

Abstract
Cell-targeting conjugates of Saporin 6, a ribosome inactivating protein (RIP), were prepared using the Saporin Ala 157 Cys mutant, a small molecule inhibitor of integrin αvβ3, and a potent cytotoxin, auristatin F (AF). The conjugates selectively and potently inhibited proliferation of tumor cells expressing the target integrins. We anticipate that the small molecule-RIP bioconjugate approach can be broadly applied using other small molecule drugs.
Keywords: Auristatin F, Conjugate, inhibitor, Integrin, Ribosome-inactivating protein (RIP), Saporin, Small molecule
Graphical Abstract
Cell-targeting conjugates of Saporin 6, prepared using a small molecule inhibitor of integrins avb3 and avb5 and Saporin A157C mutant, selectively and potently inhibited proliferation of tumor cells expressing the target integrins.
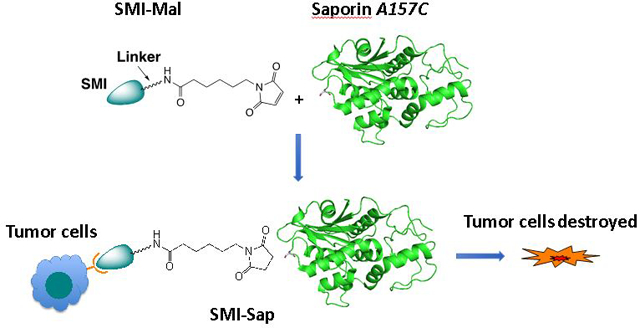
Ribosome-inactivating proteins (RIPs) are extremely potent toxins that act on both dividing and non-dividing cells through a medium size (30 kDa) enzymatic ‘A’ chain as the toxin domain.[1] They cause damage to ribosomes in an irreversible manner by removing one or more adenine residues from ribosomal RNA (rRNA) and thereby arresting protein synthesis. Most RIPs are classified as either type 1 or 2 (RIP1 and RIP2). RIP1 toxins contain only the enzymatic ‘A’ chain, whereas RIP2 toxins also possesses a cell binding lectin ‘B’ chain that mediates its cellular uptake. It is anticipated that such toxins could mediate strong therapeutic effects when they are targeted to tumor cells and selectively taken up by such cells.[2] Indeed, both RIP1 and ‘A’ chain of RIP2, upon conjugation to cell-targeting agents, such as monoclonal antibodies (Abs) or growth factors, have shown promising results in animal models. Studies with Ab-RIP or GF-RIP conjugates, however, did not progress beyond early Phase 1 Clinical Trials, because the conjugates generated immune responses and caused side effects. In addition, a large molecular size of the conjugates restricted their penetration and distribution in solid tumors.[3] In the meantime, there are indications that the side effects and immunogenicity of a RIP conjugate are minimized through PEGylation and by using humanized Abs.[4] Molecular size of a RIP conjugate is also reduced modestly by replacing a full length Ab with an Ab fragment. Nonetheless, an alternative approach that reduces the size further can offer RIP conjugates with superior efficacies. With this notion, we are developing a small molecule inhibitor (SMI)-directed approach for RIP delivery, which can afford SMI-RIP conjugates without increasing the molecular size of a RIP.
A SMI of a cell surface receptor mediates selective delivery of both the low molecular weight drug and large proteins to target cells similarly to a classical monoclonal Ab. This SMI-directed drug delivery approach can utilize potent SMIs of cancer-associated targets that are already available in large numbers.[6] Earlier, we developed SMI-cytotoxin conjugates, using a potent dual SMI of integrins αvβ3 and αvβ5, and monomethyl-auristatin (MMAE). MMAE and its analog, auristatin F (AF), are potent microtubule inhibitors and vascular damaging agents (VDA),[12] previously used in antibody-drug conjugates. Integrins αvβ3 and αvβ5 are known to overexpress in many human tumors and by endothelial cells lining the vasculature in angiogenic tumors. Both integrins have been targeted with several SMIs, Abs, SMI-drug, and SMI-bioconjugates for selective cancer therapy.[13] Our studies with SMI-MMAE conjugates have shown that the latter mediated a selective toxicity to tumor cells expressing the target integrins.[7] Separately, we prepared a series of chemically programmed Abs (cpAbs) by chemical programing of a monoclonal Ab, 38C2, using potent SMIs of integrins αvβ3, αvβ5, αvβ6, and α5β1. These cpAbs mediated a strong anti-tumor effect selectively to tumor cells expressing the respective integrin(s).[8] Along the same line, we have now prepared two SMI-RIP[5] conjugates using a RIP1 mutant, an ‘alanine 157 cysteine’ (A157C) mutant of Saporin 6 (viz., Sap-C).[9] Both conjugates are directed to integrins αvβ3 and αvβ5 using a potent and dual SMI 1 of these adhesion recpetors (Figure 1).[10] The second conjugate, SMI(AF)-Sap, has SMI 1 and AF, 2,[11] coupled to Sap through a trifunctional linker. We reasoned that AF could dissociate from the conjugate upon tumor-uptake, and enhance cell-killing effects of the SMI-Sap. Evaluation has shown that both the SMI-Sap and SMI(Sap)-AF conjugates mediated highly selective and strong toxicity to tumor cells expressing the target integrins. Additional conjugation of AF to Sap in the SMI(AF)-Sap conjugate did not further enhance the cytotoxicity of the already very potent SMI-Sap conjugate when measured in vitro, but might provide an advantage when targeting a tumor.
Figure 1.
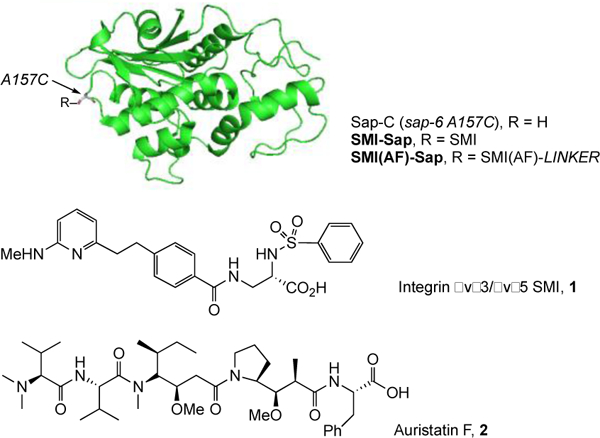
Structure of a RIP1 mutant, Saporin 6 alanine 157 cysteine (A157C), and small the molecule inhibitor of integrins αvβ3 and αvβ5, as well as the potent cytotoxin, Auristatin F used here.
Our studies started with synthesis of the two conjugates, SMI-Sap and SMI(AF)-Sap, which were prepared by reacting Sap-C with compounds 3 and 4 as shown in Scheme 1 through the maleimide-thiol coupling reaction.[14] Here, SMI 1 is coupled to maleimide function through a bi-functional linker in compound 3, and through a tri-functional linker having AF, 2, at the third end in compound 4. For the SMI-Sap and SMI(AF)-Sap production, the key intermediates 3 and 4 were prepared starting with the readily available SMIs 5a or 5b in three to five steps, (Scheme 1 and Supporting Information (SI, Scheme S–1). Separately, Sap-C was produced in E. Coli, and isolated from the culture by cation exchange chromatography and concentrating the eluants using Amicon (10 kDa MCWO), as described.[9] The residual material underwent a dithiothreitol-mediated dithiol reduction step, and was further purified by size-exclusion chromatography. Subsequently, Sap-C was reacted with intermediates 3 and 4, and the crude products were purified using FPLC to afford the desired SMI-Sap and SMI(AF)-Sap conjugates.
Scheme 1.
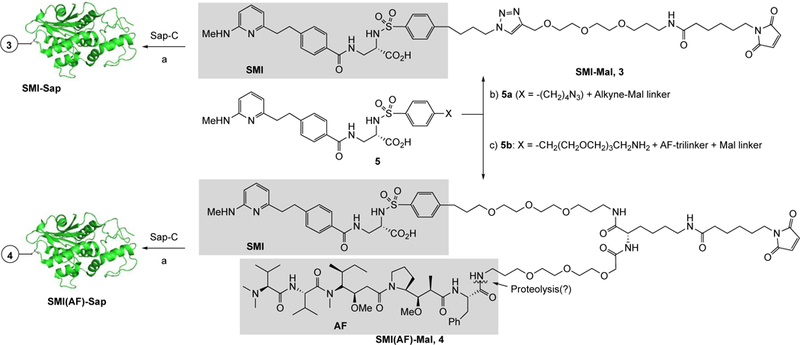
Synthesis of Sap conjugates, SMI-Sap and SMI(AF)-Sap, using Saporin 6 alanine 157 cysteine’ (Sap A157C) mutant and a small molecule inhibitor (SMI) of αvβ3/αvβ5 integrins or the SMI-Auristatin F (AF) heterodimer. Key: a) TCEP buffer, purification using FPLC, b) Cu-catalyzed alkyne-azide coupling (Cu-AAC) reaction, c) Peptide coupling reaction.
To evaluate the selectivity and cytotoxic activity of the Sap conjugates, we chose M21 and M21-L human melanoma cells and MDA-MB-435 human breast cancer cell variants. The M21 and M21-L cell lines express β1 integrins, including α5β1, but they differ from each other in the expression of αv integrins; while M21 cells express αvβ3 highly, αvβ5 integrin moderately, and αvβ8 integrin weakly, the M21-derived M21-L cells lack the αv subunit gene and therefore express neither of the target integrins.[15] In the MDA-MB-435 cell model, the control cells moderately express the β3 integrin subunit gene which was knocked down in 435β3-minus variant for comparison[16]. Both cell variants express low levels of αvβ5 and αvβ6, and many of the β1 integrins.[17] We confirmed the expression of integrins αvβ3 and αvβ5 in M21 cells and lack of αv integrins in M21-L cells, as well as binding of compound 1-derived cpAb 38C2-1 to αvβ3 and αvβ5 integrins using M21 and UCLA-P3 cells, the latter expressing αvβ5 but not αvβ3, as described previously (Figures 3). We further analyzed and confirmed that 38C2-1 bound strongly to OVCA433 cells and variants of the human colon cancer SW480 cell line, including puro control SW480, SW480 β3, and SW480 β6 cells (See Supporting Information). OVCA433 cells express integrins αvβ5 and αvβ6, and the puro control SW480 cells express αvβ5 integrin, whereas in SW480 β3 and SW480 β6 cells, the β3 and β6 integrin was experimentally over-expressed.[18] These data suggest that SMI 1 is a broad inhibitor of several αv integrins, including αvβ3, αvβ5, and αvβ6. Because there was no appreciable binding to M21-L cells, we concluded that 38C2-1 did not bind integrin α5β1 or other β1 integrins. We further argued that the two SMI-Sap and SMI(AF)-Sap conjugates might mediate toxicity to M21 cells through binding to the αvβ3 and αvβ5 integrins, to 435ScrB through all three αvβ3, αvβ5, and αvβ6 integrins, and to 435β3-minus cells through αvβ5 and αvβ6 in the absence of β3 integrin. We determined the selectivity and cytotoxic activity of our first SMI-Sap conjugate by incubating M21 and M21-L cells with the test conjugates. Unconjugated SMI 1 and the unmodified Sap-C were used for comparison. The results are shown in Figure 4A–B. Evidently, the SMI-Sap conjugate mediated cell-killing of M21 cells at a subnanomolar IC50, whereas both inhibitor 1 and Sap-C were nontoxic at up to 10 nM concentration (Figure 4A). As shown in Figure 4B, the SMI-Sap conjugate mediated toxicity to M21 cells, whereas the M21-L cells lacking the target integrins were not affected under the conditions tested (at 20 nM SMI conjugate).
Figure 3.
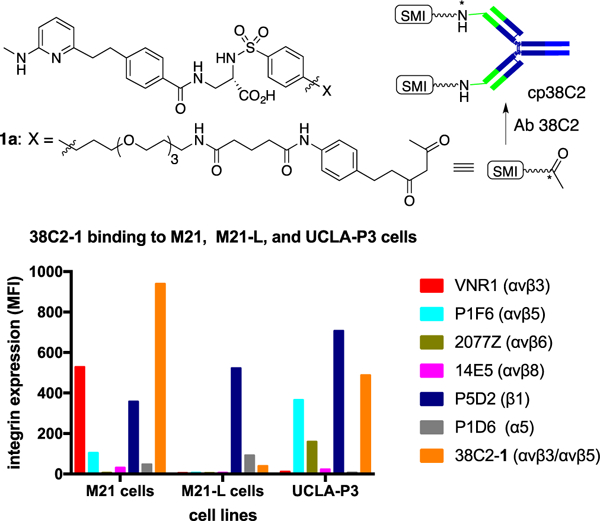
Flow cytometry analysis showing (a) the αvβ3/αvβ5 integrin expression level in M21 cells, and (b) binding of cpAb 38C2-1, prepared by chemical programming of aldolase Ab 38C2 using a diketone derivative, 1a, of a potent αvβ3/αvβ5 integrin SMI, 1, to M21 and UCLA-P3 tumor cells expressing the target integrins. APC labeled anti-mouse polyclonal Ab (at a 1:100 dilution, i.e., 10 μg/mL in FACS buffer) was used to detect the binding of the control Abs (10 μg/mL) and cpAb 38C2–1 (20 μg/mL) to cells. The y-axis gives the relative mean fluorescence intensity in linear scale, and the x-axis describes cell line name. Key: Abs used to detect integrin expression on M21, M21-L, and UCLA-P3 cells were: VNR1 (for αvβ3), P1F6 (αvβ5), 2077Z (αvβ6), 14E5 (αvβ8), P5D2 (β1), and P1D6 (α5).
Figure 4.
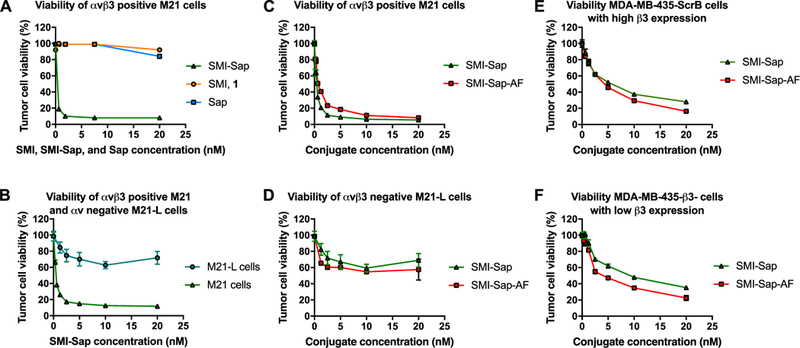
Effect of the SMI-Sap and SMI(AF)-Sap conjugates on cell viability and proliferation. SMI-Sap conjugate mediates cellular toxicity to M21 cells at a subnanomolar IC50, but not to M21-L cells (A and B). SMI 1 and Sap also show minimum toxicity to M21 cells at 20nM concentration. SMI-Sap and SMI(AF)-Sap conjugates mediate strong toxicity (sub-nanomolar IC50) to M21 cells (C) but not to M21-L cells. SMI-Sap and SMI(AF)-Sap conjugates are also active against MDA-MB-435 cell variants 435 β3- in which the β3 integrin subunit gene had been knocked down, as well as to 435 ScrB in which β3 integrin expression had been restored by transduction with the β3 wild type gene (E, F). To measure effects of the compounds and conjugates on tumor cell viability and proliferation, 5×1034 cells were plated into 24 well plates and incubated with or without compounds and conjugates at various concentration. After 72 hrs, cells were harvested and counted. Live cells were identified and counted based on trypan blue exclusion. Cell-survival assay was performed once, and there were three replicates.
In subsequent experiments, we examined and compared the two SMI-Sap and SMI(AF)-Sap conjugates using M21 and M21-L cells, and determined whether AF addition in the second conjugate amplified the effects of Sap in SMI(AF)-Sap conjugate. As shown in Figures 4C and 4D, the SMI(AF)-Sap also mediated a strong and selective toxicity to M21 cells, but the effect on M21 cells was not different from the SMI-Sap conjugate. These results document two major findings. First, the SMI clearly delivers target specificity and the constructs are stable until internalized by the target cells to enable cytotoxic activity. Second, the presence of Sap alone as the cytotoxic moiety of the conjugates is sufficient to fully execute target cell killing. Nevertheless, additional inclusion of AF in the SMI(AF)-Sap conjugate might enhance desired effects within the complexity of a tumor where one type of cytotoxic moiety might not be sufficient to achieve full target toxicity. Importantly, the unchanged efficacy between SMI-Sap and SMI(AF)-Sap in our models demonstrates that this double conjugate targets cells with equal selectivity and equal efficacy, providing strong evidence for stability and maintained targeting potency of an SMI, even in a complex multi-functional conjugate. Importantly, the in vitro data show that the SMI(AF)-Sap conjugate is not metabolized by the cells to release free AF, indicating that the selectivity is fully maintained with this conjugate based targeting mechanism. In fact, using compound 14 (AF with linker), we found that the free compound indeed caused a low toxicity to M21 cells at 20nM concentration. This unwanted effect is eliminated when the compound is included in the stable conjugate and taken up by the targeted cells only.
Finally, we examined the two SMI-Sap conjugates using the MDA-MB-435ScrB and 435b3-minus cell variants, and found that both conjugates mediated a strong cytotoxic effect (IC50 values of ~5 nM) to these cells (Figures 4E, 4F). These results indicate that on tumor cells that express multiple target integrins recognized by the SMI component of the drug conjugate, experimental knock-down of one of at least three αv containing target integrins still left sufficient recognition sites on the tumor cells for effective cell killing. This result is remarkable in view of the fact that αvβ3, the target experimentally reduced in this cell model, is the major αv integrin on MDA-MB-435 breast cancer cells. Thus, the threshold for target expression by tumor cells to be effectively eliminated by the SMI-drug conjugates is apparently remarkably low. The result demonstrates that the conjugates are exclusively selective, highly effective and the targeting mechanism very sensitive.
In summary, we have shown for the first time that a RIP1 mutant, such as Sap-6 A157C, reacts with an SMI or a heterodimer of two SMI drugs through thiol-maleimide coupling to afford bi- or tri-functional SMI-RIP1 conjugates. In this manner, we have coupled Sap-6 A157C mutant to an SMI that targets integrins αvβ3 and αvβ5, as well as an integrin SMI-AF (AF as a second SMI) heterodimer to afford SMI-Sap and the SMI(AF)-Sap conjugates, respectively. Both conjugates mediate strong and selective toxicity to tumor cells expressing the target integrins, with addition of AF conjugation to potentially convey tumor relevant cytotoxic efficacy while maintaining exquisite selective targeting. We conclude that this SMI-directed strategy for RIP delivery might be broadly applied using other RIPs and SMIs, while inclusion of second SMI drug designed to neutralize another intracellular target through a noncleavable linker is possible without compromising selectivity and cellular uptake of very potent SMI-RIP conjugates.
Supplementary Material
Figure 2.
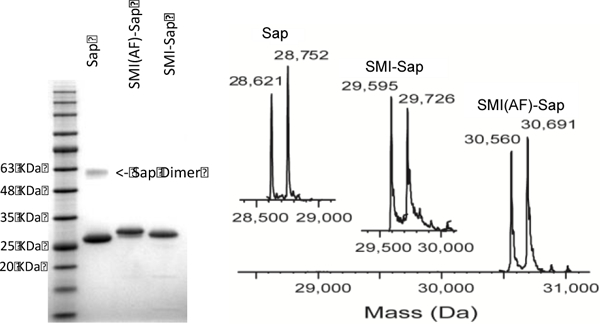
Analysis of Saporin and conjugates using (A) gel, and (B) ESI mass spectral analysis.
Acknowledgement:
We thank to Dr. Peter G. Schultz for support in preparing and isolating saporin and its conjugates, and acknowledge grant support from the NCI (R01CA120289 to SCS, R21CA198595 to BF, R01CA170140 to BF, R01CA170737 to BF) and from the BCRP CDMRP (BC 123479 to BF).
Footnotes
Electronic Supplementary Information (ESI), including synthesis of compounds 3 and 4, and production and evaluation of the SMI-Sap, SMI(AF)-Sap, and cpAb 38C2-1 available, See DOI: 10.1039/x0xx00000x
Notes and references:
- 1 ).Schrot J, Weng A and Melzig MF, Toxins, 2015, 7, 1556–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2 ).a) Polito L, Bortolotti M, Pedrazzi M and Bolognesi A, Toxins 2011, 3, 697–720. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Gilabert-Oriol R, Weng A, Mallinckrodt B, Melzig MF, Fuchs H and Thakur M, Curr Pharm Des. 2014, 20, 6584–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3 ).Bagga S, Seth D and Batra JK, J. Biol. Chem. 2003, 278, 4813–20. [DOI] [PubMed] [Google Scholar]
- 4 ).a) Pennell CA and Erickson HA, Immunol Res. 2002, 25, 177–191. [DOI] [PubMed] [Google Scholar]; b) Zheng JC, Lei N, He QC, Hu W, Jin JG, Meng Y, Deng NH, Meng YF, Zhang CJ and Shen FB, Immunopharmacol Immunotoxicol. 2012, 34, 866–73. [DOI] [PubMed] [Google Scholar]; c) Shin MC, Zhang J, David AE, Trommer WE, Kwon YM, Min KA, Kim JH and Yang VC, Control Release. 2013, 172, 169–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5 ).Collins BE; Blixt O; Han S; Duong B; Li H; Nathan JK; Bovin N; Paulson JC J Immunol. 2006, 177, 2994–3003. [DOI] [PubMed] [Google Scholar]
- 6 ).Krall N; Pretto F; Decurtins W; Bernardes GJ; Supuran CT; Neri D Angew. Chem. Int. Ed., 2014, 53, 4231–5. [DOI] [PubMed] [Google Scholar]
- 7 ).Liu Y; Bajjuri KM; Liu C; Sinha SC Mol Pharm., 2012, 9, 168–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8 ).a) Sinha SC; Das S; Li L-S; Lerner RA; Barbas CF III Nature Prot. 2007, 2, 449–56; [DOI] [PubMed] [Google Scholar]; b) Rader C Trends Biotech. 2014, 32, 186–97; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Liu Y; Goswami RK; Liu C; Sinha SC Mol Pharm., 2015, 12, 2544–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9 ).Hutchins BM; Kazane SA; Staflin K; Forsyth JS; Felding-Habermann B; Smider VV; Schultz PG Chem. Biol., 2011, 18, 299–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10 ).Seguin L; Desgrosellier JS; Weis SM; Cheresh DA Trends Cell Biol. 2015, 25, 234–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11 ).Doronina SO; Bovee TD; Meyer DW; Miyamoto JB; Anderson ME; Morris-Tilden CA; Senter PD Bioconjug. Chem. 2008, 19, 1960–3. [DOI] [PubMed] [Google Scholar]
- 12 ).Prokopiou EM; Cooper PA; Pettit GR; Bibby MC; Shnyder SD Mol Med Rep., 2010, 3, 309–13. [DOI] [PubMed] [Google Scholar]
- 13 ).Dal Corso A; Pignataro L; Belvisi L; Gennari C Curr Top Med Chem. 2016, 16, 314–29. [DOI] [PubMed] [Google Scholar]
- 14 ).Scales CW; Convertine AJ; McCormick CL Biomacromolecules. 2006, 7, 1389–92. [DOI] [PubMed] [Google Scholar]
- 15 ).Felding-Habermann B, Mueller BM, Romerdahl CA, Cheresh DA J Clin. Invest. 1992, 89, 2018–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16 ).Lorger M; Krueger JS; O’Neal M; Staflin K; Felding-Habermann B Proc. Natl. Acad. Sci. USA 2009, 106, 10666–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17 ).Taherian Aliakbar, Li Xinlei, Liu Yongqing, Thomas A Haas. BMC Cancer 2011, 11, 293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18 ).Weinreb PH, Simon KJ, Rayhorn P, Yang WJ, Leone DR, Dolinski BM, Pearse BR, Yokota Y, Kawakatsu H, Atakilit A, Sheppard D, Violette SM J. Biol. Chem. 2004, 279, 17875–7. [DOI] [PubMed] [Google Scholar]
- 19 ).Axup JY; Bajjuri KM; Ritland M; Hutchins BM; Kim CH; Kazane SA; Halder R; Forsyth JS; Santidrian AF; Stafin K; Liu Y; Tran H; Seller AJ; Biroc SL; Szydlik A; Pinkstaff J; Tian F; Sinha SC; Felding-Habermann B; Smider VV; Schultz PG Proc Natl Acad Sci U S A. 2012, 109, 16101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.