

Abstract
Standard cytotoxicity assays, which require the collection of lysates or fixed cells at multiple time points, have limited sensitivity and capacity to assess factors that influence neuronal fate. These assays require the observation of separate populations of cells at discrete time points. As a result, individual cells cannot be followed prospectively over time, severely limiting the ability to discriminate whether subcellular events, such as puncta formation or protein mislocalization, are pathogenic drivers of disease, homeostatic responses, or merely coincidental phenomena. Single-cell longitudinal microscopy overcomes these limitations, allowing the researcher to determine differences in survival between populations and draw causal relationships with enhanced sensitivity. This video guide will outline a representative workflow for experiments measuring single-cell survival of rat primary cortical neurons expressing a fluorescent protein marker. The viewer will learn how to achieve high-efficiency transfections, collect and process images enabling the prospective tracking of individual cells, and compare the relative survival of neuronal populations using Cox proportional hazards analysis.
Keywords: Neuroscience, Issue 143, Cell death, neurodegeneration, fluorescence microscopy, automation, transfection, survival analysis
Introduction
Abnormal cell death is a driving factor in many diseases, including cancer, neurodegeneration, and stroke1. Robust and sensitive assays for cell death are essential to the characterization of these disorders, as well as the development of therapeutic strategies for extending or reducing cellular survival. There are currently dozens of techniques for measuring cell death, either directly or through surrogate markers2. For example, cell death can be assessed visually with the help of vital dyes that selectively stain dead or living cells3, or by monitoring the appearance of specific phospholipids on the plasma membrane4,5,6. Measurements of intracellular components or cellular metabolites released into the media upon cellular dissolution can also be used as proxies for cell death7,8. Alternatively, cellular viability can be approximated by assessing metabolic activity9,10. Though these methods provide rapid means of assessing cell survival, they are not without caveats. Each technique observes the culture as a single population, rendering it impossible to distinguish between individual cells and their unique rates of survival. Furthermore, such population-based assays are unable to measure factors that may be important for cell death, including cellular morphology, protein expression, or localization. In many cases, these assays are limited to discrete time points, and do not allow for the continuous observation of cells over time.
In contrast, longitudinal fluorescence microscopy is a highly flexible methodology that directly and continuously monitors the risk of death on a single-cell basis11. In brief, longitudinal fluorescence microscopy enables thousands of individual cells to be tracked at regular intervals for extended periods of time, allowing precise determinations of cell death and the factors that enhance or suppress cell death. At its base, the method involves the transient transfection or transduction of cells with vectors encoding fluorescent proteins. A unique fiduciary is then established, and the position of each transfected cell in relation to this landmark allows the user to image and track individual cells over the course of hours, days, or weeks. When these images are viewed sequentially, cell death is marked by characteristic changes in fluorescence, morphology, and fragmentation of the cell body, enabling the assignment of a time of death for each cell. The calculated rate of death, determined by the hazard function, can then be quantitatively compared between conditions, or related to select cellular characteristics using univariate or multivariate Cox proportional hazards analysis1. Together, these approaches enable the accurate and objective discrimination of rates of cell death among cellular populations, and the identification of variables that significantly predict cell death and/or survival (Figure 1).
Figure 1: Schema for a typical survival experiment.
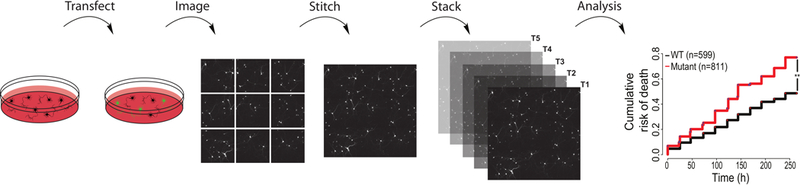
Rat cortical neurons are transfected at DIV4 using the procedure outlined in this article. Beginning 24 h post-transfection, cells are imaged at regularly spaced intervals in accordance with the specific requirements of the experiment. Images are stitched and stacked before cell death is scored, and Cox proportional hazard analysis is used to compare the risk of death between populations.
Although this method can be used to monitor survival in any post-mitotic cell type in a variety of plating formats, this protocol will describe conditions for transfecting and imaging rat cortical neurons cultured in a 96-well plate.
Protocol
All vertebrate animal work was approved by the Committee on the Use and Care of Animals at the University of Michigan (protocol # PRO00007096). Experiments are carefully planned to minimize the number of animals sacrificed. Pregnant female wild-type (WT), non-transgenic Long Evans rats (Rattus norvegicus) are housed singly in chambers equipped with environmental enrichment, and cared for by the Unit for Laboratory Animal Medicine (ULAM) at the University of Michigan, in accordance with the NIH-supported Guide for the Care and Use of Laboratory Animals. All rats were kept in routine housing for as little time as possible prior to euthanasia, consistent with the recommendations of the Guidelines on Euthanasia of the American Veterinary Medical Association and the University of Michigan Methods of Euthanasia by Species Guidelines.
1. Material Preparation
Dissect cortical neurons from embryonic day 19–20 rat pups and culture rat cortical neurons at 0.5 × 106 cells per milliliter on poly-D-lysine coated plates for 4 days in vitro, as described previously13,14,15,16,17,18,19
Prepare the plasmid DNA of interest following the steps outlined by an endotoxin-free plasmid DNA isolation kit (see Table of Materials). Quantify the resultant DNA using a spectrophotometer.
-
On in vitro day 4 (DIV4), aliquot, filter sterilize, and incubate the following media at 37 °C: 6 mL reduced serum media (RSM; e.g., OptiMEM), 25 mL neuronal basal media (NBM), 40 mL NBKY (NBM + 1 mM kynurenic acid + 10 mM MgCl2, adjusted to a pH of 7.4), 10 mL NBC (NBM + 1× neuronal cell culture supplement + 1× L-glutamine supplement + 1× Pen Strep).
NOTE: Volumes listed are sufficient for transfecting one 96-well plate. Refer to the Table of Materials for specific reagents.
2. Transfection of Rat Cortical Neurons
-
Modify the provided Example transfection sheet (see Supplemental File 1) by adjusting the plate type, plate map, number of DNAs, DNA concentration, and number of wells (green boxes).
NOTE: The total DNA sums to 0.2 μg per well, regardless of whether one (e.g., DNA A) or multiple (e.g., DNA B and C) DNA constructs are added to each well.
Working from the spreadsheet, combine the appropriate amount of RSM and DNA in one tube. Combine the appropriate amount of RSM and transfection reagent (e.g., Lipofectamine) in a separate tube.
Incubate at room temperature (RT) for 5 min.
Combine the DNA and transfection reagent RSM mixtures and incubate at RT for 20 min.
During this incubation step, use a multichannel pipette and sterile plastic troughs to wash cells 2× with 100 μL per well of NBM. Reserve the conditioned media (CM) and store at 37 °C. For this and following steps, take care to minimize the amount of time neurons are exposed to air.
Remove the NBM media and replace with 100 μL per well of NBKY.
After 20 min have passed, add 50 μL of the transfection reagent/DNA mixture dropwise to each well.
Incubate cells with the transfection reagent/DNA complexes for 20 min at 37 °C.
Rinse 2x with NBKY and replace with 100 μL of CM and 100 μL of NBC per well.
-
Successfully transfected cells should be visible by fluorescence microscopy within 16–24 h of transfection. To gauge efficiency, use a fluorescent microscope to check the transfection after overnight incubation at 37 °C.
NOTE: This technique results in an overall transfection efficiency of 5 to 10%.
3. Imaging
Place the plate on a fluorescent microscope with a motorized stage, and establish a fiduciary (e.g., a mark on the bottom of the plate) that will allow the user to align the plate each time it is imaged. Save an image of this fiduciary for reference.
Navigate to a field of interest and note the x-y coordinates relative to the fiduciary.
Focus on transfected cells expressing a fluorescent label.
-
Take fluorescent images in the appropriate channel or channels (e.g., red fluorescent protein [RFP], green fluorescent protein [GFP], 4’,6-diamidino-2-phenylindole [DAPI]), either manually or in an automated manner. By taking several images at regularly-spaced intervals, a montage of the well can be assembled during image processing (see step 4).
NOTE: The spacing depends on several factors, including magnification, the optics of the microscope, and the detector size. In general, the optical spacing between adjacent images will be between 90–95% of the size of each individual image, to allow for a small degree of image overlap and feature alignment.
Repeat this process as often as required, aligning to the original fiduciary each time. For survival analysis, imaging takes place every 6–24 h, depending on the cell type and the purpose of the experiment.
4. Image Processing
NOTE: Following image acquisition, a series of processing steps are required prior to image analysis. These include, but are not limited to, stitching, stacking, and background subtraction (Figure 1). The goal of these steps is to produce an image stack, or time series, in which cells are clearly discernible from their background and easy to follow over multiple time points. A dedicated FIJI macro (Image_Processing.ijm, see Supplemental File 2), performs basic stitching, stacking, and background subtraction. An explanation of each step and the parameters to consider when performing image processing is provided in the discussion section.
Adjust the raw data or input directory to match the formatting shown in Figure 2.
If time points are not contiguous (i.e., T1, T2, T3), rename these folders so that they are. This step is critical to ensure that the Image_Processing macro does not crash during stacking.
Double-click on the Fiji icon to open the program, then click and drag the Image_Processing macro onto the Fiji bar. This will open the macro within Fiji.
Adjust lines 2–7 of the Image_Processing macro to specify the input directory containing images, the desired output directory for stitched and stacked images, the number of imaging timepoints, number of fluorescent channels and plate format.
Determine the order in which the images were acquired. To test this, manually stitch a montage of images in FIJI by maneuvering to the Plugins drop down menu | Stitching | Grid/Collection stitching. Adjust the settings within the dropdown menus Type and Order until an accurately stitched image is produced.
Adjust the GRID_TYPE and STITCH_ORDER variables in lines 8 and 9 of the Image_Processing macro to match these selections.
-
Specify the number of images per well by adjusting line 10 in the Image_Processing macro.
NOTE: For a 2 × 2 montage of images, this line would read DIM = 2.
If background subtraction is required, adjust line 14 in the Image_Processing macro to BGSUB = true.
-
Set the rolling ball radius by adjusting line 15 in the Image_Processing macro.
NOTE: For optimal results, set the radius to at least the diameter of the largest foreground object in the image.
Click Run to start the Image_Processing macro. Once started, Image_Processing will automatically advance through stitching, stacking, and background subtraction.
Figure 2: Required file structure.

The provided FIJI macro requires that the raw data are formatted in a specific way. To utilize Image_Processing, organize the raw data as shown on the left. An example experiment and accompanying file structure is shown on the right.
5. Scoring Cell Death
NOTE: See the Discussion section for more information on scoring cell death and censoring data.
Locate the image stacks produced by the Image_Processing script. Open these in FIJI.
-
Use the point tool within FIJI to individually label each cell with a number. Pressing t after each point will add the cell identifier to the ROI (region-of-interest) Manager.
NOTE: The identifiers can be visualized by clicking the labels and show all checkboxes in the ROI Manager.
-
Progress through the timepoints in each image stack and record the timepoint when each cell either dies or needs to be censored in the file Survival_spreadsheet.csv (see Supplemental File 3).
-
Each cell occupies a single row in the spreadsheet, where a unique identifier (ID) for each cell consists of its corresponding well and ROI number within that well. tp_death is the last time point a cell is observed to be alive, while time_death represents the actual time of death in hours. For each cell, input these data. It is critical to maintain this structure for subsequent analysis using survival.R (see Supplemental File 4).
NOTE: The criteria for determining cell death are crucial and may vary depending on cell type. Three main criteria are used in the identification of dead neurons11,20 (Figure 3): loss of fluorescence intensity (e.g., Neuron 1 at 69 h), rounding of the cell body (e.g., Neuron 2 at 188 h), and the loss of neurite integrity or blebbing (e.g., Neuron 2 at 188 h).
-
-
Record the censor status of the cell in the last column.
NOTE: Here, due to the peculiar way censoring is handled by R, censored cells are marked by 0, while uncensored cells are marked by 1. Note that all cells that live to the last time point are censored, and therefore marked as 0.
Figure 3: Scoring cell death in transfected rat cortical neurons.
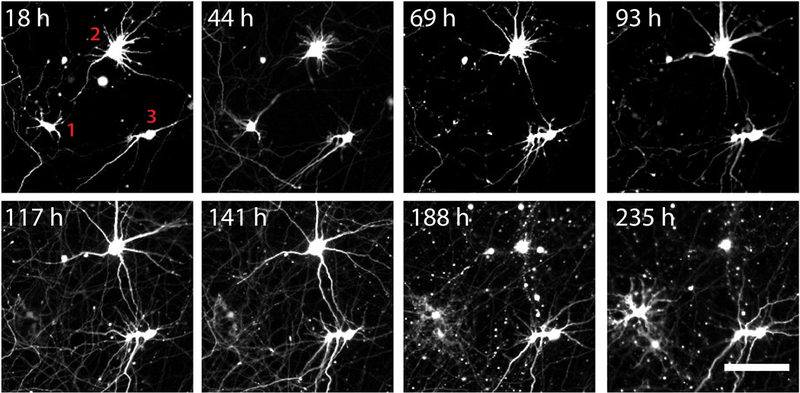
Using the methods described in this article, rat cortical neurons were transfected with a plasmid encoding the fluorescent protein mApple. Cells were then imaged approximately every 24 h, the images were stitched and stacked, and cell death scored using the criteria provided. Cell death is indicated for Neuron 1 at 69 h, as evidenced by loss of fluorescence. Neuron 2 dies at 188 h, as indicated by fragmentation of the processes and rounding of the cell body. Neuron 3 survives for the duration of the experiment. Note that some cells become visible only late in the experiment, as evidenced by the appearance of a new cell at 235 h. Only cells that are visible at the initial time of imaging are included within subsequent analyses. Scale bar = 50 μm.
6. Performing Cox Proportional Hazards Analysis and Visualizing Results
If necessary, download R studio at https://cran.r-project.org/mirrors.html.
Open R studio and double-click the icon for the survival.R script.
Place the cursor on line 2 of survival.R and click the run button in the main R studio window in order to load the survival library.
Change line 5 of the survival.R script to match the location of the file Survival_spreadsheet.csv Click on run to load the survival data as a dataframe.
Highlight lines 8 and 9 and click the run button in the R studio console window in order to perform Cox proportional hazards analysis. Results and output statistics appear in the console window of R studio.
Highlight lines 12–16 of survival.R and hit the run button in order to produce a cumulative risk of death plot, which will appear in the plots tab in R studio. This file can be saved by clicking on the export button above the plot.
If it is desirable to plot the survival data as a Kaplan-Meier curve, highlight lines 19–24 of survival.R and hit the run button.
Representative Results
Using the transfection procedure described here, DIV4 rat cortical neurons were transfected with a plasmid encoding the fluorescent protein mApple. Beginning 24 h post-transfection, cells were imaged by fluorescence microscopy every 24 h for 10 consecutive days. The resultant images were organized as indicated in Figure 2, then stitched, stacked, and scored for cell death (Figure 1). Figure 3 shows a time course for 3 representative neurons, two of which die during the course of the experiment (Neurons 1 and 2) while the third survives (Neuron 3).
Survival data were analyzed using the R script provided (survival.R), and the results summarized in Figure 4. The table generated upon running lines 7 and 8 of the survival.R code provides a summary of the Cox proportional hazards analysis. Four particularly important statistics in the table are highlighted in Figure 4A. The number in Box 1 represents the hazard ratio for the group “Mutant” relative to “WT”. Notice that the “WT” group is not listed. This is because the “WT” group serves as the reference population - the risk of death observed in all other groups is compared to that of the reference population to calculate the hazard ratio. Therefore, hazard ratios greater than 1 indicate a faster rate of death in comparison to the reference population, and values less than 1 represent a reduced rate of death. In the example provided, mutant cells display a hazard ratio of 2.2, meaning that they died 2.2x faster than WT cells. By default, R will arrange the groups in alphanumeric order, with the top group serving as the reference population. Placing numbers in front of group names is an easy way to establish the order in which they are evaluated. The values in Boxes 2 and 3 represent the p-values and 95% confidence intervals for the hazard ratios, respectively, calculated by Cox proportional hazards analysis. In Box 4, the results of the log-rank test are reported. This test evaluates whether there is a statistically significant difference in survival among populations being tested, but does not describe which groups are different from the others, and does not calculate a magnitude for the observed difference.
Figure 4: Interpretation of Cox proportional hazard analysis.
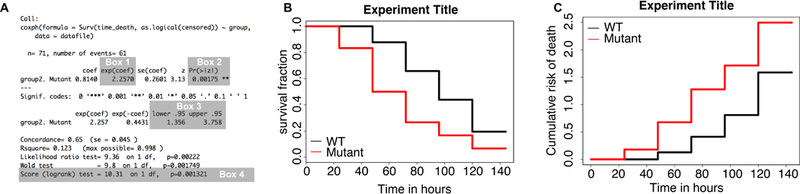
(A) The output summary includes four important statistics that are highlighted in this figure. Box 1 includes the hazard ratio of the experimental group relative to the control group, while Box 2 and Box 3 show the p-values and 95% confidence interval for each hazard ratio, respectively. Box 4 highlights the results of the log-rank test. These data are also depicted via a Kaplan-Meier curve (B) and a cumulative risk of death plot (C).
Kaplan-Meier curves (Figure 4B) are widely used in clinical trials for evaluating the effects of an intervention on patient survival. For this reason, many researchers are familiar with interpreting survival data visualized this way. In the context of single-cell survival, these plots depict the fraction of cells alive over time in each group. Rather than plotting cell survival, an alternative approach is to depict the rate of cell death in each group via a cumulative risk of death plot (Figure 4C). In most survival studies, the number of events does not follow a linear progression; rather, for a given rate of death a greater number of events is observed at earlier times. For example, in a population of 100 cells, if 20% of cells die between intervals, then 20 cells will die within the first interval, 16 during the second interval, 13 during the third interval, and so on. This logarithmic trend is conceptually easier to visualize using cumulative risk of death plots, since the y-axis represents the negative log transform of cellular survival. Alternatively, the y-axis of the cumulative risk of death plot can also be presented as % cell death, calculated as 1–1 /ecumulatlve risk of death. These plots also enable straightforward comparisons of the risk of death between populations. The magnitude of the hazard ratio reflects the slope of the cumulative risk of death plot for each population, relative to that of the reference group.
Discussion
Here, methodology to directly monitor neuronal survival on a single-cell basis is presented. In contrast to traditional assays for cell death that are limited to discrete time points and entire populations of cells, this method allows for the continuous assessment of a variety of factors such as cellular morphology, protein expression, or localization, and can determine how each factor influences cellular survival in a prospective manner.
This methodology can be modified to fit a wide array of experimental needs. The frequency and duration of imaging can be easily adjusted, and any protein of interest can be co-transfected with the fluorescent marker to model disease states or investigate protein function13,14,15,16,17,18,19. Though this article describes the optimal procedure for transfecting rat cortical neurons, the experimental schema may be applied to any post-mitotic cell type. However, the optimal transfection conditions may need to be optimized on a per-cell line basis, and substrates may need to be adjusted to prevent cells from clumping or moving too much to reliably track.
Image processing and analysis requirements will vary depending on the specific parameters of each longitudinal microscopy experiment. A brief explanation of each critical step is included below to help customize the protocol to better match an experiment’s demands.
Stitching:
If a montage of images is taken, stitching can be performed to create a single, larger image for each field of view. For most applications it is preferable to perform stitching prior to stacking. If only one image is taken per well, there is no need to perform this step.
Stacking:
Rather than tracking cells over time across separate image files, stacking can be performed to align consecutive images into a single time series, analogous to a stop frame animation. With successful fiduciary alignment, the individual frames comprising the stacked image will be closely aligned. However, if there are noticeable shifts or rotations between frames, image registration is needed. The Image_Processing macro automatically performs registration using the FIJI plugin “MultiStackReg.” This plugin helps reduce small misalignments between imaging runs. However, with significant shifts, manually cropping and realigning images may be required.
Background subtraction (optional):
One potential issue that may arise during image acquisition is uneven illumination. This will result in variations in signal intensity across an image that can confound estimates of fluorescence intensity. In these instances, intensity variations can be eliminated by background subtraction techniques. These are particularly relevant with low signal to noise ratios, where intensity shifts due to uneven illumination can be comparable in magnitude to the signal of the fluorophore itself. There are many background subtraction algorithms, several of which have associated FIJI plugins. The choice of which algorithm to use depends on the properties of the image itself and the signal being measured. Within the FIJI macro Image_Processing, the user is given the option to perform “rolling ball” background subtraction on a stacked set of images (line 14). In this method, a local background is determined for every pixel based on the average intensity of a circle surrounding that pixel. This value is then subtracted from the pixel’s initial value. The optimal value for the radius of the circle used for local background estimates will differ based upon the diameter of the largest foreground object in the image.
Scoring cell death:
For accurate comparisons between populations, it is essential that the criteria outlined above to identify dead neurons be applied consistently across the entire dataset. Furthermore, blinding the individuals scoring cell death to the experimental groups under investigation eliminates potential sources of bias. Depending on the specific criteria and their generalizability, they may be incorporated into automated algorithms for the unbiased assessment of cellular survival15,16,17,18,19.
In the context of survival analysis and other time-to-event analyses, there are three possible outcomes. First, the event (cell death) has occurred, and the time at which the event occurred is recorded. Second, the event did not occur during the time frame of observation. These observations are censored at the completion of the experiment. Third, the event could not be scored because the cell moved out of the field of view, or was obscured by nearby cells. In this case, the cell is censored when it can no longer be accurately tracked. For the first outcome, the precise timing of cell death may be difficult to determine based on the imaging interval. For instance, a cell that is alive initially but marked as dead 24 h later may have died at any point within that 24 h period. To be conservative, it is good practice to record the time of death as the last time a cell can be confidently identified as alive (left censoring).
Performing Cox proportional hazards analysis and visualizing results:
The accompanying Survival.R script enables the comparison of risk of death among populations and their statistical significance using Cox proportional hazards analysis (Figure 4A), and also plot results as either a Kaplan-Meier curve (Figure 4B) or a cumulative risk of death plot (Figure 4C). Survival analysis, Cox proportional hazards analysis, and the “survival” package in R are described in more detail by Christensen12 and at https://cran.r-project.org/web/packages/survival/survival.pdf.
By adapting the procedures outlined here, a variety of neuronal features can be related to survival. Generation of an ROI around the cell body and/or nucleus enables the user to longitudinally monitor cell size and morphology, protein expression level and localization, or the formation of subcellular structures such as puncta or protein aggregates13,14,15,16,17,18,19.Importantly, because each of these factors is observed in relation to cell death, it is possible to quantitatively determine how well individual factors predict cellular survival or death during the given time frame. Protein metabolism and cellular pathways may also be assayed by expressing fluorescent reporters that provide real-time measurements of underlying cellular physiology (e.g., gCaMP6f to assay activity). By employing this powerful approach, factors that drive cellular maintenance, function, and dysfunction can be uncovered and studied in detail, thereby inspiring new avenues of inquiry.
Supplementary Material
Acknowledgments
We thank Steve Finkbeiner and members of the Finkbeiner lab for pioneering robotic microscopy. We also thank Dan Peisach for building the initial software required for image processing and automated survival analysis. This work was funded by the National Institute for Neurological Disorders and Stroke (NINDS) R01-NS097542, the University of Michigan Protein Folding Disease Initiative, and Ann Arbor Active Against ALS.
Footnotes
Disclosures
The authors have nothing to disclose.
Video Link
The video component of this article can be found at https://www.jove.com/video/59036/
References
- 1.Lockshin RA, Zakeri Z Cell death in health and disease. Journal of Cellular and Molecular Medicine. 11, 1214–1224 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kepp O, Galluzzi L, Lipinski M, Yuan J, Kroemer G Cell death assays for drug discovery. Nature Reviews Drug Discovery. 10, 221–237 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Lemasters JJ et al. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochimica et Biophysica Acta Bioenergetics. 1366, 177–196 (1998). [DOI] [PubMed] [Google Scholar]
- 4.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. Journal of Immunological Methods. 184, 39–51 (1995). [DOI] [PubMed] [Google Scholar]
- 5.Chien KR, Abrams J, Serroni A, Martin JT, Farber JL Accelerated phospholipid degradation and associated membrane dysfunction in irreversible, ischemic liver cell injury. Journal of Biological Chemistry. 253, 4809–4817 (1978). [PubMed] [Google Scholar]
- 6.Zwaal RFA, Comfurius P, Bevers EM Surface exposure of phosphatidylserine in pathological cells. Cell and Molecular Life Sciences. 62, 971–988 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell DB, Santone KS, Acosta D Evaluation of cytotoxicity in cultured cells by enzyme leakage. Journal of Tissue Culture Methods. 6 113–116 (1980). [Google Scholar]
- 8.Moran JH, Schnellmann RG A rapid beta-NADH-linked fluorescence assay for lactate dehydrogenase in cellular death. Journal of Pharmacological and Toxicological Methods. 36, 41–44 (1996). [DOI] [PubMed] [Google Scholar]
- 9.Mossman T Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. Journal of Immunological Methods. 65 (1–2), 55–63 (1983). [DOI] [PubMed] [Google Scholar]
- 10.Guerzoni LPB et al. In Vitro Modulation of TrkB Receptor Signaling upon Sequential Delivery of Curcumin-DHA Loaded Carriers Towards Promoting Neuronal Survival. Pharmaceutical Research. 34 (2), 492–505 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Arrasate M, Finkbeiner S Automated microscope system for determining factors that predict neuronal fate. Proceedings of the National Academy of Science of the United States of America. 102, 3840–3845 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Christensen E Multivariate survival analysis using Cox’s regression model. Hepatology. 7, 1346–1358 (1987). [DOI] [PubMed] [Google Scholar]
- 13.Barmada SJ et al. Autophagy induction enhances TDP43 turnover and survival in neuronal ALS models. Nature Chemical Biology. 10, 677–685 (2014).] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barmada SJ et al. Amelioration of toxicity in neuronal models of amyotrophic lateral sclerosis by hUPF1. Proceedings of the National Academy of Science of the United States of America. 112, 7821–7826 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Malik AM et al. Matrin 3-dependent neurotoxicity is modified by nucleic acid binding and nucleocytoplasmic localization. Elife. 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Archbold HC et al. TDP43 nuclear export and neurodegeneration in models of amyotrophic lateral sclerosis and frontotemporal dementia. Scientific Reports. 8, 4606 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Green KM et al. RAN translation at C9orf72-associated repeat expansions is selectively enhanced by the integrated stress response. Nature Communications. 8, 2005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Park S-K et al. Overexpression of the essential Sis1 chaperone reduces TDP-43 effects on toxicity and proteolysis. PLOS Genetics. 13, e1006805 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gupta R et al. The Proline/Arginine Dipeptide from Hexanucleotide Repeat Expanded C9ORF72 Inhibits the Proteasome. eNeuro. 4, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Angelov B, Angelova A Nanoscale clustering of the neurotrophin receptor TrkB revealed by super-resolution STED microscopy. Nanoscale. 9 (28), 9797–9804 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.