

Abstract
Objectives
There are several types of metaphyseal chondrodysplasia and various clinical types have been differentiated. The Schmid type of metaphyseal chondrodysplasia is the most common. Diffuse metaphyseal flaring, irregularity, and growth plate widening, which are most severe in the knees, are the most striking radiological features of this disease. The Schmid type of metaphyseal dysostosis is characterized by failure of normal mineralization of the zone of provisional calcification, leading to widened physes and enlarged knobby metaphyses, effectively causing shortening of the tubular bones, splaying of the metaphyses, coxa vara, and bow legs. Orthopaedic interventions were primarily performed on the lower extremities.
Methods
Twelve children (seven girls and five boys) aged 7–10 years were enrolled in this study. Moderate short stature was a uniform feature associated with predominant involvement of the proximal femora and bow legs resulted in the development of angular deformities. A waddling gait was a consequence of coxa vara in eight children. Valgus osteotomy of the proximal femur was planned after physeal closure for the group of children with coxa vara. Hemiepiphysiodesis was performed to re‐align the genu varum in three children.
Results
Other forms of metaphyseal dysostosis were ruled based on full clinical and radiographic phenotypes, with confirmation through molecular pathology. Mutations in the COL10A1 gene located on chromosome 6q21‐q22.3 were confirmed. Re‐alignment was accomplished in our group of patients.
Conclusion
The most striking clinical features of Schmid metaphyseal chondrodysplasia which appear within the first 2–3 years of life are: moderate short limbs and short stature, a waddling gait, and increasing shortness of stature with age. The Schmid type of metaphyseal chondrodysplasia is a disorder that arises from defective type X collagen, which is typically found in the hypertrophic zone of the physes. Moderate short stature and a waddling gait associated with pain are the most common clinical presentations. Osteotomies to correct bow legs are sometimes combined with lengthening procedures. Recurrence of the deformities with growth is not uncommon; therefore, hemiepiphysiodesis or stapling might be indicated in some cases.
Keywords: Bowed legs, Metaphyseal dysostosis Schmid type, Mutations in the COL10A1, Surgical corrections
Introduction
The Schmid type is the most common and least severe type of metaphyseal chondrodysplasia; it is often mistaken for rickets or achondroplasia. It is characterized by a relatively mild–moderate form of metaphyseal chondrodysplasia resembling vitamin D deficiency rickets. Patients present in childhood with short bowed limbs, a pronounced lumbar lordosis, and a waddling gait. Radiologically, the metaphyses are flared, cupped, ragged, and splayed, mostly notable in the lower limbs. Mild hand involvement (shortening of tubular bones, metaphyseal cupping of metacarpals, and proximal phalanges) is common. The proximal femoral metaphyses is particularly irregular and splayed, associated with medial beaking. Coxa vara is present to a variable degree. In severe cases of coxa vara, a triangular bone fragment may be present on the inferior aspect of the femoral neck. Mild to moderate platyspondyly and end‐plate irregularities are not a common finding. Metaphyseal chondrodysplasia is a disorder of calcification of metaphyseal cartilage, which, according to Rubin, arises because of failure to form hypertrophic cartilage at the “ physis.” This suggests that metaphyseal dysostosis is really a chondrodystrophy and may be due to an enzymal defect1, 2, 3, 4, 5, 6, 7.
In accordance with the current literature, the incidence of Schmid chondrodysplasia has been estimated to be around three to six cases per million of population. In our clinical research, we found this figure to be false and a profound underestimation. The clinical pattern varies widely and this sort of variation is the actual cause of underestimation of the incidence of this disease. Other types of metaphyseal chondrodysplasia have been reported to occur mostly in certain races. For instance, McKusick's cartilage‐hair hypoplasia has been reported to be limited to the Amish sect. This notion is incorrect: based on our research, all forms of metaphyseal chondrodysplasia are seen in different populations. Most of the cases of the Schmid type have arisen from the study of index families, which has shown that inheritance is usually through an autosomal dominant gene8, 9, 10.
To assess children with short stature and skeletal deformities secondary to metaphyseal dysplasia, we need to understand the most common types encountered in orthopaedic practices. The base line tool of management of such children is the clinical and the radiographic phenotypic characterization. Metaphyseal chondrodysplasias are skeletal diseases involving the metaphyses of the long bones, resulting in severe disturbance in the longitudinal growth of bones, while the development of the epiphyses is preserved. The Jansen type is the most severe type of metaphyseal chondrodysplasia, in which inheritance is highly likely via an autosomal dominant pattern. Jansen metaphyseal chondrodysplasia is apparent at birth because of severe short stature (basically there is shortness of the limbs as well as their distal segments). In addition, children with Jansen type metaphyseal chondrodysplasia may have dysmorphic facial features (exophthalmic eyes and prominent superciliary arches). The other type of metaphyseal chondrodysplasia is the McKusick type, which is characterized by hallmarks of metaphyseal dysplasia, short stature, fine, sparse, blond hair, transient macrocytic anemia (and occasional hypoplastic anemia) and immunodeficiency. The median adult height in males is 131 cm and in females is 123 cm. In the lower limbs, the metaphyseal dysplasia is said to be more severe at the knee than at the proximal femur. There may be cone‐shaped epiphyses of the phalanges.
Once the clinical and radiographic phenotypes are well recognized, genotypic characterization is the confirmatory tool used to establish a definite diagnosis. The constellation of clinical procedures enables orthopaedic surgeons to draw the appropriate plan of treatment.
Orthopaedic treatment in patients with Schmid's type of metaphyseal chondrodysplasia (SMCD) is primarily confined to the lower extremities. Valgus osteotomy of the proximal femur is to be considered for children with significant coxa vara. Valgus osteotomy is needed with varus angulation of more than 120°. Usually, the entire femur in children with Schmid's syndrome is presented with a varus bow, with the clinical appearance of genu varum. In some patients, the varus alignment may improve spontaneously during childhood. Other indications for surgical correction include a triangular fragment in the inferior femoral neck and progressive deformity. If surgical realignment of genu varum is performed, distal femoral as well as proximal tibial osteotomies are usually required. In many instances, recurrence of deformity with growth is not an uncommon outcome in patients who have undergone osteotomies. Guided growth procedures, such as hemiepiphysiodesis/stapling, may improve angular deformities in some children.
Surgical planning for any type of guided growth procedure to correct skeletal deformity must simultaneously address the postoperative asymmetric growth and the amount of growth remaining. The 8‐plate is a tension band plate construct designed to allow guided growth in deformity correction. The 8‐plate is positioned on the cortex of the bone, moving the fulcrum of correction to the side of the bone, to allow more rapid correction of angular deformities. The toggling of the screws allows most of the growth plate to grow normally while preventing growth directly under the 8‐plate. Wiemann et al. compared 39 limbs with staple hemiepiphysiodesis and 24 limbs treated with 8‐plate. There was no difference between the two groups in the rate of correction or the frequency of complications. They concluded that staple hemiepiphysiodesis is as effective with respect to rates of correction and complications as the 8‐plate for guided correction of angular deformities. In addition, the rate of complications was greater in patients with pathologic physes11, 12, 13, 14, 15, 16.
Materials and Methods
The study protocol was approved by the Ethics Committee of l’hospital d'senfants de Tunis‐Service d’Orthopedie Infantile No‐99/12‐President du comite Medical (Prof. Ben Ghachem and the Ethics Committee of the Turner Scientific Research Institute, No. 3/2016, Saint‐Petersburg, Russia) and informed consent was obtained from the patients’ guardians. This study was conducted based on clinical and radiographic evaluation of a group of children and their parents/grandparents and relatives, and was carried out between 1 January 2008 to March 2016.
Clinical Phenotypic Characterizations
The clinical and the radiographic phenotype followed by genotypic correlation in the group of children was the key factor for diagnosis. Twelve children (seven girls and five boys) aged 4–10 years of different ethnic origins presented with various forms of orthopaedic abnormalities and were the indices of our study. All patients underwent comprehensive clinical and radiographic phenotypic characterization. They were enrolled through the research partnership between the author, the Hospital of Speising, Vienna, Austria, President du comite Medical‐ l’hospital d’enfants de Tunis and the Turner Scientific Research Institute, Russia. Vitamin D deficiency rickets was the first diagnosis established by the pediatricians in almost all our patients. Vitamin D and calcium were prescribed heavily; the confusion among pediatricians emerged from the fact that they considered every bow leg to be due to vitamin D deficiency rickets.
Twelve children presented with short limbs, short stature, bowed legs and waddling gait. In our group of patients, all had lower limb deformities, with most requiring orthopaedic surgeries. One patient demonstrated no deformities and normal stature at age 11 years (height, −1.2 standard deviations [SD]), while the others had severe short stature (<−3.5 SDs). All showed normal craniofacial contour bilateral genu varum and one patient had internal tibial torsion (left > right), exaggerated lumbar lordosis, limited abduction and internal rotation of hips.
The metaphyseal dysplasia is mainly predominant in the proximal femora associated with short stature. All our patients developed metaphyseal changes of the lower limbs between 3 and 6 years of age. Parents sought advice because of the apparent bowing of the lower extremities.
Radiographic Phenotypic Characterization
Radiographs showed bilateral coxa vara with gross disorganization of the proximal femoral and humeral metaphyses (i.e. metaphyseal changes were most pronounced at the hips and knees), and valgus knee (both sides) with the apex of deformity at the physeal level (deformity of femoral and tibial epiphyses and metaphyses, resulting in 25° valgus on the right side and 18° valgus on the left side). Valgus ankles had asymmetrical epiphyses (Fig. 1).
Figure 1.
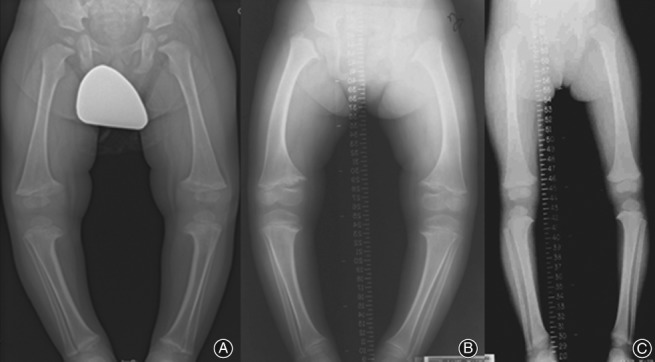
(A) Genu varum in a 5‐year‐old boy with Schmid's metaphyseal chondrodysdplasia presented with bowing of the legs; (B) genu varum in a 7‐year‐old girl with Schmid's metaphyseal chondrodysplasia; and (C) less pronounced genu varum in a 9‐year‐old girl with Schmid's metaphyseal dysostosis. All children showed widening of the physes associated with metaphyseal cupping that resembles rickets.
Metaphyses of knees, lower ends of tibia and radii were involved to a lesser extent, with mild changes at the lower ends of the ulna and at the elbows. There was middorsal scoliosis with possible deformity of the vertebral bodies on the concave side of the curve. Skull, ribs, all diaphyses and epiphyses were normal. The bone changes varied from mild sharp serrations of the metaphyses with increased density of the provisional zone of calcification to gross irregularity with flaring and fragmentation with widening of the growth plate. It was empirical to rule out nutritional vitamin D‐resistant rickets. All our patients had normal serum chemistry levels, which included blood count, Hb electrophoresis, urine analysis, urinary chromatogram, blood urea, serum electrolytes, calcium, inorganic phosphate, and alkaline phosphatase. Genetic tests showed heterozygous mutation in the COL10A1 gene on chromosome 6q21‐a22.3.
Results
Genetics
Collagen X is a short chain collagen expressed specifically by the hypertrophic chondrocytes of the cartilage growth plate during endochondral bone formation. Accordingly, COL10A1 mutations disrupt growth plate function and cause SMCD. SMCD mutations are almost exclusively located in the NC1 domain, which is crucial for both trimer formation and extracellular assembly (i.e. assembly of collagen fibers begins in the endoplasmic reticulum and is completed outside the cell). Collagen biosynthesis and assembly follow the normal pathway for a secreted protein. The collagen chains are synthesized as longer precursors called procollagens (the growing peptide chains are co‐translationally transported into the lumen of the rough endoplasmic reticulum. In the endoplasmic reticulum, the procollagen chain undergoes a series of processing reactions. First, as with other secreted proteins, glycosylation of procollagen occurs in the rough endoplasmic reticulum and Golgi complex. Galactose and glucose residues are added to hydroxylysine residues, and long oligosaccharides are added to certain asparagine residues in the C‐terminal propepetide, a segment at the C‐terminus of a procollagen molecule that is absent from mature collagen. In addition, specific proline and lysine residues in the middle of the chains are hydroxylated by membrane‐bound hydroxylases. Finally, intrachain disulfide bonds between the N‐terminal and C‐terminal propeptide sequences align the three chains before the triple helix forms in the endoplasmic reticulum13, 14, 15.
Schimd's metaphyseal chondrodysplasia has been known to show a significant phenotypic overlap with spondylometaphyseal dysplasia (SMD). SMD comprises a heterogeneous group of heritable skeletal dysplasias characterized by modification of the vertebral bodies of the spine and metaphyses of the tubular bones10. A missense mutation in the COL10A1 gene has also been shown to cause a rare spondylo‐metaphyseal chondrodysplasia Jansen type, in which apart from exhibiting a mild spinal phenotype, shares striking clinical and radiographic similarities to Schmid's metaphyseal chondrodysplasia15, 16.
Collagen X is a major constituent of the pericellular matrix of hypertrophic chondrocytes within the cartilage growth plate, and the expression of collagen X during endochondral ossification is intimately linked to the onset of cartilage calcification and extracellular matrix remodeling15, 16, 17. The absence of a functional collagen X network in mice is associated with displacement of proteoglycans, altered mineral deposition, compression of the growth plate, and hematopoietic changes15, 18, 19, 20, 21, 22.
Orthopaedic treatment is primarily confined to the lower extremities. Valgus osteotomy of the proximal femur may be indicated for children with significant coxa vara. Indications for surgical correction include a triangular fragment in the inferior femoral neck and progressive deformity11. Usually the entire femur has a varus bow, with the clinical appearance of genu varum. The varus alignment may improve spontaneously during childhood13, 23. If the femoral condyles are parallel to the floor, corrective osteotomy may not lead to improved functional results. In cases where surgical realignment of genu varum are required, distal femoral as well as proximal tibial osteotomies are usually required as well. Recurrence of deformity following osteotomies is not an uncommon manifestation. Therefore, methods including permanent and reversible hemiepiphysiodesis or stapling have been developed for modulating growth and treating genu varum via growth arrest, and may improve angular deformities in some children. The recently developed 8‐plate (eight‐plate; Orthofix, McKinney, TX, USA) can be applied to create guided growth. Stevens and Klatt performed a guided growth procedure in 14 children with rickets. Of the 53 deformities treated with staple fixation in 10 children, 45% had staple migration. Of the 15 deformities treated with 8‐plate fixation in 4 children, none had migrated. They recommended guided growth to maintain alignment to avoid later surgeries17.
When we decide to proceed with the guided growth procedure, we carefully considered the right timing for application of this procedure. On one hand, if the surgery is performed too early with respect to remaining growth, an overcorrection may develop (this can be rectified by moving the implant if the hemiepiphysiodesis or guided growth procedure is reversible. On the other hand, if the surgery is performed too late, an undercorrection or failure may be the outcome. In this case, reversal cannot be easily applied.
Surgical Corrections
Coxa vara were dealt with by bilateral proximal osteotomy; radiographic appearance after bilateral valgisation osteotomy fixed with locking plate (Fig. 2) and valgus knee (both sides) were corrected by means of guided growth technique with hemiepiphyseodesis. The plates were applied asymmetrically. On the right side, two plates (on the medial sides of distal femoral and proximal tibial physes), and on the left side, one plate (medial side of distal femoral physis) have been applied (Figs 3a, b). Clinical appearance of the lower extremities postoperatively and at the end of correction after “guided growth” using 8‐plates in a 10‐year‐old girl showed re‐alignment (Fig. 3c).
Figure 2.
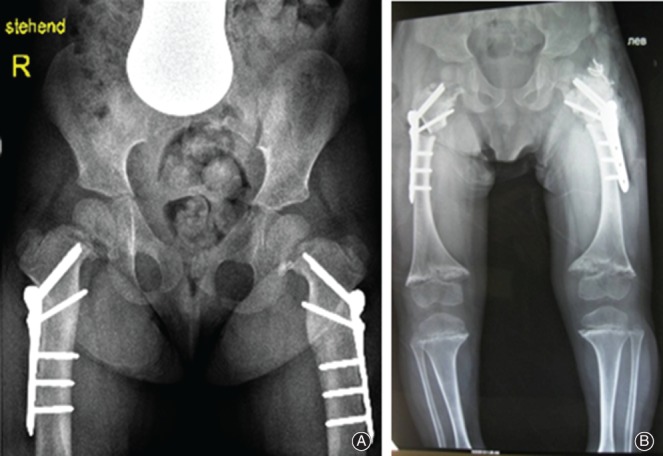
(A, B) Coxa vara were dealt with by bilateral proximal osteotomy. Radiographic appearance after bilateral valgisation osteotomy fixed with locking plate.
Figure 3.
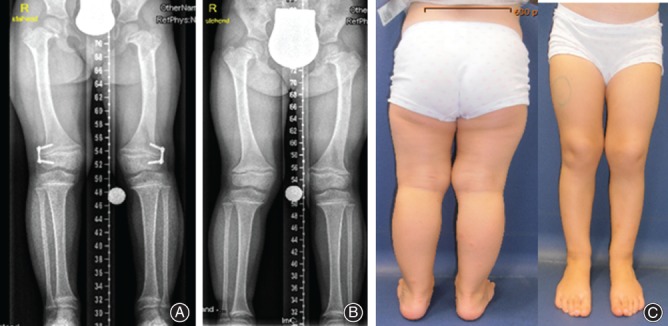
(A, B) AP radiographs show that valgus knees (both sides) were corrected by means of guided growth technique with hemiepiphyseodesis. The plates were applied asymmetrically. On the right side, two plates (on the medial sides of distal femoral and proximal tibial physes), and on the left side one plate (medial side of distal femoral physis) have been applied. (C) Clinical appearance of the lower extremities postoperatively and at the end of correction after “guided growth” using 8‐plates in a 10‐year‐old girl.
Discussion
Pitfalls of Diagnosis
Vitamin D deficiency rickets was the first diagnosis established by the pediatricians in almost all our patients. Vitamin D and calcium was heavily administered; the confusion among pediatricians emerged from the fact that they considered every bow leg to be due to vitamin D deficiency rickets. Months and even years were spent waiting for improvement, which was not achieved. No differences were noted and the bow legs persisted and became more pronounced. Skeletal changes in children with Schmid type metaphyseal dysplasia are usually not present at birth. It is a relatively mild form of metaphyseal dysplasia resembling vitamin D deficiency rickets. Skeletal changes develop with weight bearing at 3–5 years of age, at which time bowing of the legs become apparent to parents and pediatricians. Patients present in childhood with short bowed limbs, a pronounced lumbar lordosis and a waddling gait.
Results of the Diagnostic Errors
Pediatricians and physicians commonly make diagnostic errors because of the strong perception that bow legs occur either due to vitamin D deficiency rickets or due to the categorization of these children under the most common false category of idiopathic genu varum. The total omission of the clinical and radiological phenotypic characterizations adversely affects the overall management of these children. Sadly, these diagnostic errors cause parents and child tremendous anxiety and subject them to costly investigations and unnecessary treatments.
For instance, we have observed the prescription of vitamin D and calcium supplements to children and adults with bow legs as a result of diagnostic errors. The diagnosis of bow legs should be made on the child/adult as a whole and not on segmental basis (i.e. orthopaedic physicians and surgeons should not act like carpenters, repairing a broken leg of a table). It is fundamental to understand that the human body is a complex system which requires a comprehensive approach to etiological understanding.
Conclusion
Genu varum is a common deformity encountered in pediatric patients with metaphyseal dysostosis and can be associated with pain, knee instability, limitation of joint function, and a waddling gait.
Surgical correction is indicated to treat a symptomatic or progressive deformity or a vulgus thrust at the knee. The level of deformity defines the level of tibial osteotomy, and the concurrent tibial torsion which should be managed accordingly. Coxa vara is present to varying degrees. In Schmid's metaphyseal chondrodysplasia, the epiphyses are wide and the metaphyses flared, cupped and ragged, and the course of the disease is relatively benign. The head, thorax, and pelvis are not involved. The vertebral column is rarely involved in this syndrome. However, some of our patients showed anterior and posterior end‐plate irregularities and one patient manifested mild platyspondyly. The metaphyses were widened and the physes were abnormally thick.
Acknowledgment
We wish to thank Professor Hassan Gharbi, President of Ibn Zohr Institute of Radiology and Imaging studies, Tunis, for covering the expenses of investigations in three families.
Disclosure: The authors declare that they have no conflict of interest.
References
- 1. Schmid F. Beitrag zur Dysostosis enchondralis metaphysarea. Monats Kinderheilkd, 1949, 97: 393–397. [Google Scholar]
- 2. Elliott AM, Field FM, Rimoin DL, Lachman RS. Hand involvement in Schmid metaphyseal chondrodysplasia. Am J Med Genet A, 2005, 132A: 191–193. [DOI] [PubMed] [Google Scholar]
- 3. Stanley P, Sutcliffe J. Metaphyseal chondrodysplasia with dwarfism, pancreatic insufficiency and neutropenia. Pediatr Radiol, 1973, 1: 119–126. [DOI] [PubMed] [Google Scholar]
- 4. Miller SM, Paul LW. Roentgen observations in familial metaphyseal dysostosis. Radiology, 1964, 83: 665–673. [DOI] [PubMed] [Google Scholar]
- 5. Savarirayan R, Cormier‐Daire V, Lachman RS, Rimoin DL. Schmid type metaphyseal chondrodysplasia: a spondylometaphyseal dysplasia identical to the “Japanese” type. Pediatr Radiol, 2000, 30: 460–463. [DOI] [PubMed] [Google Scholar]
- 6. Rubin P. Achondroplasia versus pseudoachondroplasia. Clin N Am, 1963, 1: 621. [Google Scholar]
- 7. Wynne‐Davies R, Hall M, Apley AG. Atlas of Skeletal Dysplasia, Vol. 131 Edinburgh: Churchill Livingstone, 1985. [Google Scholar]
- 8. Jansen M. Uber atypische Chondrodystrophie (Achondroplasie) und uber eine noch nicht beschriebene angeborene Wachstumsstorung des Knochensystems: Metaphysaere Dysostosis. Zeitschr Orthop Chir, 1934, 61: 253. [Google Scholar]
- 9. McKusick VA, Eldridge R, Hostetler JA, Ruangwit U, Egeland JA. Dwarfism in the Amish. II. Cartilage‐hair hypoplasia. Bull Johns Hopkins Hosp, 1965, 116: 285–326. [PubMed] [Google Scholar]
- 10. Maroteaux P, Spranger J. The spondylometaphyseal dysplasias. A tentative classification. Pediatr Radiol, 1991, 21: 293–297. [DOI] [PubMed] [Google Scholar]
- 11. Rosenbloom AL, Smith DW. The natural history of metaphyseal dysostosis. J Pediatr, 1965, 66: 857–868. [DOI] [PubMed] [Google Scholar]
- 12. Basset GS. Orthopaedic aspects of skeletal dysplasias. Instr Course Lect, 1990, 39: 381–387. [PubMed] [Google Scholar]
- 13. Stevens PM, Klatt JB. Guided growth for pathological physes. Radiographic improvement during realignment. J Pediatr Orthop, 2008, 28: 632–639. [DOI] [PubMed] [Google Scholar]
- 14. Wiemann JM 4th, Tryon C, Szalay EA. Physeal stapling versus 8‐plate hemiepiphysiodesis for guided correction of angular deformity about the knee. J Pediatr Orthop, 2009, 29: 481–485. [DOI] [PubMed] [Google Scholar]
- 15. Wilson R, Freddi S, Chan D, Cheah KS, Bateman JF. Misfolding of collagen X chains harboring Schmid metaphyseal chondrodysplasia mutations results in aberrant disulfide bond formation, intracellular retention, and activation of the unfolded protein response. J Biol Chem, 2005, 280: 15544–15552. [DOI] [PubMed] [Google Scholar]
- 16. Chan D, Jacenko O. Phenotypic and biochemical consequences of collagen X mutations in mice and humans. Matrix Biol, 1998, 17: 169–184. [DOI] [PubMed] [Google Scholar]
- 17. Schmid TM, Popp RG, Linsenmayer TF. Hypertrophic cartilage matrix. Type X collagen, supramolecular assembly, and calcification. Ann N Y Acad Sci, 1990, 580: 64–73. [DOI] [PubMed] [Google Scholar]
- 18. Jacenko O, Roberts DW, Campbell MR, McManus PM, Gress CJ, Tao Z. Linking hematopoiesis to endochondral skeletogenesis through analysis of mice transgenic for collagen X. Am J Pathol, 2002, 160: 2019–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lachman RS, Rimoin DL, Spranger J. Metaphyseal chondrodysplasia, Schmid type. Clinical and radiographic delineation with a review of the literature. Pediatr Radiol, 1988, 18: 93–102. [DOI] [PubMed] [Google Scholar]
- 20. Ikegawa S, Nakamura K, Nagano A, Haga N, Nakamura Y. Mutations in the N‐terminal globular domain of the type X collagen gene (COL10A1) in patients with Schmid metaphyseal chondrodysplasia. Hum Mutat, 1997, 9: 131–136. [DOI] [PubMed] [Google Scholar]
- 21. Matsui Y, Kimura T, Tsumaki N, Yasui N, Ochi T. A recurrent 1992delCT mutation of the type X collagen gene in a Japanese patient with Schmid metaphyseal chondrodysplasia. Jpn J Hum Genet, 1996, 41: 339–342. [DOI] [PubMed] [Google Scholar]
- 22. Wallis GA, Rash B, Sykes B, et al Mutations within the gene encoding the alpha‐1(X) chain of type X collagen (COL10A1) cause metaphyseal chondrodysplasia type Schmid but not several other forms of metaphyseal chondrodysplasia. J Med Genet, 1996, 33: 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rosenbloom AL, Smith DW. The Natural History of Metaphyseal Dysostosis. Vol. 209 Philadelphia: WB Saunders, 1965. [DOI] [PubMed] [Google Scholar]