

Abstract
Insertions and deletions (INDELs) represent a significant fraction of inter-individual variation in the human genome yet their contribution to phenotypes is poorly understood. To confirm the quality of imputed INDELs and investigate their roles in mediating cardiometabolic phenotypes, genome-wide association and linkage analyses were performed for 15 phenotypes with 1,273,952 imputed INDELs in 1024 Mexican-origin Americans. Imputation quality was validated using whole exome sequencing with an average kappa of 0.93 in common INDELs (MAF≥5%). Association analysis revealed one genome-wide significant association signal for the Cholesterylester Transfer Protein gene (CETP) with high density lipoprotein levels (rs36229491, P=3.06×10−12); linkage analysis identified two peaks with LOD>5 (rs60560566, LOD=5.36 with insulin sensitivity (SI) and rs5825825, LOD=5.11 with adiponectin levels). Suggestive overlapping signals between linkage and association were observed: rs59849892 in the WSC Domain Containing 2 gene (WSCD2) was associated and nominally linked with SI (P=1.17×10−7, LOD=1.99). This gene has been implicated in glucose metabolism in human islet cell expression studies. In addition, rs201606363 was linked and nominally associated with low density lipoprotein (P=4.73×10−4, LOD=3.67), apolipoprotein B (P=1.39×10−3, LOD=4.64), and total cholesterol (P=1.35×10−2, LOD=3.80) levels. rs201606363 is an intronic variant of the UBE2F-SCLY fusion gene which may regulate cholesterol through selenium metabolism. In conclusion, these results confirm the feasibility of imputing INDELs from array-based SNP genotypes. Analysis of these variants using association and linkage replicated previously identified SNP signals and identified multiple novel INDEL signals. These results support the inclusion of INDELs into genetic studies to more fully interrogate the spectrum of genetic variation.
Keywords: cardiometabolic disease, genome-wide association analysis, imputation, insertion/deletions, linkage analysis
Introduction:
Insertions and deletions (INDELs) of DNA sequence contribute a significant fraction of genetic variation in the human genome [Weischenfeldt, et al. 2013]. Genetic studies have confirmed association signals between INDELs and multiple disease and non-disease related phenotypes including dietary starch consumption, autism, schizophrenia, Crohn’s disease, rheumatoid arthritis, type 1 diabetes, and obesity [Cantsilieris and White 2013; Craddock, et al. 2010; Jacquemont, et al. 2011; Malhotra and Sebat 2012; Perry, et al. 2007; Pinto, et al. 2014]. However, compared to single nucleotide polymorphism (SNP) association analyses, very few studies have included INDELs.
Traditional discovery and genotyping approaches for INDELs largely rely on polymerase chain reaction (PCR) and several other modified PCR-based techniques [Dhawan and Padh 2009]. These techniques are either expensive or inconvenient and therefore not ideal for large-scale studies [Almal and Padh 2012]. More recently, short-read DNA sequencing data from the 1000 Genomes Project (1000G) has enabled a better constructed set of INDELs across different ethnicities with enhanced size and breakpoint resolution [Mills, et al. 2011; Sudmant, et al. 2010]. The release of the 1000G reference panel has enabled researchers, for the first time, to accurately impute large numbers of INDELs from array-based genotypes [Abecasis, et al. 2012]. To further investigate the feasibility of INDEL imputation, Lu et al. characterized the linkage disequilibrium between INDELs and nearby tagging SNPs using Next Generation Sequencing (NGS) data. These results suggest a high concordance rate and correlation between INDELs and nearby SNPs of similar minor allele frequencies (MAF), which suggests ample opportunities for genome-wide association studies (GWAS) with imputed INDELs to capture additional variation across the genome.
To evaluate the quality of INDEL imputation as well as comprehensively investigate their role in cardiometabolic phenotypes, we performed genome-wide association and linkage analyses of 1,273,952 imputed INDELs in 1024 Mexican-origin Americans from the Insulin Resistance Atherosclerosis Family Study (IRASFS)[Henkin, et al. 2003]. The phenotypes included in the study were well established biomarker, diabetes and serum cholesterol measures. These phenotypes are widely used in population genetic studies and multiple significant association and linkage signals have been identified [Bowden, et al. 2010; Palmer, et al. 2015; Willer, et al. 2013]. In IRASFS, SNP association and linkage analyses were performed using these phenotypes [Hellwege, et al. 2015; Hellwege, et al. 2014a; Hellwege, et al. 2014b] yet no INDEL analysis has been conducted. Therefore, we hypothesize that INDELs can be imputed based on array-based data from GWAS and with appropriate association and linkage approaches, analysis of INDELs may identify additional novel signals and provide valuable biological insights.
Materials and Methods:
Insulin Resistance Atherosclerosis Family Study (IRASFS)
The study design, recruitment, and phenotyping for the IRASFS has been previously described [Henkin, et al. 2003]. In brief, the IRASFS was designed to investigate the genetic and environmental basis of insulin resistance and adiposity. Mexican Americans included in this cohort (N=1,417 individuals, 90 pedigrees) were recruited from clinical centers in San Antonio, TX and San Luis Valley, CO. Since the criteria for selection was based on reported family size but not on affection (diabetes) status, about 12.7% of genotyped subjects had diabetes. After removing individuals with diabetes and incomplete phenotypes, 1024 individuals from 88 pedigrees were included in this report. The study protocol was approved by the Institutional Review Board of each participating clinical and analysis site and all participants provided their written informed consent.
Genotyping and Imputation
GWAS genotyping was supported through the Genetics Underlying Diabetes in Hispanics (GUARDIAN) Consortium [Goodarzi, et al. 2014] using the Illumina OmniExpress and Omni1S arrays (Illumina Inc.; San Diego, CA, USA) in 1024 individuals as well as 13 duplicate controls. A detailed description of genotyping platforms and quality controls has been previously published [Palmer, et al. 2015]. Imputation was performed using IMPUTE2 [Howie, et al. 2009] and the 1000G phase l version 3 integrated reference panel (Cosmopolitan panel) [Abecasis, et al. 2012]. Variants were checked for confidence score (>0.90), information score (>0.50), and Mendelian errors resulting in a total of 1,273,952 high quality INDELs. INDEL annotation was performed using ANNOVAR [Wang, et al. 2010]. To validate the imputation quality, 9,370 INDELs that were captured by whole exome sequencing were analyzed for sequence-imputation concordance using the kappa statistic.
Exome sequencing
Exome sequencing was performed at Texas Biomedical Research Institute using the Illumina Nextera Exome Enrichment System in conjunction with the Illumina HiSeq 2500 sequencer. All sequence reads were passed through the Illumina Data Analysis Pipeline. All sequence reads from samples passing QC criteria were mapped to the human genome reference sequence (hg19). A detailed description of the sequencing platforms and analysis pipeline can be found in supplemental material.
Phenotypes
Phenotype acquisition and variable calculations have been previously described [Henkin, et al. 2003; Wing, et al. 2011]. Briefly, insulin sensitivity (SI) and glucose effectiveness (SG) were obtained using the frequently sampled intravenous glucose tolerance test with minimal model (MINMOD) analysis [Bergman, et al. 1985; Pacini and Bergman 1986]. Acute insulin response (AIR) was measured at 8 minutes following glucose infusion as the mean insulin increment in plasma insulin concentration above the basal concentration. Disposition Index (DI) was calculated as the product of SI and AIR. Metabolic clearance rate of insulin (MCRI) was calculated as the ratio of the insulin dose over the incremental area under the curve of insulin. Fasting plasma glucose (GFAST) and insulin (FINS) and biomarkers were measured from a fasting blood draw using standardized approaches. Homeostatic model assessment of beta-cell function and insulin resistance (HOMAB and HOMAIR, respecitvely) were computed from fasting measures using published equations [Levy, et al. 1998]. Plasma adiponectin (ADP) levels were measured by radioimmunoassay (RIA; Linco Research, St Charles, MO, USA), which uses a polyclonal anti-adiponectin antibody which recognizes trimers and higher multimers of adiponectin and includes recognition of the globular domain. Serum total cholesterol (CHOL), high density lipoprotein cholesterol (HDL), and triglycerides (TRIG) were measured on fasting blood samples by standard techniques. Low density lipoprotein cholesterol (LDL) levels were computed using Friedewald formula [Friedewald, et al. 1972]. Apolipoprotein B (APOB) was measured using immuneprecipitation.
To fulfill the distributional assumptions of conditional normality, phenotypic transformations were performed. Specifically, AIR and DI were sign-square root transformed; ADP, FINS, HOMA, MCRI, SI, HDL, CHOL, and TRIG were log transformed; LDL and APOB were square root transformed; GFAST and SG were normally distributed and therefore required no transformation. For ADP, as a missense variant G45R in the ADIPOQ gene has been previously identified to have a large impact on adiponectin levels in IRASFS [Bowden, et al. 2010], a separate analysis for ADP was performed with the adjustment for the G45R variant (rs200573126; ADP_G45R).
Statistical Analysis
Tests of associations between individual variants and quantitative traits were computed using the Wald test from the variance component model implemented in Sequential Oligogenic Linkage Analysis Routines (SOLAR) [Almasy and Blangero 1998]. Each variant was coded to an additive model based on the minor allele as the reference allele. Association was calculated adjusting for age, gender, BMI, recruitment center (San Antonio, TX or San Luis Valley, CO), and admixture estimates. Admixture estimates were calculated as described previously using maximum likelihood estimation of individual ancestries as implemented in ADMIXTURE [Alexander, et al. 2009; Gao, et al. 2015]. For family-based linkage analysis, variant specific identity-by-descent (IBD) probabilities for best-guess genotypes (likelihood>0.9) were computed using the Monte Carlo method implemented in SOLAR as described previously [Hellwege, et al. 2014a]. Genotypes with likelihood estimates less than 0.9 were zeroed to avoid Mendelian inconsistencies. This resulted in a total of 1,273,952 INDELs for analysis (N=560,367 with MAF≥0.05) [O’Connell and Weeks 1998]. Two-point linkage was performed using the variance components method implemented in SOLAR, with adjustment of age, gender, recruitment center, and BMI. Estimation of the phenotypic variance explain by SNPs and INDELs was performed using GCTA [Yang, et al. 2010; Yang, et al. 2011]. Statistical analysis of kappa value variations across categories was performed with non-parametric GLM and the enrichment of MAF of linked INDELs was performed with an exact chi-square test using SAS version 9.4 (SAS Institute, Inc, Cary, North Carolina).
Results:
Characteristics of the study individuals are shown in Table 1. Overall, individuals were predominantly female (41% male) and were overweight with an average BMI of 28.3 kg/m2. A total of 1024 individuals were included for the analysis of 15 cardiometabolic phenotypes. Overall, 1,273,952 INDELs were successfully analyzed for association and two-point linkage. Among them, 12.4% (N=157,954) were rare variants with only single or double minor allele observations and 55% (N=713,582) were low frequency variants as defined by a MAF<0.05 (Figure 1a). The INDELs were then stratified by the size of the insertion/deletion, which was defined as the difference of the length of the two alleles. As shown in Figure 1b, 53% (681,840) of the variants were single base insertions/deletions and those greater or equal to 8bp constituted less than 3% (53,245). Functional annotation suggested the majority of the INDELs were intergenic and intronic, i.e. 54% and 37%, respectively. There were only 4,582 exonic variants (less than 1%, including alternative splicing and ncRNA exonic variants) (Figure 1c). The comparison of exonic and non-exonic variants suggested no difference in MAF and a trend of enrichment in multiple bases insertion/deletions (P=0.23) in exonic variants (Figure S1).
Table 1.
Demographic characteristics of the study population.
| N | Mean ± SD | Median | |
|---|---|---|---|
| Age (years) | 1024 | 40.63±13.68 | 39.3 |
| Male (%) | 1024 | 0.41% | - |
| BMI (kg/m2) | 1024 | 28.28±5.77 | 27.52 |
| SI | 1016 | 2.14±1.86 | 1.7 |
| SG | 1016 | 0.021±0.0089 | 0.02 |
| AIR (pmol−1) | 1016 | 760.33±649.12 | 586.95 |
| DI (AIR × SI) | 1016 | 1315.91±1235.30 | 1004.79 |
| GFAST (mg/dl) | 1014 | 93.41±9.48 | 92 |
| FINS (μU/ml) | 1015 | 14.90±11.03 | 12 |
| HOMAIR | 974 | 1.67±1.04 | 1.4 |
| HOMAB | 974 | 120.76±45.62 | 113.85 |
| MCRI | 948 | 5.47±2.46 | 5.15 |
| APOB | 947 | 88.23±21.95 | 86 |
| TRIG (mg/dl) | 1012 | 118.30±82.02 | 97 |
| LDL (mg/dl) | 1004 | 109.04±30.11 | 106 |
| HDL (mg/dl) | 1012 | 43.58±12.27 | 42 |
| CHOL (mg/dl) | 1013 | 176.12±35.64 | 174 |
| ADP (μg/ml) | 940 | 13.36±6.36 | 12.49 |
Figure 1.
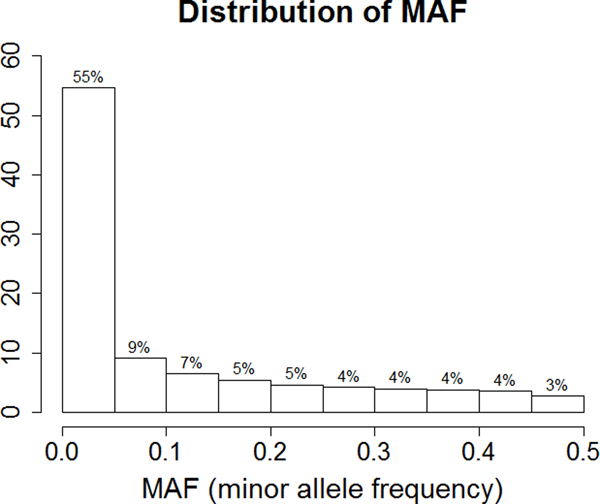
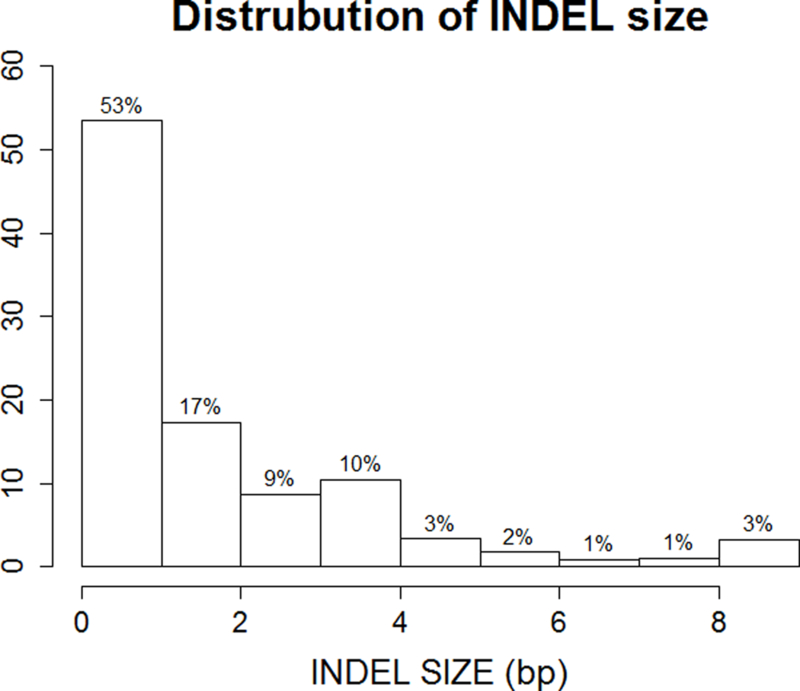
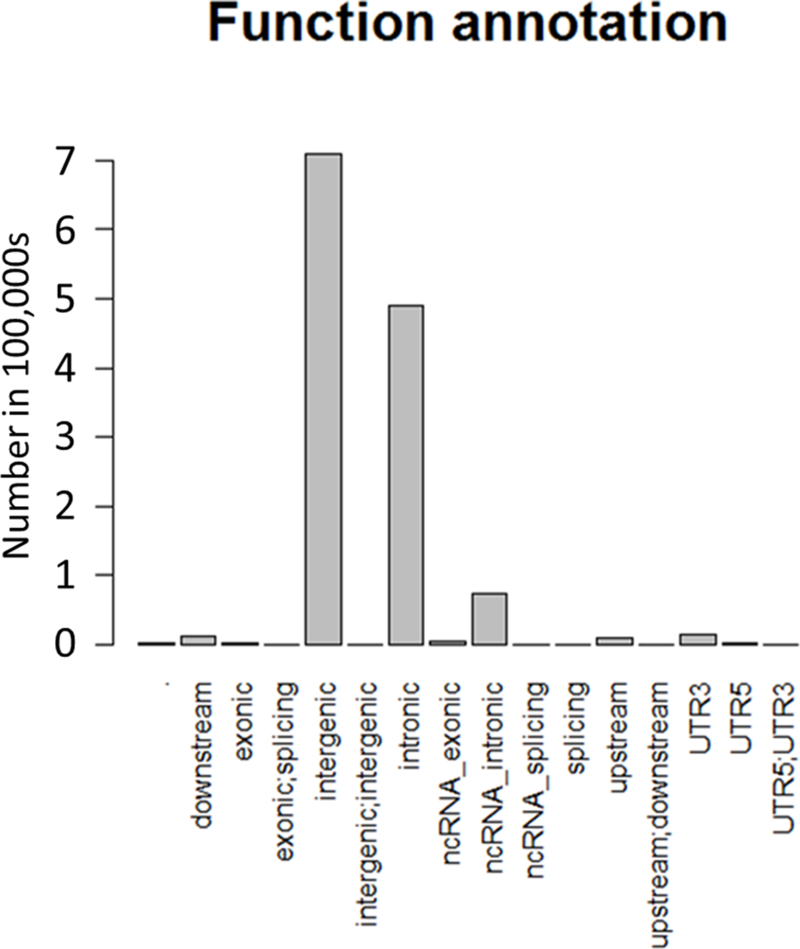
Distribution of MAF (a), INDEL size (b), and function annotations (c) for the entire INDELs dataset (N=1,273,952)
Imputation quality
Imputation quality was evaluated through comparison of genotypes from 9,370 imputed and sequenced INDELs using Cohen’s kappa statistic, which is a conservative measure of agreement over concordance rate. The results are summarized in Figure 2a. Overall, imputed genotypes were highly concordant with sequenced genotypes with an average kappa of 0.87. Most of the poorly imputed variants were of low frequency and the average kappa for common variants (MAF≥0.05) was 0.93 (Figure 2b). Further analysis revealed kappa values varied significantly based on different INDEL sizes (P=0.0006, lower kappa values for large INDEL variants) and functional annotations (P<0.001) (Figure S2).
Figure 2.
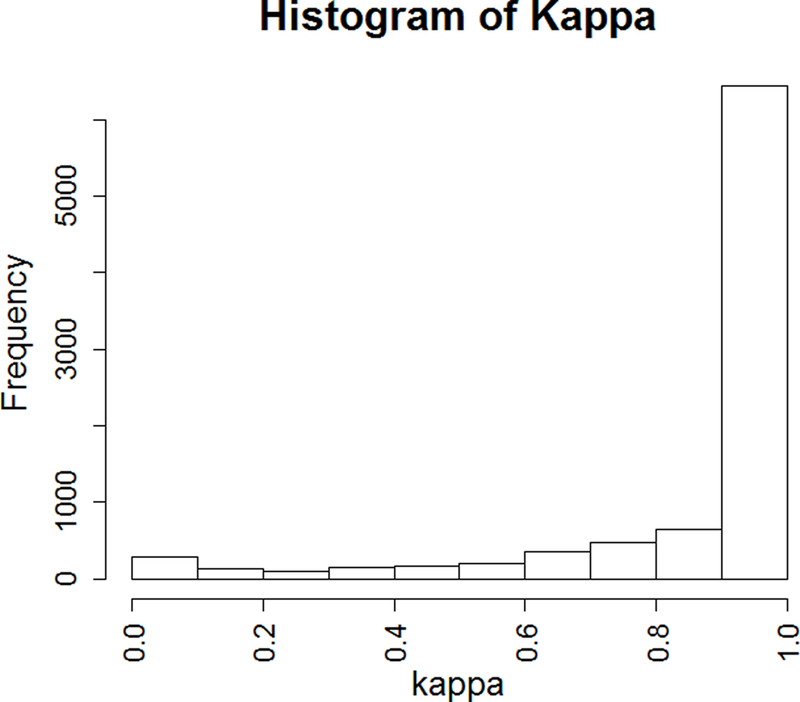
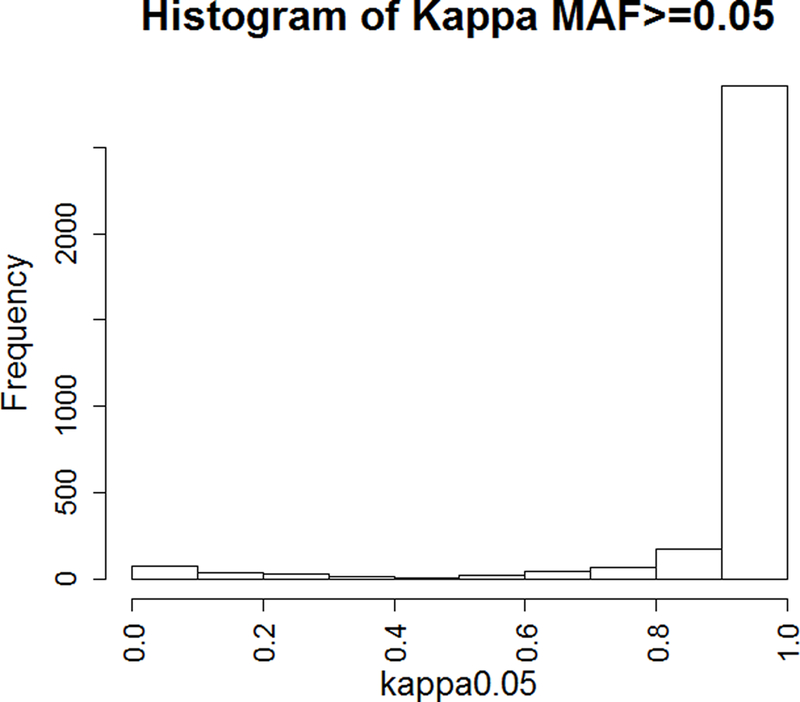
The evaluation of INDEL imputation quality using exome sequencing data. a. the distribution of kappa for all tested variants (N=9,370); b. the distribution of kappa for common (MAF≥0.05) variants (N=3,280).
Top signals from association and linkage analysis
The strongest evidence of association was observed with insertion rs36229491 with HDL levels (P=3.06×10−12, LOD=1.52, Table 2, Figure S3). This is a common variant with a MAF of 27% located 1,591 bp upstream of the cholesterylester transfer protein gene (CETP). On average, minor allele carriers have a 25% increase in plasma HDL per allele. In addition, a CETP intronic insertion rs35874588 (MAF=0.45) was also detected with a strong LOD score of 4.33 and a P value of 5.48×10-4. The two variants are poorly correlated (r2=0.05) and analysis conditioned on rs36229491 failed to abolish the rs35874588 signal, suggesting at least two independent signals exist. These results were consistent with previous findings and rs36229491 and rs35874588 were in strong LD with previously identified SNPs rs3764261 and rs5882 (r2=0.98 and 0.95, respectively) [Hellwege, et al. 2014a; Willer, et al. 2008].
Table 2.
A selection of top signals from association and linkage analysis.
| SNP | Chr:pos(hg19) | Allelesa | RAFb | function | Gene | P-value | Beta±SE | LOD | Trait |
|---|---|---|---|---|---|---|---|---|---|
| rs3832063 | 2:238944441 | A/ACT | 0.39 | ncRNA_intronic | UBE2F-SCLY | 3.50E-05 | −0.27±0.066 | 2.70 | LDL |
| rs201606363 | 2:238962655 | A/ATG | 0.43 | ncRNA_intronic | UBE2F-SCLY | 4.73E-04 | −0.23±0.065 | 3.67 | LDL |
| rs201606363 | 2:238962655 | A/ATG | 0.43 | ncRNA_intronic | UBE2F-SCLY | 1.39E-03 | −0.16±0.051 | 4.60 | APOB |
| rs201606363 | 2:238962655 | A/ATG | 0.43 | ncRNA_intronic | UBE2F-SCLY | 1.35E-02 | −0.022±0.0090 | 3.80 | CHOL |
| rs200229736 | 6:77298727 | C/CTTA | 0.22 | intergenic | IMPG1-HTR1B | 1.60E-06 | −0.11±0.024 | 2.80 | SI |
| rs201447751 | 7:67151523 | ACC/A | 0.29 | intergenic | LINC01372-LOC102723427 | 8.99E-02 | 0.037±0.022 | 5.21 | SI |
| rs149336631 | 7:67152121 | C/CTG | 0.30 | intergenic | LINC01372-LOC102723427 | 1.16E-01 | 0.034±0.022 | 5.30 | SI |
| rs60560566 | 7:67153704 | T/TG | 0.30 | intergenic | LINC01372-LOC102723427 | 1.19E-01 | 0.034±0.022 | 5.36 | SI |
| rs200122735 | 8:16588076 | TA/T | 0.39 | intergenic | MSR1-FGF20 | 4.01E-01 | 0.047±0.056 | 4.50 | APOB |
| rs201639667 | 8:16588078 | AC/A | 0.39 | intergenic | MSR1-FGF20 | 4.34E-01 | 0.044±0.056 | 4.69 | APOB |
| rs201639667 | 8:16588078 | AC/A | 0.39 | intergenic | MSR1-FGF20 | 8.04E-01 | −0.0025±0.010 | 3.70 | CHOL |
| rs138536353 | 9:9513612 | C/CAT | 0.13 | intronic | PTPRD | 8.59E-06 | −0.36±0.080 | 3.00 | APOB |
| rs5896378 | 9:10194679 | TTACA/T | 0.16 | intronic | PTPRD | 6.38E-01 | −0.033±0.070 | 3.90 | APOB |
| rs201315549 | 12:108590892 | ACCC/A | 0.02 | intronic | WSCD2 | 1.75E-06 | 0.39±0.081 | 1.79 | SI |
| rs59849892 | 12:108593020 | AT/A | 0.02 | intronic | WSCD2 | 1.20E-07 | 0.41±0.077 | 2.00 | SI |
| rs36229491 | 16:56994244 | TA/T | 0.27 | intergenic | HERPUD1-CETP | 3.06E-12 | 0.092±0.013 | 1.52 | HDL |
| rs66512242 | 16:56996645 | G/GCC | 0.46 | intronic | CETP | 3.00E-05 | 0.050±0.012 | 2.00 | HDL |
| rs12720908 | 16:57001254 | TCACA/T | 0.18 | intronic | CETP | 2.70E-05 | −0.065±0.016 | 2.01 | HDL |
| rs35874588 | 16:57009651 | TC/T | 0.45 | intronic | CETP | 5.50E-04 | 0.042±0.012 | 4.30 | HDL |
| rs5825825 | 18:65832839 | A/AT | 0.33 | intergenic | LOC643542-TMX3 | 6.13E-01 | 0.013±0.025 | 5.11 | ADP_G45R |
Reference/Other allele with minor allele on the left
Reference allele frequency based on the entire population.
For linkage analysis, two strong linkage peaks were detected with LOD scores greater than 5: rs60560566 (7q11.22, MAF=0.30) was strongly linked with SI with a LOD of 5.36 and rs5825825 (18q22.1, MAF=0.33) was strongly linked with ADP with a LOD of 5.11 after adjustment for the ADIPOQ G45R variant (rs200573126). rs60560566 marks a single base pair deletion located in a gene dessert region, i.e. no genes were identified ±400kb. rs5825825 is a single base pair insertion located intergenically between dermatan sulfate epimerase-like gene (DSEL) and thioredoxin related transmembrane protein 3 gene (TMX3). Another variant rs201639667 (MAF=0.39) was strongly linked to APOB and CHOL with LOD scores of 4.69 and 3.71, respectively. This is a single base pair insertion located between the macrophage scavenger receptor 1 gene (MSR1) and fibroblast growth factor 20 gene (FGF20). MSR1 is closely involved in macrophage-associated physiological and pathological processes and overexpression of the gene results in increased clearance of modified lipoproteins by Kupffer cells, resulting reduced VLDL levels [Herijgers, et al. 2000]. rs59849892, an intronic variant in the WSC domain containing 2 gene (WSCD2), was both associated and linked with SI (P=1.17×10−7, LOD=1.99). WSCD2 has been shown to be involved in glucose metabolism consistent with expression patterns observed in human islets [Taneera, et al. 2015]. Variant rs201606363 was strongly linked and associated with LDL (P=4.73×10−4, LOD=3.67), APOB (P=1.39×10−3, LOD=4.64), and TC (P=1.35×10−2, LOD=3.80). This common variant (MAF=0.43) is located between the ubiquitin-conjugating enzyme E2F gene (UBE2F) and the selenocysteine lyase gene (SCLY). SCLY has been shown to be involved in selenium metabolism and selenoproteins synthesis [Mihara, et al. 2000]. High serum selenium concentrations have been shown to be associated with increased serum concentrations of total and LDL cholesterol in epidemiological studies [Laclaustra, et al. 2010]. Therefore, SCLY may play an important role in this association.
Characteristics of the nominally associated and linked INDELs
To investigate whether an enrichment of a certain characteristic exists for nominally linked and associated INDELs, the dataset was parsed based on nominal association (P<0.05), association (P<0.005), nominal linkage (LOD>0.5), and linkage (LOD>1) as exemplified by TRIG and HDL (Figure S4). As shown in the figures, no enrichment was observed for INDEL size with association and linkage signals (P=0.91). For functional annotation, a slightly higher proportion of exonic variants was observed for associated and linked variants. However, no statistically significant conclusions could be drawn due to the limited number of exonic variants in this study (n=4,582) (P=0.79). In contrast, a significant enrichment of common variants (MAF≥0.05) for linked INDELs was observed (P<0.0001) but not for association (P=0.82). This observation is likely attributed to the greater power of common variants in linkage analysis.
Discussion:
In this study, INDELs wrere successfully imputed using array-based SNP genotypes in a Mexican-origin American cohort. Genome-wide association and linkage analysis were performed with cardiometabolic phenotypes using 1.27 million high quality imputed INDELs.
One key question to be asked through this series of experiments was whether INDELs could be imputed using array-based genotypes and be analyzed to complement SNPs. Thus, a comparison was performed with genotypes derived from imputation and exome sequencing data using the kappa statistic. Despite some variations in rare variants (MAF<5%), common variants reached an average kappa value of 0.93, suggesting INDELs can be reliably imputed from array-based SNP genotypes. In addition, genome-wide association and linkage analyses were successfully performed for imputed INDELs using 15 cardiometabolic phenotypes. After excluding rare variants (MAF<0.01), all models were well behaved with inflation factors ranging from 0.96–1.08 (Figure S5). Furthermore, examples of novel INDEL signals were identified and previously identified signals were successfully replicated from both association and linkage analysis as exemplified by CETP and ADIPOQ, respectively. In addition, the phenotypic variance explained by genome-wide common SNPs (N=5,650,462, MAF≥5%) increased 0.04% and 0.03% after including common INDELs (N=596,746, MAF≥5%) for HDL and TG, respectively. To further explore the coverage of imputed INDELs, a comparison was performed between imputed INDELs and exome sequencing captured INDELs. Overall, 38,122 INDELs were captured by exome sequencing and among them 9,370 were also successfully imputed based on GWAS genotypes. As shown in Figure S6, the variants missed by imputation were predominantly (92%) rare variants with MAF<0.05, which indicates good imputation coverage of common INDEL variants derived from array-based SNP genotypes. The imputation of rare INDELs suffers similarly to that of rare SNPs [Howie, et al. 2012].
Consistent with our previous findings, linkage and association tend to provide independent genetic information [Hellwege, et al. 2014a], i.e. association and linkage signals have limited overlap (Figure S5). This observation is not beyond expectation as association screens for variants having high LD with the causal variant (short range) while linkage detects variants co-segregating with the causal variant (long range). However, when the causal variant is directly tagged, both linkage and association are robustly able to detect the variant [An, et al. 2013; Bowden, et al. 2010]. Interestingly, when the causal variant is not well tagged, linkage tends to be more sensitive than association. For example, SNP rs200573126 (G45R) was previously reported as the causal variant for low adiponectin levels in IRASFS [An, et al. 2013; Bowden, et al. 2010]. This variant was not well tagged by nearby INDELs and thus, no association signals were observed in the region (Figure S7). In contrast, linkage analysis detected a LOD peak of 7.67 (rs112871276; spanning Chr3:181332042–186759808), suggesting a better sensitivity based on limited LD with the causal variant (Figure S7). In conclusion, linkage analysis is a valuable approach for genetic studies which provides information independent from association analysis.
Despite the encouraging results study limitations exist. First, the modest sample size in IRASFS (n=1024) limits the power for the association analysis of rare variants, especially given the fact that 55% of the INDELs in the study had a MAF<0.05. In addition, although good correlation and concordance rates were observed between imputed and sequenced variants, it is still possible that imputation errors impede signal discoveries, especially for rare variants which were observed to have poorer imputation quality. Third, since SOLAR identity-by-descent (IBD) calculations do not accept dosage genotypes, analysis requires a conversion of imputed dosage genotypes into best-guess. Also, to remove all Mendelian inconsistencies, all genotypes with potential errors were removed, which further dampened statistical power. Despite the rapidly developing technologies in the field, the genotyping and calling methods for short repeats in regions of complex genomic structure are still not robust [Treangen and Salzberg 2012]. Therefore, the INDELs included in this study represent an incomplete coverage of the genome. Whole genome sequencing or using a reference panel with better genome coverage for imputation would be able to address this issue.
In summary, a total of 1,273,952 INDELs were successfully imputed from array-based SNP genotypes and a genome-wide association and linkage analysis were performed. Imputation quality was partially confirmed using exome sequencing data. We identified a genome-wide significant signal for association (CETP) with HDL and two signals (four variants) reached a linkage LOD score of 5 for SI and ADP. Overall, INDEL analysis successfully confirmed previously identified signals from exome chip and GWAS [Hellwege, et al. 2014a] and evidence of suggestive novel loci was observed. Our results support that INDELs can be confidently imputed based on array-based SNP genotypes and association and linkage are complementary approaches that can be used for the analysis of imputed INDELs.
Supplementary Material
Figure 3.
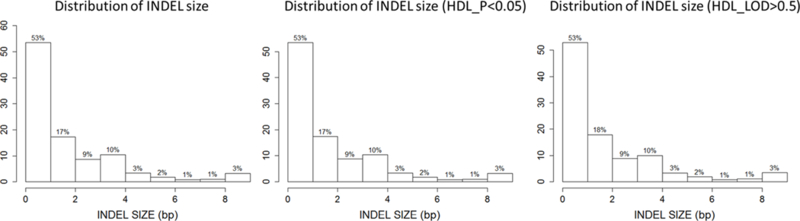
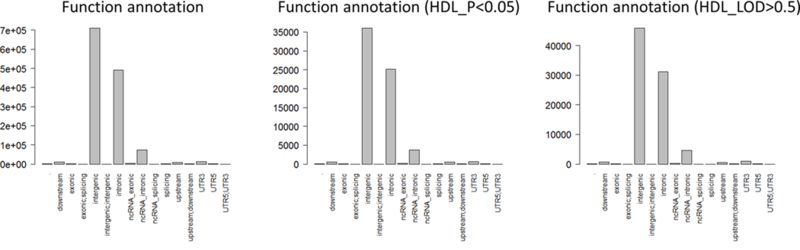
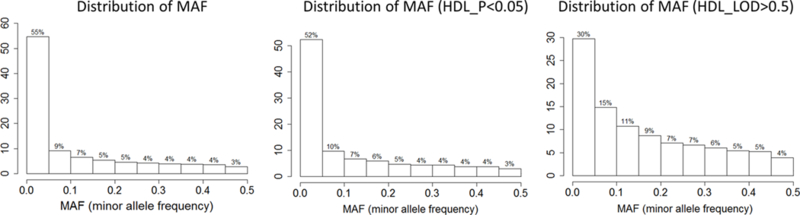
The distribution of INDELs nominally associated (P<0.05) and linked (LOD>0.5) with HDL a. INDEL size distributions, b. function annotations, c. MAF.
Acknowledgements and funding information:
This research was supported by DK097524 from the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) and HG007112 from the National Human Genome Research Institute (NHGRI). Computational resources were provided, in part, by the Wake Forest School of Medicine Center for Public Health Genomics. The authors thank the other investigators, the staff, and the participants of the studies for their valuable contributions. The provision of genotyping data was supported in part by UL1TR000124 (CTSI) and DK063491 (DRC).
Footnotes
Conflict of interest:
none
Ethical statement:
Participants included in this study were recruited from clinical centers in San Antonio, TX and San Luis Valley, CO. The Institutional Review Board of each clinical (UT Health Science Center San Antonio Review Board and Colorado Multiple Institutional Review Board, respectively) and analysis (Wake Forest School of Medicine) site approved the study protocol and all participants provided their written informed consent.
Reference
- Abecasis GR, Auton A, Brooks LD, DePristo MA, Durbin RM, Handsaker RE, Kang HM, Marth GT, McVean GA. 2012. An integrated map of genetic variation from 1,092 human genomes. Nature 491(7422):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, Lange K. 2009. Fast model-based estimation of ancestry in unrelated individuals. Genome Res 19(9):1655–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almal SH, Padh H. 2012. Implications of gene copy-number variation in health and diseases. J Hum Genet 57(1):6–13. [DOI] [PubMed] [Google Scholar]
- Almasy L, Blangero J. 1998. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet 62(5):1198–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An SS, Palmer ND, Hanley AJ, Ziegler JT, Brown WM, Haffner SM, Norris JM, Rotter JI, Guo X, Chen YD and others. 2013. Estimating the contributions of rare and common genetic variations and clinical measures to a model trait: adiponectin. Genet Epidemiol 37(1):13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman RN, Finegood DT, Ader M. 1985. Assessment of insulin sensitivity in vivo. Endocr Rev 6(1):45–86. [DOI] [PubMed] [Google Scholar]
- Bowden DW, An SS, Palmer ND, Brown WM, Norris JM, Haffner SM, Hawkins GA, Guo X, Rotter JI, Chen YD and others. 2010. Molecular basis of a linkage peak: exome sequencing and family-based analysis identify a rare genetic variant in the ADIPOQ gene in the IRAS Family Study. Hum Mol Genet 19(20):4112–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantsilieris S, White SJ. 2013. Correlating multiallelic copy number polymorphisms with disease susceptibility. Hum Mutat 34(1):1–13. [DOI] [PubMed] [Google Scholar]
- Craddock N, Hurles ME, Cardin N, Pearson RD, Plagnol V, Robson S, Vukcevic D, Barnes C, Conrad DF, Giannoulatou E and others. 2010. Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature 464(7289):713–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan D, Padh H. 2009. Pharmacogenetics: technologies to detect copy number variations. Curr Opin Mol Ther 11(6):670–80. [PubMed] [Google Scholar]
- Friedewald WT, Levy RI, Fredrickson DS. 1972. Estimation of the concentration of low-density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 18(6):499–502. [PubMed] [Google Scholar]
- Gao C, Wang N, Guo X, Ziegler JT, Taylor KD, Xiang AH, Hai Y, Kridel SJ, Nadler JL, Kandeel F and others. 2015. A Comprehensive Analysis of Common and Rare Variants to Identify Adiposity Loci in Hispanic Americans: The IRAS Family Study (IRASFS). PLoS One 10(11):e0134649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodarzi MO, Langefeld CD, Xiang AH, Chen YD, Guo X, Hanley AJ, Raffel LJ, Kandeel F, Nadler JL, Buchanan TA and others. 2014. Insulin sensitivity and insulin clearance are heritable and have strong genetic correlation in Mexican Americans. Obesity (Silver Spring) 22(4):1157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwege JN, Palmer ND, Mark Brown W, Ziegler JT, Sandy An S, Guo X, Ida Chen YD, Taylor K, Hawkins GA, Ng MC and others. 2015. Empirical characteristics of family-based linkage to a complex trait: the ADIPOQ region and adiponectin levels. Hum Genet 134(2):203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwege JN, Palmer ND, Raffield LM, Ng MC, Hawkins GA, Long J, Lorenzo C, Norris JM, Ida Chen YD, Speliotes EK and others. 2014a. Genome-wide family-based linkage analysis of exome chip variants and cardiometabolic risk. Genet Epidemiol 38(4):345–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellwege JN, Palmer ND, Ziegler JT, Langefeld CD, Lorenzo C, Norris JM, Takamura T, Bowden DW. 2014b. Genetic variants in selenoprotein P plasma 1 gene (SEPP1) are associated with fasting insulin and first phase insulin response in Hispanics. Gene 534(1):33–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henkin L, Bergman RN, Bowden DW, Ellsworth DL, Haffner SM, Langefeld CD, Mitchell BD, Norris JM, Rewers M, Saad MF and others. 2003. Genetic epidemiology of insulin resistance and visceral adiposity. The IRAS Family Study design and methods. Ann Epidemiol 13(4):211–7. [DOI] [PubMed] [Google Scholar]
- Herijgers N, de Winther MP, Van Eck M, Havekes LM, Hofker MH, Hoogerbrugge PM, Van Berkel TJ. 2000. Effect of human scavenger receptor class A overexpression in bone marrow-derived cells on lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knockout mice. J Lipid Res 41(9):1402–9. [PubMed] [Google Scholar]
- Howie B, Fuchsberger C, Stephens M, Marchini J, Abecasis GR. 2012. Fast and accurate genotype imputation in genome-wide association studies through pre-phasing. Nat Genet 44(8):955–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howie BN, Donnelly P, Marchini J. 2009. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5(6):e1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacquemont S, Reymond A, Zufferey F, Harewood L, Walters RG, Kutalik Z, Martinet D, Shen Y, Valsesia A, Beckmann ND and others. 2011. Mirror extreme BMI phenotypes associated with gene dosage at the chromosome 16p11.2 locus. Nature 478(7367):97–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laclaustra M, Stranges S, Navas-Acien A, Ordovas JM, Guallar E. 2010. Serum selenium and serum lipids in US adults: National Health and Nutrition Examination Survey (NHANES) 2003–2004. Atherosclerosis 210(2):643–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy JC, Matthews DR, Hermans MP. 1998. Correct homeostasis model assessment (HOMA) evaluation uses the computer program. Diabetes Care 21(12):2191–2. [DOI] [PubMed] [Google Scholar]
- Malhotra D, Sebat J. 2012. CNVs: harbingers of a rare variant revolution in psychiatric genetics. Cell 148(6):1223–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mihara H, Kurihara T, Watanabe T, Yoshimura T, Esaki N. 2000. cDNA cloning, purification, and characterization of mouse liver selenocysteine lyase. Candidate for selenium delivery protein in selenoprotein synthesis. J Biol Chem 275(9):6195–200. [DOI] [PubMed] [Google Scholar]
- Mills RE, Walter K, Stewart C, Handsaker RE, Chen K, Alkan C, Abyzov A, Yoon SC, Ye K, Cheetham RK and others. 2011. Mapping copy number variation by population-scale genome sequencing. Nature 470(7332):59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell JR, Weeks DE. 1998. PedCheck: a program for identification of genotype incompatibilities in linkage analysis. Am J Hum Genet 63(1):259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacini G, Bergman RN. 1986. MINMOD: a computer program to calculate insulin sensitivity and pancreatic responsivity from the frequently sampled intravenous glucose tolerance test. Comput Methods Programs Biomed 23(2):113–22. [DOI] [PubMed] [Google Scholar]
- Palmer ND, Goodarzi MO, Langefeld CD, Wang N, Guo X, Taylor KD, Fingerlin TE, Norris JM, Buchanan TA, Xiang AH and others. 2015. Genetic Variants Associated With Quantitative Glucose Homeostasis Traits Translate to Type 2 Diabetes in Mexican Americans: The GUARDIAN (Genetics Underlying Diabetes in Hispanics) Consortium. Diabetes 64(5):1853–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry GH, Dominy NJ, Claw KG, Lee AS, Fiegler H, Redon R, Werner J, Villanea FA, Mountain JL, Misra R and others. 2007. Diet and the evolution of human amylase gene copy number variation. Nat Genet 39(10):1256–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto D, Delaby E, Merico D, Barbosa M, Merikangas A, Klei L, Thiruvahindrapuram B, Xu X, Ziman R, Wang Z and others. 2014. Convergence of genes and cellular pathways dysregulated in autism spectrum disorders. Am J Hum Genet 94(5):677–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Kitzman JO, Antonacci F, Alkan C, Malig M, Tsalenko A, Sampas N, Bruhn L, Shendure J, Eichler EE. 2010. Diversity of human copy number variation and multicopy genes. Science 330(6004):641–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taneera J, Fadista J, Ahlqvist E, Atac D, Ottosson-Laakso E, Wollheim CB, Groop L. 2015. Identification of novel genes for glucose metabolism based upon expression pattern in human islets and effect on insulin secretion and glycemia. Hum Mol Genet 24(7):1945–55. [DOI] [PubMed] [Google Scholar]
- Treangen TJ, Salzberg SL. 2012. Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat Rev Genet 13(1):36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li M, Hakonarson H. 2010. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res 38(16):e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weischenfeldt J, Symmons O, Spitz F, Korbel JO. 2013. Phenotypic impact of genomic structural variation: insights from and for human disease. Nat Rev Genet 14(2):125–38. [DOI] [PubMed] [Google Scholar]
- Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, Heath SC, Timpson NJ, Najjar SS, Stringham HM and others. 2008. Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet 40(2):161–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S and others. 2013. Discovery and refinement of loci associated with lipid levels. Nat Genet 45(11):1274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing MR, Ziegler JM, Langefeld CD, Roh BH, Palmer ND, Mayer-Davis EJ, Rewers MJ, Haffner SM, Wagenknecht LE, Bowden DW. 2011. Analysis of FTO gene variants with obesity and glucose homeostasis measures in the multiethnic Insulin Resistance Atherosclerosis Study cohort. Int J Obes (Lond) 35(9):1173–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, Madden PA, Heath AC, Martin NG, Montgomery GW and others. 2010. Common SNPs explain a large proportion of the heritability for human height. Nat Genet 42(7):565–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Lee SH, Goddard ME, Visscher PM. 2011. GCTA: a tool for genome-wide complex trait analysis. Am J Hum Genet 88(1):76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.