

Abstract
Peroxisome proliferator-activated receptor gamma (PPARγ or PPARG) is a ligand-activated transcription factor belonging to the nuclear hormone receptor superfamily. It plays a master role in the differentiation and proliferation of adipose tissues. It has two major isoforms, PPARγ1 and PPARγ2, encoded from a single gene using two separate promoters and alternative splicing. Among them, PPARγ2 is most abundantly expressed in adipocytes and plays major adipogenic and lipogenic roles in the tissue. Furthermore, it has been shown that PPARγ2 is also expressed in the liver, specifically in hepatocytes, and its expression level positively correlates with fat accumulation induced by pathological conditions such as obesity and diabetes. Knockout of the hepatic Pparg gene ameliorates hepatic steatosis induced by diet or genetic manipulations. Transcriptional activation of Pparg in the liver induces the adipogenic program to store fatty acids in lipid droplets as observed in adipocytes. Understanding how the hepatic Pparg gene expression is regulated will help develop preventative and therapeutic treatments for non-alcoholic fatty liver disease (NAFLD). Due to the potential adverse effect of hepatic Pparg gene deletion on peripheral tissue functions, therapeutic interventions that target PPARγ for fatty liver diseases require fine-tuning of this gene’s expression and transcriptional activity.
Keywords: Non-alcoholic fatty liver disease (NAFLD), High fat diet (HFD), Adipogenesis, Gene expression, Peroxisome proliferator-activated receptor, gamma (PPARγ)
1. Introduction
The liver is a major organ that controls whole body lipid homeostasis by regulating lipid uptake from the circulatory system, as well as de novo synthesis and delivery of the synthesized lipids in the form of very low-density lipoprotein (VLDL) to peripheral tissues.1 Though the liver controls whole body lipid homeostasis, the white adipose tissue is considered to be the major organ for storing extra lipids. Therefore, the liver plays the aforementioned specific roles to maintain and regulate homeostatic lipid fluxes in normal conditions and is not destined to store fat for storage purposes. However, under pathophysiological conditions such as obesity and diabetes, unbalanced lipid flux in the liver results in fat accumulation. Fat accumulation is the initial and prerequisite step for the progression to the more serious non-alcoholic fatty liver disease (NAFLD), which includes non-alcoholic steatohepatitis (NASH), fibrosis, cirrhosis, and hepatocellular carcinoma.2,3 Especially, NAFLD has become an epidemic problem worldwide in recent years. Therefore, understanding the underlying molecular mechanisms of hepatic fat accumulation will be a stepping stone for potential therapeutic and preventative approaches to control the progression of NAFLD.4
The two regulatory components in hepatic lipogenesis are the transcription factors, sterol regulatory element-binding protein (SREBP) and carbohydrate responsive element-binding protein (ChREBP), which are specifically activated by insulin and glucose, respectively, through distinctive activation mechanisms.5 In this review, we will discuss another transcription regulator called peroxisome proliferator-activated receptor gamma (PPARγ or PPARG) and its role in hepatic fat accumulation in pathophysiological conditions, especially diet-induced obesity.
2. Cloning and structure of PPARγ
Peroxisome proliferator-activated receptor (PPAR) belongs to the nuclear hormone receptor superfamily, which is a group of ligand-activated transcription factors classified by the similarity in their protein structure, and is involved in adipogenesis.6–8 PPARα is the first PPAR cloned by low stringent hybridization using a mixture of oligonucleotides derived from a highly conserved region in the deoxyribonucleic acid (DNA) binding domain of several nuclear hormone receptors.9 PPARα is specifically activated by peroxisome proliferators and fatty acid (FA) derivatives. Two direct AGGTCA repeats separated by a single nucleotide (DR1) were identified as a specific DNA binding sequence of PPARα in the promoters of the genes encoding rat acyl-CoA oxidase and 3-ketoacyl-CoA thiolase, conferring a strong FA oxidation activity on the receptor.10 PPARγ was originally isolated from Xenopus and subsequently from mouse using a similar homology cloning strategy.11,12 The encoded mouse PPARγ protein has 83% homology in the DNA-binding domains and 70% homology in the ligand binding domains with the mouse PPARα protein. Due to the structural homology, PPARγ can activate PPARα target gene promoters in response to peroxisome proliferators.12 Like many other nuclear hormone receptors, PPARγ forms a heterodimer with retinoid X receptor (RXR) to bind to the consensus DR1 sequence in target gene promoters.10,13 Another isoform of PPARγ, termed PPARγ2, has been identified in a mouse adipocyte cDNA library when searching for adipocyte differentiation factor(s) that bind to the adipocyte protein 2 (aP2) enhancer region; this isoform encodes 30 additional amino acids amino-terminal to the first ATG of the earlier PPARγ, termed PPARγ1.13 In humans, PPARγ2 contains 28 additional amino acids at the N-terminal side of the ATG compared with PPARγ1.14 PPARγ2 is considered the predominant form in adipose tissues and it is mainly involved in adipocyte formation. A laborious approach identified the mouse PPARγ genomic DNA structure, revealing two separate 5’ promoters controlling the expression of the two major isoforms (Fig. 1).15 The genomic structure of the human gene is very similar to the mouse gene. Two different promoters regulated the expression of the two isoforms via alternative splicing as identified in the mouse gene.16 The separate regulatory systems produce different mRNA expression patterns of the two isoforms in mammals. Both PPARG1 and PPARG2 mRNAs are abundantly expressed in white adipose tissues and in lower levels in skeletal muscles.17 However, the PPARG2 mRNA expression is limited to the above two tissues, whereas PPARG1 mRNA has been observed in many other tissues such as heart, liver and spleen. Furthermore, the levels of PPARG2 mRNA in adipocytes and liver, two major tissues in lipid homeostasis, positively correlate with obesity and high fat diet (HFD) feeding in humans and mice, whereas the PPARG1 mRNA levels do not change under these pathophysiological and nutritional conditions.17–19 These findings strongly suggest that PPARγ2 should be the target for therapeutic and preventative approaches for metabolic disorders associated with obesity.
Fig. 1. Gene and protein structures of mouse and human PPARγ isoforms.
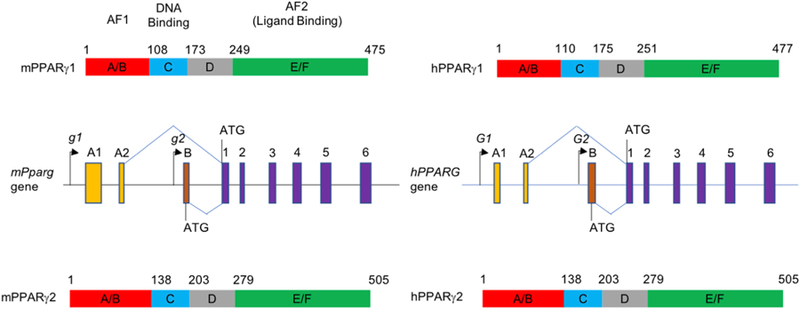
Genomic structures of mouse Pparg1 and Pparg2 (middle left), and human PPARG1 and PPARG2 (middle right) are depicted, based on the earlier reports.15,16 Purple solid rectangles are common exons for Pparg1 and Pparg2 isoforms. Orange (A1 and A2) and brown rectangles (B) are specific for Pparg1 and Pparg2 isoforms, respectively. PPARγ1 protein structures are shown at the top and PPARγ2 structures are presented at the bottom. The functional domains of mouse and human PPARγs are based on sequence alignment reported in an earlier review.20 Abbreviations: PPARγ, peroxisome proliferator-activated receptor gamma; AF, activation function.
A variety of long chain FA and peroxisome proliferators including the fibrate class of hypolipidemic drugs were reported to activate PPARγ to various degrees.12,21–23 Thiazolidinediones (TZDs), a group of anti-diabetic drugs for type 2 diabetes, as specific PPARγ agonists, were a breakthrough for the field of diabetes.24 Even though troglitazone was removed from the market due to hepatotoxicity, TZDs have become important tools that induce physiological functions of PPARγ in glucose and lipid metabolism.7,25–28
3. Adipogenic role of PPARγ
Compared with PPARg1, PPARγ2 is a potent transcription activator and adipogenic factor, because of its additional amino acids at the N-terminus.29,30 Knockdown of PPARγ in adipose tissues using the Cre-lox system led to impairment in the formation of white and brown adipocytes, emphasizing its essential roles in adipogenesis and hepatic fat accumulation.26,31 Accordingly, when PPARγ was selectively knocked down in adipocytes of adult mice by an inducible Cre-lox system, the mature PPARγ-null white and brown adipocytes died within a few days and were replaced by newly formed, normal PPARγ-positive adipocytes.32 However, these animals, with adipocyte-specific PPARγ knockout, derived from an aP2 promoter-driven Cre system, demonstrated controversial insulin sensitivity phenotypes owing to non-adipocyte deletions due to a broad tissue expression pattern of Cre recombinase driven by the promoter.33,34 These studies also demonstrated that gene encoding adiponectin (Adipoq) promoter-driven Cre system exhibited a more selective adipocyte-specific expression. The adipocyte-specific PPARγ null mice, which were generated by the Adipoq-Cre system, exhibited more significant lipoatrophy, insulin resistance and hepatomegaly, confirming a critical role of adipocyte PPARγ in fat cell formation and whole body metabolic homeostasis, which strongly agrees phenotypically with other mouse models of lipoatrophy.35–38 One of the common phenotypes of all these animals with adipocyte-specific deletion of PPARγ is an increase in hepatic triglyceride (TG) accumulation, which is accompanied by a dramatic increase in the expression of hepatic PPARγ.35 Hepatic steatosis may be a consequence of increased hepatic FA influx due to lack of adipocytes for TG storage and/or the enhanced adipogenic program by upregulated PPARγ expression. In accordance with these observations, many mouse models of obesity and diabetes developing fatty livers are associated with higher expression of PPARγ.27,36,37,39–43 More specifically, hepatocyte-specific PPARγ expression appears to be responsible for the fat accumulation. Albumin promoter driven Cre recombinase (Alb-Cre), which was mainly expressed in hepatocytes (though expression in cholangiocytes has also been reported, the expression of Pparg in the tissue has not been described), mediated hepatocyte-specific deletion of Pparg, which rescued steatotic phenotypes in various animal models.27,37,43–45 Especially, because the hepatocyte-specific Alb-Cre deletion markedly diminished the expression of Pparg2, but not Pparg1, PPARγ2 appears to be the major isoform in hepatocytes that contributes to fat accumulation.37 Consistent with these observations, many studies have reported that only the expression of Pparg2, not Pparg1, was significantly increased in the livers of animal models of obesity and hepatic steatosis, even though its expression was low in normal conditions.18,19,46,47 Moreover, Westerbacka et al.48 have reported a strong correlation between hepatic fat accumulation and increased expression of PPARG2 in human subjects. In addition, over-expression of PPARγ1 or PPARγ2 in hepatocytes led to the development of fatty liver.47,49 Interestingly, PPARγ overexpression was also accompanied by increased transcription of several inflammatory marker genes, suggesting a possible association between PPARγ-mediated fat accumulation and NASH development.47 This observation exhibited a stark contrast to the antagonism between the proinflammatory cytokine, tumor necrosis factor alpha (TNFα), and PPARγ in adipocytes.50–52 Kupffer cells (KCs), resident liver macrophages, are important components for eliminating and detoxifying microorganisms, endotoxins and xenobiotics, which are directed from the gut. KCs constitute approximately 15% of the total liver cell population accounting for 80%–90% of the tissue macro-phages.53,54 Paradoxically, when KCs are activated to protect the host from invasion of these toxic materials, they release cytokines, chemokines, and reactive oxygen species, which eventually adversely affected the liver function. An earlier study has emphasized a crucial role of KCs in the development of hepatic steatosis and insulin resistance, as depletion of KCs by gadolinium chloride (GdCl3) treatment protected rats from these metabolic disorders, which were induced by a high fat and high sucrose diet.54 A crucial component for the KCs’ role appears to be TNFα, a major proinflammatory cytokine released by macrophages. Bone marrow transplant from TNFα null donor animals or treatment with anti-TNFα antibodies improved NAFLD in diet-induced or genetically modified animal models.54–56 The expression of Pparg has been detected in macrophages and its functional role as a negative regulator of macrophage activation and cytokine production was identified.57,58 This regulation is mediated through inhibition of the transcription factors, activator protein 1 (AP1), signal transducer and activator of transcription (STAT) and nuclear factor-κ B (NF-κB), which are the major regulators of macrophage activation and TNFα synthesis. These important functions of PPARγ in KC activation and the role of KCs in hepatic steatosis links KC specific PPARγ to the development of hepatic steatosis. In animal studies, macrophage-specific Pparg gene knockout using the Cre-lox system indeed lowered TG accumulation upon HFD feeding compared with control mice. However, in this study reduction in TG accumulation was more evident in hepatocyte-specific Pparg knockout animals, suggesting that hepatocyte-specific PPARγ is the major contributor to TG accumulation.43 Hepatic stellate cells (HSCs) play an important role in the development of NAFLD as they are the major cells producing collagen, which is responsible for fibrosis when activated.59 Though PPARγ is expressed in HSCs and is involved in inhibition of HSC proliferation and activation, the role of stellate PPARγ in hepatic steatosis has not been directly addressed.60–62 Moreover, activation of HSCs is not associated with hepatic fat accumulation, at least in a subset of human patients with NAFLD.59 Nonetheless, the role of stellate PPARγ appears to be beneficial for liver function, i.e., PPARγ activation inhibits HSC activation and fibrogenesis, and, reciprocally, HSC activation negatively regulates its own PPARγ expression.59,62
4. Lipid homeostasis regulated by hepatic PPARγ
As anticipated from its adipogenic role in white adipocytes, overexpression of PPARγ1 or PPARγ2 significantly induced fat accumulation in the liver. Interestingly, in the case of PPARγ1 overexpression, significant fat accumulation was observed only in PPARα knockout liver, where FA oxidation was disrupted, while adenovirus-mediated overexpression of PPARγ2 increased hepatic fat accumulation in wide type (WT) mice fed chow within 7 days post adenovirus injection.47,49 Because the two PPARγ isoforms share the same DNA binding specificity, the difference in fat accumulation efficacy must result from the difference in their transcription activity, as PPARγ2 has 5–10-fold greater transcription activity than PPARγ1.29 On the contrary, reduction in the expression of PPARγ2, mediated by short hairpin interfering RNA (shRNA), lowered the hepatic TG levels in mice fed HFD for 4 weeks.47 In this study, Yamazaki et al.47 have analyzed the mRNA levels of genes involved in lipogenesis including Pparg and Srebp1c at different time points after HFD feedings. Though Srebp1c activation is strongly regulated by well-recognized posttranslational steps, the mRNA level of Pparg2 rose before that of Srebp1c at 4 weeks of HFD feeding containing butter (high in saturated fats), similarly to their target genes: cluster of differentiation 36 (CD36) and adipose differentiation-related protein (Adrp), and fatty acid synthase (Fasn).63–66 In most of the mouse studies with HFD feeding, PPARγ was the early-induced lipogenic transcription factor in the liver.42,65 A controversial result has been reported in another HFD feeding experiment where the mRNA levels of Srebp1c rose from 1 day after the HFD feeding.67 This different observation may result from different sources of saturated fat in the diets; the latter study used HFD containing coconut oil, while the former studies used HFD containing butter or lard. The saturated fats in coconut oil are mostly 12 carbon lauric acids, while butter and lard contains C-16 palmitic acids as the major saturated fatty acid (SFA) components, which better represents the overnutrition cause of human fatty liver.
4.1. Downstream target genes of PPARγ
Many target genes of PPARγ have been reported in adipocyte studies.13,68,69 These include Fabp4/aP2, Cebpa/C/EBPa, Cfd/Adipsin, CD36 and lipoprotein lipase (LpL), which play important roles in adipogenesis and lipogenesis. Expression of these genes was also strongly induced by PPARγ overexpression in PPARα-deficient liver, suggesting that PPARγ-mediated hepatic steatosis is caused by the conserved adipogenic properties of the transcription factor.49 These results were corroborated by results obtained from WT mouse liver transduced with an adenoviral vector containing PPARγ and from a hepatic AML-12 cell line stably expressing PPARγ.47,70 On the contrary, reduction in hepatic Pparg gene expression mediated by Alb-Cre driven-Floxed gene deletion or by acute delivery of adeno-viruses containing shRNA decreased the expression of these adipogenic and lipogenic target genes, and reduced fat accumulation in obese mouse models.27,37,47 Interestingly, while the mRNA level of Srebp1c was not reduced in PPARγ-deficient livers, its target genes such as Fasn, acetyl-CoA carboxylase (Acc) and stearoyl-Coenzyme A desaturase 1 (Scd-1) were effectively downregulated. These observations suggest that PPARγ synergistically activates these lipogenic genes possibly in association with activated SREBP1 or by inducing SREBP1 cleavage to activate these genes indirectly.71 To identify the direct transcription targets of hepatic PPARγ, Matsusue et al.72 analyzed RNA isolated from livers of ob/ob and PPARg deficient ob/ob mice using a subtractive cloning strategy. The subtractive screening identified the fat-specific protein 27 (Fsp27)/Cidec gene as one of the direct PPARγ targets containing a peroxisome proliferator response element (PPRE) in its proximal promoter region. The study focused on FSP27, which belongs to the cell death-inducing DFFA-like effector (CIDE) family and contributes to energy storage in white adipose tissues through the formation of large lipid droplets.73,74 FSP27 overexpression mediated by adenoviruses containing Fsp27 increased hepatic fat accumulation in the ob/ob-PPARg/Cre+ mice, while Fsp27 knockdown by adenoviruses expressing shRNA targeting the gene reduced hepatic fat accumulation in the ob/ob mice, indicating that FSP27 is a downstream PPARγ target responsible for hepatic fat accumulation in mice. Induction of Fsp27 expression has also been observed in an earlier study on hepatic steatosis by PPARγ overexpression.49 It seems clear that increased PPARγ expression in the liver directly or indirectly activates various genes involved not only in lipogenesis, but also in adipogenesis, thereby promoting hepatic steatosis. It has been reported that the genes involved in uptake, intracellular trafficking, esterification and storage of FAs such as Cd36, Fabp4/aP2, monoacylglycerol O-acyltransferase 1 (Mogat1), Plin2/Adrp and Fsp27 are direct targets of PPARγ.63,75 However, lipogenic genes such as Fasn, Scd-1 and Acc have not been proven as direct targets of PPARγ, yet. The upregulation of these genes by PPARγ overexpression may be mediated through SREBP1C, a direct regulator of these genes, which can be activated transcriptionally or through proteolytic cleavage.71,76 Thus, the direct role of PPARγ in the liver lipid homeostasis is to enhance FAs, either from the diet or from lipolysis of white adipose tissues, up-take them from circulation and store them in lipid droplets. Several mechanistic pathways have been implicated in hepatic lipid homeostasis: FA uptake, de novo FA synthesis, FA oxidation, and FA export via VLDL secretion.77 Dominant esterification of dietary FAs into TGs rather than esterification of newly synthesized FAs highly contributes to the hepatic fat accumulation by HFD feeding, which has been demonstrated by mass isotopomer distribution analysis following [1–13C] acetate infusion into experimental mice.78 This observation signifies an important role of PPARγ in the development of fatty liver induced by HFD.
4.2. Upstream regulators of Pparg2 gene expression
It is clear that overexpression of hepatic PPARγ leads to fat accumulation through transcriptional activation of genes responsible for lipid uptake and storage. Unraveling how Pparg expression is regulated is key to discovering therapeutic and preventative approaches for NAFLD. A recent study on PPARγ2 upregulation by HFD feeding adequately illustrated the linkage between PPARγ2 activation and diet-induced hepatic fat accumulation.79 The study using ghrelin receptor knockout mice and exogenous ghrelin infusion has demonstrated that ghrelin, a gastric hormone released during fasting, triggered the activation of mammalian target of rapamycin (mTOR) signaling and Pparg2 mRNA expression in the liver, thereby inducing lipid accumulation. The highly specific PPARγ antagonist, GW9962, blocked ghrelin-induced TG accumulation and lipogenic gene expression in cultured hepatocytes, confirming that PPARγ is the downstream target responsible for ghrelin-induced lipid accumulation. In conjunction with the finding that HFD induces expansion of ghrelin-producing cells, ghrelin signaling is an important mediator that links HFD feeding and PPARγ2-induced hepatic steatosis; however, the responsible transcription factor has not been identified or suggested in that study.80 The idea that mTOR signaling induces PPARγ2 activation is also strongly supported by the rapid induction of Pparg2 expression by insulin in adipose tissues.81 In another elaborate study, AP1 was identified as a direct regulator of Pparg2 transcription.82 AP1 is a dimeric transcription factor that forms homodimers or heterodimers with various basic leucine zipper proteins such as the JUN proto-oncogene (JUN), FOS proto-oncogene (FOS), MAF bZIP transcription factor (MAF) and activating transcription factor (ATF) subfamilies, and is involved in cell survival and proliferation in response to physiological stimuli and environmental insults.83 Using individual AP1 monomer gene gain- and loss-of-function mouse models, and transient transfection reporter assays, Hasenfuss et al. have identified that Fra1/Jun or Fra2/Jun heterodimers repressed Pparg2 gene transcription and hepatic lipid accumulation, while Fos/Jun dimers induced them. Though the study established AP1 as a direct regulator of Pparg2 and hepatic lipid homeostasis, the extracellular signaling pathways leading to the formation of specific heterodimers of AP1 proteins remain to be revealed. Several upstream regulators of Pparg2 associated with hepatic steatosis have been identified. Retinoic acid signaling activates the novel transcription repressor, hes family bHLH transcription factor 6 (HES6), which represses hepatocyte nuclear factor 4α transcription activity on the Pparg2 promoter.84–86 It is worth noting that HES1, a direct target of cyclic adenosine monophosphate (cAMP) response element binding protein, is directly implicated in the repression of Pparg1 gene expression during fasting-induced breakdown of hepatic lipids.87 Though it has not been studied in the setting of hepatic steatosis, progesterone has also been reported to directly induce Pparg2 expression through progesterone receptor in ovaries and macrophages.88,89
4.3. Two faces of hepatic PPARγ in lipid metabolism
A line of reports presented in this review suggests that PPARγ overexpression in the liver induced by HFD feeding or pathophysiological stresses leads to lipid accumulation, which is the initiation step in the development of NAFLD (Fig. 2). Blocking Pparg gene expression in the liver of HFD-fed mice reduced not only lipid accumulation, but also the expression of inflammatory genes, which is an indication of NASH progression.47 This observation was strongly supported by a study using HFD-fed Fra1-liver-specific transgenic mice with low Pparg2 gene expression, where inflammatory marker genes’ expression and serum alanine aminotransferase (ALT) levels were significantly downregulated, compared with the control counterparts.82 These observations strongly suggest that the induction of hepatic PPARγ2 expression is linked to NASH development.
Fig. 2. Upstream regulators and downstream targets of PPARγ2-mediated hepatic lipid homeostasis.
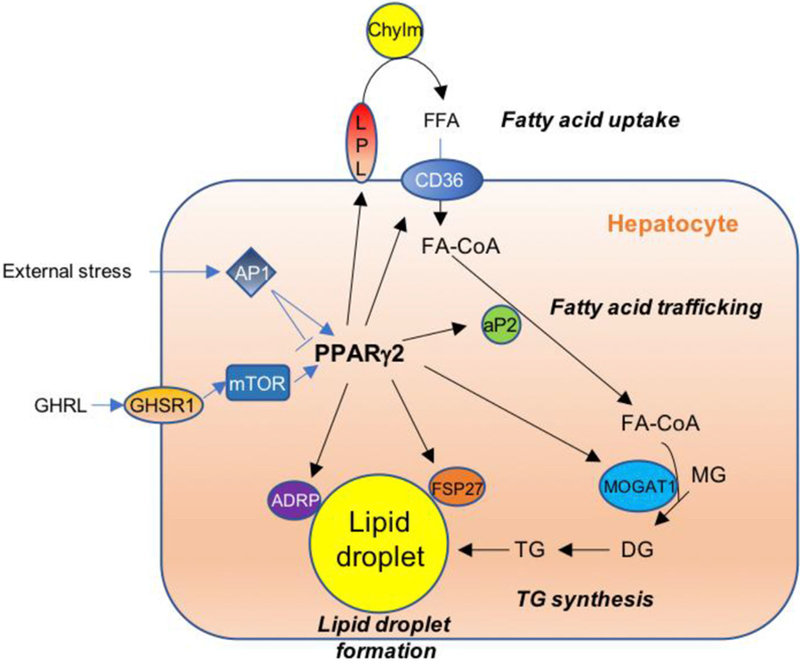
Increased ghrelin by chronic HFD consumption induces Pparg2 gene expression through mTOR signaling. Similarly, external stresses (i.e., nutritional stress) induce or inhibit Pparg2 transcription via AP1 depending on the specific heterodimer formation. Increased PPARγ2 activates downstream target genes such as LPL and CD36 for FA uptake, aP2 for intracellular FA trafficking, MOGAT1 for synthesis of diacylglycerol, and ADRP and FSP27 for lipid droplet accumulation. These targets are not major genes in hepatic TG synthesis during normal conditions. However, when PPARγ2 expression is increased by HFD feeding, these target genes contribute to lipid accumulation in the liver. Abbreviations: PPARγ2, peroxisome proliferator-activated receptor gamma 2; Chylm, chylomicron remnants; FFA, free fatty acids; FA-CoA, fatty acyl-Coenzyme A; MG, monoacylglycerol; DG, diacylglycerol; TG, triacylglycerol; GHRL, ghrelin; GHSR1, ghrelin receptor; LPL, lipoprotein lipase; CD36, cluster of differentiation 36; aP2, adipocyte protein 2; AP1, activator protein 1; mTOR, mammalian target of rapamycin; ADRP, adipose differentiation-related protein; FSP27, fat specific protein 27; MOGAT1, monoacylglycerol O-acyltransferase 1.
Considering its direct role in TG synthesis and lipid droplet formation, PPARγ may have a protective role against lipotoxicity mediated by free fatty acids (FFAs) and their derivatives.90 Especially, the promotion of TG synthesis is considered a protective pathway against SFA insults.91 While unsaturated FAs are readily incorporated into inert TGs, excess SFAs remain largely unesterified.92 These excess free SFAs are rapidly incorporated into saturated phospholipid species that can be integrated into the endoplasmic reticulum (ER) membrane bilayers.93 Because the ER membrane contains unsaturated phosphatidylcholine as the major phospholipid component, incorporation of saturated phospholipids into the membrane results in the loss of membrane fluidity and dissociation of protein folding chaperones, i.e., protein disulfide isomerase and GRP78, from the membrane, which may trigger the unfolded protein response and ER stress, a well-documented etiology of NAFLD.93–96 As Listenberger et al. have reported, promotion of TG synthesis and lipid droplet formation should protect the liver from damage induced by dietary SFAs. When whole body deletion of Pparg2 was introduced in ob/ob mice, their liver contained more ceramides, well-known mediators of NASH development, compared with the control ob/ob mice.97,98 However, because that study focused on β-cell dysfunction by lipotoxicity, the liver phenotypes were not examined. Nonetheless, this observation indicates that ablation of hepatic Pparg may result in adverse effects on other peripheral tissues, which were also manifested in the liver-specific Pparg knockout ob/ob mice.27 In NAFLD induced by a methionine-choline deficient (MCD) diet, adenovirus-mediated hepatic PPARγ overexpression protected the liver from fibrotic NASH.99 Earlier manifestations of mitochondrial membrane stiffening and down-regulation of key genes in TG synthesis in the liver of mice fed an MCD diet may explain the protection by PPARγ in this setting.100,101
5. Conclusions
Many reports point out that HFD consumption strongly induces PPARγ2 expression thereby activating downstream target genes’ expression to facilitate FA uptake, intracellular trafficking, TG synthesis and lipid droplets formation (Fig. 2). This leads to TG accumulation in the liver, an initial prerequisite step in the proposed two-hit theory of NAFLD.2,3 Hepatic PPARγ2 expression is maintained at a low level in normal conditions, which allows the liver to deliver newly synthesized and/or dietary FAs in the form of VLDL to other peripheral tissues as an energy source. The induction of hepatic PPARγ2 expression upon HFD feeding appears to protect peripheral tissues from lipotoxicity by promoting a program to store excess FFAs in the form of TGs. This would be a normal process for combating against constant fat flux in the circulation. However, ablation of hepatic PPARγ reduced not only fat accumulation, but also inflammatory genes’ expression and serum ALT levels, which are the signs of NASH development. Therefore, lowering hepatic PPARγ expression is one of the strategies for preventing NAFLD progression. To elucidate the possible linkage between HFD-induced Pparg2 expression and ghrelin signaling or stress activated AP1 transcription factor, further studies are warranted. Furthermore, clarifying the link between HFD feeding and hepatic Pparg2 gene expression will help develop therapeutic and preventative approaches for treating NAFLD. Developing selective PPARγ modulators that can fine-tune its transcriptional activity is another field that requires additional attention.102
Acknowledgements
This work was supported by USA National Institutes of Health (NIH) grant, R01DK093774 to Y.K. Lee.
Footnotes
Edited by Peiling Zhu and Genshu Wang.
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Browning JD, Horton JD. Molecular mediators of hepatic steatosis and liver injury. J Clin Invest 2004;114:147–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Day CP, James OF. Steatohepatitis: A tale of two “hits”? Gastroenterology 1998;114:842–845. [DOI] [PubMed] [Google Scholar]
- 3.Day CP, James OF. Hepatic steatosis: Innocent bystander or guilty party? Hepatology 1998;27:1463–1466. [DOI] [PubMed] [Google Scholar]
- 4.Loomba RS, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686–690. [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Viscarra J, Kim SJ, Sul HS. Transcriptional regulation of hepatic lipogenesis. Nat Rev Mol Cell Biol 2015;16:678–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barak Y, Nelson MC, Ong ES, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell 1999;4:585–595. [DOI] [PubMed] [Google Scholar]
- 7.Kubota N, Terauchi Y, Miki H, et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell 1999;4: 597–609. [DOI] [PubMed] [Google Scholar]
- 8.Rosen ED, Sarraf P, Troy AE, et al. PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro. Mol Cell 1999;4:611–617. [DOI] [PubMed] [Google Scholar]
- 9.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature 1990;347:645–650. [DOI] [PubMed] [Google Scholar]
- 10.Kliewer SA, Umesono K, Noonan DJ, Heyman RA, Evans RM. Convergence of 9-cis retinoic acid and peroxisome proliferator signalling pathways through heterodimer formation of their receptors. Nature 1992;358:771–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dreyer C, Krey G, Keller H, Givel F, Helftenbein G, Wahli W. Control of the peroxisomal beta-oxidation pathway by a novel family of nuclear hormone receptors. Cell 1992;68:879–887. [DOI] [PubMed] [Google Scholar]
- 12.Zhu Y, Alvares K, Huang Q, Rao MS, Reddy JK. Cloning of a new member of the peroxisome proliferator-activated receptor gene family from mouse liver. J Biol Chem 1993;268:26817–26820. [PubMed] [Google Scholar]
- 13.Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: Tissue-specific regulator of an adipocyte enhancer. Genes Dev 1994;8: 1224–1234. [DOI] [PubMed] [Google Scholar]
- 14.Elbrecht A, Chen Y, Cullinan CA, et al. Molecular cloning, expression and characterization of human peroxisome proliferator activated receptors gamma 1 and gamma 2. Biochem Biophys Res Commun 1996;224:431–437. [DOI] [PubMed] [Google Scholar]
- 15.Zhu Y, Qi C, Korenberg JR, et al. Structural organization of mouse peroxisome proliferator-activated receptor gamma (mPPAR gamma) gene: Alternative promoter use and different splicing yield two mPPAR gamma isoforms. Proc Natl Acad Sci U S A 1995;92:7921–7925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fajas L, Auboeuf D, Raspé E, et al. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem 1997;272: 18779–18789. [DOI] [PubMed] [Google Scholar]
- 17.Vidal-Puig AJ, Considine RV, Jimenez-Lin~an M, et al. Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids. J Clin Invest 1997;99:2416–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vidal-Puig A, Jimenez-Liñan M, Lowell BB, et al. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest 1996;97: 2553–2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Edvardsson U, Bergstro€m M, Alexandersson M, Bamberg K, Ljung B, Dahllöf B. Rosiglitazone (BRL49653), a PPARgamma-selective agonist, causes peroxisome proliferator-like liver effects in obese mice. J Lipid Res 1999;40: 1177–1184. [PubMed] [Google Scholar]
- 20.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: Nuclear control of metabolism. Endocr Rev 1999;20:649–688. [DOI] [PubMed] [Google Scholar]
- 21.Kliewer SA, Forman BM, Blumberg B, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci U S A 1994;91:7355–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell 1995;83: 813–819. [DOI] [PubMed] [Google Scholar]
- 23.Forman BM, Tontonoz P, Chen J, Brun RP, Spiegelman BM, Evans RM. 15-Deoxy-delta 12, 14-prostaglandin J2 is a ligand for the adipocyte determination factor PPAR gamma. Cell 1995;83:803–812. [DOI] [PubMed] [Google Scholar]
- 24.Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA. An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 1995;270:12953–12956. [DOI] [PubMed] [Google Scholar]
- 25.Gale EA. Troglitazone: The lesson that nobody learned? Diabetologia 2006;49: 1–6. [DOI] [PubMed] [Google Scholar]
- 26.He W, Barak Y, Hevener A, et al. Adipose-specific peroxisome proliferator-activated receptor gamma knockout causes insulin resistance in fat and liver but not in muscle. Proc Natl Acad Sci U S A 2003;100:15712–15717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Matsusue K, Haluzik M, Lambert G, et al. Liver-specific disruption of PPAR-gamma in leptin-deficient mice improves fatty liver but aggravates diabetic phenotypes. J Clin Invest 2003;111:737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutchak PA, Katafuchi T, Bookout AL, et al. Fibroblast growth factor-21 regulates PPARγ activity and the antidiabetic actions of thiazolidinediones. Cell 2012;148:556–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Werman A, Hollenberg A, Solanes G, Bjorbaek C, Vidal-Puig AJ, Flier JS. Ligand-independent activation domain in the N terminus of peroxisome proliferator-activated receptor gamma (PPARgamma). Differential activity of PPAR-gamma1 and −2 isoforms and influence of insulin. J Biol Chem 1997;272: 20230–20235. [DOI] [PubMed] [Google Scholar]
- 30.Ren D, Collingwood TN, Rebar EJ, Wolffe AP, Camp HS. PPARgamma knock-down by engineered transcription factors: Exogenous PPARgamma2 but not PPARgamma1 reactivates adipogenesis. Genes Dev 2002;16:27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones JR, Barrick C, Kim KA, et al. Deletion of PPARgamma in adipose tissues of mice protects against high fat diet-induced obesity and insulin resistance. Proc Natl Acad Sci U S A 2005;102:6207–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Imai T, Takakuwa R, Marchand S, et al. Peroxisome proliferator-activated receptor gamma is required in mature white and brown adipocytes for their survival in the mouse. Proc Natl Acad Sci U S A 2004;101:4543–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee KY, Russell SJ, Ussar S, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes 2013;62:864–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mullican SE, Tomaru T, Gaddis CA, Peed LC, Sundaram A, Lazar MA. A novel adipose-specific gene deletion model demonstrates potential pitfalls of existing methods. Mol Endocrinol 2013;27:127–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang F, Mullican SE, DiSpirito JR, Peed LC, Lazar MA. Lipoatrophy and severe metabolic disturbance in mice with fat-specific deletion of PPARg. Proc Natl Acad Sci U S A 2013;110:18656–18661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burant CF, Sreenan S, Hirano K, et al. Troglitazone action is independent of adipose tissue. J Clin Invest 1997;100:2900–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gavrilova O, Haluzik M, Matsusue K, et al. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem 2003;278: 34268–34276. [DOI] [PubMed] [Google Scholar]
- 38.Koutnikova H, Stephanopoulos TA, Watanabe M, et al. Compensation by the muscle limits the metabolic consequences of lipodystrophy in PPAR gamma hypomorphic mice. Proc Natl Acad Sci U S A 2003;100:14457–14462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chao L, Marcus-Samuels B, Mason MM, et al. Adipose tissue is required for the antidiabetic, but not for the hypolipidemic, effect of thiazolidinediones. J Clin Invest 2000;106:1221–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Memon RA, Tecott LH, Nonogaki K, et al. Up-regulation of peroxisome proliferator-activated receptors (PPAR-alpha) and PPAR-gamma messenger ribonucleic acid expression in the liver in murine obesity: Troglitazone induces expression of PPAR-gamma-responsive adipose tissue-specific genes in the liver of obese diabetic mice. Endocrinology 2000;141:4021–4031. [DOI] [PubMed] [Google Scholar]
- 41.Bedoucha M, Atzpodien E, Boelsterli UA. Diabetic KKAy mice exhibit increased hepatic PPARgamma1 gene expression and develop hepatic steatosis upon chronic treatment with antidiabetic thiazolidinediones. J Hepatol 2001;35: 17–23. [DOI] [PubMed] [Google Scholar]
- 42.Inoue M, Ohtake T, Motomura W, et al. Increased expression of PPARgamma in high fat diet-induced liver steatosis in mice. Biochem Biophys Res Commun 2005;336:215–222. [DOI] [PubMed] [Google Scholar]
- 43.Morán-Salvador E, López-Parra M, García-Alonso V, et al. Role for PPARγ in obesity-induced hepatic steatosis as determined by hepatocyte- and macrophage-specific conditional knockouts. FASEB J 2011;25:2538–2550. [DOI] [PubMed] [Google Scholar]
- 44.Yakar S, Liu JL, Stannard B, et al. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A 1999;96:7324–7329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu X, Kobayashi S, Qiao W, et al. Induction of intrahepatic cholangiocellular carcinoma by liver-specific disruption of Smad4 and Pten in mice. J Clin Invest 2006;116:1843–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang YL, Hernandez-Ono A, Siri P, et al. Aberrant hepatic expression of PPARgamma2 stimulates hepatic lipogenesis in a mouse model of obesity, insulin resistance, dyslipidemia, and hepatic steatosis. J Biol Chem 2006;281: 37603–37615. [DOI] [PubMed] [Google Scholar]
- 47.Yamazaki T, Shiraishi S, Kishimoto K, Miura S, Ezaki O. An increase in liver PPARγ2 is an initial event to induce fatty liver in response to a diet high in butter: PPARγ2 knockdown improves fatty liver induced by high-saturated fat. J Nutr Biochem 2011;22:543–553. [DOI] [PubMed] [Google Scholar]
- 48.Westerbacka J, Kolak M, Kiviluoto T, et al. Genes involved in fatty acid partitioning and binding, lipolysis, monocyte/macrophage recruitment, and inflammation are overexpressed in the human fatty liver of insulin-resistant subjects. Diabetes 2007;56:2759–2765. [DOI] [PubMed] [Google Scholar]
- 49.Yu S, Matsusue K, Kashireddy P, et al. Adipocyte-specific gene expression and adipogenic steatosis in the mouse liver due to peroxisome proliferator-activated receptor gamma1 (PPARgamma1) overexpression. J Biol Chem 2003;278:498–505. [DOI] [PubMed] [Google Scholar]
- 50.Zhang B, Berger J, Hu E, et al. Negative regulation of peroxisome proliferator-activated receptor-gamma gene expression contributes to the antiadipogenic effects of tumor necrosis factor-alpha. Mol Endocrinol 1996;10:1457–1466. [DOI] [PubMed] [Google Scholar]
- 51.Rosenbaum SE, Greenberg AS. The short- and long-term effects of tumor necrosis factor-alpha and BRL 49653 on peroxisome proliferator-activated receptor (PPAR)gamma2 gene expression and other adipocyte genes. Mol Endocrinol 1998;12:1150–1160. [DOI] [PubMed] [Google Scholar]
- 52.Okuno A, Tamemoto H, Tobe K, et al. Troglitazone increases the number of small adipocytes without the change of white adipose tissue mass in obese Zucker rats. J Clin Invest 1998;101:1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bouwens L, Baekeland M, De Zanger R, Wisse E. Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology 1986;6:718–722. [DOI] [PubMed] [Google Scholar]
- 54.Huang W, Metlakunta A, Dedousis N, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes 2010;59:347–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li Z, Yang S, Lin H, et al. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003;37: 343–350. [DOI] [PubMed] [Google Scholar]
- 56.De Taeye BM, Novitskaya T, McGuinness OP, et al. Macrophage TNF-alpha contributes to insulin resistance and hepatic steatosis in diet-induced obesity. Am J Physiol Endocrinol Metab 2007;293:E713–E725. [DOI] [PubMed] [Google Scholar]
- 57.Ricote M, Li AC, Willson TM, Kelly CJ, Glass CK. The peroxisome proliferator-activated receptor-gamma is a negative regulator of macrophage activation. Nature 1998;391:79–82. [DOI] [PubMed] [Google Scholar]
- 58.Jiang C, Ting AT, Seed B. PPAR-gamma agonists inhibit production of monocyte inflammatory cytokines. Nature 1998;391:82–86. [DOI] [PubMed] [Google Scholar]
- 59.Washington K, Wright K, Shyr Y, Hunter EB, Olson S, Raiford DS. Hepatic stellate cell activation in nonalcoholic steatohepatitis and fatty liver. Hum Pathol 2000;31:822–828. [DOI] [PubMed] [Google Scholar]
- 60.Miyahara T, Schrum L, Rippe R, et al. Peroxisome proliferator-activated receptors and hepatic stellate cell activation. J Biol Chem 2000;275: 35715–35722. [DOI] [PubMed] [Google Scholar]
- 61.Marra F, Efsen E, Romanelli RG, et al. Ligands of peroxisome proliferator-activated receptor gamma modulate profibrogenic and proinflammatory actions in hepatic stellate cells. Gastroenterology 2000;119:466–478. [DOI] [PubMed] [Google Scholar]
- 62.Galli A, Crabb DW, Ceni E, et al. Antidiabetic thiazolidinediones inhibit collagen synthesis and hepatic stellate cell activation in vivo and in vitro. Gastroenterology 2002;122:1924–1940. [DOI] [PubMed] [Google Scholar]
- 63.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell 1998;93:229–240. [DOI] [PubMed] [Google Scholar]
- 64.Chawla A, Lee CH, Barak Y, et al. PPARdelta is a very low-density lipoprotein sensor in macrophages. Proc Natl Acad Sci U S A 2003;100:1268–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Motomura W, Inoue M, Ohtake T, et al. Up-regulation of ADRP in fatty liver in human and liver steatosis in mice fed with high fat diet. Biochem Biophys Res Commun 2006;340:1111–1118. [DOI] [PubMed] [Google Scholar]
- 66.Latasa MJ, Moon YS, Kim KH, Sul HS. Nutritional regulation of the fatty acid synthase promoter in vivo: sterol regulatory element binding protein functions through an upstream region containing a sterol regulatory element. Proc Natl Acad Sci U S A 2000;97:10619–10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lin J, Yang R, Tarr PT, et al. Hyperlipidemic effects of dietary saturated fats mediated through PGC-1beta coactivation of SREBP. Cell 2005;120:261–273. [DOI] [PubMed] [Google Scholar]
- 68.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 1994;79: 1147–1156. [DOI] [PubMed] [Google Scholar]
- 69.Sugii S, Olson P, Sears DD, et al. PPARgamma activation in adipocytes is sufficient for systemic insulin sensitization. Proc Natl Acad Sci U S A 2009;106: 22504–22509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am J Physiol Endocrinol Metab 2005;288:E1195–E1205. [DOI] [PubMed] [Google Scholar]
- 71.Kim JB, Spiegelman BM. ADD1/SREBP1 promotes adipocyte differentiation and gene expression linked to fatty acid metabolism. Genes Dev 1996;10: 1096–1107. [DOI] [PubMed] [Google Scholar]
- 72.Matsusue K, Kusakabe T, Noguchi T, et al. Hepatic steatosis in leptin-deficient mice is promoted by the PPARgamma target gene Fsp27. Cell Metabol 2008;7: 302–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Danesch U, Hoeck W, Ringold GM. Cloning and transcriptional regulation of a novel adipocyte-specific gene, FSP27. CAAT-enhancer-binding protein (C/EBP) and C/EBP-like proteins interact with sequences required for differentiation-dependent expression. J Biol Chem 1992;267:7185–7193. [PubMed] [Google Scholar]
- 74.Nishino N, Tamori Y, Tateya S, et al. FSP27 contributes to efficient energy storage in murine white adipocytes by promoting the formation of unilocular lipid droplets. J Clin Invest 2008;118:2808–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lee YJ, Ko EH, Kim JE, et al. Nuclear receptor PPARγ-regulated mono-acylglycerol O-acyltransferase 1 (MGAT1) expression is responsible for the lipid accumulation in diet-induced hepatic steatosis. Proc Natl Acad Sci U S A 2012;109:13656–13661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Horton JD, Goldstein JL, Brown MS. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J Clin Invest 2002;109:1125–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fabbrini E, Sullivan S, Klein S. Obesity and nonalcoholic fatty liver disease: Biochemical, metabolic, and clinical implications. Hepatology 2010;51: 679–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oosterveer MH, van Dijk TH, Tietge UJ, et al. High fat feeding induces hepatic fatty acid elongation in mice. PLoS One 2009;4:e6066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li Z, Xu G, Qin Y, et al. Ghrelin promotes hepatic lipogenesis by activation of mTOR-PPARγ signaling pathway. Proc Natl Acad Sci U S A 2014;111: 13163–13168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Francois M, Barde S, Legrand R, et al. High-fat diet increases ghrelin-expressing cells in stomach, contributing to obesity. Nutrition 2016;32: 709–715. [DOI] [PubMed] [Google Scholar]
- 81.Rieusset J, Andreelli F, Auboeuf D, et al. Insulin acutely regulates the expression of the peroxisome proliferator-activated receptor-gamma in human adipocytes. Diabetes 1999;48:699–705. [DOI] [PubMed] [Google Scholar]
- 82.Hasenfuss SC, Bakiri L, Thomsen MK, Williams EG, Auwerx J, Wagner EF. Regulation of steatohepatitis and PPARγ signaling by distinct AP-1 dimers. Cell Metab 2014;19:84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Shaulian EK, arin M. AP-1 as a regulator of cell life and death. Nat Cell Biol 2002;4:E131–E136. [DOI] [PubMed] [Google Scholar]
- 84.Kim SC, Kim CK, Axe D, et al. All-trans-retinoic acid ameliorates hepatic steatosis in mice by a novel transcriptional cascade. Hepatology 2014;59: 1750–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Park JE, Lee M, Kim SC, Zhang Y, Hardwick JP, Lee YK. Hairy and enhancer of split 6 prevents hepatic lipid accumulation through inhibition of Pparg2 expression. Hepatol Commun 2017;1:1085–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Martinez-Jimenez CP, Kyrmizi I, Cardot P, Gonzalez FJ, Talianidis I. Hepatocyte nuclear factor 4alpha coordinates a transcription factor network regulating hepatic fatty acid metabolism. Mol Cell Biol 2010;30:565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Herzig S, Hedrick S, Morantte I, Koo SH, Galimi F, Montminy M. CREB controls hepatic lipid metabolism through nuclear hormone receptor PPAR-gamma. Nature 2003;426:190–193. [DOI] [PubMed] [Google Scholar]
- 88.Kim J, Sato M, Li Q, et al. Peroxisome proliferator-activated receptor gamma is a target of progesterone regulation in the preovulatory follicles and controls ovulation in mice. Mol Cell Biol 2008;28:1770–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yang X, Zhang W, Chen Y, et al. Activation of peroxisome proliferator-activated receptor γ (PPARγ) and CD36 protein expression: the dual pathophysiological roles of progesterone. J Biol Chem 2016;291:15108–15118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Neuschwander-Tetri BA. Hepatic lipotoxicity and the pathogenesis of nonalcoholic steatohepatitis: The central role of nontriglyceride fatty acid metabolites. Hepatology 2010;52:774–788. [DOI] [PubMed] [Google Scholar]
- 91.Listenberger LL, Han X, Lewis SE, et al. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A 2003;100: 3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Noguchi Y, Young JD, Aleman JO, Hansen ME, Kelleher JK, Cock G. Effect of anaplerotic fluxes and amino acid availability on hepatic lipoapoptosis. J Biol Chem 2009;284:33425–33436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res 2006;47:2726–2737. [DOI] [PubMed] [Google Scholar]
- 94.van der Sanden MH, Houweling M, van Golde LM, Vaandrager AB. Inhibition of phosphatidylcholine synthesis induces expression of the endoplasmic reticulum stress and apoptosis-related protein CCAAT/enhancer-binding protein-homologous protein (CHOP/GADD153). Biochem J 2003;369:643–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Spector AA, Yorek MA. Membrane lipid composition and cellular function. J Lipid Res 1985;26:1015–1035. [PubMed] [Google Scholar]
- 96.Gentile CL, Frye M, Pagliassotti MJ. Endoplasmic reticulum stress and the unfolded protein response in nonalcoholic fatty liver disease. Antioxidants Redox Signal 2011;15:505–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pagadala M, Kasumov T, McCullough AJ, Zein NN, Kirwan JP. Role of ceramides in nonalcoholic fatty liver disease. Trends Endocrinol Metab 2012;23: 365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Medina-Gomez G, Gray SL, Yetukuri L, et al. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet 2007;3:e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Nan YM, Han F, Kong LB, et al. Adenovirus-mediated peroxisome proliferator activated receptor gamma overexpression prevents nutritional fibrotic steatohepatitis in mice. Scand J Gastroenterol 2011;46:358–369. [DOI] [PubMed] [Google Scholar]
- 100.Caballero F, Fernández A, Matías N, et al. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: Impact on mitochondrial S-adenosyl-L-methionine and glutathione. J Biol Chem 2010;285: 18528–18536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline-deficient diet. J Lipid Res 2008;49:1068–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ables GP. Update on pparγ and nonalcoholic Fatty liver disease. PPAR Res 2012;2012:912351. [DOI] [PMC free article] [PubMed] [Google Scholar]