in the rat: a model for gestational diabetes mellitus. Am J Obstet Gynecol 190:858–865 [DOI] [PubMed] [Google Scholar]
- 13.Weng J, Ekelund M, Lehto M et al. (2002) Screening for MODY mutations, GAD antibodies, and type 1 diabetes associated HLA genotypes in women with gestational diabetes mellitus. Diab Care 25:68–71 [DOI] [PubMed] [Google Scholar]
- 14.Shaat N, Karlsson E, Lernmark Å et al. (2006) Common variants in MODY genes increase the risk of gestational diabetes mellitus. Diabetologia 49:1545–1551 [DOI] [PubMed] [Google Scholar]
- 15.Shaat N, Ekelund M, Lernmark Å et al. (2005) Association of the E23K polymorphism in the KCNJ11 gene with gestational diabetes mellitus. Diabetologia 48:2544–2551 [DOI] [PubMed] [Google Scholar]
- 16.Shaat N, Lernmark Å, Karlsson E et al. (2007) A variant in the transcription factor 7-like 2 (TCF7L2) gene is associated with an increased risk of gestational diabetes mellitus. Diabetologia 50:972–979 [DOI] [PubMed] [Google Scholar]
- 17.Freinkel N (1980) Banting Lecture 1980. Of pregnancy and progeny. Diabetes 29:1023–1035 [DOI] [PubMed] [Google Scholar]
- 18.Gunton JE, Kulkarni RN, Yim S et al. (2005) Loss of ARNT/HIF1beta mediates altered gene expression and pancreatic-islet dysfunction in human type 2 diabetes. Cell 122:337–349 [DOI] [PubMed] [Google Scholar]
- 19.Tomita S, Sinal CJ, Yim SH, Gonzalez FJ (2000) Conditional disruption of the aryl hydrocarbon receptor nuclear translocator (Arnt) gene leads to loss of target gene induction by the aryl hydrocarbon receptor and hypoxia-inducible factor 1alpha. Mol Endocrinol 14:1674–1681 [DOI] [PubMed] [Google Scholar]
- 20.Zhang L, Macia L, Turner N et al. (2009) Peripheral neuropeptide Y Y1 receptors regulate lipid oxidation and fat accretion. Int J Obes (London) 34:357–373 [DOI] [PubMed] [Google Scholar]
- 21.Ferrannini E (1988) The theoretical bases of indirect calorimetry: a review. Metabolism 37:287–301 [DOI] [PubMed] [Google Scholar]
- 22.Kulkarni RN, Bruning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR (1999) Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 96:329–339 [DOI] [PubMed] [Google Scholar]
- 23.Cheng K, Ho K, Stokes R et al. (2010) Hypoxia-inducible factor-1 α regulates β-cell function in mouse and human islets. J Clin Investig 120:2171–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johnen H, Lin S, Kuffner T et al. (2007) Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat Med 13:1333–1340 [DOI] [PubMed] [Google Scholar]
- 25.Seidell JC, Muller DC, Sorkin JD, Andres R (1992) Fasting respiratory exchange ratio and resting metabolic rate as predictors of weight gain: the Baltimore Longitudinal Study on Aging. Int J Obes Relat Metab Disord 16:667–674 [PubMed] [Google Scholar]
- 26.Stride A, Shepherd M, Frayling TM, Bulman MP, Ellard S, Hattersley AT (2002) Intrauterine hyperglycemia is associated with an earlier diagnosis of diabetes in HNF-1alpha gene mutation carriers. Diab Care 25:2287–2291 [DOI] [PubMed] [Google Scholar]
- 27.Jaquet D, Tregouet DA, Godefroy T et al. (2002) Combined effects of genetic and environmental factors on insulin resistance associated with reduced fetal growth. Diabetes 51:3473–3478 [DOI] [PubMed] [Google Scholar]
- 28.Muhlhausler BS, Adam CL, Findlay PA, Duffield JA, McMillen IC (2006) Increased maternal nutrition alters development of the appetite-regulating network in the brain. FASEB J 20:1257–1259 [DOI] [PubMed] [Google Scholar]
- 29.Plagemann A, Harder T, Melchior K, Rake A, Rohde W, Dorner G (1999) Elevation of hypothalamic neuropeptide Y-neurons in adult offspring of diabetic mother rats. NeuroReport 10:3211–3216 [DOI] [PubMed] [Google Scholar]
- 30.Marra M, Scalfi L, Contaldo F, Pasanisi F (2004) Fasting respiratory quotient as a predictor of long-term weight changes in non-obese women. Ann Nutr Metab 48:189–192 [DOI] [PubMed] [Google Scholar]
- 31.Zurlo F, Lillioja S, Esposito-Del Puente A et al. (1990) Low ratio of fat to carbohydrate oxidation as predictor of weight gain: study of 24-h RQ. Am J Physiol 259:E650–E657 [DOI] [PubMed] [Google Scholar]
- 32.Thomas CD, Peters JC, Reed GW, Abumrad NN, Sun M, Hill JO (1992) Nutrient balance and energy expenditure during ad libitum feeding of high-fat and high-carbohydrate diets in humans. Am J Clin Nutr 55:934–942 [DOI] [PubMed] [Google Scholar]
- 33.Sainsbury A, Cooney GJ, Herzog H (2002) Hypothalamic regulation of energy homeostasis. Best Pract Res Clin Endocrinol Metab 16:623–637 [DOI] [PubMed] [Google Scholar]
- 34.Kennaway DJ (2002) Programming of the fetal suprachiasmatic nucleus and subsequent adult rhythmicity. Trends Endocrinol Metab 13:398–402 [DOI] [PubMed] [Google Scholar]
- 35.Gill SR, Pop M, Deboy RT et al. (2006) Metagenomic analysis of the human distal gut microbiome. Science 312:1355–1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Turnbaugh PJ, Hamady M, Yatsunenko T et al. (2009) A core gut microbiome in obese and lean twins. Nature 457:480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuda M, Aono M, Moriga M, Okuma M (1991) Centrally administered NPY stimulated gastric acid and pepsin secretion by a vagally mediated mechanism. Regul Pept 35:31–41 [DOI] [PubMed] [Google Scholar]
- 38.Crowther CA, Hiller JE, Moss JR, McPhee AJ, Jeffries WS, Robinson JS (2005) The effect of gestational diabetes mellitus on pregnancy outcomes. N Engl J Med 352:2477–2486 [DOI] [PubMed] [Google Scholar]
- 39.HAPO-Study Cooperative Research Group (2008) Hyperglycemia and adverse pregnancy outcomes. N Engl J Med 358:1991–2002 [DOI] [PubMed] [Google Scholar]