

Abstract
Translation elongation is a highly coordinated, multistep, multi-factor process that ensures accurate and efficient addition of amino acids to a growing nascent-peptide encoded in the sequence of translated mRNA. While translation elongation is heavily regulated by external factors, there are clear evidences that mRNA and nascent-peptide sequences control elongation dynamics, determining both sequence and structure of synthesized proteins. Advances in methods have driven experiments that revealed the basic mechanisms of elongation as well as the mechanisms of regulation by mRNA and nascent-peptide sequences. In this review, we highlight how mRNA and nascent-peptide elements manipulate the translation machinery to alter the dynamics and pathway of elongation.
Keywords: Protein synthesis, Ribosome, Recoding, Translation control, mRNA, Nascent-peptide chain
1. Introduction
Translation is the end-point of genetic information transfer, where the nucleotide sequence of the messenger RNA (mRNA) dictates the sequence and structure of the nascent protein. The mechanism of protein synthesis is conserved across the domains of life and is driven by the ribosome, a two-subunit RNA-protein machine. Thus, fundamental insights into how the ribosome and the broader translation machinery function have been and will be crucial to understand gene expression. During translation, ribosomes assemble at the start site marked by a set of three consecutive nucleotides (a “codon”), adds one amino acid encoded by the next codon (“decodes”), moves to the next non-overlapping codon (“translocates”), and repeats the decoding and translocation steps until a stop codon reaches the decoding center of the ribosome. Repeats of decoding and translocation events are referred to as the elongation phase of translation, where the nascent peptide is elongated by amino acids to build a functional protein.
Our view of translation elongation has evolved over the past decades. In a simplistic model, decoding and translocation events are metrically repeated to produce a nascent protein faithful to the codon sequence within the mRNA. Yet, this model insufficiently accounted for numerous instances where mRNA sequence fails to predict the sequence of the resulting protein. Studies of these special cases of elongation further revealed that local elongation dynamics are influenced by the surrounding mRNA sequences, which create competing elongation pathways that may alter the definition of individual codons. Re-definition of codons due to dynamic interactions with neighboring mRNA elements are collectively referred to as ‘recoding’ (1). Recoding is pervasive throughout the domains of life and is especially enriched in viruses. Thus, the static and deterministic view of elongation must be adjusted to include the statistical and dynamic process of recoding. A dynamic view of translation is poised to integrate recent developments in other relevant fields of biology, such as the emerging field of epitranscriptomics, where mRNA can be chemically modified to alter translation.
An enriched genetic code emerges after accounting for the dynamic nature of translation. Various stretches of nucleotide sequences within the mRNA (“mRNA elements”) regulate the rhythm of its own translation elongation, which in turn dictate the protein structure and function. mRNA elements may cooperatively interact with the translation machinery and alter protein synthesis in a programmed manner. For example, mRNA structures within the coding region have been implicated in recruiting translation factors to aid alternative decoding or disrupt translocation to enhance recoding phenomena (2, 3). In addition, the nature of the newly-synthesized protein (“nascent-peptide elements”) also affects translation, which adds yet another layer of control (4, 5). Nascent-peptide elements together with the mRNA elements can alter translocation or decoding dynamics during recoding (6, 7). This review aims to provide a framework for how various mRNA and nascent-peptide elements affect the dynamics of protein synthesis.
Our richer view of dynamic translation has been driven by improved biochemical, genetic, structural, biophysical and computational methods. We will first discuss the technological developments that have led to our current model of translation elongation. We will then review how known mRNA and nascent-peptide elements perturb the dynamics of translation elongation both in decoding and in translocation. Finally, we will discuss how mRNA and nascent-peptide elements work cooperatively to define the decoding pathway in specific cases of recoding.
2. Current methods to study dynamics of translation elongation
The molecular mechanism of translation elongation involves various dynamic structural rearrangements and their coupled chemical reactions. Over the last fifty years, numerous structural, biophysical and biochemical tools have been developed to observe these different aspects of translation. Integrating data from various methods, a detailed molecular mechanism of translation elongation cycle has been constructed.
2.1. Structural methods
Structural methods continue to be fundamental in delineating the molecular basis of translation. X-ray crystallography has provided high-resolution views of translational components, including ribosomes. NMR spectroscopy is used to determine structural dynamics of ribosomal domains (8) and nascent protein chains (9), and recently whole ribosomes using solid state methods (10–13), but is hindered by fundamental limitations in solution for large (>500 kDa) systems. Recent advances in cryogenic electron microscopy (cryoEM) have yielded much excitement in the field of translation by providing high-resolution information about translational intermediates and larger complexes that have been previously inaccessible by other structural methods.
X-ray crystallography exposed the first atomic-resolution structures of the translation complex in various functional states. The high resolution (2–3.5Å) of ribosomal crystallography arises from the constructive interference of scattered x-rays generated by a regular ordering of biomolecules within a crystal lattice (14), which creates strong x-ray diffraction patterns. The diffraction patterns are used to derive 3-dimensional electron densities and, with molecular modeling, the structures of biomolecules are determined. X-ray crystallography has been used to ascertain dozens of high-resolution structures of both bacterial and eukaryotic translation complexes in various functional states. Techniques for crystallization have advanced to allow ligands such as mRNA, tRNA and various translation factors to be stably bound to the ribosome during or after crystallization (14). These structures have revolutionized our understanding of translation, and provide a molecular blueprint for subsequent engineering of translational components (14). However, high-resolution is achieved by molecular ordering, thus eliminating intrinsic macromolecular heterogeneity. Packing of translational complexes within a crystal lattice may induce non-biological contacts or prevent crystallization of larger assemblies. X-ray crystallography also proves to be difficult in solving unstable intermediates of biological processes that are too transient to be captured and ordered during crystallization.
CryoEM avoids many of pitfalls of x-ray crystallography: sample heterogeneity is well tolerated, ribosomes can be prepared in a wide-range of near-physiological conditions prior to imaging, and ribosome complexes, which are embedded in vitreous ice rather than forced into a crystal lattice, retain biological contacts. These factors coupled with recent advances in cryoEM hardware and software have made cryoEM a powerful method for ribosome structural studies, achieving a near-atomic resolution of intermediate states of translation (15–17). The ensemble of structures in a sample represents the most populated, stable intermediates along the reaction pathway. Similar to x-ray crystallography, cryoEM does not provide dynamic information, but different structural classes can be ordered post-hoc based on structural similarity (17). Time-resolved cryoEM techniques that uses microfluidic chips and environmental chambers for sample preparation rather than the standard sample preparation, which includes mixing, pipetting onto the grid, and plunge-freezing, can be used to capture sub-second intermediates (18, 19). The combination of cryoEM with time-resolved methods will be a strong driver of future structural work on translation.
2.2. Bulk kinetics
The static views of molecular architecture provided by the structural methods require a temporal axis. Bulk kinetics (also referred to as ensemble kinetics) based on stopped-flow or quench-flow approaches allow for the determination of rates of chemical reactions with high (millisecond to second) temporal resolution. In a typical bulk kinetics experimental scheme, two solutions are rapidly mixed for a set short period of time, and a fluorescence signal (stopped-flow) or reaction quenching and product concentration (quench-flow) is measured as a function of time. A reaction must be initiated such that all reactants begin their chemical journey simultaneously. However, during multistep or repetitive processes, a bulk collection of molecules can rapidly desynchronize, obfuscating the presence of intermediate states. The usual detection method for quench-flow involves measuring radio-labeled chemicals such as an amino acid or GTP in the reaction quenched at different time points (20). Unlike many chemical transformations, structural changes may be complex and reversible, requiring real-time measurements not accessible using the quench-flow approach. In the stopped-flow apparatus, conformational changes during a reaction can be measured via fluorescence or light-scattering changes in real time (21). These kinetic measurements can be fit to reaction mechanism model from which the kinetic parameters for the biochemical reactions can be extrapolated. Measuring kinetics of multi-step reactions requires careful formulation of kinetic models, which involves fitting multiple parameters for each reaction step to limited experimental time-points.
Recent advances in the bulk kinetics methods have resolved rate constants and processivity factors for multi-step chemical reactions, such as penta-peptide formations (22, 23). Measuring kinetics of multi-step reactions requires careful formulation of kinetic models, which involves fitting multiple parameters for each reaction step to limited experimental time-points. This has been done either by global fitting of multiple parameters (22) or by deriving necessary kinetic reactions to model the process (23). Tracking changes of elongation kinetics during the translation of multiple codons is a crucial feature in studying different mRNA and nascent-peptide elements during translation, where the kinetic effect may occur over multiple cycles of elongation.
2.3. Single-molecule methods
Single-molecule methods probe the dynamics of translation due to their ability to detect and sort structural and compositional heterogeneity and observe multiple parallel pathways. Using relatively simple surface functionalization methods, individual biomolecules can be immobilized on an optically transparent surface for prolonged tracking. Single-molecule method provides a tracking of translation over multiple codons without complications arising from complex kinetic models, which is extremely useful in studying mRNA and nascent peptide elements. Improvements over the last two decades in camera and dye technologies coupled to advances in image analysis methods have led to the explosion of single-molecule fluorescence microscopy methods, allowing detection of temporal changes in composition and conformation of single biomolecules during translation (24–30). The development of optical and magnetic trapping technologies has enabled direct measurement of forces and mechanical stability of molecular complexes (31). Combination of fluorescence and force methods will be a powerful tool to study translation, but is still technically challenging (32).
Single-molecule fluorescence methods applied to translation led to the identification of key intrinsic dynamic features of the ribosome and its ligands. To track the composition of translational complexes, fluorophores are covalently attached to tRNAs, translation factors, and ribosome, through numerous methods (26). Observing conformational changes requires pairs of conjugated fluorophores to be proximally located within a typical dynamic range of 20–80 Å to observe single-molecule Förster resonance energy transfer (smFRET) between the two or more dyes. Based on the efficiency of FRET, the distance changes between the dye-labeling sites can be detected. Interpreting single-molecule fluorescence data requires specific care to avoid artifacts arising from the experimental setup, which is well out-lined in the classic review by the Ha group (33). A combination of multiple smFRET-pairs and direct labeling of translational factors has been used to correlate conformational and compositional changes during translation in real time (34–36).
2.4. Ribosome profiling (RiboSeq)
Novel DNA and RNA sequencing-based techniques have revolutionized biological measurement. Ribosome profiling (RiboSeq) was specifically developed to study translational activity over the entire transcriptome (37–39). The method utilizes the ability of the ribosome to protect actively translated portions of mRNA from RNase degradation, thereby defining, with nucleotide resolution, the position of the ribosome on that mRNA by sequencing the ribosome protected segments (RPS) of mRNA. These data are compared with sequencing data on the entire transcriptome in parallel, to discriminate transcriptional control effects from observed translational control effects. By using the number of sequencing reads and individual mRNA expression levels as standards, RiboSeq exposes global translational dynamics with a single-base resolution.
Notably, RiboSeq is highly sensitive in detecting unexpected and alternative potential protein products synthesized by translating previously unannotated coding regions. Further, subsets of ribosomes can be purified based on their location within a cell or interaction with another factor, revealing different translational profiles within the same cell (40). However, each step in the sample preparation workflow can introduce experimental biases or artifacts. Antibiotic treatments prior to harvesting can bias against rare codons, and lead to artificial accumulation of reads at initiation sites (41). The experimental procedure used to generate the deep-sequencing library can also introduce bias (42). Despite such pitfalls, RiboSeq can uncover much about the translational dynamics from the whole-organism to the level of specific cell types (43). Such sequencing-based approaches will continue to have a large impact on analysis of genome-wide biophysics during translation and other processes.
3. General overview of translation
The ribosome spatially and temporally coordinates molecular interactions during translation, which can be organized into four phases of initiation, elongation, termination, and recycling. During initiation, the small (30S in bacteria, 40S in eukaryotes; referred to as 30S) and large (50S in bacteria, 60S in eukaryotes; referred to as 50S) ribosomal subunits assemble at the start codon to begin protein synthesis via elongation. Elongation ends when the ribosome reaches a stop codon, whereby a protein factor for termination is recruited to release the synthesized nascent polypeptide from the translation complex. The post-termination complex is then disassembled immediately during recycling. While the initiation, termination, and recycling phases of translation have been shown to be heavily regulated to control the quality and abundance of nascent proteins synthesized from translation (44–49), this review will focus on presenting kinetic and structural schematics of each elongation cycle, where its regulation may lead to different structures and functions of nascent proteins. Mechanisms of how mRNA and nascent-peptide elements regulate protein synthesis will be discussed in the context of schematics presented here.
3.1. Elongation cycle: Decoding
During each elongation cycle, the conformation and composition of the translational complex evolve temporally through several structures (14, 16) (Figure 1). Kinetic parameters for each structural transition are well tuned to provide both processivity and fidelity, which is achieved through proofreading steps that monitor the correct codon-anticodon pairing (50, 51). The elongation cycle begins with decoding, where the cognate tRNA is accommodated into the aminoacyl-acceptor tRNA site (A site) for the peptidyl-transfer reaction (14, 16, 20, 52, 53) with the peptidyl-tRNA site (P site) bound peptidyl-tRNA. During the elongation cycle, tRNAs assume various hybrid conformation within the ribosome, such as A/P state, where the first letter denotes its contact to the 30S, and the second letter denotes its contact to the 50S.
Figure 1.

Schematic of translation elongation. (Top) For an addition of one amino acid to the growing nascent peptide chain, the conformation and composition of the ribosome cycles through two global states: First, the A-site codon is decoded via (i) initial selection and (ii) proofreading by the incoming aminoacyl-tRNA in complex with EF-Tu and GTP. Proofreading ends with the transfer of the nascent peptide to the amino acid on the accommodated tRNA, which is followed by global conformational changes and translocation (with the help of EF-G complexed with GTP), displaying the next codon in the A site.
Results from different methods have been essential to understand the process of initial tRNA selection (Figure 2). Bulk kinetics were crucial in constructing early models of decoding (20, 52, 53), which are now supported by observations made using other methods such as cryoEM (16, 54) and single-molecule fluorescence microscopy (24). Collectively, these studies have revealed that decoding begins when aminoacyl-tRNA (aa-tRNA) samples the T site of the ribosome (Figure 1; (i) Decoding: Initial Selection) as a ternary complex (TC) with a GTP molecule and an elongation factor (EF-Tu in bacteria, eEF1A in eukaryotes; referred to as EF-Tu here), which increases the affinity of tRNA to the ribosome (55). The anticodon of the sampling tRNA initially does not base pair with the A-site presented codon (16, 54), but the tRNA dynamically deforms to form codon-anticodon helix, placing the tRNA in the A*/T state (14). Dynamic formation of the codon-anticodon helix stabilizes tRNA TC binding to the ribosome (20, 24, 52, 53). This is energetically favored for the cognate pairing, which serves to discriminate the non-cognate and near-cognate species kinetically before tRNA TC dissociates from the T site (20, 24, 52, 53). Structural methods such as NMR (56) and x-ray crystallography (57, 58) have identified that the correct formation of the codon-anticodon helix leads to activation of the universally conserved monitoring bases (A1492, A1493 and G530 of 16S rRNA within the 30S ribosomal subunit), which dynamically flip toward the codon-anticodon helix and form multiple hydrogen bonds with the 2’-OH and other conserved chemical groups of the correctly formed helix. The proper interaction among monitoring bases and the codon-anticodon helix positions tRNA TC into the A/T state, providing optimal stereochemistry for the hydrolysis of GTP within the TC through interaction with the sarcin-ricin loop in the 50S ribosomal subunit (58). Recent computational work has shown that the energy landscape of initial selection leading to GTP-hydrolysis is markedly different between cognate and near-cognate tRNA bound complexes, where selectivity for the cognate substrate is amplified in multiple steps (20, 52, 53, 59). The irreversible GTP-hydrolysis marks the end of the initial selection phase of decoding, and subjects the tRNA in the A/T state to additional proofreading steps.
Figure 2.
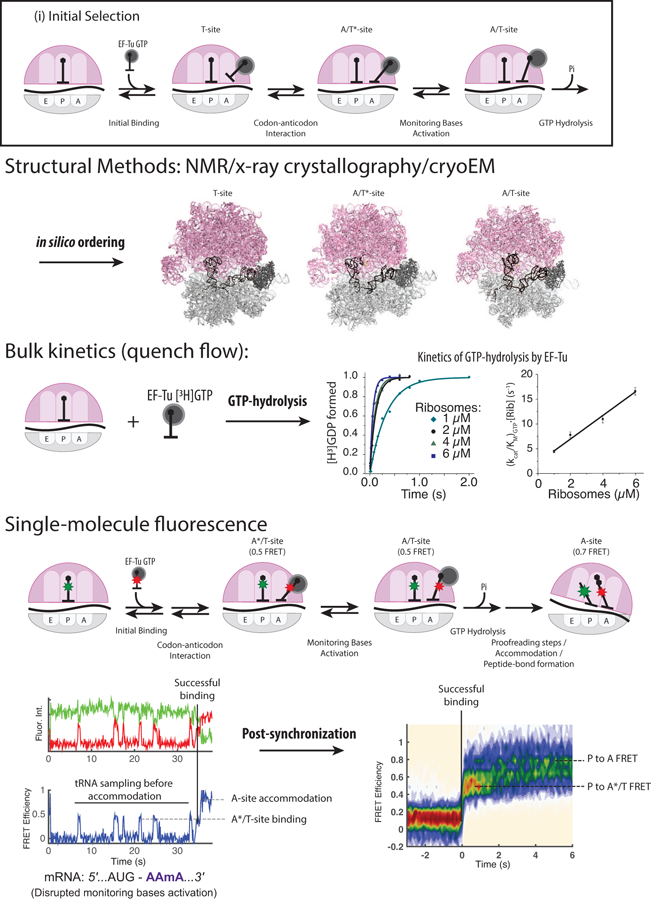
Different methods used to study translation. The current model of translation elongation has advanced through combining results from different methods to construct a coherent model. Structural methods such as NMR, x-ray crystallography, and cryoEM provided the structural context of elongation mechanism at the atomic level. Bulk kinetics methods provide the dynamics links between different biochemical and structural states. Single-molecule fluorescence methods bridge structure and dynamics further by observing both simultaneously. Structures shown here were adapted from (16) (PDB entry 5UYK, 5UYL and 5UYM). Bulk kinetics and single-molecule data were adapted from Choi et al (in press).
Clever experimental designs using the existing techniques have further elucidated the pathways of translational fidelity through proofreading steps (Figure 1; (ii) Decoding: Proofreading). The proofreading mechanism uses the same stability of codon-anticodon interaction and its interaction with monitoring bases to discriminate kinetically against non-cognate tRNA species. Recently, bulk kinetics experiments using engineered tRNAs have suggested that upon GTP-hydrolysis, the on-pathway event of EF-Tu•GDP dissociation kinetically competes with the dissociation of EF-Tu•GDP•aa-tRNA, which results in a tRNA rejection pathway (60). Here, the correct stereochemistry among the monitoring bases and the codon-anticodon helix favors the EF-Tu•GDP dissociation pathway over the EF-Tu•GDP•aa-tRNA rejection pathway (60, 61), serving as the first stage of proofreading, although the involved structural mechanism has not been elucidated. Upon EF-Tu•GDP dissociation, aa-tRNA is subjected to the second stage of proofreading, where the on-pathway event of the aa-tRNA accommodation to the A site competes with the dissociation of aa-tRNA from the A/T-state. The A-site accommodation places the amino acid moiety of the aa-tRNA in a correct stereochemistry in the peptidyl transferase center (PTC) of the large ribosomal subunit, which then catalyzes a rapid transfer of the peptide from the P-site tRNA to the amino acid (62).
3.2. Elongation cycle: Translocation
The peptidyl-transfer reaction after tRNA accommodation induces multiple large-scale conformational changes in the translational complex (63), creating a correct substrate for translocation (Figure 1; (iii) Translocation) (35). Transfer of the nascent peptide to the A-site tRNA allows a movement of the acceptor stems of the P-site and A-site tRNAs into the exit (E) and P sites of the large subunit, respectively (64). These new tRNA conformations are defined as hybrid states, denoted as P/E and A/P states. In addition to the tRNA movements, peptide bond formation is also followed by a rapid counterclockwise rotation of 30S subunit by 3–10° with respect to 50S subunit, resulting in a rotated-state conformation (65, 66). The rotated-state conformation is the cognate substrate of translocation, catalyzed by an elongation factor (EF-G in bacteria and eEF2 in eukaryotes; referred to as EF-G here). EF-G interacts with the ribosome to stabilize the tRNAs in the hybrid state (21, 67–70). Ribosome binding activates the GTPase of EF-G for GTP hydrolysis (71), leading to its conformational change catalyzing translocation (67–69).
Translocation is by definition dynamic and many of the intermediate states involved are transient. As such, dynamic methods—bulk kinetics, single-molecule approaches, multiple structures by cryoEM, computational studies—have been essential to outline the mechanism of translocation (21, 34, 72–75). Using molecular dynamics simulations, different translocation intermediate structures from cryoEM have been temporally ordered to build a structural pathway of translocation (76). Based on these results, combinations of multiple single-molecule fluorescence signals or fluorescence signal with the measured kinetics from bulk methods have produced detailed models of translocation (21, 34, 74, 75). During translocation, the head of the 30S subunit swivels counterclockwise with respect to the body by 18–21° followed by a rapid relaxation back, coupled to disengagement of the mRNA and tRNAs from the decoding center (72, 73, 77). These changes are also accompanied by a clockwise rotation of the 30S subunit relative to 50S subunit that places the ribosome back into the non-rotated intersubunit conformation (35, 78, 79). EF-G then dissociates from the non-rotated post-translocation complex, with tRNAs occupying the P and E sites (80).
After translocation, the dissociation kinetics of deacylated tRNA from the E site determines the structural pathway following translocation. The post-translocation complex is in the non-rotated conformation with a vacant A site (80), where tRNAs can sample and decode the next codon (25, 81). The decoding process is not allosterically affected by the presence or the absence of the E-site tRNA (25, 81–83). However, upon peptidyl-transfer reaction, deacylated tRNA needs to be moved to the P/E hybrid state for translocation. Thus, E-site occupancy, perhaps due to slow E-site tRNA dissociation kinetics, would preclude adoption of the tRNA hybrid conformation until the E-site tRNA dissociates (36). The E-site tRNA dissociation kinetics are sensitive to intracellular ionic strength (81, 84), temperature and tRNA identity (36, 85), and possibly modulated by additional elongation factors, such as eEF3 in fungi (86, 87) or its bacterial homolog, EF4. More work is required to understand the detailed role of the E-site in elongation.
4. mRNA elements altering translation elongation
Beyond their intrinsic capacity to encode proteins, mRNAs possess numerous regulatory elements that impact the rate of protein synthesis at multiple stages. We will focus on the increasingly prominent role the mRNA plays in controlling the rate of elongation. We will cover how i) modifications to a single nucleotide, ii) codon usage, and iii) larger regulatory elements within the mRNA alter translation elongation.
4.1. Nucleotide modifications
A large number of chemical modifications – 163 distinct modifications cataloged to date (88) – have been detected in RNAs. In particular, the last several years have yielded a vast expansion of data that document “RNA epigenetics” (89) and the mRNA “epitranscriptome” (90) – the dynamic co- or post-transcriptional addition of modifications to the nucleotides within mRNAs. The epitranscriptome field has moved rapidly, and several excellent reviews have recently been published that highlights recent discoveries around mRNA modifications and the techniques used to identify them (91–94). Modified nucleotides within mRNA have been shown to affect all aspects of RNA biology— mRNA splicing, export, localization, and stability, as well as multiple stages of translation, including elongation (91, 93).
Chemical modifications that occur on nucleotides within the mRNA coding region can be divided into three broad categories (Figure 3). First, hydrogen-bonding groups on the nucleotide base can be modified, thereby altering its ability to base pair with incoming tRNA. Second, the nucleotide base can be modified outside the Watson-Crick edge of the base, without alteration to its hydrogen-bonding ability. Third, the ribose backbone can be modified, which may affect the tertiary structure of mRNA or RNA-specific recognition mechanisms. Each type of modification is expected to induce different effects during decoding, thereby suggesting new ways to regulate the dynamics of translation elongation. In this section, we focus on three known mRNA modifications – methylation of adenosine at the N6 position (N6-methyladenosine, m6A), the isomerization of uracil (pseudouridine, Ψ), and ribose methylation at the 2’ position (2′-O-methylation).
Figure 3.

Example of chemical modifications in mRNA. Among hundreds of known chemical modifications of RNA bases, several modifications occur within the coding region of mRNA can be classified into three categories. First, modifications occurring in the Watson-crick edge of RNA bases that disrupt base-pairing of mRNA (m6A and m1A), and hinder secondary structure formation or codon-anticodon interaction. Second, modifications occurring outside the Watson-Crick edges of the base (Ψ), which do not alter base-pairing ability of the modified base, but could distort the base-pairing of nearby RNA bases. Third, modifications on the ribose of RNA disrupt its interaction with rRNA during decoding (2′-O-methylation).
m6A is the most common internal mRNA modification, where one of two N6 hydrogens that participates in the base-pairing is modified to a methyl group (Figure 3). Tens of thousands of m6A sites have been defined in thousands of mRNAs from human, mouse, yeast, plant, and bacterial models (95, 96). Substitution of adenosine nucleotides (A) to m6A within the open-reading frame of mRNAs inhibits translation elongation directly (97). While m6A and U form canonical base pairs, they are thermodynamically less stable in duplexes than canonical A-U base pairs (98). While m6A base-pairs with the corresponding U in the tRNA anti-codon in the ribosomal A site, m6A at any of the three positions in a codon disrupts decoding, with the greatest kinetic inhibition at the first nucleotide (99). These data suggest that translation kinetics for particular mRNAs in bacteria and eukaryotes could be modulated by dynamic m6A (de)modifications. While less common in coding regions (100, 101), N1-methyladenosine (m1A) also substantially inhibits tRNA selection and accommodation (102), possibly by preventing codon-anticodon interactions.
The isomerization of uridine to Ψ is the most abundant internal RNA modification, which rearranges atoms outside the Watson-Crick edge (Figure 3). In the ribosome, Ψ nucleotides are present in the peptidyl transferase and decoding centers, where they enhance translation efficiency and fidelity (103, 104). In mRNA, stop codons (UGA, UAA or UAG) with Ψ as the first nucleotide suppress translation termination and allow read-through of premature termination codons in vitro and in vivo (105, 106). Structural analyses of bacterial 30S ribosomal subunits revealed that the presence of a Ψ-A Watson-Crick base pair at the first position of a codon-anticodon interaction distorts the conformation of the decoding center (105), allowing typically prohibited non-cognate tRNAs to decode the modified stop codon via the Hoogsteen base pairing. Yet, among hundreds to thousands of Ψ found within the coding regions of eukaryotic mRNAs (107, 108), modifications of stop codons were extremely rare. How sense codons are affected by the presence of Ψ remains unclear and is an exciting avenue for future studies.
Lastly, 2′-O-methylation is an emerging mRNA modification in eukaryotes. Chemically, by replacing the reactive 2’-OH group with an unreactive O-methyl group, 2′-O-methylation decreases the susceptibility of the nucleotide to nucleolytic attack (i.e. hydrolysis) and stabilizes RNA helices (109). The 2’-OH of RNA is a critical contact point in interactions that are specific for RNA over DNA. Thus, 2′-O-methylation may have substantial impact on protein-RNA, RNA-RNA, and DNA-RNA interactions. Thousands of 2′-O-methylation sites were recently identified within human mRNAs, predominately enriched in the coding region (110). Intriguingly, 2′-O-methylation stalls translation elongation at the modified nucleotide during decoding similar to m6A, with the greatest impact when the codon is 2′-O-methylated at position 2 (in press) (97). A primary cause of the stall is the disruption of the conserved rRNA monitoring bases, which interact with the mRNA 2′-O-moieties for decoding. The distortion of the decoding site leads to both increased rejection of the cognate tRNAs during the initial tRNA selection and subsequent proofreading phase of translation elongation, manifesting as a long stall prior to a successful decoding event. While the functional role of such a pause has not yet uncovered, both m6A and 2′-O-methylation in mRNA showcase how the rate of translation elongation can be widely tuned by chemical modifications to mRNA.
4.2. Codon choice alters decoding kinetics and the pathway of elongation
Each amino acid is encoded by up to six synonymous codons and their usage frequencies vary. In many instances, codon usage is optimally balanced with the abundance of the corresponding aa-tRNAs that recognize them (111, 112). Indeed, inclusion of non-optimal codons inhibits elongation and destabilizes the mRNA (113, 114). The abundance of aa-tRNAs is modulated during cell proliferation and differentiation (115), and in development (116), cancer (117, 118), and other human diseases (119), which together appears to play an important role in regulating the rate of protein synthesis. Thus, seemingly silent DNA mutations that do not impact the encoded amino acid may result in a switch from an optimal to non-optimal codon (or vice versa), altering the abundance or function of the protein.
Synonymous codon usage has a direct impact on the kinetics of decoding and translocation. The rate of aa-tRNA binding is dictated by both the codon present in the A site and its abundance in a cell (120). The usage of rare codons with low concentrations of corresponding aa-tRNAs thus leads to elongation pauses during the decoding phase. While the slow decoding rate induced by rare codons may lead to decreased mRNA stability in cells (113, 114), it may also allow domains of the nascent proteins to co-translationally fold properly; rare codons are especially enriched at the end of protein secondary structures (121). The decoding rate of the A-site codon may be further affected by the codon present in the P site, as permutations of synonymous codons at two adjacent positions contribute to the overall rate of elongation (122). In tandem, the dissociation kinetics of deacylated tRNA from the E site may differ across synonymous codons. Slow E-site tRNA dissociation retains the ribosome in a conformation that is refractory to translocation, where its lifetime depends on the rate of tRNA dissociation (36). Dissociation kinetics of the E-site tRNA are dependent on the tRNA species (36, 85), and may differ among synonymous codons. We speculate that, together with tRNA abundance, the differential usage of synonymous codons in the A, P and E site of the ribosome may lead to non-uniform rates of elongation across an mRNA, which awaits full elucidation.
4.3. Stable RNA structures within mRNA
Information encoded in mRNAs occurs in a three-dimensional context. mRNAs can dynamically fold into complex secondary and tertiary structures that may serve as regulatory elements via recruiting factors that recognize RNA structures, disrupting translocation during translation, or modulating more complex translational events on the ribosome.
mRNA structure can be a ligand for translation factors. The incorporation of the 21st amino-acid, selenocysteine, relies on an RNA-binding protein that specifically recognizes an mRNA stem-loop. A specialized elongation factor (SelB in bacteria, eEFSec-SBP2 in eukaryotes; referred to as SelB) complexed with selenocystenyl-tRNA (Sec-tRNASec) and GTP binds to an mRNA stem-loop – the selenocysteine insertion sequence (SECIS) (54). This interaction facilitates timely delivery of Sec-tRNASec ternary complex to the ribosomal A site, resulting in the recoding of the UGA stop codon. SECIS is an excellent example of how an mRNA structure and its interaction with an RNA-binding protein can alter decoding.
mRNA structures need to be unfolded prior to decoding by the ribosome. During translation, the single-stranded mRNA is threaded through the 5–6 nucleotide-long entrance channel that is composed of three ribosomal proteins (S3, S4 and S5 in bacteria) (123, 124). The relatively narrow width of the mRNA entrance channel prohibits RNA duplexes from entering. Therefore, structured mRNA elements need to be unfolded approximately two codons before it reaches the A site for decoding (125), possibly during translocation step. The unfolding of mRNA structure requires free energy, likely powered by EF-G, which hydrolyzes GTP to catalyze translocation. The proposed model of ribosome translocation on an mRNA involves conformational change of EF-G, which primes the mRNA-tRNA complex for translocation (75, 126). However, the mechanism of translocation through an mRNA hairpin is still debated; while the force generated during the EF-G conformational change may be used directly for helicase activity (75), it has also been suggested that translocation can capture the breathing of the base of mRNA structure (31). Regardless of its helicase mechanism, mRNA structures have been known to slow down elongation, and used to enhance recoding efficiency in many cases (2, 22, 127).
mRNA sequences may base-pair with the rRNA to tether itself to the ribosome. The role of mRNA-rRNA base-pairing is a key aspect of bacterial translation initiation and integral to certain recoding phenomena, such as −1/+1 programmed ribosomal frameshifting (128). However, the mechanism of how mRNA-rRNA base-pairing interactions alters the dynamics of translation elongation have been debated. Early in the study of translation in bacteria, a stretch of mRNA sequences named the Shine-Dalgarno sequence (SD-sequence) was recognized as an important feature of translation initiation, guiding the position of the start codon on the ribosome by base-pairing with the small ribosomal subunit. Its effect during elongation surfaced during the study of programmed ribosomal frameshifting, where it has been hypothesized to “push” or “pull” mRNA to aid frameshifting in both directions. Yet, recent reports have shown that the effect of the SD-sequence on translation elongation kinetics may be minimal (129). Therefore, the mechanism of how the SD-sequence and similar mRNA-rRNA interactions affects translation and enhances recoding phenomena needs further elucidation.
5. Control of Translation Elongation by Nascent-Peptide Elements
In addition to mRNA elements-mediated regulation, the amino acid sequence of the nascent protein chain also regulates translation (4, 5). Amino acid side-chain geometries and chemistries influence substrate positioning within the PTC (5). In the context of the nascent peptide, amino acids can interact with the ribosomal exit tunnel (a ~100 Å-long conduit that bridges the PTC and the solvent environment), resulting in changes in the rate of translation elongation (4). In this section, we will discuss the mechanisms of amino acid sequence-induced ribosomal stalling and its basis in regulating molecular mechanisms such as ribosomal secretion signal and antibiotic resistance.
5.1. Proline-mediated translation stall
The rate of peptidyl transfer for each codon varies depending on how efficient a particular amino acid is as a P-site peptidyl donor and an A-site peptidyl acceptor (130). Proline is the most striking example, since it is the only N-alkylated and cyclic proteinogenic amino acid. As the A-site peptidyl acceptor, the transfer reaction to proline is much slower compared with that of other amino acids like phenylalanine. The higher pKa of the α-N group brought about by the alkylation slows down the rate-limiting peptidyl transfer reaction to an A-site proline (131, 132). Recent ribosomal crystal structures with Pro-tRNA analog in the A site revealed unfavorable positioning of the substrate in the PTC (133), which likely further contributes to the slow peptidyl transfer rate of proline. The unfavorable positioning of proline may also explain its poor activity as the peptidyl donor in the ribosomal P site (134). The low reactivity of proline in both A and P sites results in an amplified inhibition of translating consecutive proline (135, 136).
Proline residues can therefore potentially regulate the rate of protein synthesis. The stalling caused by stretches of proline residues can be alleviated by a special elongation factor (EF-P in bacteria and eIF5A in eukaryotes; referred to as EF-P here) (134, 137, 138), by decreasing the activation energy for formation of the next peptide bond via a favorable entropy change (139). Ribosome profiling results revealed significant ribosomal stalling events at the poly-Pro regions in mRNAs when EF-P was knocked out (140), reflecting the direct effect of EF-P/poly-Pro on mRNA translation. The recent cryoEM structure of EF-P bound to the stalled ribosome suggested that EF-P stabilizes conformation of the nascent peptide and tRNAs to allow efficient translation of poly-Pro regions (141). Combined with cellular levels of EF-P, stretches of proline residues directly affect the expression levels of the corresponding proteins, especially those related in the synthesis of key components of the translation machinery; this may in turn act as an indirect regulator for the translation of other mRNAs or global protein synthesis (142, 143).
5.2. Interaction within the nascent peptide exit tunnel affects translation elongation
Unlike proline residues, which only require interaction with the PTC to induce translational stalling, there are longer nascent peptide sequences that can arrest translating ribosomes through specific interactions with the constricted region of the ribosomal exit tunnel (4). One of the most extensively studied examples of nascent chain-induced stalling is secM/secA ribosomal secretion signaling pathway in bacteria (144). Expression of the SecA protein is coupled to the cellular protein secretion pathway via a negative feedback mechanism, where inefficiencies in the secretion pathway involving SecA induce increased translation of the secA gene. The mechanism of such modulation involves the translation of a secretory protein SecM, which acts to monitor the efficiency of protein export. SecM includes a nascent-peptide sequence that induces a stall in translation, which is relieved when the SecM-stalled ribosomes are docked to the Sec translocon on the cell membrane via the SecA-mediated protein secretion pathway. The secA initiation site is located in the 3’-UTR of secM and, under normal conditions, is structurally sequestered away from ribosomal binding (Figure 4). The prolonged translation stalling on SecM re-arranges mRNA structures to expose the initiation site of the secA gene and thus prompts the synthesis of SecA protein.
Figure 4.
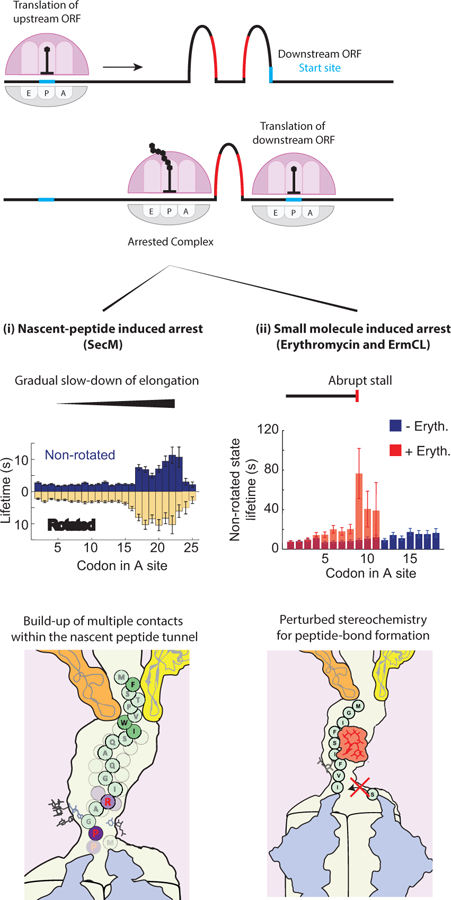
Interactions within the nascent-peptide exit tunnel (NPET) modulate translation dynamics. Various nascent-peptide elements are known to stall translation of the upstream open-reading frame (ORF) at a precise location to induce rearrangements of mRNA folding and expose the previously sequestered translation start-site of the downstream ORF. The stall can be induced by nascent-peptide elements only, some of which have been shown to slow-down elongation gradually by building multiple contacts within the NPET as the nascent peptide is extended. In addition, interactions with a small molecule can cause an abrupt stall of translation at one codon. Single-molecule data and graphics of NPET were adapted from (148) and (153).
Biochemical and genetic studies have identified a 17-aa peptide in SecM, F150XXXXWIXXXXGIRAGP166 (144), that is sufficient for stalling translating ribosomes at Gly165 (145). Structural studies by cryoEM showed how interactions between the SecM-stalling peptide and the ribosomal exit tunnel affect positioning of substrates in the PTC and slow down the peptidyl transfer reaction between Gly165 and Pro166 (146). Disruption in the PTC and interaction within the nascent peptide exit tunnel may affect translocation as well as the peptidyl transfer reaction. Using cryoEM, Zhang et al. captured not only the inactive PTC state that disfavored the accommodation of the incoming A-site tRNA and peptide bond formation, but also another state where the translocation was impaired after successful peptide-bond formation (147). Recent single molecule studies uncovered the real-time dynamic nature of this stalling phenomenon, which involves prolonged decoding and translocation at several positions on SecM (148). This work suggests that residues in SecM interact cooperatively with the ribosome to induce a gradual slow-down of translation elongation. The gradual slow-down of elongation suggests multiple interactions between the nascent peptide and the ribosome, which evolve over the course of translating a SecM-stalling peptide (Figure 4).
Other peptide sequences with moderate effects on ribosomal stalling may be used more pervasively to regulate biomolecular processes. To date, many translation stalling peptide sequences have been identified (4). New methods to identify peptide sequences that induce translational stalling continue to be developed: using bioinformatics, Navon et al. identified underrepresented short-peptide sequences that indeed stall translation (149). Through genetic selection, Tanner et al. evolved a novel peptide sequence, FXXYXIWPP, that impairs peptidyl transfer causing translating ribosomes to stall (150). How different nascent peptide elements are used to induce perturbation in decoding and/or translocation kinetics in prokaryotic and eukaryotic elongation still needs further clarification.
5.3. Sequence-context dependent action of antibiotics
Many antibiotics target key functions of translation by interacting with active sites within the ribosome. Certain classes of antibiotics specifically impede the peptidyl-transfer reaction (151), whereas others, such as macrolides like erythromycin, bind to the ribosomal exit tunnel and cause ribosomal stalling and abortive translation (152). Initial studies suggested that erythromycin acted as a plug in the ribosome exit tunnel blocking the passage of the nascent peptide. However, recent results indicate that the erythromycin mechanism of action relies on the interplay between the nascent peptide, the tunnel and the drug itself, where the susceptibility to drug depends on the nascent peptide sequence (153, 154). Similarly, actions of different antibiotics such as chloramphenicol and linezolid have also been shown to depend on the distinctive nascent peptide sequence, leading to stable accommodation events of incoming tRNA but preventing peptide-bond formation on the stalling site only (unpublished results) (155). Bound antibiotics likely interact with nascent-peptide residues to disrupt the PTC, which affects the accommodation dynamics of the next A-site tRNA and results in abortive translation.
Translational inhibition is blocked through mechanisms of antibiotic resistance. For erythromycin, the basis for the regulation of resistance is very similar to the translational attenuation principle as described above for that of the secM-secA gene regulation. The activation of the macrolide resistance gene ermC relies on translational stalling on the upstream ermCL gene to upregulate protein synthesis (156). The ermCL gene harbors a specific nascent peptide sequence that induces a strong translational arrest in the presence of the erythromycin (Figure 4) (152, 153, 157). The stalled ribosome induces a rearrangement of mRNA structures to expose the initiation site of the downstream ermC gene and up-regulates the expression level of the ErmC methyltransferase (Figure 4) (156). The translation of ermC confers antibiotic resistance by dimethylation of A2058 of 23S rRNA (158), which likely disrupts the binding of the erythromycin to the ribosome. Similarly, a strong chloramphenicol-dependent stall-inducing nascent peptide sequence is present in the catA86L or cmlAL genes to upregulate expression of the downstream catA86 or cmlA genes to confer resistance to chloramphenicol (159). A nascent-peptide sequence can also confer antibiotic resistance for translating individual proteins, where bound antibiotics are evicted through interactions with the growing nascent chain (153, 160). Although distinct interactions of the nascent chain, the exit tunnel and the small molecules are employed in different drug actions, these studies underscore how such interactions can modulate translation dynamics. Therefore, a dynamic view of translation is necessary to understand the mechanisms of antibiotics and the development of resistance.
6. Recoding
The existence of multiple mRNA and nascent-peptide elements that affect translation elongation at various steps highlights the flexibility of the translation machinery to utilize different signals encoded in the mRNA. Individually, the major effect of mRNA and nascent-peptide elements is to modulate the rate of translation elongation. However, combinations of multiple effects may break the linearity of elongation and bifurcate its pathway to produce two different proteins in a tuned ratio from the same mRNA, referred to as “recoding” events. Across the domains of life, different genes utilize recoding signals to increase the density of their genetic information or to regulate the production of a functional protein. In recoding, the normal elongation pathway competes with the recoded pathway, and determining the exact branch point of pathways is critical in elucidating the mechanism of specific recoding phenomenon. Different branching points involved in the various recoding phenomena are starting to be exposed, occurring either during translocation or decoding steps, or combination of both in the extreme case (Figure 5).
Figure 5.

Mechanisms of recoding events involving disrupted translocation and decoding.
A. −1 Frameshifting is likely to occur during translocation, impeded by mRNA structure. Translocation into the structured region of mRNA leads to multiple futile bindings of EF-G (shown from smFRET experiments), which may hydrolyze GTP to translocate the codon-anticodon and unfold the mRNA structure simultaneously. The EF-G-bound state may be susceptible to the frameshifting prior to translocation.
B. +1 Frameshifting may occur during inefficient decoding, where the A-site substrate in the +1 frame (aa-tRNA ternary complex) may compete with the A-site substrate in the original frame (release factors or another aa-tRNA ternary complex). Intermediate structures and dynamics of +1 frameshifting have yet to be revealed.
C. The first stage of bypassing involves impeded decoding by the folding of the nascent-peptide chain within the exit tunnel, which allows mRNA to fold into the vacant A-site and weaken codon-anticodon interaction in the P site, facilitating the take-off of the ribosome from the current P-site codon. Shown structures were adapted from (180).
D. After take-off, landing of the ribosome during bypassing may involve several mRNA structures that limit the translocation of the taken-off ribosome. Repeated EF-G binding during landing has been observed in smFRET experiments. Yet the role of EF-G during landing is still unclear.
6.1. The repurposing of translocation for −1 frameshifting
Programmed ribosomal −1 frameshifting (referred to as “−1 frameshifting”) is the best-studied recoding phenomenon to date. During −1 frameshifting, the open reading frame of the mRNA contains a programmed “slippery sequence”: A sequence of nucleotides where translation can resume in the original frame (0-frame) or in another shifted one base in the 5’ direction (−1-frame). Translation of a −1 frameshifting signal thus produces two different proteins that share the same N-terminal sequence with a fixed stoichiometry. The most well-known usage of −1 frameshifting is in human immunodeficiency virus (HIV), where a −1 frameshifting signal is used to produce Gag and Gag-Pol proteins at a pre-determined ratio (1). Regulating protein expressions through −1 frameshifting enriches information density of genomes and likely benefits viruses such as HIV, where the fitness of viruses is linked to the compactness of its genome. New instances of −1 frameshifting are still being discovered in viruses, bacteria and eukaryotes, and its usage may be much higher than anticipated, perhaps stimulated by external elements such as RNA-binding proteins (161), microRNAs (162) and antibiotics (163).
Translation normally requires ribosomes to maintain their original reading frame throughout elongation. Errors resulting from the spontaneous shifting of the reading frame are energetically costly, resulting in the production of unwanted proteins that may be harmful to the cell. The error rate for the spontaneous shifting has been estimated as 10−5 (164, 165), which is an order of magnitude smaller than the 10−4 rate of missense errors (substitution of a single amino-acid) (166, 167). Therefore, −1 frameshifting signals that result in 30–70% efficiency present a controlled way to circumvent an error prevention mechanism utilized during normal elongation (1–3).
The set of factors involved in −1 frameshifting is continually expanding and includes both mRNA and nascent-peptide elements, as well as external factors. Among them, the slippery sequence within the mRNA is essential. They typically conform to a tetrameric or heptameric sequence (N–NNX–XXZ or X–XXX–XXZ, where N, X and Z denotes a nucleotide, and a dash denotes separation of codons in the original reading frame) that allows cognate tRNA pairing in two frames. The heptameric sequence generally yields higher frameshifting efficiency than that of the tetrameric sequence, indicating that it can involve up to two tRNAs (168).
However, the slippery sequence alone does not induce highly efficient −1 frameshifting, usually less than 5% (1–3, 169, 170). Downstream mRNA structures are most often required for highly efficient −1 frameshifting of up to 50–70% (1–3, 169, 170). The involved structures are strikingly varied, from a simple stem-loop to a pseudoknot to long-range RNA folding that span distances of thousands of nucleotide bases within a single mRNA molecule (1–3). The stability of the RNA structures positively correlates with −1 frameshifting efficiency (170). Moreover, microRNAs (162) and RNA-binding proteins (161) enhance −1 frameshifting efficiency, very likely by increasing the stability of mRNA structure. In addition to the stability of the mRNA structure, its position relative to the slippery sequence also affects −1 frameshifting. Highly-efficient frameshifting systems frequently contain 5–6 nucleotides between the 3’ end of the slippery sequence and the 5’ base of the mRNA structure. Deviation from this distance in either direction decreases frameshifting efficiency (127, 169).
The mechanism of −1 frameshifting is emerging from a combination of different methods. Through studies of individual mRNA structures used in −1 frameshifting, it is clear that they act to induce a pause during translation elongation. The pause most likely occurs during translocation (22, 171, 172), requiring multiple EF-G bindings to catalyze translocation on the slippery sequence coupled with the simultaneous unfolding of the mRNA structure (Figure 5A). Indeed, the distance of 5–6 nucleotides between the slippery sequence and the mRNA structure matches the length of the mRNA entrance channel (123, 124). Furthermore, this distance induces a substantially longer translocation pause than the same structure placed 7 nucleotides away from the slippery sequence (Unpublished data) (125).
Within the translation elongation cycle, translocation poses a unique opportunity for −1 frameshifting. During translocation, the ribosome needs to transfer tRNA-mRNA complexes from one position (A/P and P/E) to another (P and E) (73). This transition must disrupt all existing interactions between the tRNA-mRNA complex and the ribosome, specifically on the 30S ribosomal subunit. Thus, at this point in translation, the reading frame is particularly vulnerable to spontaneous shifts. The slippery sequence may act to lower the energy barrier for such spontaneous shifts towards one direction in the translocation intermediate state, whereas the mRNA structure may act to increase the lifetime of such a state (171, 172). The transition state may require binding of EF-G and its conformational change after GTP hydrolysis (75), which may be necessary to weaken the tRNA-mRNA/ribosome interaction (77). This −1 frameshifting mechanism suggests that it repurposes futile translocation cycles catalyzed by EF-G to unfold the mRNA structure to lower the energy barrier for −1 frameshifting.
The link between repeated translocation attempts on the slippery sequence and −1 frameshifting explains the use of different factors to enhance −1 frameshifting efficiency. The nascent-peptide signal can also disrupt translocation and induce multiple translocation attempts by EF-G (148), which may act in a similar manner as the mRNA structure to increase −1 frameshifting efficiency. The presence of antibiotics such as telithromycin may increase the efficiency of −1 frameshifting by mimicking such nascent-peptide signals (153, 163). On poly-A mRNA sequences, repeated A–AAA–AAA sequences can act as slippery sequences, and multiple translocations from one slippery sequence to the next may explain the high occurrence of frameshifting at these locations that signal aberrant translation happening in the untranslated region (173, 174). Finally, the energy barrier for spontaneous frameshifting on the slippery sequence not only depends on the composition of the slippery sequence itself, but also on external elements. Particularly, the internal SD-sequence may induce a force to alter its free energy barrier height and facilitate −1 frameshifting (169). Taken together, the −1 frameshifting cassette repurposes the translocation force, which is used to unfold mRNA structures or disengage interactions within the nascent peptide exit tunnel, to redefine the reading frame of the actively translating ribosome.
6.2. Disruption of decoding by mRNA used in +1 frameshifting
While −1 frameshifting illustrates the use of mRNA and nascent-peptide elements to induce recoding by disrupting translocation, the mechanism of other recoding phenomena, such as +1 frameshifting and bypassing, may exploit delayed decoding. Programmed +1 frameshifting was first identified in the prfB gene of E. coli, which encodes for release factor 2 (RF2) that recognizes UGA and UAA stop codons for translation termination. However, the RF2 sequence is encoded in the +1 reading frame (shifted one base toward the mRNA 3’ direction) respect to its initiation site, requiring +1 frameshifting to bypass the internal UGA stop codon in the 0-frame. In fact, the +1 frameshifting occurs at a slippery sequence (CUU-UGA) next to the stop codon, which allows cognate pairing of tRNALeu (with AAG anticodon) in two frames (128). In addition to the slippery sequence, a nearby strong SD-sequence placed enhances +1 frameshifting efficiency as well. Yet, the strongest modulator of +1 frameshifting efficiency on prfB is the intracellular concentration of RF2 itself (175), where its high concentration inhibits +1 frameshifting efficiency. This suggests that the absence of the cognate substrate (aa-tRNA or RF2) in the A site may make the ribosome more susceptible to +1 frameshifting. The mechanism of prfB +1 frameshifting presents a negative feedback system to control the concentration of RF2, where its absence promotes +1 frameshifting to synthesis more copies of RF2. While the report of +1 frameshifting occurrence in other system has been sparse, recent reports suggested that +1 frameshifting occurs pervasively in some of Euplotes species, where the stop codon again signals +1 or +2 frameshifting instead of termination, possibly related to kinetic competition between 0-frame and +1- or +2-frame A-site substrates (Figure 5B) (176, 177).
6.3. Disruption of decoding and translocation used in bypassing
Translational bypassing is a spectacular and striking example of recoding, involving an interplay of mRNA structure and nascent peptide chain interactions. During bypassing observed in gene 60 of T4 bacteriophage, a population of the translating ribosomes is detached from the codon-anticodon interaction (“take-off”) to hop a stretch of nucleotides in the coding region, and resume translation at a downstream matching codon (“landing”) (6, 178). Among the factors utilized to enhance bypassing, the nascent peptide sequence has been suggested to serve dual roles during bypassing (Figure 5C). First, its interaction with the nascent peptide exit tunnel may prevent peptidyl-tRNA from dissociation once the codon-anticodon interaction has been disrupted during take-off. Second, the nascent peptide may disrupt the decoding dynamics similar to the SecM sequence (179) in order to keep the ribosomal A site vacant. Recent cryoEM and single-molecule studies have shown that a small mRNA structure can form within the vacant A site (Figure 5C) (179, 180), which has been suggested to disrupt the codon-anticodon interaction prior to take-off. The A site codon is the UAG stop codon, which may prolong the vacancy in the A site similar to +1 frameshifting.
Once the ribosome has broken codon-anticodon interaction in the P site during the take-off, it needs to translocate to find the correct landing site. Prior to landing, the ribosome is likely to be in the rotated-like state, observed using smFRET methods, where EF-G-catalyzed translocation is inhibited (Figure 5D) (179). In addition to the nascent peptide element and mRNA structure forming in the A site, bypassing may involve two more mRNA structures, which may act to bookend the slippage of ribosomes after take-off (179, 181). However, it is less likely that these mRNA structures disrupt translocation to enhance bypassing efficiency, dissimilar to what has been observed in −1 frameshifting. The rotated-like state is resolved once the ribosome finds its correct landing site, where normal elongation resumes (179). The different use of the same mRNA and nascent-peptide elements observed across different recoding phenomena underlines the flexibility of the translational machinery, and the enrichment of genetic codes during the protein synthesis.
7. Conclusions and Perspectives
Myriad ways of controlling translation elongation dynamics have been revealed during the past decades, with more surely to be uncovered. This review has focused on how the core substrate and product of translation – mRNA and protein – can act as modulators of the overall process. The molecular and dynamic complexity of translation will continue to challenge our ability to capture mechanistic detail with atomic precision. However, the promising confluence of structural, dynamic and genomic and proteomic methods bodes well for our ability to unravel this complexity. Technological advances in instrumentation sensitivity and computational power underlies this progress; understanding the central role of translation in health and disease requires it.
Literature Cited:
- 1.Atkins JF, Gesteland RF, eds. 2010. Recoding: Expansion of Decoding Rules Enriches Gene Expression New: Springer Verlag [Google Scholar]
- 2.Caliskan N, Peske F, Rodnina MV. 2015. Changed in translation: mRNA recoding by −1 programmed ribosomal frameshifting. Trends Biochem. Sci 40(5):265–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Atkins JF, Loughran G, Bhatt PR, Firth AE, Baranov PV. 2016. Ribosomal frameshifting and transcriptional slippage: From genetic steganography and cryptography to adventitious use. Nucleic Acids Res 44(15):7007–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito K, Chiba S. 2013. Arrest Peptides: Cis -Acting Modulators of Translation. Annu. Rev. Biochem 82(1):171–202 [DOI] [PubMed] [Google Scholar]
- 5.Wilson DN, Arenz S, Beckmann R. 2016. Translation regulation via nascent polypeptide-mediated ribosome stalling. Curr. Opin. Struct. Biol 37:123–33 [DOI] [PubMed] [Google Scholar]
- 6.Weiss RB, Huang WM, Dunn DM. 1990. A nascent peptide is required for ribosomal bypass of the coding gap in bacteriophage T4 gene 60. Cell 62(1):117–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meydan S, Klepacki D, Karthikeyan S, Margus T, Thomas P, et al. 2017. Programmed Ribosomal Frameshifting Generates a Copper Transporter and a Copper Chaperone from the Same Gene. Mol. Cell 65(2):207–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mustoe AM, Brooks CL, Al-Hashimi HM. 2014. Hierarchy of RNA Functional Dynamics. Annu. Rev. Biochem 83(1):441–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Javed A, Christodoulou J, Cabrita LD, Orlova EV. 2017. The ribosome and its role in protein folding: Looking through a magnifying glass. Acta Crystallogr. Sect. D Struct. Biol 73(6):509–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nygaard R, Romaniuk JAH, Rice DM, Cegelski L. 2017. Whole Ribosome NMR: Dipolar Couplings and Contributions to Whole Cells. J. Phys. Chem. B 121(40):9331–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gelis I, Vitzthum V, Dhimole N, Caporini MA, Schedlbauer A, et al. 2013. Solid-state NMR enhanced by dynamic nuclear polarization as a novel tool for ribosome structural biology. J. Biomol. NMR 56(2):85–93 [DOI] [PubMed] [Google Scholar]
- 12.Kurauskas V, Crublet E, Macek P, Kerfah R, Gauto DF, et al. 2016. Sensitive proton-detected solid-state NMR spectroscopy of large proteins with selective CH 3 labelling: application to the 50S ribosome subunit. Chem. Commun 52(61):9558–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barbet-Massin E, Huang CT, Daebel V, Hsu STD, Reif B. 2015. Site-specific solid-state NMR studies of “trigger factor” in complex with the large ribosomal subunit 50S. Angew. Chemie - Int. Ed 54(14):4367–69 [DOI] [PubMed] [Google Scholar]
- 14.Voorhees RM, Ramakrishnan V. 2013. Structural basis of the translational elongation cycle. Annu. Rev. Biochem 82:203–36 [DOI] [PubMed] [Google Scholar]
- 15.Liu Z, Gutierrez-Vargas C, Wei J, Grassucci RA, Sun M, et al. 2017. Determination of the ribosome structure to a resolution of 2.5 Å by single-particle cryo-EM. Protein Sci 26(1):82–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loveland AB, Demo G, Grigorieff N, Korostelev AA. 2017. Ensemble cryo-EM elucidates the mechanism of translation fidelity. Nature 546(7656):113–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer N, Neumann P, Konevega AL, Bock LV., Ficner R, et al. 2015. Structure of the E. coli ribosome–EF-Tu complex at <3 Å resolution by Cs-corrected cryo-EM. Nature 520(7548):567–70 [DOI] [PubMed] [Google Scholar]
- 18.Chen B, Kaledhonkar S, Sun M, Shen B, Lu Z, et al. 2015. Structural Dynamics of Ribosome Subunit Association Studied by Mixing-Spraying Time-Resolved Cryogenic Electron Microscopy. Structure 23(6):1097–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaikh TR, Yassin AS, Lu Z, Barnard D, Meng X, et al. 2014. Initial bridges between two ribosomal subunits are formed within 9.4 milliseconds, as studied by time-resolved cryo-EM. Proc. Natl. Acad. Sci 111(27):9822–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson M, Bouakaz E, Lovmar M, Ehrenberg M. 2008. The Kinetics of Ribosomal Peptidyl Transfer Revisited. Mol. Cell 30(5):589–98 [DOI] [PubMed] [Google Scholar]
- 21.Belardinelli R, Sharma H, Caliskan N, Cunha CE, Peske F, et al. 2016. Choreography of molecular movements during ribosome progression along mRNA. Nat. Struct. Mol. Biol 23(4):342–48 [DOI] [PubMed] [Google Scholar]
- 22.Caliskan N, Katunin VI, Belardinelli R, Peske F, Rodnina MV. 2014. Programmed −1 frameshifting by kinetic partitioning during impeded translocation. Cell 157(7):1619–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang J, Kwiatkowski M, Forster AC. 2015. Kinetics of Ribosome-Catalyzed Polymerization Using Artificial Aminoacyl-tRNA Substrates Clarifies Inefficiencies and Improvements. ACS Chem. Biol 10(10):2187–92 [DOI] [PubMed] [Google Scholar]
- 24.Blanchard SC, RLG Jr, Kim HD, Chu S, Puglisi JD. 2004. tRNA selection and kinetic proofreading in translation. Nat. Struct. Mol. Biol 11(10):1008–14 [DOI] [PubMed] [Google Scholar]
- 25.Uemura S, Aitken CE, Korlach J, Flusberg B a, Turner SW, Puglisi JD. 2010. Real-time tRNA transit on single translating ribosomes at codon resolution. Nature 464(7291):1012–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marshall RA, Aitken CE, Dorywalska M, Puglisi JD. 2008. Translation at the single-molecule level. Annu. Rev. Biochem 77:177–203 [DOI] [PubMed] [Google Scholar]
- 27.Frank J, Gonzalez RL. 2010. Structure and dynamics of a processive Brownian motor: the translating ribosome. Annu. Rev. Biochem 79:381–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang Y, Qin H, Kudaravalli RD, Kirillov SV., Dempsey GT, et al. 2007. Single-molecule structural dynamics of EF-G-ribosome interaction during translocation. Biochemistry 46(38):10767–75 [DOI] [PubMed] [Google Scholar]
- 29.Munro JB, Altman RB, O’Connor N, Blanchard SC. 2007. Identification of Two Distinct Hybrid State Intermediates on the Ribosome. Mol. Cell 25(4):505–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fei J, Kosuri P, MacDougall DD, Gonzalez RL. 2008. Coupling of Ribosomal L1 Stalk and tRNA Dynamics during Translation Elongation. Mol. Cell 30(3):348–59 [DOI] [PubMed] [Google Scholar]
- 31.Qu X, Wen J-D, Lancaster L, Noller HF, Bustamante C, Tinoco I. 2011. The ribosome uses two active mechanisms to unwind messenger RNA during translation. Nature 475(7354):118–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Comstock MJ, Ha T, Chemla YR. 2011. Ultrahigh-resolution optical trap with single-fluorophore sensitivity. Nat. Methods 8(4):335–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roy R, Hohng S, Ha T. 2008. A practical guide to single-molecule FRET. Nat. Methods 5(6):507–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wasserman MR, Alejo JL, Altman RB, Blanchard SC. 2016. Multiperspective smFRET reveals rate-determining late intermediates of ribosomal translocation. Nat. Struct. Mol. Biol 23(4):333–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen J, Petrov A, Tsai A, O’Leary SE, Puglisi JD. 2013. Coordinated conformational and compositional dynamics drive ribosome translocation. Nat. Struct. Mol. Biol 20(6):718–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi J, Puglisi JD. 2017. Three tRNAs on the ribosome slow translation elongation. Proc. Natl. Acad. Sci 114(52):13691–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. 2009. Genome-Wide Analysis in Vivo of Translation with Nucleotide Resolution Using Ribosome Profiling. Science (80-. ) 324(5924):218–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brar GA, Weissman JS. 2015. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat. Rev. Mol. Cell Biol 16(11):651–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McGlincy NJ, Ingolia NT. 2017. Transcriptome-wide measurement of translation by ribosome profiling. Methods 126:112–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jan CH, Williams CC, Weissman JS. 2015. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science) 346(6210):1257521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bartholomäus A, Del Campo C, Ignatova Z. 2016. Mapping the non-standardized biases of ribosome profiling. Biol. Chem 397(1):23–35 [DOI] [PubMed] [Google Scholar]
- 42.Van Dijk EL, Jaszczyszyn Y, Thermes C. 2014. Library preparation methods for next-generation sequencing: Tone down the bias. Exp. Cell Res 322(1):12–20 [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez C, Sims JS, Hornstein N, Mela A, Garcia F, et al. 2014. Ribosome Profiling Reveals a Cell-Type-Specific Translational Landscape in Brain Tumors. J. Neurosci 34(33):10924–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen J, Choi J, O’Leary SE, Prabhakar A, Petrov A, et al. 2016. The molecular choreography of protein synthesis: translational control, regulation, and pathways. Q. Rev. Biophys 49:e11. [DOI] [PubMed] [Google Scholar]
- 45.Simms CL, Thomas EN, Zaher HS. 2017. Ribosome-based quality control of mRNA and nascent peptides. Wiley Interdiscip. Rev. RNA 8(1): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Buskirk AR, Green R. 2017. Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Philos. Trans. R. Soc. B Biol. Sci 372(1716):20160183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Joazeiro CAP. 2017. Ribosomal Stalling During Translation: Providing Substrates for Ribosome-Associated Protein Quality Control. Annu. Rev. Cell Dev. Biol 33(1):annurev-cellbio-111315–125249 [DOI] [PubMed] [Google Scholar]
- 48.Brandman O, Hegde RS. 2016. Ribosome-associated protein quality control. Nat. Struct. Mol. Biol 23(1):7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Defenouillère Q, Fromont-Racine M. 2017. The ribosome-bound quality control complex: from aberrant peptide clearance to proteostasis maintenance. Curr. Genet 63(6):997–1005 [DOI] [PubMed] [Google Scholar]
- 50.Ninio J 1975. Kinetic amplification of enzyme discrimination. Biochimie 57(5):587–95 [DOI] [PubMed] [Google Scholar]
- 51.Hopfield JJ. 1974. Kinetic Proofreading: A New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc. Natl. Acad. Sci 71(10):4135–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gromadski KB, Rodnina MV. 2004. Kinetic determinants of high-fidelity tRNA discrimination on the ribosome. Mol. Cell 13(2):191–200 [DOI] [PubMed] [Google Scholar]
- 53.Gromadski KB, Daviter T, Rodnina MV. 2006. A uniform response to mismatches in codon-anticodon complexes ensures ribosomal fidelity. Mol. Cell 21(3):369–77 [DOI] [PubMed] [Google Scholar]
- 54.Fischer N, Neumann P, Bock LV., Maracci C, Wang Z, et al. 2016. The pathway to GTPase activation of elongation factor SelB on the ribosome. Nature 540(7631):80–85 [DOI] [PubMed] [Google Scholar]
- 55.Rodnina MV, Gromadski KB, Kothe U, Wieden HJ. 2005. Recognition and selection of tRNA in translation. FEBS Lett 579(4 SPEC. ISS.):938–42 [DOI] [PubMed] [Google Scholar]
- 56.Yoshizawa S, Fourmy D, Puglisi JD. 1999. Recognition of the Codon-Anticodon Helix by Ribosomal RNA. Science (80-. ) 285(5434):1722–25 [DOI] [PubMed] [Google Scholar]
- 57.Ogle JM, Brodersen DE, Clemons WM, Tarry MJ, Carter AP, Ramakrishnan V. 2001. Recognition of Cognate Transfer RNA by the 30S Ribosomal Subunit. Science (80-. ) 292(5518):897–902 [DOI] [PubMed] [Google Scholar]
- 58.Ogle JM, Murphy FV, Tarry MJ, Ramakrishnan V. 2002. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell 111(5):721–32 [DOI] [PubMed] [Google Scholar]
- 59.Satpati P, Sund J, Åqvist J. 2014. Structure-based energetics of mRNA decoding on the ribosome. Biochemistry 53(10):1714–22 [DOI] [PubMed] [Google Scholar]
- 60.Ieong K-W, Uzun Ü, Selmer M, Ehrenberg M. 2016. Two proofreading steps amplify the accuracy of genetic code translation. Proc. Natl. Acad. Sci. U. S. A, p. 201610917. [DOI] [PMC free article] [PubMed]
- 61.Noel JK, Whitford PC. 2016. How EF-Tu can contribute to efficient proofreading of aa-tRNA by the ribosome. Nat. Commun 7(May):1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beringer M, Rodnina MV. 2007. The Ribosomal Peptidyl Transferase. Mol. Cell 26(3):311–21 [DOI] [PubMed] [Google Scholar]
- 63.Ratje AH, Loerke J, Mikolajka A, Brünner M, Hildebrand PW, et al. 2010. Head swivel on the ribosome facilitates translocation by means of intra-subunit tRNA hybrid sites. Nature 468(7324):713–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moazed D, Noller HF. 1989. Intermediate states in the movement of transfer RNA in the ribosome. Nature 342(6246):142–48 [DOI] [PubMed] [Google Scholar]
- 65.Frank J, Agrawal RK. 2000. A ratchet-like inter-subunit reorganization of the ribosome during translocation. Nature 406(6793):318–22 [DOI] [PubMed] [Google Scholar]
- 66.Valle M, Zavialov A, Sengupta J, Rawat U, Ehrenberg M, Frank J. 2003. Locking and Unlocking of Ribosomal Motions. Cell 114(1):123–34 [DOI] [PubMed] [Google Scholar]
- 67.Brilot AF, Korostelev AA, Ermolenko DN, Grigorieff N. 2013. Structure of the ribosome with elongation factor G trapped in the pretranslocation state. Proc. Natl. Acad. Sci. U. S. A 110(52):20994–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Pulk A, Cate JHD. 2013. Control of ribosomal subunit rotation by elongation factor G. Science 340(6140):1235970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tourigny DS, Fernández IS, Kelley AC, Ramakrishnan V. 2013. Elongation factor G bound to the ribosome in an intermediate state of translocation. Science 340(6140):1235490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sharma H, Adio S, Senyushkina T, Belardinelli R, Peske F, Rodnina MV. Kinetics of Spontaneous and EF-G-Accelerated Rotation of Ribosomal Subunits. Cell Rep [DOI] [PubMed]
- 71.Rodnina MV, Savelsbergh A, Katunin VI, Wintermeyer W. 1997. Hydrolysis of GTP by elongation factor G drives tRNA movement on the ribosome. Nature 385:37–41 [DOI] [PubMed] [Google Scholar]
- 72.Guo Z, Noller HF. 2012. Rotation of the head of the 30S ribosomal subunit during mRNA translocation. Proc. Natl. Acad. Sci 109(50):20391–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou J, Lancaster L, Donohue JP, Noller HF. 2014. How the ribosome hands the A-site tRNA to the P site during EF-G–catalyzed translocation. Science (80-. ) 345(6201):1188–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adio S, Senyushkina T, Peske F, Fischer N, Wintermeyer W, Rodnina MV. 2015. Fluctuations between multiple EF-G-induced chimeric tRNA states during translocation on the ribosome. Nat. Commun 6(May):1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chen C, Cui X, Beausang JF, Zhang H, Farrell I, et al. 2016. Elongation factor G initiates translocation through a power stroke. Proc. Natl. Acad. Sci 113(27):201602668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bock LV, Blau C, Schröder GF, Davydov II, Fischer N, et al. 2013. Energy barriers and driving forces in tRNA translocation through the ribosome. Nat. Struct. Mol. Biol 20(12):1390–96 [DOI] [PubMed] [Google Scholar]
- 77.Liu G, Song G, Zhang D, Zhang D, Li Z, et al. 2014. EF-G catalyzes tRNA translocation by disrupting interactions between decoding center and codon–anticodon duplex. Nat Struct Mol Biol 21(9):817–24 [DOI] [PubMed] [Google Scholar]
- 78.Marshall RA, Dorywalska M, Puglisi JD. 2008. Irreversible chemical steps control intersubunit dynamics during translation. Proc. Natl. Acad. Sci. U. S. A 105(40):15364–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ermolenko DN, Majumdar ZK, Hickerson RP, Spiegel PC, Clegg RM, Noller HF. 2007. Observation of Intersubunit Movement of the Ribosome in Solution Using FRET. J. Mol. Biol 370(3):530–40 [DOI] [PubMed] [Google Scholar]
- 80.Gao Y, Selmer M, Dunham CM, Weixlbaumer A, Kelley AC, Ramakrishnan V. 2009. The Structure of the Ribosome with Elongation Factor G Trapped in the Posttranslocational State. Science (80-. ) 326(October):694–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chen C, Stevens B, Kaur J, Smilansky Z, Cooperman BS, Goldman YE. 2011. Allosteric vs. spontaneous exit-site (E-site) tRNA dissociation early in protein synthesis. Proc. Natl. Acad. Sci 108(41):1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Semenkov YP, Rodnina MV, Wintermeyer W. 1996. The “allosteric three-site model” of elongation cannot be confirmed in a well-defined ribosome system from Escherichia coli. Proc. Natl. Acad. Sci 93(22):12183–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Petropoulos AD, Green R. 2012. Further in vitro exploration fails to support the allosteric three-site model. J. Biol. Chem 287(15):11642–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burkhardt N, Jünemann R, Spahn CM, Nierhaus KH. 1998. Ribosomal tRNA binding sites: three-site models of translation. Crit. Rev. Biochem. Mol. Biol 33(2):95–149 [DOI] [PubMed] [Google Scholar]
- 85.Fei J, Bronson JE, Hofman JM, Srinivas RL, Wiggins CH, Gonzalez RL. 2009. Allosteric collaboration between elongation factor G and the ribosomal L1 stalk directs tRNA movements during translation. Proc. Natl. Acad. Sci 106(37):15702–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Triana-Alonso FJ, Chakraburtty K, Nierhaus KH. 1995. The elongation factor 3 unique in higher fungi and essential for protein biosynthesis is an E site factor. J. Biol. Chem 270(35):20473–78 [DOI] [PubMed] [Google Scholar]
- 87.Pech M, Karim Z, Yamamoto H, Kitakawa M, Qin Y, Nierhaus KH. 2011. Elongation factor 4 (EF4/LepA) accelerates protein synthesis at increased Mg2+ concentrations. Proc. Natl. Acad. Sci 108(8):3199–3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Machnicka MA, Milanowska K, Oglou OO, Purta E, Kurkowska M, et al. 2017. MODOMICS: A database of RNA modification pathways - 2017 update. Nucleic Acids Res 41(D1):1–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.He C 2010. Grand Challenge Commentary: RNA epigenetics? Nat. Chem. Biol 6(12):863–65 [DOI] [PubMed] [Google Scholar]
- 90.Saletore Y, Meyer K, Korlach J, Vilfan ID, Jaffrey S, Mason CE. 2012. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol 13(10):175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Roundtree IA, Evans ME, Pan T, He C. 2017. Dynamic RNA Modifications in Gene Expression Regulation. Cell 169(7):1187–1200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Li X, Xiong X, Yi C. 2016. Epitranscriptome sequencing technologies: decoding RNA modifications. Nat. Methods 14(1):23–31 [DOI] [PubMed] [Google Scholar]
- 93.Lewis CJT, Pan T, Kalsotra A. 2017. RNA modifications and structures cooperate to guide RNA–protein interactions. Nat. Rev. Mol. Cell Biol 18(3):202–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Helm M, Motorin Y. 2017. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat. Rev. Genet 18(5):275–91 [DOI] [PubMed] [Google Scholar]
- 95.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485(7397):201–6 [DOI] [PubMed] [Google Scholar]
- 96.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell 149(7):1635–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hoernes TP, Clementi N, Faserl K, Glasner H, Breuker K, et al. 2016. Nucleotide modifications within bacterial messenger RNAs regulate their translation and are able to rewire the genetic code. Nucleic Acids Res 44(2):852–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Roost C, Lynch SR, Batista PJ, Qu K, Chang HY, Kool ET. 2015. Structure and thermodynamics of N6-methyladenosine in RNA: A spring-loaded base modification. J. Am. Chem. Soc 137(5):2107–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Choi J, Ieong K-W, Demirci H, Chen J, Petrov A, et al. 2016. N6-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat. Struct. Mol. Biol 23(2):110–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li X, Xiong X, Wang K, Wang L, Shu X, et al. 2016. Transcriptome-wide mapping reveals reversible and dynamic N1-methyladenosine methylome. Nat. Chem. Biol 12(5):311–16 [DOI] [PubMed] [Google Scholar]
- 101.Dominissini D, Nachtergaele S, Moshitch-Moshkovitz S, Peer E, Kol N, et al. 2016. The dynamic N1-methyladenosine methylome in eukaryotic messenger RNA. Nature 530(7591):441–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.You C, Dai X, Wang Y. 2017. Position-dependent effects of regioisomeric methylated adenine and guanine ribonucleosides on translation. Nucleic Acids Res 45(15):9059–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ge J, Yu YT. 2013. RNA pseudouridylation: New insights into an old modification. Trends Biochem. Sci 38(4):210–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.King TH, Liu B, McCully RR, Fournier MJ. 2003. Ribosome structure and activity are altered in cells lacking snoRNPs that form pseudouridines in the peptidyl transferase center. Mol. Cell 11(2):425–35 [DOI] [PubMed] [Google Scholar]
- 105.Fernández IS, Ng CL, Kelley AC, Wu G, Yu Y-T, Ramakrishnan V. 2013. Unusual base pairing during the decoding of a stop codon by the ribosome. Nature 500(7460):107–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Karijolich J, Yu Y-T. 2011. Converting nonsense codons into sense codons by targeted pseudouridylation. Nature 474(7351):395–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schwartz S, Bernstein DA, Mumbach MR, Jovanovic M, Herbst RH, et al. 2014. Transcriptome-wide mapping reveals widespread dynamic-regulated pseudouridylation of ncRNA and mRNA. Cell 159(1):148–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Carlile TM, Rojas-Duran MF, Zinshteyn B, Shin H, Bartoli KM, Gilbert WV. 2014. Pseudouridine profiling reveals regulated mRNA pseudouridylation in yeast and human cells. Nature 515(7525):143–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Cummins LL, Owens SR, Risen LM, Lesnik EA, Freier SM, et al. 1995. Characterization of fully 2’-modified oligoribonucleotide hetero- and homoduplex hybridization and nuclease sensitivity. Nucleic Acids Res 23(11):2019–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Dai Q, Moshitch-Moshkovitz S, Han D, Kol N, Amariglio N, et al. 2017. Nm-seq maps 2′-O-methylation sites in human mRNA with base precision. Nat. Methods 14(7):695–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Novoa EM, Ribas de Pouplana L. 2012. Speeding with control: Codon usage, tRNAs, and ribosomes. Trends Genet 28(11):574–81 [DOI] [PubMed] [Google Scholar]
- 112.Richter JD, Coller J. 2015. Pausing on Polyribosomes: Make Way for Elongation in Translational Control. Cell 163(2):292–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, et al. 2015. Codon optimality is a major determinant of mRNA stability. Cell 160(6):1111–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Boël G, Letso R, Neely H, Price WN, Wong K-H, et al. 2016. Codon influence on protein expression in E. coli correlates with mRNA levels. Nature 529(7586):358–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gingold H, Tehler D, Christoffersen NR, Nielsen MM, Asmar F, et al. 2014. A Dual Program for Translation Regulation in Cellular Proliferation and Differentiation. Cell 158(6):1281–92 [DOI] [PubMed] [Google Scholar]
- 116.Schmitt BM, Rudolph KLM, Karagianni P, Schmitt BM, Rudolph KLM, et al. 2014. High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA − tRNA interface. Genome Res 24:1797–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Pavon-Eternod M, Gomes S, Geslain R, Dai Q, Rosner MR, Pan T. 2009. tRNA over-expression in breast cancer and functional consequences. Nucleic Acids Res 37(21):7268–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Goodarzi H, Nguyen HCB, Zhang S, Dill BD, Molina H, Tavazoie SF. 2016. Modulated expression of specific tRNAs drives gene expression and cancer progression. Cell 165(6):1416–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kirchner S, Ignatova Z. 2014. Emerging roles of tRNA in adaptive translation, signalling dynamics and disease. Nat. Rev. Genet 16(2):98–112 [DOI] [PubMed] [Google Scholar]
- 120.Buhr F, Jha S, Thommen M, Mittelstaet J, Kutz F, et al. 2016. Synonymous Codons Direct Cotranslational Folding toward Different Protein Conformations. Mol. Cell 61(3):341–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pechmann S, Frydman J. 2012. Evolutionary conservation of codon optimality reveals hidden signatures of cotranslational folding. Nat. Struct. Mol. Biol 20(2):237–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gamble CE, Brule CE, Dean KM, Fields S, Grayhack EJ. 2016. Adjacent Codons Act in Concert to Modulate Translation Efficiency in Yeast. Cell 166(3):679–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yusupova GZ, Yusupov MM, Cate JHD, Noller HF. 2001. The path of messenger RNA through the ribosome. Cell 106(2):233–41 [DOI] [PubMed] [Google Scholar]
- 124.Jenner LB, Demeshkina N, Yusupova G, Yusupov M. 2010. Structural aspects of messenger RNA reading frame maintenance by the ribosome. Nat. Struct. Mol. Biol 17(5):555–60 [DOI] [PubMed] [Google Scholar]
- 125.Chen C, Zhang H, Broitman SL, Reiche M, Farrell I, et al. 2013. Dynamics of translation by single ribosomes through mRNA secondary structures. Nat. Struct. Mol. Biol 20(5):582–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Liu T, Kaplan A, Alexander L, Yan S, Wen J-D, et al. 2014. Direct measurement of the mechanical work during translocation by the ribosome. Elife 3:e03406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kontos H, Napthine S, Brierley I. 2001. Ribosomal pausing at a frameshifter RNA pseudoknot is sensitive to reading phase but shows little correlation with frameshift efficiency. Mol. Cell. Biol 21(24):8657–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Farabaugh PJ. 1996. Programmed translational frameshifting. Annu. Rev. Genet 30(1):507–28 [DOI] [PubMed] [Google Scholar]
- 129.Mohammad F, Woolstenhulme CJ, Green R, Buskirk AR. 2016. Clarifying the Translational Pausing Landscape in Bacteria by Ribosome Profiling. Cell Rep 14(4):686–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wohlgemuth I, Brenner S, Beringer M, Rodnina MV. 2008. Modulation of the rate of peptidyl transfer on the ribosome by the nature of substrates. J. Biol. Chem 283(47):32229–35 [DOI] [PubMed] [Google Scholar]
- 131.Johansson M, Ieong K-W, Trobro S, Strazewski P, Åqvist J, et al. 2011. pH-sensitivity of the ribosomal peptidyl transfer reaction dependent on the identity of the A-site aminoacyl-tRNA. Proc. Natl. Acad. Sci. U. S. A 108(1):79–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pavlov MY, Watts RE, Tan Z, Cornish VW, Ehrenberg M, Forster AC. 2009. Slow peptide bond formation by proline and other N-alkylamino acids in translation. Proc. Natl. Acad. Sci. U. S. A 106(1):50–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Melnikov S, Mailliot J, Rigger L, Neuner S, Shin B, et al. 2016. Molecular insights into protein synthesis with proline residues. EMBO Rep 17(12):1776–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Doerfel LK, Wohlgemuth I, Kothe C, Peske F, Urlaub H, Rodnina MV. 2013. EF-P Is Essential for Rapid Synthesis of Proteins Containing Consecutive Proline Residues. Science (80-. ) 339(6115):85–88 [DOI] [PubMed] [Google Scholar]
- 135.Starosta AL, Lassak J, Peil L, Atkinson GC, Virumäe K, et al. 2014. Translational stalling at polyproline stretches is modulated by the sequence context upstream of the stall site. Nucleic Acids Res 42(16):10711–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Peil L, Starosta AL, Lassak J, Atkinson GC, Virumae K, et al. 2013. Distinct XPPX sequence motifs induce ribosome stalling, which is rescued by the translation elongation factor EF-P. Proc. Natl. Acad. Sci 110(38):15265–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gutierrez E, Shin BS, Woolstenhulme CJ, Kim JR, Saini P, et al. 2013. eif5A promotes translation of polyproline motifs. Mol. Cell 51(1):35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ude S, Lassak J, Starosta AL, Kraxenberger T, Wilson DN, Jung K. 2013. Translation Elongation Factor EF-P Alleviates Ribosome Stalling at Polyproline Stretches 339(January):82–86 [DOI] [PubMed] [Google Scholar]
- 139.Doerfel LK, Wohlgemuth I, Kubyshkin V, Starosta AL, Wilson DN, et al. 2015. Entropic contribution of elongation factor P to proline positioning at the catalytic center of the ribosome. J. Am. Chem. Soc 137(40):12997–6 [DOI] [PubMed] [Google Scholar]
- 140.Woolstenhulme CJ, Guydosh NR, Green R, Buskirk AR. 2015. High-Precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep 11(1):13–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Huter P, Arenz S, Bock LV., Graf M, Frister JO, et al. 2017. Structural Basis for Polyproline-Mediated Ribosome Stalling and Rescue by the Translation Elongation Factor EF-P. Mol. Cell 68(3):515–527.e6 [DOI] [PubMed] [Google Scholar]
- 142.Nam D, Choi E, Shin D, Lee EJ. 2016. tRNAPro-mediated downregulation of elongation factor P is required for mgtCBR expression during Salmonella infection. Mol. Microbiol 102(2):221–32 [DOI] [PubMed] [Google Scholar]
- 143.Elgamal S, Katz A, Hersch SJ, Newsom D, White P, et al. 2014. EF-P Dependent Pauses Integrate Proximal and Distal Signals during Translation. PLoS Genet 10(8): [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Nakatogawa H, Ito K. 2002. The ribosomal exit tunnel functions as a discriminating gate. Cell 108(5):629–36 [DOI] [PubMed] [Google Scholar]
- 145.Muto H, Nakatogawa H, Ito K. 2006. Genetically Encoded but Nonpolypeptide Prolyl-tRNA Functions in the A Site for SecM-Mediated Ribosomal Stall. Mol. Cell 22(4):545–52 [DOI] [PubMed] [Google Scholar]
- 146.Bhushan S, Hoffmann T, Seidelt B, Frauenfeld J, Mielke T, et al. 2011. SecM-stalled ribosomes adopt an altered geometry at the peptidyl transferase center. PLoS Biol 9(1):e1000581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Zhang J, Pan X, Yan K, Sun S, Gao N, Sui SF. 2015. Mechanisms of ribosome stalling by SecM at multiple elongation steps. Elife 4:1–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tsai A, Kornberg G, Johansson M, Chen J, Puglisi JD. 2014. The dynamics of SecM-induced translational stalling. Cell Rep 7(5):1521–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Navon SP, Kornberg G, Chen J, Schwartzman T, Tsai A, et al. 2016. Amino acid sequence repertoire of the bacterial proteome and the occurrence of untranslatable sequences. Proc. Natl. Acad. Sci. U. S. A 113(26):7166–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Tanner DR, Cariello DA, Woolstenhulme CJ, Broadbent MA, Buskirk AR. 2009. Genetic identification of nascent peptides that induce ribosome stalling. J. Biol. Chem 284(50):34809–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vázquez-Laslop N, Mankin AS. 2014. Triggering Peptide-Dependent Translation Arrest by Small Molecules: Ribosome Stalling Modulated by Antibiotics. In Regulatory Nascent Polypeptides, ed Ito K, Chiba S, pp. 165–86
- 152.Arenz S, Meydan S, Starosta AL, Berninghausen O, Beckmann R, et al. 2014. Drug sensing by the ribosome induces translational arrest via active site perturbation. Mol. Cell 56(3):446–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Johansson M, Chen J, Tsai A, Kornberg G, Puglisi JD. 2014. Sequence-dependent elongation dynamics on macrolide-bound ribosomes. Cell Rep 7(5):1534–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Arenz S, Bock LV., Graf M, Innis CA, Beckmann R, et al. 2016. A combined cryo-EM and molecular dynamics approach reveals the mechanism of ErmBL-mediated translation arrest. Nat. Commun 7(May):12026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Marks J, Kannan K, Roncase EJ, Klepacki D, Kefi A, et al. 2016. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc. Natl. Acad. Sci. U. S. A 113(43):12150–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Weisblum B 1995. Erythromycin resistance by ribosome modification. Antimicrob. Agents Chemother 39(3):577–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Vazquez-Laslop N, Thum C, Mankin AS. 2008. Molecular Mechanism of Drug-Dependent Ribosome Stalling. Mol. Cell 30(2):190–202 [DOI] [PubMed] [Google Scholar]
- 158.Weisblum B 1995. Insights into erythromycin action from studies of its activity as inducer of resistance. Antimicrob. Agents Chemother 39(4):797–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lovett PS. 1996. Translation attenuation regulation of chloramphenicol resistance in bacteria - A review. Gene 179(1):157–62 [DOI] [PubMed] [Google Scholar]
- 160.Kannan K, Vázquez-Laslop N, Mankin AS. 2012. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell 151(3):508–20 [DOI] [PubMed] [Google Scholar]
- 161.Napthine S, Ling R, Finch LK, Jones JD, Bell S, et al. 2017. Protein-directed ribosomal frameshifting temporally regulates gene expression. Nat. Commun In press:1–11 [DOI] [PMC free article] [PubMed]
- 162.Belew AT, Meskauskas A, Musalgaonkar S, Advani VM, Sulima SO, et al. 2014. Ribosomal frameshifting in the CCR5 mRNA is regulated by miRNAs and the NMD pathway. Nature 512(7514):265–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Gupta P, Kannan K, Mankin AS, Vázquez-Laslop N. 2013. Regulation of Gene Expression by Macrolide-Induced Ribosomal Frameshifting. Mol. Cell 52(5):629–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Atkins JF, Elseviers D, Gorini L. 1972. Low activity of beta-galactosidase in frameshift mutants of Escherichia coli. Proc. Natl. Acad. Sci 69(5):1192–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Jørgensen F, Kurland CG. 1990. Processivity errors of gene expression in Escherichia coli. J. Mol. Biol 215(4):511–21 [DOI] [PubMed] [Google Scholar]
- 166.Parker J 1989. Errors and alternatives in reading the universal genetic code. Microbiol. Rev 53(3):273–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Kramer EB, Farabaugh PJ. 2007. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA 13(1):87–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Sharma V, Prère MF, Canal I, Firth AE, Atkins JF, et al. 2014. Analysis of tetra-and hepta-nucleotides motifs promoting-1 ribosomal frameshifting in Escherichia coli. Nucleic Acids Res 42(11):7210–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Larsen B, Wills NM, Gesteland RF, Atkins JF. 1994. rRNA-mRNA base pairing stimulates a programmed −1 ribosomal frameshift. J. Bacteriol 176(22):6842–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Larsen B, Gesteland RF, Atkins JF. 1997. Structural probing and mutagenic analysis of the stem-loop required for Escherichia coli dnaX ribosomal frameshifting: programmed efficiency of 50%. J. Mol. Biol 271(1):47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Kim H-K, Liu F, Fei J, Bustamante C, Gonzalez RL, Tinoco I. 2014. A frameshifting stimulatory stem loop destabilizes the hybrid state and impedes ribosomal translocation. Proc. Natl. Acad. Sci. U. S. A 111(15):5538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim HK, Tinoco I. 2017. EF-G catalyzed translocation dynamics in the presence of ribosomal frameshifting stimulatory signals. Nucleic Acids Res 45(5):2865–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Koutmou KS, Schuller AP, Brunelle JL, Radhakrishnan A, Djuranovic S, Green R. 2015. Ribosomes slide on lysine-encoding homopolymeric A stretches. Elife 2015(4):1–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Arthur L, Pavlovic-Djuranovic S, Smith-Koutmou K, Green R, Szczesny P, Djuranovic S. 2015. Translational control by lysine-encoding A-rich sequences. Sci. Adv 1(6):e1500154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Donly BC, Edgar CD, Adamski FM, Tate WP. 1990. Frameshift autoregulation in the gene for Escherichia coli release factor 2: Partly functional mutants result in frameshift enhancement. Nucleic Acids Res 18(22):6517–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Lobanov AV, Heaphy SM, Turanov A a, Gerashchenko MV, Pucciarelli S, et al. 2016. Position-dependent termination and widespread obligatory frameshifting in Euplotes translation. Nat. Struct. Mol. Biol 24(November):585–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wang R, Xiong J, Wang W, Miao W, Liang A. 2016. High frequency of +1 programmed ribosomal frameshifting in Euplotes octocarinatus. Sci. Rep 6(February):21139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Huang WM, Ao S, Casjens S, Orlandi R, Zeikus R, et al. 1988. A Persistent Untranslated Sequence Within Bacteriophage T4 DNA Topoisomerase Gene 60. Science 239(4843):1005–12 [DOI] [PubMed] [Google Scholar]
- 179.Chen J, Coakley A, O’Connor M, Petrov A, O’Leary SE, et al. 2015. Coupling of mRNA Structure Rearrangement to Ribosome Movement during Bypassing of Non-coding Regions. Cell 163(5):1267–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Agirrezabala X, Samatova E, Klimova M, Zamora M, Gil-carton D, et al. 2017. Ribosome rearrangements at the onset of translational bypassing. Sci. Adv 3:e1700147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Samatova E, Konevega AL, Wills NM, Atkins JF, Rodnina MV. 2014. High-efficiency translational bypassing of non-coding nucleotides specified by mRNA structure and nascent peptide. Nat. Commun 5:4459. [DOI] [PubMed] [Google Scholar]