

Abstract
Background:
The importance of B lymphocytes to present antigens for antibody production is well documented. In contrast, very little is known about their capacity to influence CD4+ T-cell activation during a primary or secondary response to allergens.
Objective:
Using mouse models of asthma, we investigated the role of B cells as antigen-presenting cells in priming and maintenance of TH cell responses.
Methods:
Mice were immunized through the intranasal route with house dust mite (HDM) extract derived from Dermatophagoides pteronyssinus. B cells were depleted in HDM-sensitized animals to investigate the importance of B cells in maintenance of the allergic response. B cells were depleted before HDM sensitization to investigate the role of B cells in T-cell priming; furthermore, HDM sensitization was performed in mice with MHC class II expression restricted to the B-cell lineage.
Results:
We found that B cells serve as potent antigen-presenting cells ex vivo and restimulate in vivo–primed HDM-specific TH cells. HDM antigens were taken up by B cells independently of B-cell receptor specificity, indicating that HDM uptake and antigen presentation to CD4+ T cells is not restricted to rare B cells carrying HDM-specific B cell receptors. B-cell depletion before HDM challenge in HDM-sensitized mice resulted in a dramatic reduction of allergic response, indicating the role of B cells in amplification of TH2 responses. In contrast, HDM sensitization of mice in which MHC class II expression was restricted to B cells revealed the inability of these cells to prime TH2 responses but highlighted their unexpected role in priming TH1 and TH17 responses.
Conclusion:
Collectively, these data reveal new mechanisms leading to initiation and exacerbation of the allergic response that might have implications for designing new therapeutic strategies to combat HDM allergy.
Keywords: Mouse model of asthma, house dust mite, B cells, antigen presentation, initiation of allergic response, maintenance of allergic response
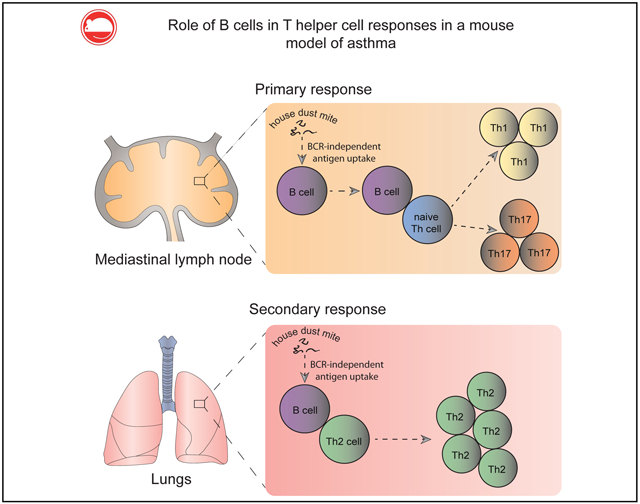
GRAPHICAL ABSTRACT
Allergy is a major problem in industrialized countries nowadays, and its prevalence is on the increase. According to the World Health Organization, asthma is estimated to affect around 300 million persons worldwide, with 250,000 deaths per year caused by the disease.1 The hallmark of this disorder is a strong TH2 response with upregulated levels of IL-4, IL-5, and IL-13, which lead to enhanced IgE and IgG1 production and cell recruitment to the site of allergen entry. Antibodies of the IgE class, by binding to high-affinity FcεRI on mast cells and basophils, cause immediate hypersensitivity reactions on antigen re-exposure. This reaction is mediated by preformed mediators (eg, histamine, serine proteases, carboxypeptidase A, and proteoglycans), newly sensitized lipid mediators (eg, leukotriene [LT] C4, LTD4, and LTE4), and cytokines/chemokines2 that act in concert with responses by other cell types and lead to exaggerated immune response and tissue damage.
The majority of mouse studies concerning allergic responses have used ovalbumin (OVA) as a model antigen. The main advantages of these approaches came with development of T-cell receptor (TCR)–transgenic OT-I and OT-II mice, which carry OVA-specific CD8+ and CD4+ T cells, respectively. These tools opened several new avenues to explore immune responses, such as tracking of OVA-specific T-cell migration or differentiation. Also, use of OVA narrowed immune responses to a single antigen and allowed easy isolation of high numbers of antigen-specific cells, creating a “neat” system to interpret experimental data. Early work using the OVA model provided evidence that B cells are not necessary for allergy development,3-5 and subsequent work showed that dendritic cells (DCs) play a nonredundant role in maintenance of allergic responses in vivo.6 However, although allergies to OVA can occur in real life, they are far rarer than allergies to other environmental antigens, such as birch pollen, cockroach, or house dust mites (HDMs). In contrast to these physiologically relevant allergens, OVA is a single protein, does not display enzymatic activity, and does not carry microbial load. Therefore B-cell responses to OVA might not reflect those to complex, clinically relevant allergens. For these reasons, studies using the latter have recently started to emerge, leading to important insights into the mechanisms behind allergic inflammation. For example, by using HDM extracts, central roles for epithelial cells, pulmonary DCs, and type 2 innate lymphoid cells in orchestrating allergic sensitization have been unraveled.7 In addition, the role of B cells in HDM-induced responses has been re-evaluated.8-10 However, although some studies indicate that B cells are necessary during HDM sensitization but dispensable during challenge,9 others indicate that B cells are dispensable for T-cell priming but necessary for effector CD4+ T-cell expansion.10
In this study we evaluated the role of B cells as antigen-presenting cells (APCs) in primary and recall responses by using experimental models of HDM-induced allergy. We found that in contrast to the OVA model, B cells display a unique capacity to take up HDM antigens in vivo independently of their B-cell receptor (BCR) specificity. Also, they acted as potent APCs ex vivo and efficiently restimulated in vivo–primed HDM-specific effector/memory TH cells. By depleting B cells before HDM challenge in HDM-sensitized mice, we observed a pronounced reduction in the allergic response, manifested by reduced numbers of effector/memory TH cells, eosinophils, and neutrophils in the lungs, as well as impaired capacity of effector/memory TH cells in the lungs to secrete TH2-associated cytokines on ex vivo restimulation. By depleting B cells before HDM sensitization, we observed that they could induce TH1 rather than TH2 responses. Finally, with the use of a mouse strain in which MHC class II expression was restricted to the B-cell linage, we provide evidence that B cells are capable of initiating TH1 and TH17 but not TH2 responses against HDM in vivo.
Overall, our data are consistent with previous reports underlining the importance of B cells in driving allergic inflammation. Additionally, we provide new evidence on the dual role of B cells depending on the stage of allergic reaction, which might help explain some of the controversies in the field.
METHODS
Mice
C57BL/6 and BALB/c inbred mice (wild-type [WT]) were purchased from Harlan Italy Srl (Udine, Italy) and Envigo RMS Srl (Udine, Italy), respectively. B-MHC-II and Flox mice were provided by Dr Gregory Wu (Washington University, St Louis, Mo) and have been described previously.11 MD4 (002595) mice were obtained from the Jackson Laboratory (Bar Harbor, Me). All mice were bred and maintained under specific pathogen-free conditions. Six- to 8-week-old female mice were used for experiments. They were treated in accordance with guidelines of the Swiss Federal Veterinary Office, and the experiments were approved by the Dipartimento della sanità e della socialità of Canton Ticino.
Antigens and immunizations
HDM extract from Dermatophagoides pteronyssinus (Greer Laboratories, Lenoir, NC) and endotoxin-free OVA protein (Hyglos, Bernried am Starnberger See, Germany) were resuspended in PBS (Sigma-Aldrich, St Louis, Mo). Low-molecular-weight compounds, such as peptides, were excluded from the HDM extract with use of PD-10 desalting columns (GE Healthcare, Fairfield, Conn). Before intranasal administration, mice were anesthetized with isoflurane (4% in air) for 5 minutes and treated with 20 μg of HDM resuspended in 20 μL of PBS. As a negative control, 20 μL of PBS was administered. Solutions were applied on days 0, 7, 8, 9, 10, and 11, and mice were killed on day 14. Alternatively, mice were immunized on days 0, 11, 12, and 13 and killed on day 14. One hundred micrograms of HDM in 33 μL of PBS was used to study priming of T-cell responses. As a negative control, 33 μL of PBS was applied. Mice were immunized on days 0 and 1 and killed on day 7.
In some experiments mAb (clone 18B12) against murine CD20 was introduced (250 μg administered intravenously plus 130 μg administered intranasally) 2 days before or 9 days after HDM sensitization to deplete B lymphocytes. As a control, isotype-matched control antibody against human CD20 (clone 2B8) was administrated in the same manner. In some experiments HDM or OVA proteins were labeled with the Alexa Fluor (AF) 647 Labeling Kit (Invitrogen, Carlsbad, Calif), eluted with PBS, and administered intranasally at a dose of 20 μg.
CD4+ T-cell transfer
Spleens and mesenteric lymph nodes (Mes-LNs) were collected from naive WT C57BL/6 mice and smashed through a 40-μm nylon cell strainer (Falcon; Thermo Fisher Scientific, Waltham, Mass) to obtain a homogenous suspension. CD4+ T cells were isolated with the CD4+ T Cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany), according to the manufacturer’s instructions. Cell purity was confirmed by using flow cytometry and was always greater than 97%. Cells (107) were injected intravenously into Flox and B-MHC-II mice 15 days before HDM immunization to reconstitute the CD4+ T-cell compartment.
Bronchoalveolar lavage fluid, lungs, and lymph node collection
Bronchoalveolar lavage (BAL) for cytokine measurement was performed with 1 mL of PBS. BAL fluid was spun down, and supernatants were collected and stored at −20°C until further processing. Lungs were perfused with 10 mL of PBS before collection, finely cut with scissors, and digested for 1 hour at 37°C in a solution of Collagenase D (Sigma-Aldrich) at a concentration of 2 mg/mL and DNAse I (Sigma-Aldrich) at a concentration of 0.1 mg/mL in PBS. This was followed by smashing of lung pieces through a 40-μm nylon cell strainer. Cell suspensions were washed twice with MACS buffer before downstream applications. Mediastinal lymph nodes (MLNs) were collected, smashed through a 40-μm nylon cell strainer, washed once with MACS buffer, and used for downstream applications.
Cell sorting
For sorting of lung CD4+ T cells, B cells, and DCs, lung cell suspensions were incubated with anti-CD4 microbeads (clone L3T4; Miltenyi Biotec), anti-CD19 beads (Miltenyi Biotec), and anti-CD11c microbeads (Miltenyi Biotec) and isolated with LS columns (Miltenyi Biotec), according to the manufacturer’s guidelines. This was followed by sorting on a FACSAria III cell sorter. Activated CD4+ T cells were sorted as CD4+CD44+CD11c−Siglec-F−. Lung B cells were sorted as CD19+B220highCD11c−CD4−. DCs were sorted as CD11c+Siglec-F−CD4−. For sorting cells from MLNs, cell suspensions were stained directly with antibody cocktail and sorted on a FACSAria III cell sorter. Total CD4+ T cells were sorted as CD4+B220−CD8−CD11c−, and DCs were sorted as CD11c+B220−CD8−CD4−.
Flow cytometry
The following antibodies or streptavidin coupled to biotin, peridinin-chlorophyll-protein–Cy5.5, fluorescein isothiocyanate, AF488, allophycocyanin, AF647, Pacific Blue, Pacific Orange, allophycocyanin-Cy7, phycoerythrin, and phycoerythrin-Cy7 were purchased from BioLegend (San Diego, Calif), eBioscience (San Diego, Calif), BD Biosciences (San Jose, Calif), or Invitrogen (Carlsbad, Calif): CD19 (clone 6D5), B220 (RA3-6B2), CD3 (145-2C11), CD4 (RM4-5), CD11c (N418), CD8α (53-6.7), CD45.1 (A20), CD45.2 (104), CD44 (IM7), CXCR5 (2G8), MHC class II (M5/114.15.2), Siglec-F (E50-2440), CD11b (M1/70), CCR3 (TG14/CCR3), Gr-1 (RB6-8C5), CD80 (16-10A1), CD86 (GL1), CD40 (3/23), OX40 ligand (RM134L), CD43 (S7), CD5 (53-7.3), IgM (R6-60.2), and IgD (11-26c). Dead cells were excluded with 7-aminoactinomycin D (eBioscience). Flow cytometry was performed on either a FACSCanto II or BD Fortessa (BD Biosciences), and data were analyzed with FlowJo software (TreeStar, Ashland, Ore).
Ex vivo experiments
For evaluation of B-cell capacity to restimulate in vivo–primed T cells, 20,000 lung CD4+CD44+ T cells were cocultured with 20,000 APCs in the presence or absence of HDM at a concentration of 40 μg/mL or in the presence of titrated doses of HDM (40, 13.3, 4.44, and 1.48 μg/mL). In experiments evaluating the capacity of B cells to restimulate in vivo–primed T cells in the absence of exogenous antigens, 100,000 lung B cells from HDM-immunized mice or 100,000 lung B cells from naive mice were cocultured with 100,000 lung CD4+CD44+ T cells from HDM-immunized mice. In experiments with B-cell depletion before challenge, 500,000 total lung cells were plated in the presence of HDM (40 μg/mL) for 4 days. In the same experiments 10,000 lung CD4+CD44+ T cells were cocultured with 10,000 lung DCs in the presence of HDM (40 μg/mL). In experiments addressing CD4+ T-cell priming, 10,000 CD4+ T cells from MLNs or 10,000 lung CD4+ T cells were cocultured with 3,000 MLN DCs or lung DCs, respectively. In some experiments T cells were labeled with carboxyfluorescein succinimidyl ester (CFSE) before plating. CFSE (Thermo Fisher) labeling was performed at a concentration of 2 μmol/L in 1% FBS (Gibco, Thermo Fisher) for 7 minutes at 37°C. Cells were subsequently washed twice and plated. In each ex vivo experiment, cells were washed and cultured in RPMI medium supplemented with 10% FBS (Gibco, Thermo Fisher), 1% nonessential amino acids, 1% sodium pyruvate, 1% penicillin/streptomycin, 2 mmol/L glutamine (all from Life Technologies), and 0.1% 2-mercaptoetanol (Lubio Science, Lucerne, Switzerland). All cultures were incubated in a 96-well round-bottom plates (Costar, Cambridge, Mass) at 37°C in a 5% CO2 atmosphere for 4 days, and the supernatant was collected for analysis. In some experiments proliferation of T cells was assessed on a FACSCanto II or BD Fortessa based on CFSE dilution.
Cytokine and immunoglobulin measurement
Cytokine measurements were performed with the Mouse Th1/Th2/Th17/Th22 13plex Multiplex Kit (eBioscience). Alternatively, Ready-Set-Go kits for IL-4, IL-5, IL-10, IL-13, IL-17, and IFN-γ (eBioscience) were used. In all cases protocols were according to the manufacturer’s guidelines. HDM-specific immunoglobulin levels were measured by using ELISA. Ninety-six-well half-area plates (Costar) were coated with 10 μg/mL HDM in PBS overnight at 4°C. Plates were blocked for 2 hours at room temperature in 1% BSA (Sigma-Aldrich) in PBS and serial dilutions of sera incubated for 2 hours at room temperature. Alkaline phosphate–labeled anti-mouse IgE (South-ernBiotech, Birmingham, Ala) were incubated for 1 hour at room temperature, followed by addition of pNPP substrate (Sigma-Aldrich). Absorbance was measured at 405-nm wavelength with the use of microplate reader (Molecular Devices, Sunnyvale, Calif).
OVA model of asthma
The protocol of OVA-induced asthma was adapted from Swedin et al.12 Briefly, mice were sensitized on days 0 and 7 through intraperitoneal injection of 10 μg of OVA protein in 1 mg of ImjectAlum (Thermo Fisher) and challenged with 20 μg of OVA protein in PBS on days 14, 15, and 16. Mice were killed for experiments on day 17.
Statistics
Data were plotted and analyzed with Prism 6 software (GraphPad Software, La Jolla, Calif). When comparing 2 groups, the unpaired 2-tailed Student t test (for data displaying Gaussian distribution) or Mann-Whitney test (non-Gaussian distribution) was used. When comparing more than 2 groups, 1-way ANOVA (Gaussian distribution) or the Kruskal-Wallis test (non-Gaussian distribution) with correction for multiple comparisons (Tukey test) was used. Data distribution was assessed by using the D’Agostino-Pearson omnibus normality test. Graphs show means ± SEMs.
RESULTS
B cells are present in the lungs at steady state and after HDM immunization and outnumber DCs
In this study we used a mouse model of allergic sensitization in which BALB/c mice are repeatedly exposed to allergens administered intranasally. Mice were treated with PBS or HDM on days 0, 7, 8, 9, 10, and 11 and killed on day 14 (referred to hereafter as HDM-sensitized mice and PBS-treated mice) to analyze the abundance of the B-lymphocyte population in the lungs at steady state and on allergen sensitization. Lungs were collected and processed for flow cytometric analysis. B cells constituted 20% to 25% of CD45+ cells in the lungs of both groups of mice (Fig 1, A). On HDM sensitization, the total number of cells in the lungs increased, as did the number of B cells (Fig 1, B), which is consistent with other models of allergic inflammation.10,13,14 Importantly, both in PBS-treated and HDM-sensitized mice, B cells outnumbered DCs. The majority of B cells present in the lungs were CD19+CD43− B-2 cells, whereas CD19highCD43+ B-1 cells15 constituted around 8% of total B cells (Fig 1, C and D). Interestingly, most B-2 cells were of the naive phenotype (IgD+IgMlo) both in PBS-treated mice and mice sensitized with HDM (Fig 1, E and F). Consistent with their localization in nonlymphoid tissue, lung B cells expressed reduced levels of CXCR5 compared with circulating, splenic, or MLN B cells (Fig 1, G and H).
FIG 1.

B cells are present in the lungs and outnumber DCs. A and B, BALB/c mice were sensitized intranasally with HDM or treated with PBS as a control on days 0, 7, 8, 9, 10, and 11 and analyzed on day 14. Representative fluorescence-activated cell sorting (FACS) plots of B cells among total lung CD45+ cells (Fig 1, A) and total numbers of Bcells (CD45+CD19+B220+) and DCs (CD45+CD11c+Siglec-F−) in lungs(mean ± SEM, n = 4 mice per group; Fig 1, B). C and D, Representative FACS plots of B-1 cells (CD19hiCD43+) among total B cells (CD45+CD19+B220+) in blood, spleen, and lungs of a naive mouse (Fig 1, C) and cumulative data of percentage (mean ± SEM) of B-1 and B-2 cells from 8 mice (Fig 1, D). E and F, Representative FACS plots of IgM/IgD staining of lung B cells from PBS- or HDM-treated mice (Fig 1, E) and cumulative data (mean ± SEM) from 7 PBS-treated and 13 HDM-treated animals (Fig 1, F). IgM−IgD− B cells consisted mainly of IgG+ cells (around 73% in the PBS group and 78% in the HDM group). Additionally, around 19% of IgM−IgD− B cells in both groups were IgA+1 B cells. We found virtually no IgE+ B cells at steady state or on HDM immunization, whereas IgG1+ B cells were present at steady state and increased in frequency on allergic inflammation (9% and 22%, respectively). G and H, Surface CXCR5 expression on B cells isolated from the indicated organs of naive mice. Representative FACS plot (Fig 1, G) and cumulative data (mean ± SEM) from 8 mice (Fig 1, H) are shown. Data are representative of at least 3 independent experiments. *P < .05 and ****P < .0001.
Lung B cells efficiently capture HDM antigens in vivo independently of their BCR specificity
To assess whether B cells residing in the lungs could efficiently capture HDM antigens, we sensitized mice with HDM on days 0, 11, and 12 and challenged them on day 13 with fluorescently labeled HDM (HDM-AF647). Twenty-four hours after challenge, 3.4% to 5.2% of lung B cells stained positively for HDM-AF647 (Fig 2, A and B). In the same samples a large proportion of lung DCs (7.4% to 21.1%) but only a few lung CD4+ T cells (0.3% to 1.5%) stained positive. Given the high abundance of B cells in the lungs, total numbers of HDM-AF647+ B cells and HDM-AF647+ DCs were comparable. Uptake of HDM was observed also in B cells from MD4 mice, which carry rearranged heavy and light chain transgenes encoding for a hen egg lysozyme–specific mAb (Fig 2, C and D),16 indicating that this process did not rely on BCR specificity. However, lung B cells from BALB/c mice were less efficient in taking up fluorescently labeled OVA (OVA-AF647) compared with lung DCs, even when coinjected with unlabeled HDM (Fig 2, E-G), indicating that efficient antigen internalization by B cells is a property characteristic of the antigenic features of HDM extract but not OVA.
FIG 2.

Lung B cells efficiently capture HDM antigens independently of their BCR specificity. A and B, BALB/c mice were sensitized intranasally with HDM on days 0, 11, and 12 and received AF647-labeled HDM on day 13. Percentages of HDM-AF647+ B cells, DCs, and CD4+ T cells in the lungs were measured on day 14. Representative fluorescence-activated cell sorting (FACS) plots (Fig 2, A) and cumulative percentages and total cell numbers (mean ± SEM) from 5 mice (Fig 2, B) are shown. C and D, C57BL/6 or BCR-transgenic MD4 mice (on C57BL/6 background) were treated intranasally with HDM or HDM-AF647 on day 0 and were analyzed 1 day later. Representative FACS plots (Fig 2, C) and percentages and geometric mean fluorescence intensity (GMFI; mean ± SEM) of lung HDM-AF647+ B cells from 4 mice per group (Fig 2, D) are shown. E and F, BALB/c mice were treated intranasally with OVA-AF647 on day 0 and analyzed 1 day later. Representative FACS plots (Fig 2, E) and percentages and total numbers of OVA-AF647+ cells (mean ± SEM) in lungs from 4 animals (Fig 2, F) are shown. G, BALB/c mice were treated intranasally with HDM-AF488 or OVA-AF647 plus unlabeled HDM and analyzed 1 day later. Shown is the percentage of fluorescence-positive cells (mean ± SEM) in lungs from 3 (HDM) and 4 (OVA+HDM) animals. Data are representative of at least 2 independent experiments. **P < .01 and ***P < .001.
B cells serve as potent APCs ex vivo
Given the efficiency of B cells in HDM uptake, we investigated whether they could serve as APCs for effector/memory T cells. Mice were sensitized with HDM on days 0, 7, 8, 9, 10, and 11, and lungs were collected on day 14. Lung CD4+CD44+ T cells were isolated and cultured with lung B cells (HDM B cells) or lung DCs (HDM DCs) from HDM-sensitized mice or splenic B cells from PBS-treated mice (CTRL B cells) in the absence or presence of HDM extract. In these experiments we excluded the potentially confounding presence of peptides in the HDM extract through size exclusion chromatography. Interestingly, B cells from different sources were equally efficient in presenting HDM antigens and in inducing cytokine production by CD4+CD44+ T cells, and their efficacy was similar (in the case of IL-4) or even higher (in the case of IL-5 and IL-13) than that of lung DCs (Fig 3, A). However, in contrast to DCs, B cells were unable to restimulate effector CD4+ T cells to produce IL-17 (Fig 3, A).
FIG 3.

B cells efficiently present HDM antigens to in vivo–primed CD4+CD44+ T cells. A, BALB/c mice were sensitized intranasally with HDM or treated with PBS as a control on days 0, 7, 8, 9, 10, and 11 and analyzed on day 14. Lung B cells and DCs from HDM-treated mice (HDM B cells and HDM DCs, respectively) and splenic B cells from PBS-treated mice (CTRL B cells) were isolated and cocultured with lung CD4+CD44+ T cells from HDM-treated mice in the absence or presence of HDM (40 μg/mL). Graphs show cytokine concentrations in the day 4 culture supernatants measured by means of ELISA. Data are means ± SEMs. B, Splenic B cells or DCs from PBS-treated mice (splenic CTRL B cells and splenic CTRL DCs, respectively) or lung B cells from PBS- or HDM-treated mice (lung CTRL B cells and lung HDM B cells, respectively) were cocultured with lung CD4+CD44+ T cells from HDM-treated mice in the presence of different HDM concentrations. Graphs show cytokine concentrations in the day 4 culture supernatants measured by means of ELISA. IL-21 levels were less than the limit of detection. C and D, To assess whether lung B cells exposed to HDM in vivo carry HDM peptide–MHC complexes on the surface for presentation to T cells, BALB/c mice were treated with HDM or PBS on days 0, 11, 12, and 13, and lung B cells were isolated 24 hours later (day 14). B cells were directly cocultured with CFSE-labeled lung CD4+CD44+ T cells. Representative fluorescence-activated cell sorting (FACS) plots showing the CFSE profile of proliferating T cells (Fig 3, C) and concentrations of IL-5 and IL-13 measured by means of ELISA (Fig 3, D). IL-4, IL-17, and IFN-γ levels were below the limit of detection. E, BALB/c mice were sensitized intraperitoneally with OVA/alum on days 0 and 7 and challenged with OVA protein intranasally on days 14, 15, and 16. On day 17, lung CD4+CD44+ T cells were isolated and cocultured with lung DCs or lung B cells from OVA-treated mice or splenic B cells from PBS-treated mice in the presence of different concentrations of OVA protein. Supernatants were collected 4 days later, and cytokine concentrations were measured by means of ELISA. IFN-γ and IL-21 levels were less than the limit of detection. i.n., Intranasal; i.p., intraperitoneal. Data are representative of at least 3 (Fig 3, A-D) or 2 (Fig 3, E) independent experiments. *P < .05 and **P < .01.
To better evaluate the efficiency of B cells in HDM antigen presentation, we set up cocultures by using different concentrations of HDM. Similarly, as in Fig 3, A, B cells from lungs of HDM-sensitized mice and lungs or spleens of PBS-treated mice were equally efficient in presenting HDM antigens to CD4+CD44+ T cells, and their efficacy was similar (in the case of IL-4) or greater (in the case of IL-5 and IL-13) than that of splenic DCs. However, they were unable to restimulate T cells to produce IL-17 and less efficient at restimulating T cells to produce IFN-γ (Fig 3, B).
We then asked whether lung B cells from HDM-sensitized mice capture enough antigen in vivo for presentation to CD4+CD44+ T cells. Mice were sensitized with HDM on days 0, 11, 12, and 13, and total lung cells were isolated on day 14 and cocultured with HDM-primed CFSE-labeled CD4+CD44+ T cells in the absence of exogenous antigen. As shown in Fig 3, C and D, lung B cells from HDM-immunized mice were able to induce proliferation and cytokine production by CD4+CD44+ T cells, whereas B cells from PBS-treated mice were not. These data indicate that lung B cells captured sufficient amounts of HDM antigens in vivo to be able to restimulate in vivo–primed CD4+CD44+ T cells.
Finally, to investigate whether these potent antigen-presenting capacities are restricted to HDM, we used a well-established OVA model of allergic asthma.12 Mice were sensitized with OVA/alum through intraperitoneal injection on days 0 and 7; challenged intranasally with OVA protein on days 14, 15, and 16; and analyzed on day 17. As depicted in Fig 3, E, both lung and splenic B cells were far less efficient than DCs in restimulating in vivo–primed CD4+CD44+ T cells in the OVA model. These data indicate that efficacy of B cells in restimulating in vivo–primed CD4+CD44+ T cells is a property characteristic of the HDM but not OVA model of allergic asthma.
Lung B cells upregulate MHC class II and CD86 on HDM immunization
Because B cells displayed efficient antigen-presenting capacity ex vivo, we wondered whether they upregulate MHC class II and costimulatory molecule expression on HDM treatment in vivo. On HDM sensitization (days 0, 11, 12, and 13), around one third of total lung B cells upregulated their MHC class II expression (Fig 4, A and B). Furthermore, these MHC class IIhi B cells had significantly higher levels of CD86, but not CD80, OX40 ligand, or CD40, compared with MHC class IIint B cells from PBS- or HDM-treated mice (Fig 4, B). Intriguingly, HDM treatment reduced surface levels of inducible costimulator ligand (ICOSL) on lung B cells (Fig 4, B).
FIG 4.

Lung B cells upregulate MHC class II and CD86 on HDM sensitization. A and B, BALB/c mice were sensitized with HDM or treated with PBS as a control on days 0, 11, 12, and 13 and analyzed on day 14. Fig 4, A, Representative fluorescence-activated cell sorting (FACS) plots of MHC class II expression by total B cells (left) and CD86 expression (right) by total B cells (in PBS-treated mice) or MHC class IIint and MHC class IIhi B cells (in HDM-treated mice). Fig 4, B, Geometric mean fluorescent intensities of MHC class II and different costimulatory molecules (geometric mean fluorescence intensity [GMFI], mean ± SEM) from 5 mice per group. C, BALB/c mice were treated as in Fig 4, A. Lung B cells were restimulated with HDM or phorbol 12-myristate 13-acetate (PMA)/ionomycin in the presence of brefeldin A for 6 hours and analyzed by using intracellular cytokine staining. Graphs illustrate percentages of cytokine-producing B cells (mean ± SEM) from 5 PBS-treated and 5 HDM-treated mice. D, Plasma was collected from HDM- and PBS-treated mice, and HDM-specific IgE, total IgG, and IgG1 levels were measured by means of ELISA. The graph shows ODs (mean ± SEM, n = 5). Data are representative of 4 (Fig 4, A) or 2 (Fig 4, B, -D) independent experiments. **P < .01 and ****p < .0001.
Because HDM sensitization results in a potent TH2 response, we wondered whether lung B cells can contribute directly to the TH2 cytokine milieu in our model. B cells have been shown to produce cytokines in various experimental models. Based on their cytokine secretion profile, they are classified into 3 main subsets: regulatory B cells (secreting IL-10 and TGF-β), Be1 cells (secreting IFN-γ, IL-12, and TNF-α), and Be2 cells (secreting IL-4, IL-6, IL-13, and TNF-α).17 To assess cytokine production by B cells from HDM-sensitized mice, we restimulated total lung cells with phorbol 12-myristate 13-acetate/ionomycin or HDM extract for 6 hours and analyzed cytokine production by using intracellular cytokine staining on CD19+B220+ B cells by means of flow cytometry. As summarized in Fig 4, C, lung B cells were unable to produce IL-4, IL-6, IL-13, or IL-10.
To study cytokine production after a longer restimulation period, CD19+B220+ B cells were sorted from lungs of PBS-treated or HDM-sensitized mice and restimulated with HDM, phorbol 12-myristate 13-acetate/ionomycin, or α-IgM/CpG for 3 days. Consistent with intracellular cytokine staining, lung B cells from HDM-immunized mice were unable to secrete IL-4, IL-6, IL-13, or IL-10 (data not shown).
Collectively, these results demonstrate that lung B cells do not differentiate into the Be2 subset on HDM sensitization and do not directly contribute to TH2 cytokine milieu in the lung tissue. Also, they did not secrete non–TH2-related cytokines in our model, such as GM-CSF, as has been shown for the innate response activator B cells (data not shown).18
We next assessed whether B cells contribute to HDM inflammation by secreting increased levels of HDM-specific IgE or IgG. Therefore we treated mice intranasally with PBS or HDM on days 0, 11, 12, and 13 and isolated plasma on day 14. We found no upregulation of HDM-specific IgE at this time point (Fig 4, D), which is consistent with data obtained by others19,20 and most likely reflects a relatively short period of immunization. Similar to IgE, we found no upregulation of HDM-specific IgG1 or IgG in our model.
Taken together, we conclude that in the model used lung B cells upregulate MHC class II and CD86 on HDM treatment, but their production of either cytokines or HDM-specific IgE is less than the detection limit.
Depletion of B cells before challenge reduces the allergic response
To investigate the contribution of B cells to allergic response in the challenge phase of HDM immunization, we depleted B cells in mice previously sensitized to HDM by using α-CD20 antibody. In these experiments all mice were treated with HDM intranasally on day 0 and either treated with α-CD20 antibody on day 9 and challenged intranasally with HDM on days 11, 12, and 13 (α-CD20-HDM group) or treated with isotype control antibody (Ig Ctrl) on day 9, followed by challenges with HDM (Ig Ctrl-HDM group) or PBS (Ig Ctrl-PBS group; Fig 5, A). α-CD20 treatment dramatically reduced B-cell numbers in the lungs (Fig 5, B). Enumeration of the lung immune infiltrate showed that mice in the α-CD20–HDM group had fewer CD4+CD44+ T cells, eosinophils, and neutrophils compared with the Ig Ctrl-HDM group, which reflected reduced allergic inflammation in the tissue (Fig 5, B).
FIG 5.

Depletion of B cells before challenge reduces the allergic response. A, Schematic representation of the experimental set-up. BALB/c mice were sensitized with HDM on day 0 and injected with α-CD20 or isotype-matched antibody on day 9 (α-CD20 and Ig Ctrl). Mice were challenged on days 11, 12, and 13 with PBS or HDM and analyzed on day 14. B, Absolute numbers of T cells (CD45+CD3+CD4+CD44+), eosinophils (CD45+CD11clo/−Siglec-F+CCR3+CD11bintGr-1int) and neutrophils (CD45+CD11bhiGr-1hiCD11c−Siglec-F−) in lungs were quantified by using flow cytometry. C, Lung CD4+CD44+ T cells were restimulated with HDM in cocultures with lung DCs for 4 days and cytokine production was measured by means of ELISA. IL-21 levels were less than the limit of detection. D, BAL fluid was collected on day 14, and cytokine production was measured by means of ELISA. IL-21 levels were less than the limit of detection. Data are pooled from 4 independent experiments. Graphs show the mean ± SEM. *P < .05, **P < .01, ***P < .001, and ****p < .0001.
We then sorted CD4+CD44+ T cells and plated them in a coculture with HDM-pulsed DCs. CD4+CD44+ T cells from the α-CD20 group secreted reduced levels of TH2 cytokines compared with their Ig Ctrl counterparts. Importantly, their response was not skewed into a different subset because these cells secreted lower amounts of IL-10 and IFN-γ, whereas IL-17 production was not significantly altered (Fig 5, C). Finally, α-CD20 treatment resulted in reduced concentrations of cytokines, particularly IL-4 and IL-13 in BAL fluid (Fig 5, D).
The strain of mice used in these experiments (BALB/c) is prone to a strong TH2 response, unlike another common laboratory strain, C57BL/6. To investigate whether perpetuation of allergic inflammation by B cells is a feature restricted to allergy-prone strains, we repeated the experiments with C57BL/6 mice. Interestingly, we obtained similar results as in the case of BALB/c mice. Numbers of effector/memory CD4+ T cells, eosinophils, and neutrophils were reduced on α-CD20 treatment (Fig 6, A). Also, the CD4+CD44+ T-cell response after α-CD20 treatment was impaired in a similar manner as in BALB/c mice. We observed reduced production of TH2 cytokines (IL-4, IL-5, and IL-13) after 4 days of coculture with DCs in the presence of HDM. Also, there were lower levels of IL-10 and IFN-γ, whereas IL-17 levels were not changed (Fig 6, B).
FIG 6.

Depletion of B cells before challenge reduces the allergic response in C57BL/6 mice. A, C57BL/6 mice were treated as in Fig 5, A, and absolute numbers of CD4+CD44+ T cells, eosinophils, and neutrophils in lungs were quantified as in Fig 5, B. B, Lung CD4+CD44+ T cells were restimulated with HDM in cocultures with lung DCs for 4 days. Supernatants were collected, and cytokine production was measured by using ELISA. IL-21 levels were less than the detection limit. Data are pooled from 3 independent experiments. Graphs show the mean ± SEM. *P < .05, **P < .01, and ****P < .0001.
Depletion of B cells before priming results in reduced capacity of CD4+ T cells to produce IFN-γ but not IL-4 or IL-13
To evaluate the importance of B cells in shaping CD4+ T-cell responses at the time of priming, we set up a protocol to deplete B cells before sensitization with HDM. For this, mice received α-CD20 or Ig Ctrl antibody 2 days before HDM administration. On day 7 after immunization, we collected their lungs and evaluated the numbers of eosinophils, neutrophils, and effector TH cells (experimental setup shown in Fig 7, A). As depicted in Fig 7, B, anti-CD20 treatment resulted in decreased numbers of eosinophils and neutrophils but not effector TH cells in lung tissue. Additionally, we collected MLNs and sorted CD4+ T cells and DCs. CD4+ T cells were labeled with CFSE and cocultured with DCs in the presence of HDM (Fig 7, A); on day 4, proliferation was assessed by using CFSE dilution. As shown in Fig 7, C, B-cell depletion before priming did not impair the ability of CD4+ T cells to proliferate on ex vivo restimulation. Also, secretion of TH2 cytokines (IL-4 and IL-13) was not changed (Fig 7, D). However, B-cell depletion before priming resulted in reduced ability of CD4+ T cells to produce IFN-γ.
FIG 7.

Depletion of B cells before priming with HDM results in reduced numbers of eosinophils and neutrophils in the lungs but does not influence the capacity of MLN TH cells to secrete IL-4 or IL-13. A, Experimental set-up of the experiment. C57BL/6 mice were treated with α-CD20 or isotype control antibody (Ig Ctrl) on day −2, treated with HDM or PBS intranasally on days 0 and 1, and analyzed on day 7. Lungs were collected, and numbers of eosinophils, neutrophils, and effector TH cells were evaluated. Additionally, CD4+ T cells were isolated from MLNs, labeled with CFSE, and cocultured with MLN DCs in the presence of HDM for 4 days. B, Enumeration of eosinophils, neutrophils, and effector TH cells in lung tissue. C, Proliferation, as assessed by means of CFSE dilution, was measured by using flow cytometry. Representative fluorescence-activated cell sorting (FACS) plots and cumulative data from 4 mice per group. D, Supernatants from cocultures were collected on day 4, and cytokine levels were measured by means of ELISA. IL-5, IL-17, and IL-21 levels were less than the limit of detection. Data are representative (Fig 7, B and C) or pooled (Fig 7, C) from 2 (Fig 7, B) or 4 (Fig 7, C and D) independent experiments. Graphs show the mean ± SEM. *P < .05 and ***P < .001.
Collectively, these results demonstrate a dual feature of B cells in this model. On the one hand, they can induce granulocyte recruitment into the lung tissue and priming of TH1 responses, but on the other hand, they are dispensable for priming of TH2 responses and recruitment/proliferation of effector TH cells.
B cells are able to initiate CD4+ T-cell responses on HDM priming
Next, we asked whether B cells are sufficient to prime naive T cells and induce TH1 differentiation or other responses, such as TH17 or regulatory T responses. To address this question, we took advantage of a mouse strain in which MHC class II expression is restricted to the B-cell lineage (B-MHC-II).11 The parental strain of these mice carries a floxed polyadenylation stop sequence in the IAβ locus, making this strain MHC class II deficient. However, by crossing it to CD19-cre mice, it is possible to restore MHC class II expression in B cells and obtain B-MHC-II strain after the second generation. Because of a lack of MHC class II expression in thymic epithelial cells, both flox mice and B-MHC-II mice have a severe reduction in peripheral CD4+ T cells. To correct for this, we first restored the CD4+ T-cell compartment in flox mice and B-MHC-II mice by injecting 107 polyclonal CD4+ T cells from naive WT C57BL/6 mice, as previously described (Fig 8, A).21 Flox and B-MHC-II mice were sensitized by means of intranasal administration of HDM on days 0 and 1. CD4+ T cells were isolated from MLNs and lungs on day 7, labeled with CFSE, and cocultured with WT DCs from PBS-treated mice in the presence of HDM for 4 days. As depicted in Fig 8, B and lung CD4+ T cells from B-MHC-II mice proliferated extensively in response to HDM restimulation ex vivo. Despite this, they were unable to secrete TH2-associated cytokines (IL-4, IL-5, and IL-13). However, they produced high levels of IL-17 and IFN-γ (Fig 8, C).
FIG 8.

B cells are able to prime CD4+ T cells in response to HDM. A, Experimental set-up. WT C57BL/6 mice were sensitized with HDM or treated with PBS on days 0 and 1 and analyzed on day 7 (left). B-MHC-II and Flox mice (both on C57BL/6 background) received intravenous injection of 107 polyclonal CD4+ T cells from WT C57BL/6 mice before HDM sensitization to reconstitute the CD4+ T-cell compartment. Mice were subsequently sensitized with HDM on days 0 and 1 and analyzed on day 7 (right), i.n., Intranasal. B, MLNs or lung CD4+ T cells were labeled with CFSE and cocultured with MLN DCs in the presence of HDM for 4 days. Proliferation was assessed by means of CFSE dilution. C, Cytokine concentrations in the day 4 culture supernatants were measured by using ELISA. IL-21 levels in both data sets and IL-5 levels in the MLN data set were less than the limit of detection. Data are representative (Fig 8, B) or pooled (Fig 8, C) from 3 independent experiments. Graphs show the mean ± SEM. *P < .05, **P < .01, ***P < .001, and ****P < .0001.
Collectively, these data indicate that B cells are able to prime HDM-specific naive CD4+ T cells in vivo. However, they are unable to induce TH2 differentiation, but instead, prime naive T cells to become IL-17– and IFN-γ–producing cells.
DISCUSSION
The field of antigen presentation in initiation and propagation of allergic responses has been dominated by DCs. It is now generally believed that this cell type plays a nonredundant role in this process.22 However, this conclusion is largely based on OVA models of allergy,3-6 which might not reflect responses to complex, physiologically relevant allergens. By using the latter, recent studies have pointed to the role of B cells as APCs in allergic responses.9,10 Also, the cross-talk between B cells and DCs in patients with helminth infections has been highlighted recently.8
In this study, using an HDM model of asthma, we have shown that in contrast to the OVA model, B cells efficiently take up antigens in vivo and serve as potent APCs ex vivo. This is in line with a recent report showing that B cells can stimulate Der p 1–specific TH cells to produce IL-5 and IL-13 in vitro.10
Interestingly, the ability of B cells to acquire HDM antigens did not rely on the specificity of the B-cell receptor because B cells with irrelevant surface immunoglobulin were equally efficient. Similar results have been obtained recently by using papain.23 This is somewhat surprising because only antigen-specific B cells are traditionally considered potent at antigen internalization. The mechanisms behind the efficient HDM uptake by B cells remain to be determined, but it is possible that internalization requires binding of 1 or more HDM antigens to a surface receptor.
The most striking observation of our study was that depletion of B cells before challenge had pronounced effects on reduction of allergic response. These included reduced numbers of effector/memory TH cells, eosinophils, and neutrophils, as well as impaired capacity of CD4+ CD44+ T cells to secrete cytokines on ex vivo restimulation. Importantly, the effects of B-cell depletion were observed in 2 different laboratory mouse strains: BALB/c and C57BL/6. This is in line with a conclusion reached in an article by Dullaers et al,10 in which they pointed to a crucial role for B cells in optimal proliferation and activation of TH2 cells in MLNs. Given the fact that B cells produce neither cytokines nor HDM-specific IgE in our model but act as efficient APCs, it is most likely that it is abrogation of sufficient TH cell stimulation by antigen-presenting B cells that confers the observed effects. Nevertheless, the existence of other mechanisms cannot be excluded. For example, the cross-talk between B cells and DCs in induction of TH2 responses on nematode infection has been reported previously.24 In this model secretion of lymphotoxin by B cells drove CXCL13 expression in the MLNs and controlled colocalization of B cells and CXCR5+ DCs. Importantly, its disruption led to a reduced development of IL-4–producing T cells. Whether similar cross-talk between B cells and DCs or B cells and other cell types in the lungs could play a role in our model of asthma remains to be investigated.
Depletion of B cells used in the above experiments is reminiscent of the B-cell depletion therapy used in human patients with various cancers and autoimmune disorders. The rationale behind the use of α-CD20 therapy in the treatment of autoimmune diseases has been the role B cells play in the production of autoantibodies. However, in some cases clinical improvement did not correlate with decreased autoantibody levels, pointing to the existence of additional factors playing a role in the therapy.25-28 For example, a study was conducted on patients with atopic eczema who had increased levels of IgE specific to various allergens, including birch and grass pollens, cat dander, and HDMs. All of the patients receiving α-CD20 therapy showed a remarkable improvement by 4 to 8 weeks after treatment, which included reduced expression levels of IL-5 and IL-13 in CD4+ T cells. Importantly, however, the levels of allergen-specific IgE were not changed.28 This example high-lights that the mechanism unraveled in our work might also be relevant for human patients with HDM allergies.
The conclusion that B-cell depletion strongly reduces ongoing allergic inflammation might be somewhat surprising because it has been proposed that DCs are necessary and sufficient to promote ongoing allergic inflammation.6 In this study authors used diphtheria toxin–based depletion of CD11c+ cells specifically before the challenge phase of immunization. However, to study allergic inflammation, they used an OVA model of asthma that, as underlined throughout our study, greatly differs from the HDM model. This drawback of the above work has been corrected by Plantinga et al,29 who used an HDM model of asthma to study the role of DCs in initiation and maintenance of TH2 immunity. In this study the authors sensitized and challenged Flt3l-deficient mice, which show a strong reduction in conventional DC numbers. Interestingly, they observed lower numbers of eosinophils, lymphocytes, and neutrophils in BAL fluid on HDM sensitization and 5 consecutive challenges. However, this was only the case in a low-dose model of HDM immunization (sensitization with 1 μg of HDM and challenge with 10 μg of HDM) but not in the high-dose model (sensitization with 100 μg of HDM and challenge with 100 μg of HDM), indicating that the conventional DC requirement might depend on the dose of antigen used. Unfortunately, the capacity of total lung cells or CD4+ T cells to produce cytokines was not reported. Therefore direct evidence for the importance of DCs for TH cell responses is missing.
A caveat of the above study is use of gene knockout mice to investigate responses during the challenge phase of immunization. Based on this approach, it is impossible to conclude whether the priming or the challenge phase of the response had been affected. Indeed, the impaired TH2 priming upon HDM sensitization might have resulted in a reduced influx of effector cells upon challenge. Hence the conclusion about the role of DCs specifically in the secondary response cannot be reached. This issue should be clarified.
Our data showing reduced allergic response on B-cell depletion before HDM challenge but not before sensitization are in contrast with findings from Ballesteros-Tato et al,9 who showed that B cells are necessary for the development of allergic inflammation during the sensitization phase but are dispensable during the challenge phase of the response. The reasons for this discrepancy is not clear, although it might be due to differences in the experimental approach. B cell–deficient mice (μMT) might differ from B cell–depleted WT mice in terms of organization and cellular composition of the lymphoid organs, lungs, or both and in T-cell function and distribution.30-32 Furthermore, antibody deficiency in μMT mice might have shaped the composition of the intestinal and lung microbiota in a different way than in WT animals, which might result in major physiologic differences.33 Alternative explanations include different immunization protocols used, composition of HDM extract, or both.
Finally, our study revealed an unexpected capacity of B cells to initiate CD4+ T-cell responses from naive precursors. The experimental approach relied on a mouse strain in which MHC class II expression is restricted to the B-cell lineage. This approach offers some advantages compared with others. It did not rely on the use of BCR-transgenic mice or antigen-specific B-cell transfer into naive hosts,34,35 avoiding a nonphysiologic excess of antigen-specific B cells at the time of priming. It also allowed us to directly assess whether B cells can prime CD4+ T-cell responses from naive precursors or only expand DC-primed effector CD4+ T cells still residing in the lymph node. Finally, it enabled us to dissect the capacity of B cells to prime TH cell responses from their necessity to do so because redundancy in antigen presentation was not a relevant factor in this system. These 2 last issues are in contrast to studies using B cell–deficient mice or mice with MHC class II expression abrogated specifically in the B-cell linage.3-5,36-39
Our conclusion that B cells can initiate priming of CD4+ T-cell responses in a system in which clonality of B cells is not artificially skewed most likely derives from the fact that B cells do not have to rely on rare antigen-specific BCRs to capture HDM. In experimental approaches relying on model antigens, such as OVA, such a notion would be less expected. Indeed, using the B-MHC II strain of mice, Barnett et al21 did not observe initiation of TH cell differentiation on OVA immunization.
Unexpectedly, we observed that B cells can initiate CD4+ T-cell differentiation into IFN-γ– and IL-17–producing cells. Classically, TH1/TH17 differentiation is perceived as a process requiring strong stimulation, including MHC class II–peptide–TCR interaction (signal 1), costimulation (signal 2), and cytokine signaling (signal 3). B cells, in contrast to DCs, do not secrete polarizing cytokines in response to HDM and express lower levels of costimulatory molecules. Therefore their ability to prime TH2 or regulatory T responses, if any, would be suspect. However, this was not the case: B cells were unable to induce polarization of IL-4/IL-13– or IL-10–secreting TH cells but efficiently primed IL-17 and IFN-γ responses. It is most likely that there are other cell types, such as DCs activated by HDM, that provide cytokines necessary for the development of these subsets. Although MHC class II deficient, they can capture antigens in the tissue, mature, and migrate into the lymph nodes, where they secrete cytokines. Subsequently, by diffusing across the lymphoid tissue, these cytokines in trans can influence CD4+ T-cell priming by B cells. This scenario requires further investigation.
The observation that sensitization to HDM induces only TH1/TH17 responses in B-MHC-II mice while a TH2/TH1 phenotype predominates in WT animals exemplifies the heterogeneity of TH cell fates and the complexity behind their regulation. Previously, such a phenomenon has been demonstrated in our laboratory, where TH cells specific to Candida albicans antigens bearing the same TCRs but belonging to different subsets were detected.40 Evaluation of whether TCR specificity in the TH2 subset of WT and TH1/TH17 subsets of B-MHC-II mice overlaps would be an important step to further support this concept and underline plasticity of the TH cell fate. Going one step further and defining molecular cues leading to this heterogeneity is the next challenge to undertake.
In conclusion, this study highlighted an important role of B cells in propagation and possibly initiation of TH cell responses to HDM, a complex and physiologically relevant allergen, and might pave the way for further discoveries in this field, as well as for development of new strategies to combat HDM allergy.
We thank David Jarrossay for cell sorting and helpful suggestions, Silvia Preite and Camilla Basso for discussion, and Luana Perlini, Enrica Mira Cató, Andrea D’Ercole, Ghassan Bahnan, and Toma Kobkyn for animal care.
Key messages.
B cells acquire HDM antigens independently of their BCR specificity and act as efficient APCs ex vivo.
B cells play an important role in amplification of TH2 responses in vivo, and their depletion leads to reduction of the allergic response.
Although they are unable to initiate TH2 responses on their own, B cells efficiently prime naive CD4+ T-cell precursors to differentiate into TH1 and TH17 subsets.
Acknowledgments
This work was in part funded by the Swiss National Science Foundation (grant n. PDAMP3_137087 and grant n. PDFMP3_137127).
Disclosure of potential conflict of interest: F. Sallusto’s institution received grant PDAMP3_137087 and grant PDFMP3_137127 from the Swiss National Science Foundation, and T. Wypych personally received a travel grant from the European Federation of Immunological Societies (EFIS) for this work. G. F. Wu’s institution received grant R01NS083678 from the National Institutes of Health for this work. Dr Sallusto personally received consultancy fees from HUMABS BioMed SA for other works. A. Lanzavecchia declares that he has no relevant conflicts of interest.
Abbreviations used
- AF
Alexa Fluor
- APC
Antigen-presenting cell
- BAL
Bronchoalveolar lavage
- BCR
B-cell receptor
- CFSE
Carboxyfluorescein succinimidyl ester
- DC
Dendritic cell
- HDM
House dust mite
- LT
Leukotriene
- MLN
Mediastinal lymph node
- OVA
Ovalbumin
- TCR
T-cell receptor
REFERENCES
- 1.Bousquet J, Khaltaev N, editors. Global surveillance, prevention and control of chronic respiratory diseases: a comprehensive approach. Geneva: World Health Organization; 2007. [Google Scholar]
- 2.Stone KD, Prussin C, Metcalfe DD. IgE, mast cells, basophils, and eosinophils. J Allergy Clin Immunol 2010;125(suppl 2):S73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Korsgren M, Erjefalt JS, Korsgren O, Sundler F, Persson CG. Allergic eosinophil-rich inflammation develops in lungs and airways of B cell-deficient mice. J Exp Med 1997;185:885–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacLean JA, Sauty A, Luster AD, Drazen JM, De Sanctis GT. Antigen-induced airway hyperresponsiveness, pulmonary eosinophilia, and chemokine expression in B cell-deficient mice. Am J Respir Cell Mol Biol 1999;20:379–87. [DOI] [PubMed] [Google Scholar]
- 5.Hamelmann E, Takeda K, Schwarze J, Vella AT, Irvin CG, Gelfand EW. Development of eosinophilic airway inflammation and airway hyperresponsiveness requires interleukin-5 but not immunoglobulin E or B lymphocytes. Am J Respir Cell Mol Biol 1999;21:480–9. [DOI] [PubMed] [Google Scholar]
- 6.van Rijt LS, Jung S, Kleinjan A, Vos N, Willart M, Duez C, et al. In vivo depletion of lung CD11c+ dendritic cells during allergen challenge abrogates the characteristic features of asthma. J Exp Med 2005;201:981–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammad H, Lambrecht BN. Barrier epithelial cells and the control of Type 2 immunity. Immunity 2015;43:29–40. [DOI] [PubMed] [Google Scholar]
- 8.Leon B, Ballesteros-Tato A, Lund FE. Dendritic cells and B cells: unexpected partners in Th2 development. J Immunol 2014;193:1531–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ballesteros-Tato A, Randall TD, Lund FE, Spolski R, Leonard WJ, Leon B. T follicular helper cell plasticity shapes pathogenic T helper 2 cell-mediated immunity to inhaled house dust mite. Immunity 2016;44:259–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dullaers M, Schuijs MJ, Willart M, Fierens K, Van Moorleghem J, Hammad H, et al. House dust mite-driven asthma and allergen-specific T cells depend on B cells when the amount of inhaled allergen is limiting. J Allergy Clin Immunol 2017;140:76–88.e7. [DOI] [PubMed] [Google Scholar]
- 11.Archambault AS, Carrero JA, Barnett LG, McGee NG, Sim J, Wright JO, et al. Cutting edge: conditional MHC class II expression reveals a limited role for B cell antigen presentation in primary and secondary CD4 T cell responses. J Immunol 2013;191:545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swedin L, Ellis R, Kemi C, Ryrfeldt A, Inman M, Dahlen SE, et al. Comparison of aerosol and intranasal challenge in a mouse model of allergic airway inflammation and hyperresponsiveness. Int Arch Allergy Immunol 2010;153:249–58. [DOI] [PubMed] [Google Scholar]
- 13.Beier KC, Hutloff A, Lohning M, Kallinich T, Kroczek RA, Hamelmann E. Inducible costimulator-positive T cells are required for allergen-induced local B-cell infiltration and antigen-specific IgE production in lung tissue. J Allergy Clin Immunol 2004;114:775–82. [DOI] [PubMed] [Google Scholar]
- 14.Lindell DM, Berlin AA, Schaller MA, Lukacs NW. B cell antigen presentation promotes Th2 responses and immunopathology during chronic allergic lung disease. PLoS One 2008;3:e3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baumgarth N The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol 2011;11:34–46. [DOI] [PubMed] [Google Scholar]
- 16.Goodnow CC, Crosbie J, Adelstein S, Lavoie TB, Smith-Gill SJ, Brink RA, et al. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 1988;334:676–82. [DOI] [PubMed] [Google Scholar]
- 17.Lund FE. Cytokine-producing B lymphocytes-key regulators of immunity. Curr Opin Immunol 2008;20:332–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science 2012; 335:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Coquet JM, Schuijs MJ, Smyth MJ, Deswarte K, Beyaert R, Braun H, et al. Interleukin-21-producing CD4(+) T cells promote type 2 immunity to house dust mites. Immunity 2015;43:318–30. [DOI] [PubMed] [Google Scholar]
- 20.Birrell MA, Van Oosterhout AJ, Belvisi MG. Do the current house dust mite-driven models really mimic allergic asthma? Eur Respir J 2010;36:1220–1. [DOI] [PubMed] [Google Scholar]
- 21.Barnett LG, Simkins HM, Barnett BE, Korn LL, Johnson AL, Wherry EJ, et al. B cell antigen presentation in the initiation of follicular helper T cell and germinal center differentiation. J Immunol 2014;192:3607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambrecht BN, Hammad H. Dendritic cell and epithelial cell interactions at the origin of murine asthma. Ann Am Thorac Soc 2014;11 (suppl 5): S236–43. [DOI] [PubMed] [Google Scholar]
- 23.Dwyer DF, Woodruff MC, Carroll MC, Austen KF, Gurish MF. B cells regulate CD4+ T cell responses to papain following B cell receptor-independent papain uptake. J Immunol 2014;193:529–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leon B, Ballesteros-Tato A, Browning JL, Dunn R, Randall TD, Lund FE. Regulation of T(h)2 development by CXCR51 dendritic cells and lymphotoxin-expressing B cells. Nat Immunol 2012;13:681–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Looney RJ, Anolik JH, Campbell D, Felgar RE, Young F, Arend LJ, et al. B cell depletion as a novel treatment for systemic lupus erythematosus: a phase I/II dose-escalation trial of rituximab. Arthritis Rheum 2004;50:2580–9. [DOI] [PubMed] [Google Scholar]
- 26.Kavanaugh A, Rosengren S, Lee SJ, Hammaker D, Firestein GS, Kalunian K, et al. Assessment of rituximab’s immunomodulatory synovial effects (ARISE trial). 1: clinical and synovial biomarker results. Ann Rheum Dis 2008;67:402–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol 2006;180:63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simon D, Hosli S, Kostylina G, Yawalkar N, Simon HU. Anti-CD20 (rituximab) treatment improves atopic eczema. J Allergy Clin Immunol 2008;121: 122–8. [DOI] [PubMed] [Google Scholar]
- 29.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, et al. Conventional and monocyte-derived CD11b(+) dendritic cells initiate and maintain T helper 2 cell-mediated immunity to house dust mite allergen. Immunity 2013;38:322–35. [DOI] [PubMed] [Google Scholar]
- 30.Ngo VN, Cornall RJ, Cyster JG. Splenic T zone development is B cell dependent. J Exp Med 2001;194:1649–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baumgarth N, Jager GC, Herman OC, Herzenberg LA. CD4+ T cells derived from B cell-deficient mice inhibit the establishment of peripheral B cell pools. Proc Natl Acad Sci U S A 2000;97:4766–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hondowicz BD, An D, Schenkel JM, Kim KS, Steach HR, Krishnamurty AT, et al. Interleukin-2-dependent allergen-specific tissue-resident memory cells drive asthma. Immunity 2016;44:155–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shulzhenko N, Morgun A, Hsiao W, Battle M, Yao M, Gavrilova O, et al. Crosstalk between B lymphocytes, microbiota and the intestinal epithelium governs immunity versus metabolism in the gut. Nat Med 2011;17:1585–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Constant SL. B lymphocytes as antigen-presenting cells for CD4+ T cell priming in vivo. J Immunol 1999;162:5695–703. [PubMed] [Google Scholar]
- 35.Rodriguez-Pinto D, Moreno J. B cells can prime naive CD4+ T cells in vivo in the absence of other professional antigen-presenting cells in a CD154-CD40-dependent manner. Eur J Immunol 2005;35:1097–105. [DOI] [PubMed] [Google Scholar]
- 36.Sunshine GH, Jimmo BL, Ianelli C, Jarvis L. Strong priming of T cells adoptively transferred into scid mice. J Exp Med 1991;174:1653–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ronchese F, Hausmann B. B lymphocytes in vivo fail to prime naive T cells but can stimulate antigen-experienced T lymphocytes. J Exp Med 1993;177:679–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Epstein MM, Di Rosa F, Jankovic D, Sher A, Matzinger P. Successful T cell priming in B cell-deficient mice. J Exp Med 1995;182:915–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Topham DJ, Tripp RA, Hamilton-Easton AM, Sarawar SR, Doherty PC. Quantitative analysis of the influenza virus-specific CD4+ T cell memory in the absence of B cells and Ig. J Immunol 1996;157:2947–52. [PubMed] [Google Scholar]
- 40.Becattini S, Latorre D, Mele F, Foglierini M, De Gregorio C, Cassotta A, et al. T cell immunity. Functional heterogeneity of human memory CD4(+) T cell clones primed by pathogens or vaccines. Science 2015;347:400–6. [DOI] [PubMed] [Google Scholar]