

Abstract
Canonical FGFs activate FGF receptors (FGFR) via paracrine or autocrine mechanisms, in a process requiring cooperation with heparan sulfate proteoglycans that function as co-receptors for FGFR activation1,2. By contrast, endocrine FGFs (FGF 19, 21, and 23) are circulating hormones that regulate critical metabolic processes in a variety of tissues3,4. FGF19 regulates bile acid synthesis and lipogenesis, while FGF21 stimulate insulin sensitivity, energy expenditure and weight loss5. Endocrine FGFs signal through FGFRs in a manner that requires Klothos, which are cell surface proteins with tandem glycosidase domains3,4. Here we describe the crystal structures of free and ligand-bound β-Klotho extracellular regions, revealing the molecular mechanism underlying the specificity of FGF21 towards β-Klotho and demonstrating how FGFR is activated in a Klotho-dependent manner. β-Klotho serves as a primary “Zip code”-like receptor for FGF21 with an FGFR functioning as a catalytic subunit that mediates intracellular signaling. Our structures also show how a sugar cutting enzyme (glycosidase) has evolved to become a specific receptor for hormones that regulate metabolic processes including lowering of blood sugar. Finally, we describe a superior agonistic variant of FGF21 and present structural insights offering development of novel therapeutics for diseases linked to endocrine FGFs.
To elucidate the mechanism of action of β-Klotho in cell signaling via FGF21 stimulation we used X-ray crystallography to determine the structure of free and ligand bound extracellular region of human β-Klotho (sKLB) (Extended Data Fig.1, Methods). The overall structure of sKLB (2.2 Å resolution, Extended Data Table 1) features two tandem glycoside hydrolase-like (GH) domains, D1 (residues 53–507) and D2 (residues 521–968), that are connected by an unstructured and flexible linker (Fig. 1a). Each GH domain can be recognized by multiple repeats of alternating β-sheet (S) and α-helix (H) that define the (β/α)8 fold (Extended Data Fig. 2a). The structure of KLBD1 (1.7 Å resolution, Extended Data Table 1), shown in Fig. 1b, is virtually identical to the structure of D1 in the context of sKLB (with overall Cα RMSD of 0.48 Å). Four loop regions in the structure of sKLB containing potential N-glycosylation sites could not be modeled due to poor electron density: a loop between H0 and S1 (residues 63–73), a loop between H1b and H1c (residues 119–125), a loop between S9 and H9a (residues 538–574), and the C-terminus (residues 968–983) of the protein (Extended Data Fig. 2a). With the exception of the C-terminus, these loops are depicted in the sKLB structures as dashed lines (Fig. 1a).
Figure 1 |. Crystal structure of extracellular domain of β-Klotho.
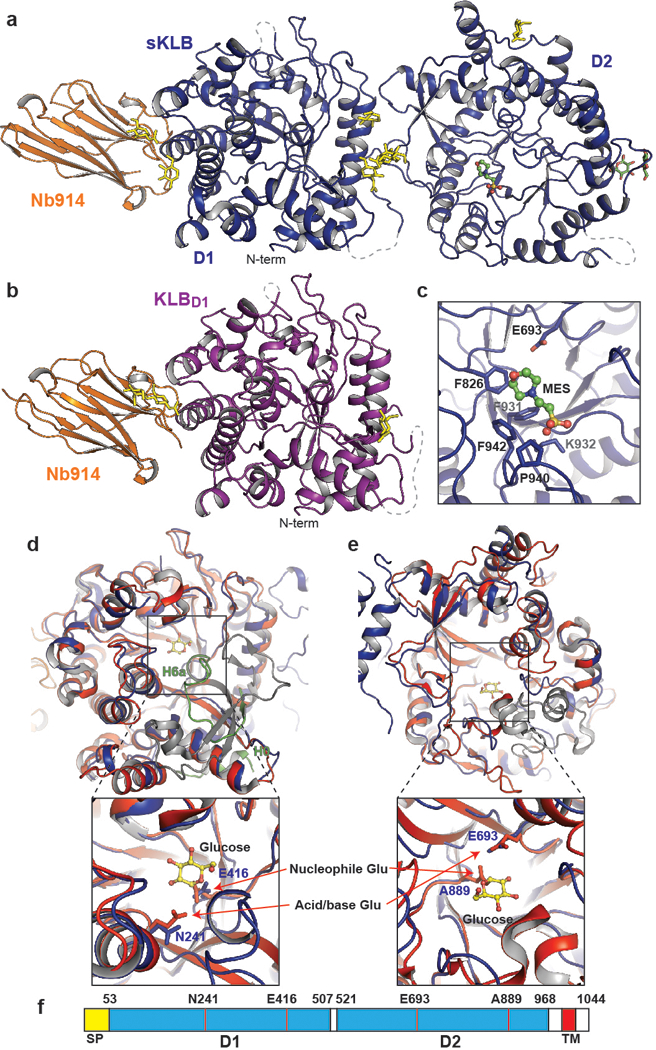
a, b, Structures of (a) sKLB (blue) and (b) KLBD1 (purple) in complex with nanobody Nb914 (orange) are shown as ribbon representation. Glycans attached to asparagine side-chains are shown as yellow sticks, glucose molecules are shown as green sticks, and MES molecule is shown as ball-and-stick representation. Regions that do not show significant electron density are drawn with grey dashed lines. c, Side chain atoms of amino acids in sKLB interacting with MES molecule are shown as sticks. Also indicated is the location of E693 which is ~6 Å apart from bound MES molecule. d, e, The structure of human cytosolic β-glucosidase (red, PDB: 2ZOX) is superimposed with D1 (d) and D2 (e) of sKLB (blue) with overall α-carbon RMSDs of 1.08 Å and 1.39 Å, respectively. Regions in sKLB that are different from β-glucosidase are colored in green and regions in β-glucosidase that are different from sKLB are colored in grey. A glucose molecule bound to β-glucosidase is shown as ball-and-stick representation in yellow. Superimposition of D1 and D2 to reveal locations of “catalytic” glutamates. Note that one of the two catalytic glutamates from each of sKLB domains is replaced by an asparagine (for D1) or an alanine (for D2). f, Diagram of β-Klotho highlighting the locations of the residues corresponding to the “catalytic” glutamates in D1 and D2 of β-Klotho.
Superimposing the structure of human cytosolic β-glucosidase (PDB: 2ZOX) on the structures of each of the two GH domains in sKLB domains gives Cα RMSDs of 1.08 Å for D1 and 1.39 Å for D2, demonstrating the strong similarity of both D1 and D2 to the glycoside hydrolase family-1 (GH1) of enzymes (Fig. 1d and 1e). The GH1 family of proteins are enzymes that hydrolyze glycosidic linkages between carbohydrate moieties (http://www.cazy.org/GH1.html) through a double-replacement mechanism mediated by two conserved glutamates located in their active site6. Intriguingly, one of the two catalytic glutamates is replaced by another amino acid in each of the sKLB domains (Fig. 1d-f). The first glutamate in D1 is replaced by N241, whereas the second glutamate in D2 is replaced by A889, indicating that neither GH domain in β-Klotho can function as an active glycoside-hydrolase enzyme. Structural alignment using the Dali server7 indicates that GH1 and GH5 members exhibit high structural similarities to each of the GH domains of sKLB, implying a common evolutionary origin. Although the overall structures of the GH domains in sKLB are very similar to GH1 family enzymes, the two sKLB GH domains exhibit important structural features that set them apart from the GH1 family of enzymes.
The pocket in D1 that corresponds to the substrate-binding region in GH1 family enzymes is largely occluded by a short helix, H6a (Fig. 1d and Extended Data Fig 3a). Moreover, a helix-turn-strand element (H6a-turn-S6b) in this region, specific to β-Klotho D1 (green in Fig. 1d), provides part of the FGF21 binding site (see below) and is quite distinct from the strand-helix-strand element in corresponding regions of cytosolic β-glucosidase (grey in Fig. 1d). Other features unique to β-Klotho include a short helix, H0 (Fig. 1d and Extended Data Fig. 3b), which begins with the first amino acid following the sKLB signal sequence (F53). This helix interacts with H5a, H6b and S5b, mostly through hydrophobic interactions, and precedes a disordered loop that is followed by the core structural elements of the (β/α)8 fold. Interestingly, E416, the remaining “catalytic” residue in D1 is located at the bottom of the substrate binding pocket (Fig. 1d and Extended Data Fig. 3a) and the orientation of the side chain of E416 is identical to the orientation of the side chain of the corresponding nucleophilic E373 residue of human cytosolic β-glucosidase.
The pocket in D2 that corresponds to the substrate binding pocket in GH1 family enzymes is not occluded by an alpha-helix in the D2 domain, but is instead accessible and occupied by a MES molecule from the crystallization buffers (Fig. 1c). Interestingly, the morpholine ring of MES interacts with aromatic rings from three phenylalanines, F931, F826, and F942 (Fig. 1c), which also play a role in the interaction of sKLB with its ligands (see below). The D2 pocket is accessible in part because of the existence of a disordered region between S9 and H9a (Extended Data Fig. 2a), resulting in formation of a more groove-like feature in this domain rather than the pocket that accommodates substrate in the active site of GH1 family members. The amino acid sequence and the length of this region vary significantly among GH1 family members. The inter-domain interface of sKLB is comprised of an extensive network of both hydrophobic and polar interactions (Extended Data Fig. 3c) encompassing a buried surface area8 of about 680 Å2.
Next, we determined the structure of sKLB in complex with C-terminal tail (CT) of FGF21 (FGF21CT) at 2.6 Å resolution (Fig. 2a and Extended Data Table 1). The final model contains amino acids P186 to S209 from FGF21CT bound to sKLB (Extended Data Fig. 4a-c) and exhibited clear electron density for FGF21CT lying across the middle of sKLB (Fig. 2b). FGF21CT binds to an elongated interface that spans D1 and D2 of sKLB with no influence on the structure of either individual domain as judged by α-carbon RMSDs of 0.33 and 0.49 Å for D1 and D2, respectively, when overlaid on the unoccupied sKLB structure. A small change in the inter-domain angle9 of 6˚ was seen upon FGF21CT binding to sKLB (Extended Data Fig. 4d). The FGF21CT-binding region on sKLB is located on the opposite side of the molecule from the linker that connects D1 and D2. Flexibility of the linker may contribute to the inter-domain dynamic properties that enable complex formation with ligands and FGFRs. Intriguingly, the sKLB:FGF21CT structure shows two distinct binding sites for two different regions of the peptide. Site 1 is located on D1, and site 2 is located in D2 with a distance of 30 Å between the two sites.
Figure 2 |. Crystal structure of sKLB bound to FGF21CT reveal two distinct binding sites.
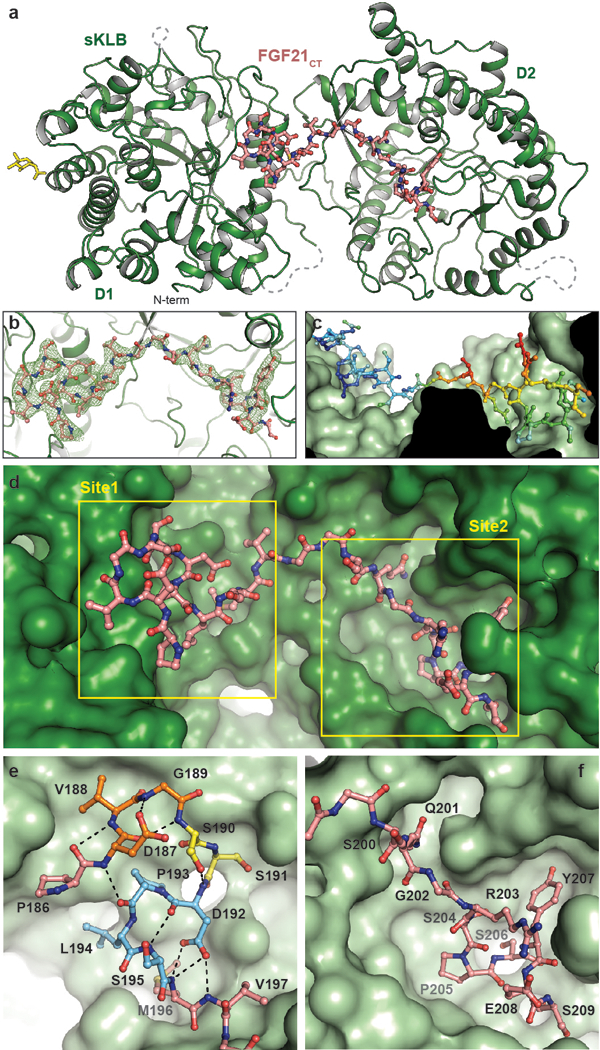
a, The structure of sKLB (green) in complex with FGF21CT (salmon) is shown as ribbon and ball-and-stick representation. N-linked glycans are shown as yellow sticks. Nb914 is omitted for clarity. Regions that do not exhibit significant electron densities are shown as grey dashed lines. b, FGF21CT binding site showing |Fo|-|Fc| omit map contoured at 3.0 σ for FGF21CT. c, Surface of sKLB interacting with FGF21CT are color-coded according to the B-factor values, ranging from 52.76 Å2 (blue) to 103.63 Å2 (red). d, Surface representation of sKLB (green) highlighting two binding sites, site 1 and site 2 of FGF21CT (salmon, ball-and-stick). e, Site 1 forms a series of internal hydrogen bonds (black dashed lines) through three consecutive turns (orange, yellow, and light blue), creating a structural element that binds to D1 of sKLB. f, Site 2 interacts with pseudo-substrate binding region of D2 of sKLB.
Site 1 on sKLB D1 engages amino acids P186-V197 of FGF21CT, primarily through hydrophobic interactions (Fig. 2e, Extended Data Fig. 4a-b). Site 1 involves a surface created on D1 by H6a, H7, the loop between S6b and H6b, and the loop between S7 and H7. Most strikingly, the region of the bound peptide ligand that associates with site 1 adopts an unusually compact and rigid structure through the formation of several well-defined turns (Fig. 2e), as follows: (1) D187-V188-G189-S190 form a type I β-turn (orange in Fig. 2e) through hydrogen bonding of the carboxyl oxygen of D187 with the backbone nitrogen of G189 and of the backbone carbonyl of D176 with the backbone amide of S190; (2) S190-S191-D192 form an ST turn (yellow in Fig. 2e) through hydrogen bonding of the S190 hydroxyl with the backbone amide of D192; (3) D192-P193-L194-S195 (light blue in Fig. 2e) form a type I β-turn (or an Asx turn that resembles a Schellman loop) through hydrogen bonding of the side chain carboxyl of D192 with the M196 and V197 backbone amides and of the D192 backbone carbonyl with the backbone amide of S195. These consecutive turns also support a long-range hydrogen bond between the D187 backbone amide and the P193 carbonyl. These intramolecular interactions cooperate to form a well-defined structural element that makes multiple specific contacts with sKLB, burying a relatively large surface area of 606 Å2.
Site 2 interactions with FGF21CT contrasts starkly with site 1 interactions, comprising a network of inter-molecular interactions of the sort typically observed between proteins and short peptides (Fig. 2f and Extended Data Fig. 4a-b). Interestingly, residues 200–209 of the FGF21CT peptide project into what would be the substrate-binding site occupied by glycosides that D2 of sKLB would hydrolyze if it were an active GH1 enzyme (Fig. 3 and see below). It is also noteworthy that the sequence of this part of FGF21CT (S-Q-G-R-S-P-S-Y-A-S) is rich (50%) in residues with side-chain hydroxyl groups, suggesting the possibility that this region of FGF21 may indeed mimic a glycoside substrate. Given these characteristics, a fascinating feature of site 2 is the interaction between the side-chain carboxyl group of E693 in sKLB with hydroxyl groups of both S204 and S206 in FGF21CT (Fig. 3d). E693 corresponds to one of the two conserved “catalytic” glutamates, and would function as a general acid/base catalyst in the Koshland double-displacement reaction of glycoside hydrolases (whereas the potential nucleophilic glutamate is replaced by alanine in D2).
Figure 3 |. Comparison of β-glucosidase and β-Klotho structures; evolution of a sugar cutting enzyme into a receptor for endocrine FGF.
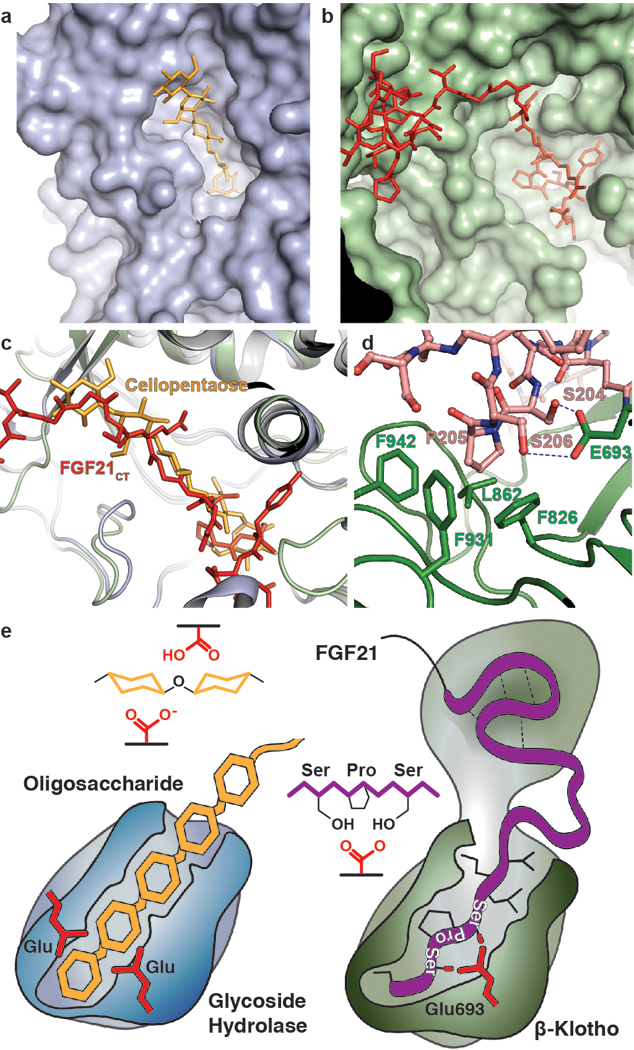
a, b, The structure of (a) rice β-glucosidase (light blue, surface presentation) in complex with cellopentaose (orange, stick presentation) (PDB: 3F5K) and (b) site 2 of sKLB (pale green, surface presentation) in complex with FGF21CT (red, stick representation). Cellopentaose binds to the active site of β-glucosidase and FGF21CT binds to the corresponding pseudo-substrate binding site of β-Klotho. c, Superimposition of the structures of cellopentaose-bound rice β-glucosidase and FGF21CT-bound sKLB. d, E693 of β-Klotho makes contacts with S-P-S motif of FGF21 via interaction with hydroxyl moieties of serines mimicking sugar hydroxyls in their interaction with glutamates in the catalytic site of β-glucosidase. e, Schematic diagram comparing the substrate-binding pocket including the two glutamates required for glycoside hydrolase activity and the ligand-binding pocket of β-Klotho depicting interactions between E693 with the S-P-S motif.
Amino acids 198–200 of FGF21CT, which connect the ligand binding site 1 and site 2, do not make significant contacts with sKLB. In addition, the electron densities in omit maps (Fig. 2b) and B-factors (Fig. 2c) suggest that this region is flexible. This conclusion is consistent with identification of an enzyme that cleaves FGF21 in this region, and is known to abolish its binding to β-Klotho11–16. As this region of FGF21 is flexible and potentially accessible for proteolysis, cleavage between the site 1 and site 2 binding regions could represent a mechanism of termination of FGF21 signaling by targeted proteolysis.
The crystal structure of sKLB bound to FGF21CT reveals how the basic framework of a glycoside hydrolase has evolved to become a specific receptor for endocrine FGFs. The β-glucosidase family of glycoside hydrolases catalyzes the hydrolysis of disaccharides as well as longer oligosaccharides, and several crystal structures of β-glucosidases in complex with oligosaccharide substrates such as cellotetraose (P. polymyxa BglB, PDB: 2Z1S) or cellopentaose (O. sativa BGlu1, PDB: 3F5K) have been determined17,18. Superimposition of the crystal structures of substrate-bound β-glucosidases with the structure of sKLB in complex with FGF21CT shows that the backbone of residues 200–209 from FGF21CT aligns very well with the location of oligosaccharides that occupy the catalytic pocket of β-glucosidases (Fig. 3a-c). As mentioned above, the mode of interaction between the hydroxyls of S204 and S206 from FGF21CT and the “catalytic” glutamate in D2 of sKLB, together with the hydrophobic interactions involving P205, are highly reminiscent of substrate interactions seen for the glycoside hydrolases6 suggesting that this is a pseudo-substrate like interaction (Fig. 3d). Oligosaccharide substrates bound to this catalytic glutamic acid in β-glucosidases active sites lie in precisely the same position as the S204-P205-S206 motif of FGF21 bound to site 2 of sKLB. In addition, the residues in sKLB that form hydrophobic interactions with P205 of FGF21, i.e., F826, F931, and F942, align well with the corresponding hydrophobic residues in β-glucosidases. These unexpected similarities indicate that the substrate-binding region of glycoside hydrolases evolved to recognize a sugar-mimicking S-P-S motif in FGF21 (Fig. 3e). As FGF19 also binds specifically to β-Klotho, it is not surprising that FGF19 also contains a S211-P212-S213 motif at its C-terminus (Extended Data Fig. 5), whereas FGF23 (which does not bind to β-Klotho) has no such sequence. Further studies on how FGF23 recognizes α-Klotho should provide guidance for development of new treatments of metabolic disorders caused by impaired phosphate homeostasis19, and information on a unique evolutionary pathway that this family of proteins might have taken.
We next analyzed the binding affinities of FGF21, wild type and various mutants in C-terminal tail of FGF21 towards sKLB to explore the contributions of different amino acids in FGF21 that take part in the interface of ligand-occupied sKLB structure. We also investigated the effects of mutations in β-Klotho’s two FGF21CT-binding sites on the ability of FGF21 to stimulate FGFR1 activation in transfected L6 myoblasts. These experiments validated the ligand binding interfaces identified in the occupied sKLB structure and demonstrated that FGF21CT binds in cooperative manner to both site 1 and site 2 in β-Klotho. Full details about these experiments are presented in Extended Data Fig. 6, 7, and Supplementary Discussion.
As endocrine FGFs play important roles in the control of metabolic processes, a variety of approaches have been utilized to develop novel therapeutics13,20–25. We reasoned that it should be possible to enhance the potency of FGF21 by introducing mutations into its C-terminal tail that strengthen interactions with β-Klotho. We introduced L194F mutation to increase hydrophobic interactions with neighboring amino acids in site 1 of β-Klotho and a R203W mutation to replace cation-pi interactions between R203 in FGF21 and H646 in site 2 of β-Klotho with pi-pi interactions. We found that R203W/L194F-mutated FGF21 (FGF21WF) bound to sKLB over 10-fold more tightly than wild-type FGF21, with a KD of 3.4 ± 1.2 nM (Fig. 4a) with enhanced ability to stimulate FGFR1c autophosphorylation and MAP kinase stimulation in L6 cells co-expressing β-Klotho and FGFR1c (Fig. 4b and Extended Data Fig. 8).
Figure 4 |. Structure-based engineering of a superior analogue of FGF21 and the mechanism of endocrine FGF signaling.
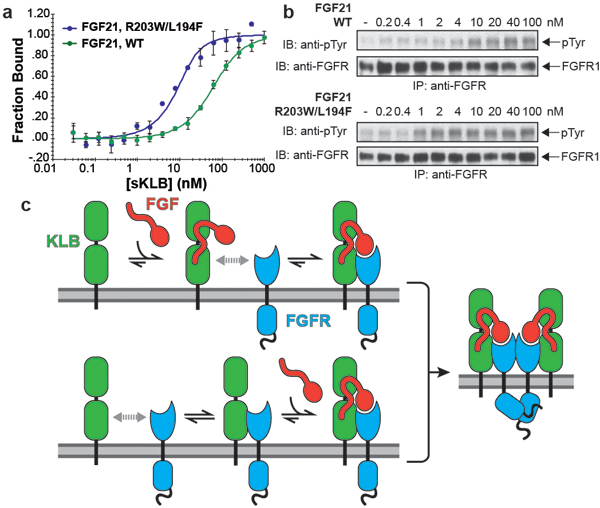
a, b, Enhanced binding affinity (a) and bioactivity (b) of an FGF21 mutant. MST binding measurements of FGF21 carrying a double L194F/R203W mutations in FGF21CT reveal approximately 10-fold increase in binding affinity to sKLB with a KD of 3.4 ± 1.3 nM and approximately 10-fold enhanced potency for stimulation of FGFR1c tyrosine phosphorylation. The dots and error bars for each graph in panel a denote means and variations of ΔFnorm (n = 3 independent samples). Individual experimental data are plotted in Supplementary Fig. 2. c, A ‘Zip code’-like mechanism for β-Klotho dependent FGF21 stimulation of FGFR1c. In the cell membrane of unstimulated cells β-Klotho and FGFR1c monomers are in equilibrium with FGFR/β-Klotho heterodimers. Due to reduced dimensionality, the binding of FGF21 to β-Klotho via FGF21 C-tail and bi-valent binding of the FGF core of FGF21 to two FGFR1c molecules will shift the equilibrium towards formation of a FGF21/FGFR1c/β-Klotho ternary complexes, resulting in stimulation of tyrosine kinase activity and cell signaling via FGFR1c. In addition, β-Klotho functions as a primary high affinity receptor for FGF21 and FGFR1c functions as a catalytic subunit that mediate receptor dimerization and intracellular signaling.
These experiments show that, rather than serving as an alternative co-receptor for FGFR1c activation by endocrine FGFs, β-Klotho functions as the primary high affinity receptor for FGF21. Importantly, we show that Klotho proteins function as specific ‘Zip code’-like signals for targeting FGF21 (or two other endocrine FGFs) to cells and tissues where they mediate their cellular responses by activating members of the FGFR families. The scheme presented in Fig. 4c depicts a model for how FGF21 binding to β-Klotho allows it to activate a β-Klotho/FGFR complex to promote cell signaling. In the model, FGFR1c and β-Klotho monomers exist in equilibrium with FGFR/β-Klotho hetero-dimers in the membrane. With a KD of ~1 μM for binding of the FGFR1c extracellular region to sKLB (Extended Data Fig. 6b), a substantial portion of FGFR1c and β-Klotho will be associated with one another at levels around 10,000 copies per cell. FGF21 binds with high affinity, KD = 43.5 nM, (Extended Data Fig. 6a) either to β-Klotho monomers or to preexisting β-Klotho/FGFR1c hetero-dimers. With FGF21 thus tethered via its C-terminal tail to β-Klotho monomers and/or β-Klotho/FGFR1c heterodimers, all three components are reduced to two dimensions at the membrane and the weak (but demonstrable) affinity of FGF21’s FGF-core for FGFR1c is sufficient to drive formation of the activated ternary FGF21/FGFR1c/β-Klotho complex via a reduced dimensionality effect on the bivalent binding of FGF to two FGFR molecules26. In this model, β-Klotho functions as a primary high affinity receptor for FGF21, whereas FGFR1c functions as a catalytic subunit that mediates receptor dimerization and intracellular signaling.
The crystal structure of sKLB bound to FGF21CT also provides clear views of how the two GH domains of β-Klotho have been ‘repurposed’ in evolution to specifically recognize FGF21. Comparing the structures of substrate-bound β-glucosidases to the second GH domain of FGF21CT-bound β-Klotho reveals how the active-site of an enzyme specialized in cutting sugars has evolved to become a specific and high-affinity cell-surface receptor for circulating hormones that regulate critical metabolic processes including lowering of blood sugars; this may not be a coincidence. Intriguingly, the C-terminus of FGF21 appears to present a structural mimic of an oligosaccharide. The similarities between FGF21 and FGF19 indicate that the specificity of the two hormones towards β-Klotho and their modes of action are very similar (Extended Data Fig. 5). Differences in the cellular responses to these two endocrine FGFs are likely to be determined by the altered binding preferences of the two ligands for the different FGFRs.
METHODS
Plasmid construction.
cDNA that encode for either amino acids 30–983 (sKLB) or 30–522 (KLBD1) of human β-Klotho (KLB) were amplified together with the tobacco etch virus (TEV) protease cleavage site and linker of four Gly residues. The resulting sequence was subcloned into a modified pCEP4 vector (Thermo Fisher Scientific Inc.) which contains sequence for Fc region of human IgG1. The expression vector for C-terminal HA-tagged KLB was generated by subcloning the gene of full-length KLB together with the HA-tag sequence into a pBABE vector. All plasmids of KLB mutants were generated by following standard site-directed mutagenesis protocol using a plasmid containing WT C-terminal HA-tagged KLB.
Expression and purification of sKLB and KLBD1.
HEK293-EBNA cells were cultured in a humidified incubator with 5% CO2 at 37˚C in DMEM (Thermo Fisher Scientific Inc.) containing 10% Fetal Bovine Serum (FBS), 100 U/mL Penicillin-Streptomycin, and 250 μg/mL G-418. The plasmids were transfected into HEK293-EBNA cells with the Lipofectamine 2000 (Thermo Fisher Scientific Inc.) and selected by treatment with 200 μg/mL of hygromycin B (Thermo Fisher Scientific Inc.) for 2–3 weeks. Cells stably expressing sKLB-Fc or KLBD1-Fc were expanded in Hyperflasks (Corning Inc.), and the media was changed to DMEM with 5% FBS when cell confluency had reached about 70%. After 7 days, the medium was collected after centrifugation at 5,000 ×g and filtration through 0.2 μm membrane. 15 μM swainsonine (Cayman Chemical) was added to the medium of cultured cells when preparing proteins for crystallization.
Media harvested from the cells expressing sKLB-Fc or KLBD1-Fc were incubated with recombinant Protein A Sepharose 4B (Thermo Fisher Scientific Inc.) overnight at 4˚C. The resin was washed with 50 column volumes of PBS and the protein was eluted from the resin using 0.1 M Glycine-HCl, pH 3.5 and immediately neutralized with 0.1 M Tris, pH 7.4. The eluted protein was incubated with recombinant TEV protease for 2 hours at room temperature to cleave the C-terminal Fc-tag, followed by incubation with recombinant Protein A Sepharose 4B for 30 min. at 4˚C to remove Fc-tag and undigested protein. The protein was then subjected to a cation exchange chromatography (Mono S 5/50 GL, GE Healthcare) using 20 mM sodium phosphate buffer at pH 7.0 (for sKLB) or at pH 6.5 (for KLBD1) and purified using a linear salt gradient. The elution fractions containing sKLB or KLBD1 were pooled, concentrated, and subjected to Superdex 200 Increase 10/300 GL (GE Healthcare) size exclusion chromatography column pre-equilibrated with 20 mM HEPES, 150 mM NaCl, pH 7.0. The eluted fractions containing sKLB or KLBD1 were pooled, concentrated, flash-frozen, and stored at −80˚C until further use. For crystallization of sKLB, two potential N-glycosylation sites, Asn308 and Asn611, were mutated to glutamine. The mutations were introduced to the sKLB-Fc plasmid by standard QuikChange site-directed mutagenesis. The expression and purification of mutant sKLB was identical to those used for WT sKLB. Typical yield of sKLB, after complete purification, is 1–2 mg per 1 liter of media from the cells stably expressing sKLB.
Expression and purification of recombinant FGF21, GST-FGF21CT, and FGFR1cD2D3.
DNA sequence that encodes for human FGF21 amino acid 29–209 harboring three mutations, L126R, P199G, and A208E, was codon-optimized for E.coli expression and synthesized (Blue Heron Biotech, LLC.). After cloning into a pET28a vector (Novagen), the plasmid was transformed into BL21-Gold (DE3) competent cells. Transformants were grown in LB medium containing 50 μg/mL kanamycin, shaking at 240 rpm at 37˚C. When the A600 of the samples reached 0.6, the bacteria were induced with 1 mM IPTG for 4 hours at 37˚C. The bacterial cell pellet, collected by centrifugation at 5,000 ×g at 4˚C, was lysed in 20 mM sodium phosphate buffer, 500 mM NaCl, 5% glycerol, at pH 7.8 using EmulsiFlex-C3 homogenizer (Avestin, Inc.), followed by centrifugation at 20,000 ×g for 30 min. at 4˚C. The supernatant containing N-terminal His6-tagged FGF21 was supplemented with 10 mM imidazole and incubated with Ni-NTA agarose (Qiagen) for 1 hr at 4˚C. The resin was washed with 20 column volume of lysis buffer containing 10 mM imidazole, and the protein was eluted from the resin with lysis buffer containing 300 mM imidazole. The protein solution was injected into HiLoad 26/600 Superdex 200 (GE Healthcare) size exclusion chromatography column equilibrated with 20 mM HEPES, 900 mM NaCl at pH 7.5. The eluted fractions containing FGF21 were pooled, concentrated to about 1.5 mg/mL, flash-frozen, and stored at −80˚C until further study. For generating GST-FGF21CT, DNA sequence encoding amino acids 169–209 of FGF21 was cloned into pGEX-4T-1 vector (GE Healthcare), and the plasmid was transformed into BL21-Gold (DE3) competent cells (Agilent Technologies). Transformants were grown in LB media containing 100 μg/mL ampicillin at 37˚C until A600 reached 0.6, and induced with 1 mM IPTG for 4 hours at 37˚C. Bacteria cells were collected, lysed in PBS using EmulsiFlex-C3 homogenizer (Avestin, Inc.), and centrifuged at 20,000 ×g for 30 minutes at 4˚C. The supernatant containing GST-FGF21CT was incubated with Glutathione Sepharose 4B (GE Healthcare) pre-equilibrated with PBS, for 1 hour at 4˚C. The beads were washed with 50 column volume of PBS and the protein was eluted with 20 mM HEPES, 150 mM NaCl, 10 mM reduced glutathione, pH 7.3. The protein solution containing GST-FGF21CT was then dialyzed against 20 mM HEPES, 150 mM NaCl before flash-freezing and storage at −80˚C. A peptide corresponding to the C-terminal region of FGF21 containing amino acids 174–209 with two substitutions, P199G and A208E, was synthesized and purified by the Tufts University Core Facility. The ligand binding region of FGFR1c was expressed in E.coli as an insoluble fraction. The protein was refolded and purified as previously described26.
Expression and purification of Nb914.
The plasmid harboring C-terminal His6-tagged Nb914 was transformed into E.coli strain WK6, and grown in TB media containing 0.1% glucose, 2 mM MgCl2, and 100 μg/ml ampicillin at 37˚C until the A600 of the sample was 1.2, and then induced with 1 mM IPTG for 4 hours. Cells were harvested and the periplasmic fraction was extracted using the modified osmotic shock protocol27. The periplasmic extract containing Nb914 was supplemented with 10 mM imidazole and incubated with Ni-NTA agarose (Qiagen) for 1 hour at 4˚C. The beads were washed with 50 column volumes of PBS containing 10 mM imidazole, and Nb914 was eluted from the resin with PBS containing 300 mM imidazole. The eluted fraction containing Nb914 were concentrated and injected into HiLoad 26/600 Superdex 200 (GE Healthcare) size exclusion chromatography column pre-equilibrated with PBS at pH 7.0. Purified Nb914 at concentration of 10 mg/mL was flash-frozen, and stored at −80˚C until further study.
Crystallization, X-ray diffraction data collection, and structure determination.
Purified sKLB or KLBD1 was mixed with Nb914, concentrated, and injected into Superdex 200 Increase 10/300 GL (GE Healthcare) size exclusion chromatography column pre-equilibrated with 20 mM HEPES, 150 mM NaCl, pH 7.0. Eluted fractions containing the complex were pooled, concentrated to 7 mg/mL, and screened for crystallization using Mosquito Crystal liquid handler (TTP Labtech). 96-well plates were incubated and imaged at 20˚C using Rock Imager 1000 (Formulatrix). sKLB in complex with Nb914 gave rod-shaped crystals when mixed with equal volume of well solution containing 14% PEG4000, 0.1 M MES, pH 6.0 and equilibrated for 10–15 days using the hanging drop vapor diffusion method. The crystals were cryopreserved by gradually transferring crystals to the mother liquor supplemented with 30% glucose before being flash-frozen in liquid nitrogen. KLBD1 in complex with Nb914 gave plate-like crystals when mixed with equal volume of well solution containing 30% PEG1000, 0.1 M HEPES pH 7.5 and equilibrated for 4–6 days using the hanging drop vapor diffusion method, which were directly flash-frozen in liquid nitrogen. For sKLB in complex with Nb914 and FGF21CT, FGF21CT was dissolved in 14% PEG4000, 0.1 M MES, pH 6.0 and added to the drop that contains sKLB. Addition of FGF21CT immediately caused deformation in most of the crystals. Crystals that stayed intact were gradually transferred into the artificial mother liquor, supplemented with 30% glucose and 50 μM FGF21CT before being flash-frozen in liquid nitrogen. X-ray diffraction data was collected at the beamlines BL-14 at the Stanford Synchrotron Radiation Lightsource, SLAC National Accelerator Laboratory, CA (for KLBD1 and sKLB) and 24-ID-E at the Advanced Photon Source, Argonne, IL (for sKLB in complex with FGF21CT). The diffraction data sets were processed using HKL200028 and XDS29. Initial phases for the data set for KLBD1 in complex with Nb914 were calculated by molecular replacement with PHASER30 using the coordinates of the cytosolic β-glucosidase (PDB: 2ZOX) and the coordinates of a nanobody that exhibits highest sequence identities with Nb914 (PDB: 5IMK, chain B) as the search models. Refinement was iteratively performed using PHENIX31 followed by manual model building using COOT32. The final coordinates of KLBD1 in complex with Nb914 was then used as a search model for the data set of sKLB in complex with Nb914, together with the coordinates for the KLBD1 as a search model for D2 of sKLB. Then the model was iteratively built and refined for sKLB. As for the data set for sKLB in complex with Nb914 and FGF21CT, initial phase information was obtained by molecular replacement using the final coordinates of sKLB in complex with Nb914, which were divided into two models each containing the coordinates for D1 with Nb914 and D2, and searched independently. Iterative cycles of refinement and rebuilding of sKLB model improved the phase, resulting significant electron densities for FGF21CT. Subsequently, the model for FGF21CT was manually built based on the |Fo|-|Fc| map followed by the final refinement cycles. The data collection and refinement statistics are summarized in Table S1. All the figures containing the structures were generated using the PyMOL Molecular Graphics System, Version 1.8 (Schrödinger, LLC.).
MicroScale Thermophoresis (MST) measurements.
All MST measurements were performed using the Monolith NT.115Pico instrument (NanoTemper Technologies) with Monolith NT.115 MST Premium Coated Capillaries. Purified FGF21 was fluorescently labeled using the Monolith Protein Labeling Kit RED-NHS (NanoTemper Technologies) according to the instruction provided by the manufacturer. Samples for binding affinity measurements of FGF21 to sKLB were prepared by mixing 35 nM of fluorescently labeled FGF21 (fl-FGF21) with a series of concentrations, ranging from 0.03 nM to 1000 nM, of purified sKLB in 20 mM HEPES, 150 mM NaCl, pH 7.0, 0.05% Tween-20, 1 mg/mL BSA. The thermophoretic movements of fl-FGF21 in each sample were monitored (LED 20%, IR laser 20%) and the normalized fluorescence intensities (FNorm), defined as Fhot/Fcold (where Fcold and Fhot refer to the fluorescence intensities averaged over 1s period before IR laser is on and 29 s after IR laser is on, respectively), for each samples were plotted against the concentrations of sKLB. For the competition assays, the thermophoresis of fl-FGF21 was measured for samples where the concentration of fl-FGF21 and sKLB mixture was kept constant with the concentrations of GST-FGF21CT varying from 2.1 nM to 35000 nM. All the data were analyzed with the MO.Affinity Analysis software (NanoTemper Technologies) provided by the manufacturer.
Surface Plasmon Resonance (SPR) measurements.
All SPR experiments were performed using a BIAcore T100 instrument (GE Healthcare) at 25 ˚C (Keck Foundation Biotechnology Resource Laboratory, Yale University) using HPBS+ buffer (GE Healthcare). Anti-GST antibody (GE Healthcare) was immobilized on a CM5 sensor chip using the instructions provided with the antibody, followed by capturing 50 RU of GST-FGF21CT. Subsequently, using the single-cycle kinetics method, a series concentration of sKLB, ranging from 25.6 nM to 1000 nM, was injected onto the surfaces with 360 s of association period, followed by the dissociation period of 1200 s. The binding kinetics were evaluated using BIAevaluation software (GE Healthcare).
Cell-based activity assays.
L6 cells stably co-expressing WT FGFR1c together with either WT β-Klotho or a variety of β-Klotho mutants, were grown in DMEM supplemented with 10% FBS, 100 U/mL Penicillin-Streptomycin, 0.1 mg/ml hygromycin and 1 μg/ml puromycin. Cells were starved overnight in DMEM with 0.5% FBS and stimulated for 10 minutes at 37˚C with either FGF1 or FGF21 at concentrations of 5 nM and 25 nM, respectively. Cells were then lysed and subjected to immunoprecipitation with anti-FGFR1 antibody followed by SDS-PAGE. The samples were then subjected to immunoblotting with either anti-phosphotyrosine (pTyr), anti-β-Klotho or anti-FGFR1 antibodies.
Statistics and Reproducibility
All of the immunoblots and binding affinity measurements presented in this work was repeated at least 3 times with similar results.
Data Availability
Coordinates and structure factors for the KLBD1:Nb914, sKLB:Nb914 and sKLB:FGF21CT:Nb914 complexes are deposited in the Protein Data Bank with the accession codes 5VAK, 5VAN, and 5VAQ, respectively.
Extended Data
Extended Data Figure 1 |. Expression, purification and crystallization of β-Klotho extracellular domain.
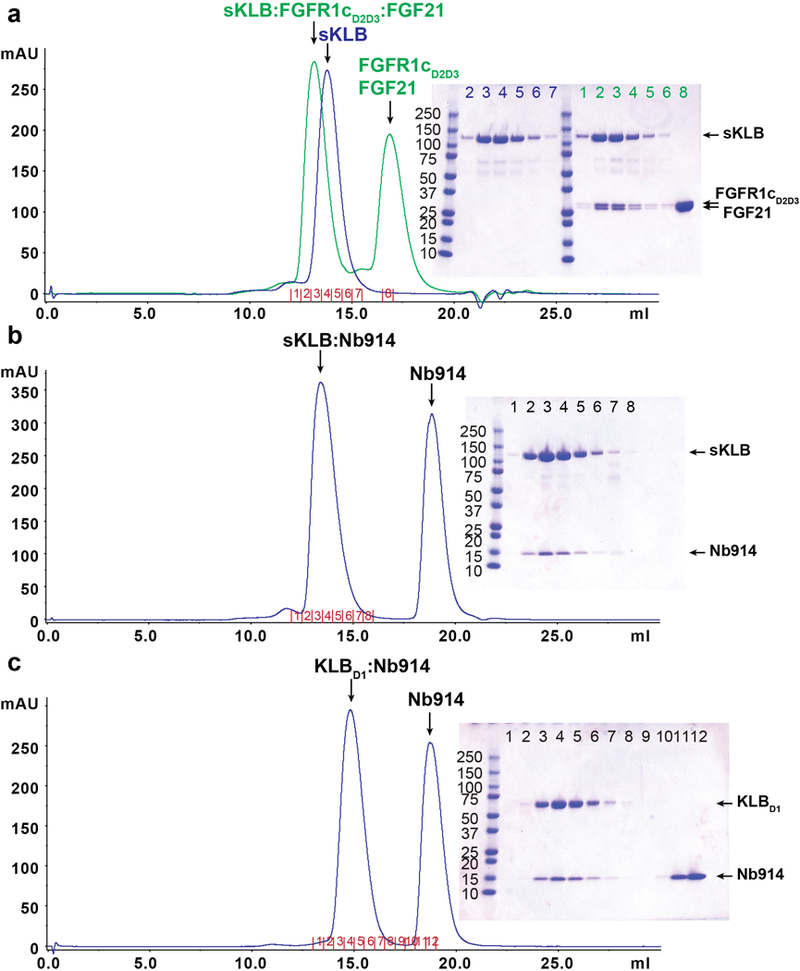
a-c, Size exclusion chromatography profiles and corresponding Coomassie-stained SDS-PAGE gels of (a) sKLB:FGFR1cD2D3: FGF21 ternary complex (green) or sKLB alone (blue), (b) sKLB in complex with Nb914, and (c) KLBD1 in complex with Nb914. The chromatograms and the SDS-PAGE gels shown are representatives of at least 3 independent preparations with similar results. A secreted protein composed of the extracellular domain of KLB fused to the Fc region of human IgG1 was produced by HEK293-EBNA cells. Following purification using a protein-A agarose resin the KLB-Fc fusion protein was subjected to proteolytic cleavage. sKLB was further purified using ion exchange and size exclusion chromatography. Multiple crystallization trials with the ternary complex formed by sKLB, FGF21 and FGFR1cD2D3 (a, green) failed to yield diffraction quality crystals. However, a preparation of sKLB bound to a nanobody, Nb914 (b), yielded crystals that diffracted X-rays to a resolution of 6–8 Å, and these were further improved by mutating two of the eleven potential N-glycosylation sites in sKLB (N308 and N611) to glutamines. The resulting crystals of an sKLB:Nb914 complex diffracted to a resolution of 2.2 Å. We also crystallized KLBD1 in complex with Nb914 (c), and collected data to a resolution of 1.7 Å. The structure of KLBD1 was first solved by molecular replacement using the coordinates of a structure of human cytosolic β-glucosidase (PDB: 2ZOX) and the coordinates of a nanobody structure (PDB: 5IMK, chain B) as search models. The structure of sKLB was subsequently determined by molecular replacement using the KLBD1 coordinates as a search model.
Extended Data Figure 2 |. Domain diagram of sKLB structure and the location of cysteine residues.
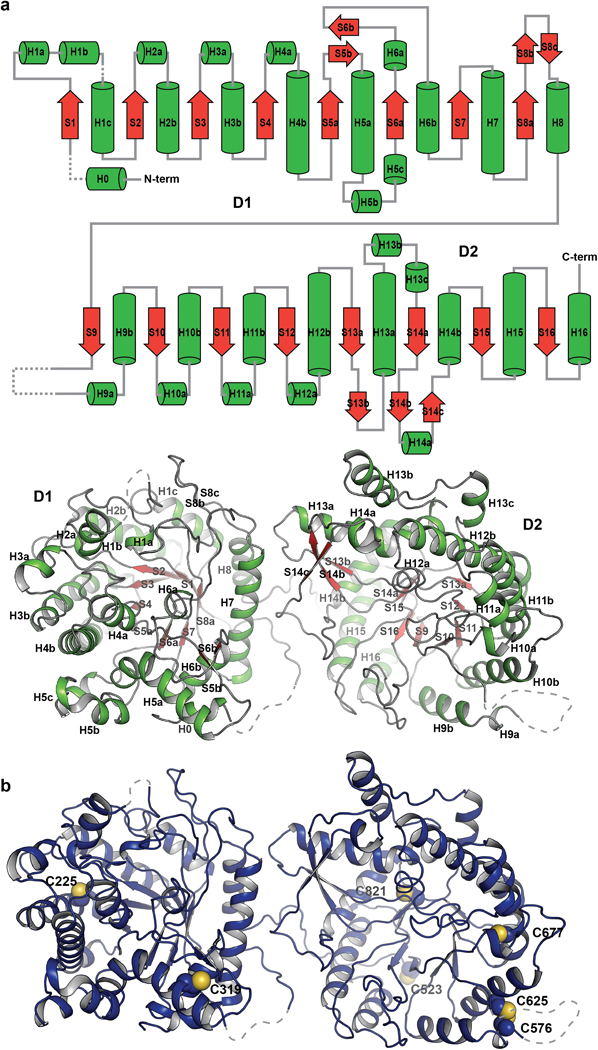
a, Secondary structure elements are designated, i.e., H for helix (green) and S for sheet (red), by numbers based on the principal elements for (β/α)8 fold. Disordered loops that are not modeled in the structure are depicted with dashed lines.
b, Seven of the 10 cysteines in the extracellular region were successfully modeled in the sKLB structure. With the exception of the disulfide bond between C576 and C625, the structure shows that these cysteines are reduced and do not form disulfide bridges. Moreover, determination of the distances between each pair of cysteines indicates that most are too far apart to form intra-molecular disulfide bonds. It cannot be ruled out, however, that C976 located in the C terminal region of sKLB (which could not be modeled due to weak electron density in this region) may form a disulfide bond with nearby C523. There is no evidence for formation of inter-molecular disulfide bonds between β-Klotho and the closely associated FGFR, FGF19 or FGF21 proteins whose cysteines all form well characterized intramolecular disulfide bonds. The functional consequences of the presence of reduced cysteines in β-Klotho are currently unknown.
Extended Data Figure 3 |. Unique structural features of sKLB.
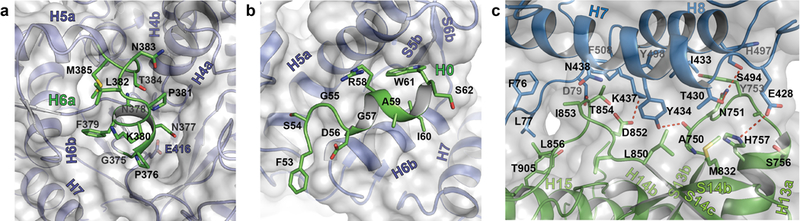
a, Interaction of H6a (green) with the pseudo-substrate binding pocket in D1 of sKLB. Glu416, the ‘catalytic’ glutamic acid residue in D1, located on the bottom of the pocket is also highlighted. b, Interaction of H0 (green) with the nearby structural elements in D1 of sKLB. c, Interface between D1 (skyblue) and D2 (green) of sKLB highlighting amino acids and structural elements as well as polar interactions (red dotted lines) between the domains.
Extended Data Figure 4 |. Detailed of interactions between sKLB and FGF21CT and conformational changes upon ligand binding.
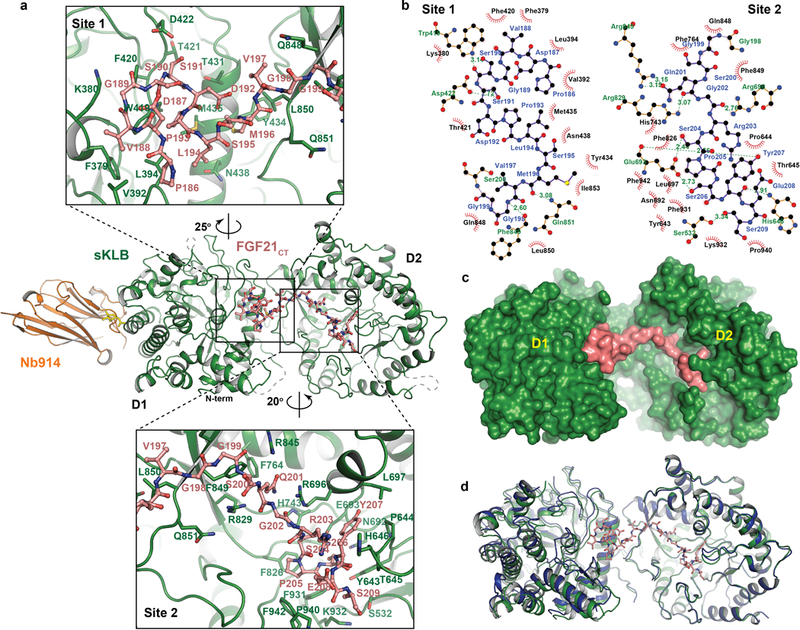
a, Amino acid residues interacting between sKLB (green) and FGF21CT (salmon) in site 1 and site 2 areas are indicated.
b, Diagram of amino acid-specific interactions between sKLB and FGF21CT within site 1 and site 2. The figure was generated using Ligplot+33.
c, Structure of sKLB (green) in complex with FGF21CT (salmon) shown as a surface representation.
d, Structure of ligand-free sKLB (blue) is overlaid onto the structure of sKLB (green) bound to FGF21CT (salmon, ball-and-stick).
Extended Data Figure 5 |. Amino acid sequence alignments of C-terminal regions of human FGF19 and FGF21.

Residues D-P, which are critical in maintaining multi-turn elements, are highlighted in light blue, and the sugar-mimicking motif, S-P-S, is highlighted in yellow. The sequence alignment reveals close sequence similarity between the C-terminal tails of FGF21 and FGF19 which is consistent with the similar binding characteristics of FGF21 and FGF19 and their isolated C-terminal regions to β-Klotho. Importantly, the sugar-mimicking motif in FGF21, S205-P206-S207, is conserved in FGF19 (S211-P212-S213). Also highlighted is the sequence D192-P193 in the region of FGF21CT that binds to site-1 of β-Klotho by stabilizing intramolecular hydrogen bonds that maintain a turn in the bound configuration of FGF21CT. This sequence is conserved in FGF19 (D198-P199), suggesting that similar intramolecular interactions responsible for mediating consecutive turns in FGF19CT may also similarly bind to site-1 of β-Klotho. Moreover, since many of the intramolecular interactions within FGF21CT bound to β-Klotho take place between main chain atoms (as observed in typical β-turn structures), only few key amino acid sequences such as D198-P199 may be sufficient for generating a similar multi-turn elements in FGF19CT as observed in the crystal structure of FGF21CT bound to β-Klotho.
Extended Data Figure 6 |. Validation of FGF21 binding interface to β-Klotho by ligand binding and cell stimulation experiments.
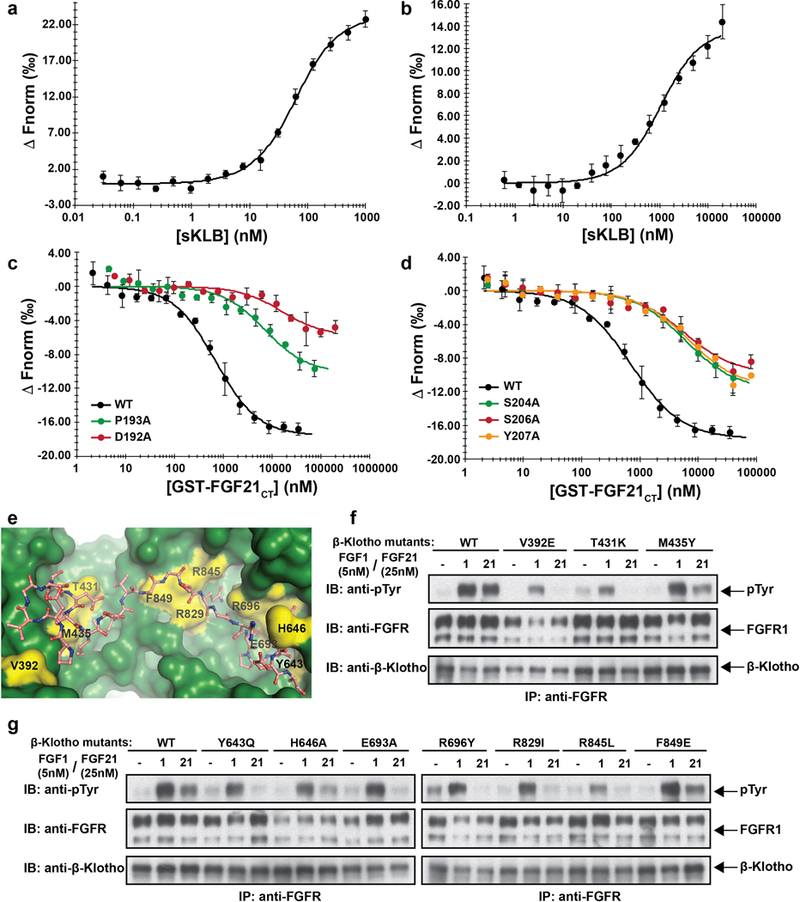
a, b, MST-based binding affinity measurements of (a) FGF21 to sKLB, and (b) FGFR1cD2D3 to sKLB, yielding KD = 43.5 ± 5.0 nM and KD = 940 ± 176 nM, respectively. c, d, MST-based competition assay with GST-FGF21CT containing mutations in either (c) site 1-interacting region or (d) site 2-interacting region. IC50 values for WT, 704 ± 96 nM; D192A, 15900 ± 6210 nM; P193A, 7160 ± 2350 nM; S204A, 5990 ± 1040 nM; S206A, 5560 ± 1590 nM; Y207A, 6630 ± 1570 nM.
The dots and error bars for each graph in panels a-d denote means and variations of ΔFnorm (n = 3 independent samples). Individual experimental data are plotted in Supplementary Fig. 2.
e, Location of mutated amino acid residues (yellow) in sKLB (green) occupied by FGF21 (salmon) that were analyzed in panels f and g. f, g, Stably transfected L6 cells co-expressing FGFR1c together with WT or β-Klotho mutants were stimulated with either FGF21 or FGF1 (control) and analyzed for FGFR1c activation by monitoring tyrosine phosphorylation of FGFR1c. Lysates of ligand stimulated or unstimulated cells were subjected to immunoprecipitation with anti FGFR1 antibodies followed by immunoblotting with either anti-pTyr or anti-FGFR1 antibodies.
Extended Data Figure 7 |. β-Klotho is required for FGFR1c-mediated signaling induced by FGF21.
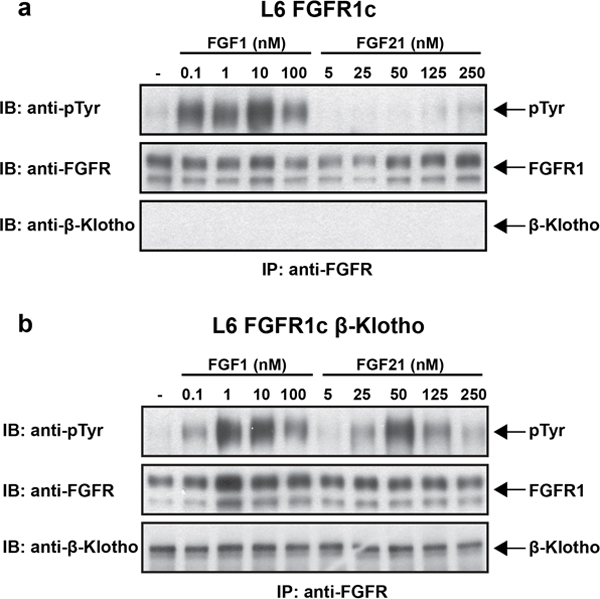
a, b, L6 cells expressing either FGFR1c alone (a) or FGFR1c together with β-Klotho (b) were stimulated with various concentrations of FGF1 or FGF21 and phosphotyrosine (pTyr) levels of FGFR are monitored by immunoprecipitation with anti FGFR1 antibodies followed by immunoblotting with anti-pTyr antibodies.
Extended Data Figure 8 |. MAP kinase stimulation induced by WT FGF21 or by the FGF21WF mutant.
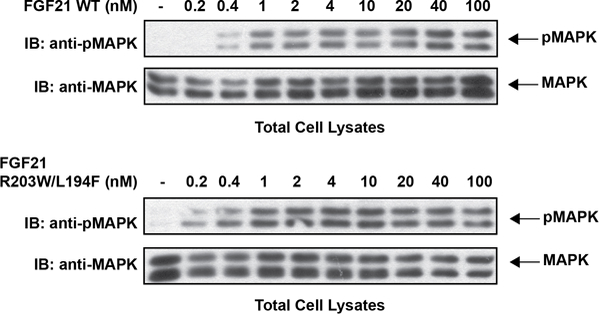
L6 cells co-expressing β-Klotho and FGFR1c were stimulated with wild-type FGF21 (upper panel) or FGF21WF (lower panel) and phosphorylation levels of MAP kinase in cell lysates were monitored.
Extended Data Figure 9 |. MST data with individual data points.
Figures containing each data are indicated.
Extended Data Table 1 |.
Crystallographic data collection and refinement statistics
| KLBD1:Nb914 | sKLB:Nb914 | sKLB:Nb914:FGF21CT | |
|---|---|---|---|
| Data collection | |||
| Space group | C2 | P212121 | P212121 |
| Cell dimensions | |||
| a, b, c (A) | 229.43, 49.35, 54.31 | 48.68, 144.07, 215.61 | 48.65, 145.49, 213.83 |
| α, β, γ (°) | 90, 100.22, 90 | 90, 90, 90 | 90, 90, 90 |
| Resolution (Å) | 41.27–1.70 (1.76–1.70) | 47.49–2.20 (2.28–2.20) | 60.14–2.61 (2.70–2.61) |
| Rmerge | 0.0546 (0.441) | 0.109 (0.880) | 0.0905 (1.289) |
| CC1/2 (%) | 99.9 (86.4) | 99.7 (76.3) | 99.7 (43.1) |
| <1/σ> | 20.36 (2.87) | 20.13 (2.71) | 13.12 (1.03) |
| Completeness (%) | 98 (87) | 100 (97) | 98 (96) |
| Redundancy | 4.2 (3.4) | 7.1 (6.9) | 3.9 (3.3) |
| Refinement | |||
| Resolution (Å) | 41.27–1.70 | 47.49–2.20 | 60.14–2.61 |
| No. of reflections used | 65178 | 77784 | 46521 |
| Rwork / Rfree (%) | 17.16 / 19.64 | 18.62 / 21.06 | 19.11 / 22.89 |
| No. of atoms | |||
| Protein | 4453 | 7731 | 7874 |
| Ligands | 42 | 125 | 13 |
| Waters | 271 | 112 | 0 |
| Average B-factors | |||
| Protein | 26.11 | 43.00 | 63.42 |
| Ligands | 45.67 | 65.37 | 68.90 |
| Waters | 30.97 | 39.59 | n/a |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.006 | 0.008 | 0.012 |
| Bond angle (°) | 0.82 | 0.95 | 1.30 |
Supplementary Material
Acknowledgments
The NSLS-SSRL is supported by P41GM111244, P41GM103393, DE-SC0012704 and by DE-AC02–76SF00515. We also thank NE-CAT (P41 GM103403) and APS (DE-AC02–06CH11357) and NIH grant 1S10OD018007. J.S. thank INSTRUCT, ESFRI, FWO) for financial support and Iman Aboutaleb for the technical assistance.
Footnotes
The authors declare no competing financial interests.
Supplementary Information is available in the online version of the paper.
REFERENCES
- 1.Eswarakumar VP, Lax I & Schlessinger J Cellular signaling by fibroblast growth factor receptors. Cytokine Growth Factor Rev 16, 139–149, doi:10.1016/j.cytogfr.2005.01.001 (2005). [DOI] [PubMed] [Google Scholar]
- 2.Belov AA & Mohammadi M Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol 5, doi:10.1101/cshperspect.a015958 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogawa Y et al. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc Natl Acad Sci U S A 104, 7432–7437, doi: 10.1073/pnas.0701600104 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Urakawa I et al. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 444, 770–774, doi: 10.1038/nature05315 (2006). [DOI] [PubMed] [Google Scholar]
- 5.Owen BM, Mangelsdorf DJ & Kliewer SA Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol Metab 26, 22–29, doi: 10.1016/j.tem.2014.10.002 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koshland DE STEREOCHEMISTRY AND THE MECHANISM OF ENZYMATIC REACTIONS. Biological Reviews 28, 416–436, doi: 10.1111/j.1469-185X.1953.tb01386.x (1953). [DOI] [Google Scholar]
- 7.Holm L & Rosenstrom P Dali server: conservation mapping in 3D. Nucleic Acids Res 38, W545–549, doi: 10.1093/nar/gkq366 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Krissinel E & Henrick K Inference of macromolecular assemblies from crystalline state. J Mol Biol 372, 774–797, doi: 10.1016/j.jmb.2007.05.022 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Hayward S & Lee RA Improvements in the analysis of domain motions in proteins from conformational change: DynDom version 1.50. J Mol Graph Model 21, 181–183 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Laskowski RA & Swindells MB LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51, 2778–2786, doi: 10.1021/ci200227u (2011). [DOI] [PubMed] [Google Scholar]
- 11.Yie J et al. FGF21 N-and C-termini play different roles in receptor interaction and activation. FEBS Lett 583, 19–24, doi: 10.1016/j.febslet.2008.11.023 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Micanovic R et al. Different roles of N-and C-termini in the functional activity of FGF21. J Cell Physiol 219, 227–234, doi: 10.1002/jcp.21675 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Hecht R et al. Rationale-Based Engineering of a Potent Long-Acting FGF21 Analog for the Treatment of Type 2 Diabetes. PLoS One 7, e49345, doi: 10.1371/journal.pone.0049345 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhen EY, Jin Z, Ackermann BL, Thomas MK & Gutierrez JA Circulating FGF21 proteolytic processing mediated by fibroblast activation protein. Biochem J 473, 605–614, doi: 10.1042/BJ20151085 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dunshee DR et al. Fibroblast Activation Protein Cleaves and Inactivates Fibroblast Growth Factor 21. J Biol Chem 291, 5986–5996, doi: 10.1074/jbc.M115.710582 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coppage AL et al. Human FGF-21 Is a Substrate of Fibroblast Activation Protein. PLoS One 11, e0151269, doi: 10.1371/journal.pone.0151269 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chuenchor W et al. The structural basis of oligosaccharide binding by rice BGlu1 beta-glucosidase. J Struct Biol 173, 169–179, doi: 10.1016/j.jsb.2010.09.021 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Isorna P et al. Crystal structures of Paenibacillus polymyxa beta-glucosidase B complexes reveal the molecular basis of substrate specificity and give new insights into the catalytic machinery of family I glycosidases. J Mol Biol 371, 1204–1218, doi: 10.1016/j.jmb.2007.05.082 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Degirolamo C, Sabba C & Moschetta A Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat Rev Drug Discov 15, 51–69, doi: 10.1038/nrd.2015.9 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Kharitonenkov A et al. Rational design of a fibroblast growth factor 21-based clinical candidate, LY2405319. PLoS One 8, e58575, doi: 10.1371/journal.pone.0058575 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang Z et al. A better anti-diabetic recombinant human fibroblast growth factor 21 (rhFGF21) modified with polyethylene glycol. PLoS One 6, e20669, doi: 10.1371/journal.pone.0020669 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang J et al. Development of a novel long-acting antidiabetic FGF21 mimetic by targeted conjugation to a scaffold antibody. J Pharmacol Exp Ther 346, 270–280, doi: 10.1124/jpet.113.204420 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Foltz IN et al. Treating diabetes and obesity with an FGF21-mimetic antibody activating the betaKlotho/FGFR1c receptor complex. Sci Transl Med 4, 162ra153, doi: 10.1126/scitranslmed.3004690 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Luo J et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci Transl Med 6, 247ra100, doi: 10.1126/scitranslmed.3009098 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Kolumam G et al. Sustained Brown Fat Stimulation and Insulin Sensitization by a Humanized Bispecific Antibody Agonist for Fibroblast Growth Factor Receptor 1/betaKlotho Complex. EBioMedicine 2, 730–743, doi: 10.1016/j.ebiom.2015.05.028 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schlessinger J et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell 6, 743–750 (2000). [DOI] [PubMed] [Google Scholar]
- 27.Pardon E et al. A general protocol for the generation of Nanobodies for structural biology. Nat Protoc 9, 674–693, doi: 10.1038/nprot.2014.039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Otwinowski Z & Minor W Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 29.Kabsch W Xds. Acta Crystallogr D Biol Crystallogr 66, 125–132, doi: 10.1107/S0907444909047337 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCoy AJ et al. Phaser crystallographic software. J Appl Crystallogr 40, 658–674, doi: 10.1107/S0021889807021206 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Adams PD et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221, doi: 10.1107/S0907444909052925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Emsley P, Lohkamp B, Scott WG & Cowtan K Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66, 486–501, doi: 10.1107/S0907444910007493 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Extended Data References
- 33.Laskowski RA & Swindells MB LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model 51, 2778–2786, doi:10.1021/ci200227u (2011). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Coordinates and structure factors for the KLBD1:Nb914, sKLB:Nb914 and sKLB:FGF21CT:Nb914 complexes are deposited in the Protein Data Bank with the accession codes 5VAK, 5VAN, and 5VAQ, respectively.