

Abstract
Schizophrenia (SZ) is a severe psychotic disorder that is highly heritable and common in the general population. The genetic heterogeneity of SZ is substantial, with contributions from common, rare, and de novo variants, in addition to environmental factors. Large genome-wide association studies have detected many variants that are associated with SZ, yet the pathways by which these variants influence risk remain largely unknown. SZ is also clinically heterogeneous, with patients exhibiting a broad range of deficits and symptom severity that vary over the course of illness and treatment, which has complicated efforts to identify risk variants. However, the underlying brain dysfunction forms a more stable trait marker that quantitative neurocognitive and neurophysiological endophenotypes may be able to objectively measure. These endophenotypes are less likely to be heterogeneous than the disorder and provide a neurobiological context to detect risk variants and underlying pathways among genes associated with SZ diagnosis. Furthermore, many endophenotypes are translational into animal model systems, allowing for direct evaluation of the neural circuit dysfunctions and neurobiological substrates. We review a selection of the most promising SZ endophenotypes, including prepulse inhibition, mismatch negativity, oculomotor antisaccade, letter-number sequencing, and continuous performance tests. We also highlight recent findings from large consortia that suggest the potential role of genes, particularly in the neuregulin and glutamate pathways, in several of these endophenotypes. Although endophenotypes require additional time and effort to assess, the insight into the underlying neurobiology that they provide may ultimately reveal the underlying genetic architecture for SZ and suggest novel treatment targets.
Keywords: endophenotype, schizophrenia, genetics, neurocognition, neurophysiology
SCHIZOPHRENIA: A GENETICALLY AND PHENOTYPICALLY COMPLEX DISORDER
Clinical Course and Presentation
Schizophrenia (SZ) is a severe and persistent psychotic disorder with a population prevalence of nearly 1% [1]. The profound clinical heterogeneity of SZ is reflected in its broad range of deficits and symptom severity. The positive symptoms associated with SZ include hallucinations, delusions, disorganized speech, and disorganized behavior, whereas the negative symptoms include diminished facial and emotional expression, poverty of speech, lack of motivation, an inability to experience pleasure, and social withdrawal. To receive a diagnosis of SZ, two or more of these symptoms must be consistently present for at least 1 month and must be severe enough to cause social and/or occupational dysfunction and neglect of hygiene. Due to the extreme heterogeneity in individual symptom profiles, Eugen Bleuler aptly described SZ as the “Group of Schizophrenias” in 1911, only 3 years after he first coined the term “schizophrenia” [2].
The onset of SZ most commonly occurs in late adolescence or early adulthood [3] and is generally chronic and unremitting, often requiring multiple hospitalizations over the course of a lifetime [4]. Although about 25% of patients with SZ will experience a full recovery with medication [5], most patients require long-term care and never return to their previous level of functioning. Given the severity of symptoms, about 40% of SZ patients attempt suicide and 5% succeed [6].
The treatment of SZ is complex. Antipsychotic medications, which target the positive symptoms, are expensive, are only partially effective, and have troublesome side effects that, when paired with the lack of insight that is characteristic of SZ, often result in non-compliance [7,8]. In fact, the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) study, which investigated the effectiveness of five commonly used antipsychotics in a sample of 1460 patients with SZ, found that about 50% of patients discontinue their medications by 6 months and that 74% of patients discontinue their medications by 18 months [9]. Thus, it is imperative that we increase our understanding of the underlying etiology of SZ to facilitate the development of novel pharmacological agents that can target specific symptoms in this heterogeneous disorder.
The Genetics of Schizophrenia
Twin and multiplex family studies dating back to the 1940s have produced a range of heritability estimates for SZ. These family-based estimates of what one might describe as broad-sense heritability take into account the role of rare variants and were summarized in a widely quoted 2003 meta-analysis of SZ as a heritability estimate of 81% [10,11]. In contrast, single-nucleotide polymorphism (SNP)–estimated heritability is calculated by way of population-based genome-wide association studies (GWAS) of common, additive genetic variation, and such studies, which do not take rare variants into account, have suggested that SZ heritability may be closer to 60–70% [10,12,13]. Note that when discussing the heritability of specific SZ endophenotypes later in this review, we are generally using broad-sense heritability.
Although sporadic, non-familial cases of SZ are often observed, family history remains the strongest and best replicated risk factor for developing SZ [14]. Compared with the ~1% prevalence in the general population, relatives of an individual with SZ have a significantly increased risk of developing the disorder—that is, the closer the relationship between a relative and an individual with SZ, the more genes that are shared and the higher the risk, which ranges from 2 to 4% for the second-degree relatives of individuals with SZ to 10 to 15% for the first-degree relatives of individuals with SZ [10]. Still, the concordance rate for monozygotic (MZ) twins, who share their entire genome, is only 45%. These studies support a strong role of genetics in SZ susceptibility but also suggest that environmental and other risk factors are involved.
Identifying the genetic variants captured by these heritability estimates has been challenging. Early efforts to map the genes contributing to SZ risk through family-based linkage and association studies implicated many chromosomal regions and suggested plausible candidate genes [15–19]. However, there have been difficulties in narrowing the source of the signal within linkage regions and in identifying functional variants within the implicated genes, and these difficulties have been compounded by inconsistent replication of findings from different samples. These issues are largely due to the fact that SZ is a complex disorder that results from the interaction of many genetic and non-genetic factors [1,20]. As such, there is no clear one-to-one relationship between genotype and phenotype. This is evident, for example, in family studies, wherein unaffected relatives often share risk variants with their affected family members. This reduced penetrance also affects case-control studies, as some portion of clinically unaffected individuals are likely carriers of SZ risk genes without expressing the phenotype. Heterogeneity also complicates gene identification, as different genes (i.e., genetic heterogeneity) or different variants of the same gene (i.e., allelic heterogeneity) may contribute to disease risk. Finally, sporadic, or non-familial, cases of SZ likely have different underlying genetic architecture than familial cases, yet both sporadic and familial SZ cases are combined in case-control studies of SZ. Such factors cause a significant reduction in the power of genetic studies, which has, in part, been addressed through more recent technological advances and the use of increasingly larger samples.
THE MODERN ERA OF GENOMICS IN SCHIZOPHRENIA
Genome-Wide Association Studies (GWAS)
The field of psychiatric genetics shifted to GWAS in 2007 with the introduction of microarray-based genotyping and the culmination of efforts to map the structure of the human genome. Such studies rely on allele frequency differences for SNPs in large samples of unrelated SZ cases and controls to implicate genomic regions containing risk genes. Although early SZ GWAS suffered from relatively small samples, combining results across studies has begun to produce replicable findings. For example, in a GWAS of 479 SZ cases and 2937 controls, followed by replication testing of 12 suggestive loci in 16,726 subjects, O’Donovan and colleagues identified ZNF804A as a possible susceptibility gene for SZ [21]. An initial GWAS of 2681 cases and 2653 controls from the Molecular Genetics of Schizophrenia (MGS) sample by Shi and colleagues found no loci reaching genome-wide significance, yet the meta-analysis of 8008 cases and 19,077 controls identified 7 SZ-associated SNPs on chromosome 6p22.1, which represents the extended major histocompatibility complex (MHC) region [22]. Likewise, an initial GWAS by Stefansson and colleagues in 2663 cases and 13,498 controls produced no significant findings, yet follow-up genotyping and meta-analysis implicated the MHC and variants in the neurogranin (NRGN) and transcription factor 4 (TCF4) genes [23]. Another study by the International Schizophrenia Consortium confirmed the association to the MHC region and demonstrated that common genetic variants in aggregate may account for >30% of the risk for SZ [24].
More recently, the formation of the Psychiatric Genomics Consortium (PGC) as the largest consortium in the history of psychiatry has enabled mega-analyses of SZ. In a landmark publication of ~37,000 cases and ~113,000 controls, the PGC identified 108 distinct regions that met genome-wide significance, 83 of which had not been previously reported [25]. These findings included many genes that are biologically plausible candidates, such as DRD2; several genes related to glutamate signaling; and the MHC region, yet the associated variants collectively only account for 3.4% of the risk for SZ. A considerable proportion of the observed heritability is thus not detectable in these very large studies of unrelated SZ cases and controls [26,27].
Next-Generation Sequencing (NGS) Studies
Another strategy in the genetic investigation of SZ has been the application of next-generation sequencing (NGS) methods that were pioneered in cancer research. Contrary to the GWAS focus on common variants, these methods seek to identify copy number variants (CNVs) and single-nucleotide variants (SNVs) that occur in less than 0.1% of the general population but that have elevated frequencies and functional roles in SZ. CNVs associated with SZ are generally large, recurrent deletions or deletions encompassing regions that are highly damaging and that consequently have large effects on risk, with odds ratios ranging from 2 to 60. Studies have collectively identified SZ-associated recurrent CNVs in several chromosomal regions (e.g., 1q21.1, 3q29, 7q11.2, 15q13.3, 16p11.2, and 22q11), all of which are also associated with autism and intellectual disability, suggesting a shared genetic architecture between these disorders [28–32]. These CNVs tend to involve neurodevelopmental pathways, and genes in the neuregulin and glutamate pathways are disproportionately disrupted in SZ [29,32,33]. Although a recent study by the PGC, which represents the largest genome-wide analysis of CNVs for any psychiatric disorder, confirmed the presence of SZ-associated CNVs in these regions, the identified loci were present in only 1.4% of the 21,094 SZ cases and account for only ~0.85% of the heritability of SZ [33].
Whole-exome sequencing (WES) studies attempt to identify SNVs in the coding sequence of genes that may contribute to risk for SZ by examining frequency differences between cases and controls through mutational burden analyses. The extremely high rate of background mutation complicates such studies, which to date have been underpowered and have generally failed to provide insight into the mechanism of illness. For example, in a WES study of 166 cases and 307 controls with follow-up genotyping of novel, potentially damaging variants in an independent sample of 2617 cases and 1800 controls, no variant reached study-wide significance [34]. Another WES of 2536 cases and 2543 controls with a focus on gene-based tests of disruptive variants similarly failed to find significance, although this study importantly confirmed a polygenic contribution of very rare (<1/10,000), damaging mutations to SZ risk [35]. Likewise, exome-sequencing studies in subsets of SZ subjects have been somewhat informative. For example, in an exome-sequencing study that investigated subjects with sporadic SZ, Gulsuner and colleagues found that de novo loss-of-function mutations occur more frequently in cases with sporadic SZ than in controls [36]; more recently, the findings in this study were supported by Yang and colleagues [37].
Gene-Expression Studies
The human genome is made up of DNA that is organized into 20,000 to 25,000 genes. Nearly every human cell contains the same genes, but different cells and tissue have different patterns of gene expression. These differences in gene expression may contribute to diseases like SZ.
Before each gene is expressed, the DNA must first be copied, or transcribed, into messenger RNA (mRNA). These transcriptions, which are collectively referred to as a transcriptome, control the function of cells. Several groups have explored the use of peripheral blood gene-expression signatures [38] and used transcriptomes to investigate the biological consequences of SZ-associated genes; these studies have suggested a potential role for ARC, NMDA receptors, synapse and immunity dysfunction, and CNS development in SZ [39].
Epigenetic Studies
Epigenetic “marks” in DNA change how genes are expressed in the process of DNA transcription to RNA and subsequent translation to protein. Environmental factors such as smoking, maternal starvation, and in utero viral infection can interact with genes via processes such as methylation and histone modification to change gene expression in the developing brain. These functional changes can be stable and have long-lasting chromatin modifications that regulate both local and genome-wide gene activity.
DNA methylation is a normal biochemical process whereby atoms are added to DNA as part of gene expression, but environmental factors can lead to the formation of differentially methylated genetic regions. Some studies have demonstrated an enrichment of such changes in SZ-related gene networks [40]. For example, one study showed that SZ-associated methylation differences were observed at 923 cytosine-phosphate-guanine (CpG) islands and that some of the replicated differentially methylated positions overlapped with top-ranked SZ regions from GWAS [41].
Nucleosomal histone modifications contribute to genome organization and function, with various histone methylation and acetylation marks (including H3-trimethyl-lysine 4 [H3K4me3] and H3-acetyl-lysine-27 [H3K27ac]) serving as key regulators for active gene promoters and enhancers [42]. Studies of histone modification are ongoing in SZ [43], and publicly accessible resources are available, including results from the dorsolateral prefrontal cortex and anterior cingulate cortex of SZ brains and UCSC browser visualizations for cell-specific maps (see http://www.psychencode.org/ or https://www.synapse.org/#!Synapse:syn4566010). Given the relative ease that histone posttranslational modifications can be observed in nearly all somatic tissues, this type of epigenetic information could serve as a useful marker if an association is found between histone modifications and SZ.
Summary
Although SZ is highly heritable, identifying risk genes has proven difficult due to the complex nature of the disorder. Large-scale GWAS employing a case-control design have detected many common variants associated with SZ diagnosis. However, genome-wide significant variants account for only a small portion of the overall risk, the effect size of each individual allele is very small, and the pathways by which these variants impact risk remain largely unknown. Studies of rare variation have shed light on possible underlying pathways, yet CNVs and SNVs account for only a small percentage of SZ cases. Likewise, although emerging technologies in gene-expression and epigenetic studies may ultimately be fruitful, the challenge of translating genetic findings to precision medicine in SZ still persists.
THE VALUE OF DEEP PHENOTYPING IN SCHIZOPHRENIA RESEARCH
A key limitation faced by researchers in SZ is that the heterogeneous clinical phenotype complicates the diagnosis, therefore diluting the genetic signal. To address this issue, Gottesman and Shields introduced the concept of “endophenotypes” in SZ [44], and the application of endophenotypes to the study of SZ has become known as “deep phenotyping.” These state-independent traits are associated with illness, are observed in unaffected family members at a higher rate than in the general population, and co-segregate with illness in families [44]. To be useful in genetic studies, an endophenotype must also be significantly heritable, reliably measured, and stable over time [45,46]. Given that endophenotypes fall within the genotype-to-phenotype pathway of the disease process, they form a critical link between genetic risk variants and the more direct lower-level biological processes that contribute to observable symptoms [47,48]. In other polygenic, complex conditions, like hypercholesterolemia, endophenotypes (i.e., cholesterol) have been particularly useful in helping to characterize the underlying biological dysfunction and to develop targetable treatments [49].
Endophenotypes are quantitative in nature and reflect a continuous distribution of trait values in the general population, consistent with polygenic theories of SZ [50–52]. Under this scenario, as detailed in Figure 1, individuals carrying non-penetrant risk variants do not diminish a linkage signal, as with an analysis of diagnosis. Indeed, SZ patients, unaffected relatives, and controls provide a range of scores and are thus informative regarding the underlying liability. Therefore, by using endophenotypes to measure the underlying liability to SZ as a quantitative trait, we gain significant power and increased resolution compared to analyses of diagnosis, which use a somewhat subjective threshold to dichotomize a continuous trait [53,54]. For a disorder like SZ, which has a prevalence of ~1% and heritability of 60–80%, the analysis of a quantitative endophenotype is expected to be approximately 100-fold more efficient than the analysis of the categorical diagnosis, which translates to a 10-fold increase in power [53]. The analysis of an endophenotype thus requires a substantially smaller sample size to achieve comparable power to an analysis of SZ diagnosis. By enriching for individuals with higher liability to SZ, who presumably carry more risk alleles, family-based designs provide a further increase in power [55].
Figure 1.
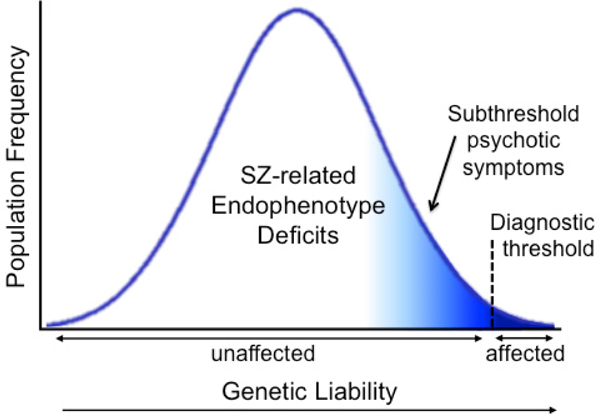
The liability threshold model. Endophenotypes capture the full spectrum of information in liability distribution, whereas diagnosis dichotomizes the distribution into “affected” and “unaffected” according to a threshold defined by subjective symptom profiles.
NEUROPHYSIOLOGICAL AND NEUROCOGNITIVE ENDOPHENOTYPES IN SCHIZOPHRENIA
Neurocognitive dysfunction is a hallmark of SZ, leading to impaired quality of life and poor functional outcome [56–58]. As such, neurocognitive deficits are endorsed by the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) and the Food and Drug Administration (FDA) as targets for the development of new treatments for SZ [59,60]. Data from the North American Prodrome Longitudinal Study (NAPLS) has further demonstrated that measures of neurocognitive functioning in SZ may have predictive utility in identifying clinically high-risk subjects who will convert to psychosis [61,62]. Some researchers have suggested that the neurocognitive deficits in attention, learning, and memory that characterize SZ may arise from a primary impairment in the filtering or “gating” of sensory information [63]. According to this hypothesis, an inability to filter out irrelevant external stimuli leads to the psychotic symptoms (e.g., hallucinations, delusions) and cognitive disorganization observed in SZ. Therefore, studying an array of endophenotypes that range from largely automatic (i.e., preattentive) neurophysiological measures to highly volitional (i.e., attentive) neurocognitive measures may provide a window into the biological processes underlying SZ. Examples of several promising endophenotype paradigms are discussed below and shown in Figure 2.
Figure 2.
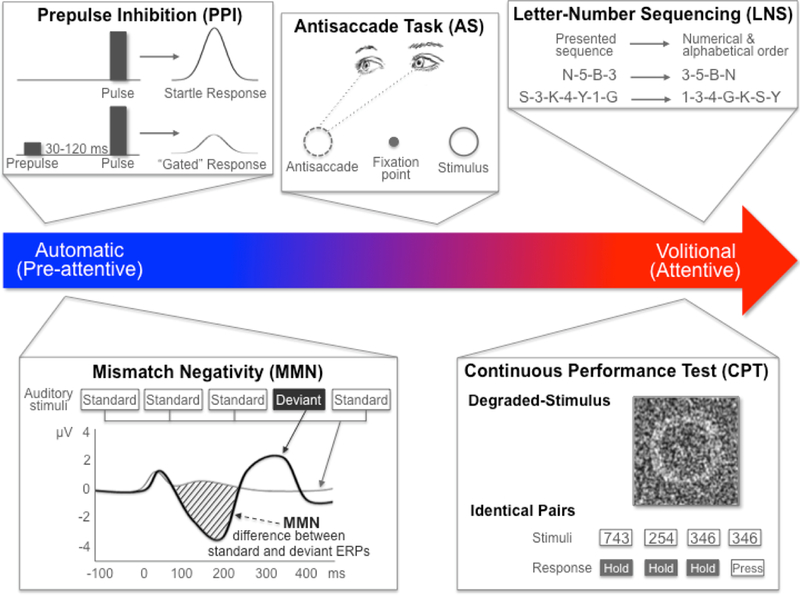
The range of SZ-related endophenotypes from automatic to volitional. PPI and MMN are neurophysiological endophenotypes representing automatic, or preattentive, processes that can be elicited with no active participation from the test subject. PPI is measured as the percent inhibition (i.e., gating) of the startle reflex in response to a weak prestimulus. MMN is measured as the difference between the waveforms for a standard stimulus ERP and a deviant stimulus ERP. Neurocognitive endophenotypes like CPT and LNS are volitional, or attentive, and require active participation from the subject. CPT measures sustained attention as the signal/noise discrimination ratio (d’) of blurred target stimuli (in the DS-CPT) or pairs of identically presented stimuli (in the CPT-IP). LNS measures working memory as the correct reordering of a verbally presented list of intermixed numbers and letters. AS is measured as the ratio of correct antisaccades (i.e., looking in the opposite direction of the presented stimulus) to total interpretable saccades (i.e., eye movements in any direction). Although AS is a neurophysiological measure, subjects are required to actively participate and control their response. AS: antisaccade; CPT: continuous performance test; DS: degraded stimulus; ERP: event-related potential; IP: identical pairs; LNS: letter-number sequencing; MMN: mismatch negativity; PPI: prepulse inhibition; SZ: schizophrenia.
Many neurophysiological and neurocognitive endophenotype measures exhibit medium-to-large deficits in SZ, exhibit moderate-to-substantial stability, and are independent of clinical state [64–67]. Here, we focus on three widely used neurophysiological, inhibitory endophenotypes that have been shown to rank highly across such parameters: prepulse inhibition, mismatch negativity, and oculomotor antisaccade [64]. Given the large body of literature describing neurocognitive measures and SZ, we describe a few representative tests with a focus on working memory and attention. Reviews of additional endophenotype paradigms that have been used in genomic and clinical-outcome studies of SZ [64–67], including neuroimaging studies [68,69], can be found elsewhere. Figure 3 demonstrates the large effect sizes of some potentially promising endophenotypes, which are described below.
Figure 3.
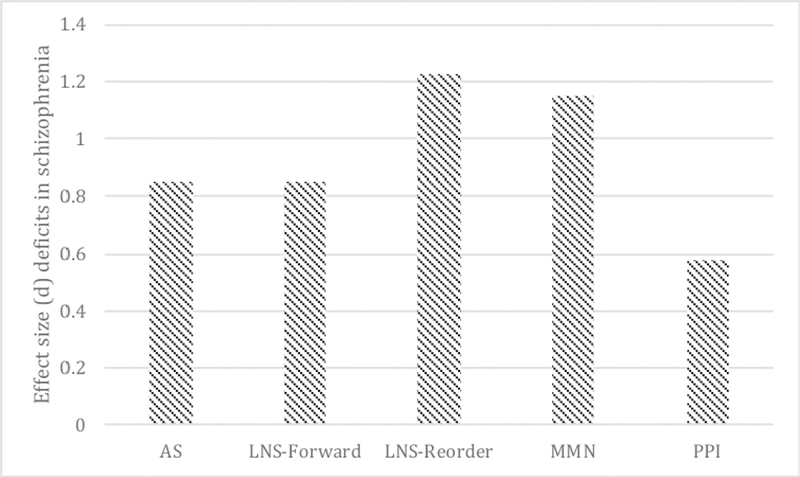
Effect sizes (Cohen’s d) of potential SZ endophenotypes as analyzed by Light and colleagues [64]. AS: antisaccade; LNS: letter-number sequencing; MMN: mismatch negativity; PPI: prepulse inhibition.
Prepulse Inhibition (PPI)
PPI of the acoustic startle reflex is a measure of sensorimotor gating that may reflect the biological processes underlying gating impairments in SZ. Compared to healthy controls, patients with SZ fail to inhibit their startle response (i.e., eye blinking) when they hear a startle-eliciting stimulus (i.e., a pulse) after hearing a weaker or softer stimulus that precedes the pulse by 30-to-120 ms (i.e., the prepulse). There is a consistent body of literature demonstrating robust PPI deficits in SZ that are relatively stable across time [64,66,70,71], despite the emergence and nearly ubiquitous use of atypical antipsychotics, which enhance PPI and make deficits more difficult to detect and quantify [72,73]. PPI deficits are also observed in unaffected relatives of SZ patients and are significantly heritable in SZ families (45–50%), suggesting that PPI may be a marker of risk for SZ rather than a consequence of illness [66,74–76]. Furthermore, while all SZ patients demonstrate comparable deficits in PPI regardless of family history of illness, the broad-sense heritability of PPI deficits is substantially increased in families with a higher genetic vulnerability to SZ (47%) compared to that observed when family history for illness is not considered (29%) [77].
Mismatch Negativity (MMN)
MMN is a component of the event-related potential (ERP) that is elicited when an infrequently presented “deviant” stimulus occurs in a sequence of repetitive “standard” auditory stimuli [78]. The deviant sound can differ in one or more perceptual features, such as duration, frequency, or intensity. MMN refers to the difference between the waveforms for the standard stimulus ERP and the deviant stimulus ERP and usually peaks between 150 and 250 ms after the presentation of the deviant stimulus [79]. Like PPI, MMN can be elicited regardless of whether a subject is paying attention to the sequence, and it is thus considered an index of preattentive sensory processing. Reduced MMN amplitude is one of the most robust findings in SZ [80], with a large effect size of about 1.0 that is relatively unaffected by medication and remains stable across time and fluctuations in clinical symptoms [64,81–84]. MMN is also 68% heritable and is present in individuals at genetic risk for developing SZ [85–90]. MMN is also associated with psychosocial functioning and has shown promise for predicting the onset of psychosis in high-risk populations [91–95]. Finally, we note that the P3a, another measure of early auditory information processing, is a promising endophenotype that is often measured in tandem with MMN [64,84,96].
Oculomotor Antisaccade (AS)
The AS task is a widely used electrophysiological measure of inhibitory failure, which is an important cause of the cognitive and clinical abnormalities found in SZ [97]. This task requires participants to fixate on a central target and respond to a peripheral cue by looking in the opposite direction at the same distance; it is measured as the ratio of correct to total interpretable saccades (0 to 1). Compared to healthy controls, SZ patients display significant deficits on an overlap version of the AS task, with a large effect size of about 1.0 [64,98,99]. Studies have also found that AS performance, latency, and gain are stable measures of pathophysiological disturbances in SZ [100,101]. In addition, large family and twin studies have shown that the AS task has a high heritability of 42–57% [74,102] with documented deficits in first-degree relatives of SZ patients [103,104]. These data suggest that AS performance represents a useful endophenotype for SZ.
Letter-Number Sequencing (LNS)
Working memory is the ability to maintain and manipulate the internal representation of a stimulus in transient memory [105]. Deficits in working memory have been extensively documented and are profoundly affected and persistent in SZ [106] and in relatives of patients with SZ [107]. These deficits are thought to reflect the fundamental cognitive disturbances that are associated with SZ [108]. A subtest of the well-standardized and normed Wechsler Memory Scale III [109], the LNS is a prototypical measurement of verbal working memory information storage with manipulation. Administration of this task involves the verbal presentation of a series of intermixed letters and numbers, increasing in difficulty from 2 stimuli to 8 stimuli as subjects advance in the task. In the reorder condition of the LNS, subjects repeat the numbers in ascending order followed by the letters in alphabetical order, which provides an assessment of executive functioning and working memory. The LNS can also be administered in the forward condition, wherein subjects simply repeat the numbers and letters in the order presented, which provides an assessment of transient online storage and retrieval. SZ patients typically perform worse (i.e., obtain lower scores) than controls for both conditions, but larger differences are usually observed for the reorder condition. The LNS ranks highly among neurocognitive endophenotypes, as it demonstrates large SZ-related deficits and longitudinal stability [64]. The LNS is also highly heritable, both individually (39%) and as part of a composite measure of working memory (27%) [74,110].
Continuous Performance Test (CPT)
Deficits in attention were included in Bleuler’s first descriptions of SZ, and such deficits remain a key nosological feature of the disorder, as well as a primary area of emphasis in SZ research [2,65]. More recent studies of endophenotypes in SZ have narrowed in on deficits in sustained focused attention, which are most commonly measured using CPTs. These tests typically take 15 min to administer and require subjects to monitor a series of target stimuli (e.g., letters, digits, shapes) for 30–100 ms between periods of vigilance during which no stimuli are presented. Several different versions of the CPT are available to assess various aspects of sustained focused attention and vigilance. For example, the Degraded Stimulus CPT (DS-CPT) is associated with a high perceptual load and involves the identification and response to highly blurred, black and white target stimuli [111,112], whereas the Identical Pairs version of the CPT (CPT-IP) is associated with a working memory load and requires the subject to respond each time a string of 3 or 4 digits is presented twice in a row [113,114]. SZ patients consistently show large performance deficits on CPTs, with meta-analyses reporting a mean effect size of 1.18 [115]. These deficits are typically not affected by psychotropics [116,117] and are relatively persistent across time and clinical status [113,118]. Deficits in CPTs are also reliably detected in the unaffected relatives of SZ patients, with moderate heritability in healthy families (39–49%) [113] and higher heritability observed in SZ families (38–79%) [74,119,120].
GENETIC STUDIES OF ENDOPHENOTYPE DEFICITS IN SCHIZOPHRENIA
Despite the increasing use of endophenotypes in the study of SZ, few large-scale genetic studies of endophenotype deficits in SZ have been published. In the largest genome-wide study of PPI to date, investigators from the Learning on Genetics of SZ Spectrum (LOGOS) identified and replicated common variants in the NGF and CALN1 genes that reached genome-wide significance [121]. Although this study only included healthy males, polygenic risk scores calculated based on the PGC SZ cohort showed that an increased risk for SZ was associated with PPI deficits. Subsequent meta-analyses have offered additional evidence to support an abnormal neurotrophin profile in SZ, with the observance of decreased peripheral blood NGF levels in SZ patients [122], and have highlighted the polygenicity of PPI, suggesting the relevance of specific variants in COMT, GRIK3, PRODH, and TCF4 [123]. A recent GWAS of pursuit initiation and AS error rate conducted by the Bipolar-Schizophrenia Network on Intermediate Phenotypes (B-SNIP) consortium implicated genes involved in nuclear trafficking and gene silencing (IPO8), axonal guidance and synaptic specificity (PCDH12), transduction of nerve signals (NRSN1), retinal degeneration (LMO7), and synaptic glutamate release (SH3GL2) in the regulation of oculomotor inhibition [124].
Other studies have examined the relationship between neurocognitive functioning and polygenic risk for SZ in large, community-based samples. For example, one study found that higher risk for SZ was associated with lower neurocognitive functioning in a very large sample of community-dwelling adults from the UK Biobank (N = 112,151) [125]. These findings are consistent with those of the Avon Longitudinal Study of Parents and Children (ALSPAC), who found that higher risk for SZ was associated with lower IQ [126]. Similarly, the Cognitive Genomics Consortium (COGENT) reported a genetic correlation of −0.17 between general cognitive function and SZ in their population-based sample of >35,000 individuals [127]. Although these studies suggest that a portion of the genetic risk for SZ may be mediated through genetic effects on neurocognitive functioning, the reliance on assessments of IQ and global cognitive summaries in population-representative samples provides little insight into the neurobiological mechanisms that may mediate the genetic overlap between cognitive functioning and SZ. Large genetic studies of neurocognitive deficits in the context of SZ are thus needed to identify the genes contributing to this shared genetic architecture.
The Consortium on the Genetics of Schizophrenia (COGS) has conducted candidate gene and linkage analyses of many SZ-related endophenotypes, including those reviewed here. The COGS results suggest that the genes contributing to the specific endophenotypes reviewed here converge on a functional gene network, which is summarized in Figure 4. In association studies that genotyped 130 families using a custom genotyping array that evaluated 94 candidate genes for SZ, the COGS identified 71 SNP-endophenotype associations that involved 28 genes and 1 or more of the endophenotypes discussed in this review and that were strong enough to satisfy an experiment-wide significance level of 0.05, after accounting for linkage disequilibrium, endophenotype correlations, family structure, gene size, and multiple testing. Furthermore, each of the associated genes shown in Figure 4 contained at least one SNP with an a posteriori chance of ≥84% of being a true finding of association, indicating that the observed associations most likely represent true positive results. Additionally, these studies identified 8 genes (CTNNA2, ERBB4, GRID2, GRIK3, GRIK4, NOS1AP, NRG1, and RELN) with pleiotropic associations across multiple endophenotypes in the COGS sample, as well as in an independent case-control study [128–130]. These 8 genes are also associated with at least two of the endophenotypes that are the focus of this review and span neurophysiological and neurocognitive domains. Linkage analyses of a larger sample of 296 COGS families using a genome-wide SNP array identified many regions of at least suggestive evidence for linkage, with several candidate and other genes of interest located beneath the linkage peaks, including ADORA2A, ATXN7, CSMD1, GRIK2, GRIN2B, GRIN3A, HTR2A, and SLC6A3 [131].
Figure 4.
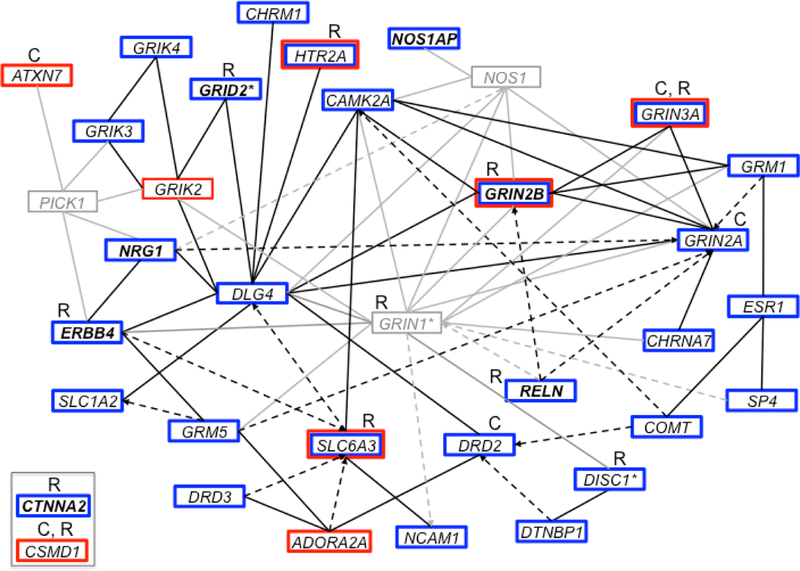
Ingenuity Pathway Analysis of the genes identified through association and linkage analyses of the endophenotypes discussed in this review. Genes are represented as nodes with solid lines representing direct protein-protein interactions and dashed arrows representing more indirect effects on expression, activation, or inhibition. Genes and interactions in black show association (blue box) or linkage (red box) with the endophenotypes listed in this review in the COGS study, whereas those in gray represent other interacting genes. Genes in a bold font are associated with at least two endophenotypes from this review and span neurophysiological and neurocognitive domains. Genes identified as associated with SZ diagnosis by GWAS of common variants are indicated with a “C,” whereas those identified by studies of rare and de novo variation are indicated with an “R.” *GRIN1 showed evidence of both association and linkage with other neurocognitive endophenotypes in the COGS study, and both DISC1 and GRID2 showed evidence of linkage with neurophysiological endophenotypes in the COGS study. Note that CSMD1 and CTNNA2 were prominent genes in these analyses, with external support from studies of SZ diagnosis, but do not directly connect into the defined network. COGS: Consortium on the Genetics of Schizophrenia; GWAS: genome-wide association study; SZ: schizophrenia.
Studies of common, rare, and de novo variation in SZ offer independent support for many of the genes implicated through linkage or association studies of SZ-related endophenotypes. For example, a recent large GWAS of SZ diagnosis conducted by the PGC and a GWAS of neurocognitive endophenotypes for SZ identified associations with ATXN7, CSMD1, DRD2, GRIN2A, GRIN3A, and GRM3 [132,133]. Other studies have shown a significant burden of rare variants in GRIN1, GRIN3A, and SLC1A2 in SZ patients [35,134], as well as a disproportionate disruption of genes in the neuregulin and glutamate pathways [29]. Studies of de novo variation have implicated CSMD1, CTNNA2, DBH, DISC1, GRID2, GRIN2B, HTR2A, RELN, and SLC6A3 in SZ and related disorders [135–143].
As Figure 4 demonstrates, genetic analyses of both SZ-related endophenotypes in family-based samples and large SZ case-control studies implicate glutamatergic neurotransmission and synaptic plasticity pathways in mediating SZ susceptibility, which is consistent with the glutamate hypothesis and an abundance of evidence implicating NRG1-ErbB4 signaling in SZ [144–146]. For a comprehensive list of genes that have been associated with SZ, see the summary of genetic findings compiled by Ren and colleagues [147].
TRANSLATIONAL STUDIES OF SCHIZOPHRENIA-ASSOCIATED DEFICITS
Cross-species investigations are important for the development of novel therapeutic interventions in SZ. For this reason, the endophenotype strategy has been advocated by the National Institute of Mental Health as part of the Research Domain Criteria (RDoC) framework [148]. Several endophenotypes are amenable to translational studies, which use intrinsic or induced deficits in animal models to mimic those observed in humans and thereby provide important insight into the neurobiological processes underlying SZ.
PPI is one example of a SZ-related endophenotype that can be reliably measured across species to understand the biology of brain-based inhibitory mechanisms [149]. PPI deficits in rats can be induced through the administration of dopamine (DA) agonists like apomorphine (APO). In one study of two rat strains displaying varying sensitivity to APO, 104 genes were found to significantly differ in their expression of the nucleus accumbens, which represents a control center for PPI. Many of these genes correlated with PPI deficits and were clustered within the DA receptor-signaling, synaptic long-term potentiation (involving glutamate transmission), and inositol phosphate metabolism pathways, suggesting that these pathways may substantially influence PPI and may therefore represent therapeutic targets [150]. By examining these findings in the context of human genetic studies, we can begin to piece together how various pathways may be involved in each specific endophenotype. For example, of the 12 human genes associated with PPI-related deficits in SZ patients and their families, 8 show significant differences in gene expression related to PPI in rats [128–130,150,151].
Some neurocognitive endophenotypes have also been investigated in animal models. Given the hypothesis that neurocognitive dysfunction in SZ results from impaired stimulus filtering and that this attentional impairment may relate to reduced expression of muscarinic acetylcholine receptors in SZ, some investigators have suggested that mice treated with scopolamine, a muscarinic receptor antagonist, may provide a model of attentional dysfunction in SZ. Using a five-choice version of the CPT (5C-CPT) developed specifically for rodents, the investigators found that mice treated with scopolamine exhibited significantly impaired attention that was characterized by reduced target detection and increased impulsive responding [152]. These findings are comparable to those observed in humans with SZ using a reverse-translated version of the 5C-CPT, which was designed to minimize demands on perceptual, visual learning, processing speed, or working memory functions and which has been validated against the CPT-IP [152]. Given that the FDA has endorsed neurocognitive deficits as a key treatment target, these and other related findings hold promise to facilitate the development of pro-cognitive therapeutics that may improve functioning in SZ patients.
In the future, investigations of the transcriptome, DNA methylation, and histone modification may also have translatable potential. These kinds of studies represent a promising area for SZ research, but such methods have not yet been entirely incorporated into SZ endophenotype studies. It is likely that the integration of these gene expression and epigenetic resources with endophenotype-related animal models may be informative in future studies.
CONCLUSIONS AND FUTURE DIRECTIONS
SZ is a clinically heterogeneous disorder in which patients exhibit a broad range of neurobiological deficits and symptom severity, and this profound heterogeneity has complicated efforts to identify genetic risk variants. The use of SZ-related endophenotypes, rather than diagnosis, in genetic studies offers several advantages that may help address these complications and facilitate the identification of genetic risk variants and aberrant molecular pathways [153–155]. Traditional case-control studies collapse the substantial phenotypic variation in SZ into a single, categorical diagnosis. However, recent studies have shown that phenotypic specificity is actually more important than a large sample size for detecting true genetic associations in genetic studies of complex diseases, including SZ [156,157]. Although the use of large samples remains important in SZ research, endophenotypes can provide the needed phenotypic specificity, as well as increased biological relevance. For a complex disorder like SZ, the genetic signal from studies that use endophenotypes rather than diagnosis should thus be stronger, more direct, and associated with fewer variants, each of larger individual effect. As each endophenotype reflects a discrete neurobiological function or pathway, they also may be useful in parsing the complex etiology of SZ and reducing the associated heterogeneity. Finally, by capitalizing on the inherent power advantages of a quantitative trait, the analysis of an endophenotype requires a substantially smaller sample size, nearly 10-fold, to achieve comparable power to an analysis of SZ diagnosis. Although assessing endophenotypes may incur an additional burden with regard to recruitment and testing, the potential payoff suggests that a deep phenotyping strategy is well worth the effort.
Genetic analyses of endophenotypes can also supplement traditional analyses of SZ diagnosis, providing additional biological insight into these results. One simple way to utilize endophenotypes is to inform existing large-scale GWAS of SZ diagnosis [132]. Using enrichment and polygenic scoring methods, we can assess how much of the common genetic risk is shared between SZ and a particular endophenotype [24,158]. In addition to estimating the overall genetic correlation between a given endophenotype and SZ, we can also investigate whether SNPs that increase the risk for SZ overlap more than we would expect by chance with SNPs that result in deficits in, for example, PPI or neurocognition.
Endophenotypes are also ideally suited to multivariate phenotype methods, which can be used to construct individual phenotypic profiles and to identify subgroups of similar SZ patients [159]. Methods that can simultaneously incorporate both phenotype and genomic information may prove particularly useful in this regard [160]. The identified subgroups can then be correlated with clinical factors, such as illness severity, clinical course, level of functioning, and medication response, to establish individualized treatment protocols. By investigating the relationship of clinical, genetic, and environmental factors to subgroups defined by endophenotypes, which reflect the underlying neurobiological dysfunctions, we may gain a better understanding of Bleuler’s “Group of Schizophrenias.”
The endophenotype strategy thus complements large-scale case-control efforts, providing the necessary biological insight to facilitate precision medicine in SZ. Ultimately, we may be able to distill the number of endophenotypes to a smaller robust subset that accounts for a majority of the variance between cases and controls [161]. Using endophenotypes to dig deeper, going beyond the broad clinical diagnostic category of SZ, will facilitate the identification of pathways leading from genetic variation to brain dysfunction. A better understanding of the genetic substrates underlying deficits in key domains defined by SZ-related endophenotypes will provide a molecular roadmap to guide the development of novel pharmacological interventions aimed at improving real-world functioning, as well as information to guide precision-based treatment decisions.
FUNDING
This material is based upon work supported in part by Merit Review Award I01 BX002241 from the US Department of Veterans Affairs Biomedical Laboratory Research and Development Service at the VA Puget Sound Health Care System. The authors were previously supported by R01 MH065571 and R01 MH065558 from the National Institute of Mental Health.
Footnotes
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
REFERENCES
- 1.Karayiorgou M, Gogos JA. A turning point in schizophrenia genetics. Neuron. 1997. November;19(5):967–79. doi: S0896-6273(00)80390-6 [DOI] [PubMed] [Google Scholar]
- 2.Bleuler E Dementia Praecox or the Group of Schizophrenias. Zinkin J trans. New York (US): International Universities Press; 1911. [Google Scholar]
- 3.Sham PC, MacLean CJ, Kendler KS. A typological model of schizophrenia based on age at onset, sex and familial morbidity. Acta Psychiatr Scand. 1994. February;89(2):135–41. [DOI] [PubMed] [Google Scholar]
- 4.Messias EL, Chen CY, Eaton WW. Epidemiology of schizophrenia: review of findings and myths. Psychiatr Clin North Am. 2007. September;30(3):323–38. doi: 10.1016/j.psc.2007.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jobe TH, Harrow M. Long-term outcome of patients with schizophrenia: a review. Can J Psychiatry. 2005. December;50(14):892–900. doi: 10.1177/070674370505001403 [DOI] [PubMed] [Google Scholar]
- 6.Lopez-Morinigo JD, Ramos-Rios R, David AS, Dutta R. Insight in schizophrenia and risk of suicide: a systematic update. Compr Psychiatry. 2012. May;53(4):313–22. doi: 10.1016/j.comppsych.2011.05.015 [DOI] [PubMed] [Google Scholar]
- 7.Bitter I, Feher L, Tenyi T, Czobor P. Treatment adherence and insight in schizophrenia. Psychiatr Hung. 2015;30(1):18–26. [PubMed] [Google Scholar]
- 8.Tessier A, Boyer L, Husky M, Bayle F, Llorca PM, Misdrahi D. Medication adherence in schizophrenia: The role of insight, therapeutic alliance and perceived trauma associated with psychiatric care. Psychiatry Res. 2017. November;257:315–21. doi: 10.1016/j.psychres.2017.07.063 [DOI] [PubMed] [Google Scholar]
- 9.Jakubovski E, Carlson JP, Bloch MH. Prognostic subgroups for remission, response, and treatment continuation in the Clinical Antipsychotic Trials of Intervention Effectiveness (CATIE) trial. J Clin Psychiatry. 2015. November;76(11):1535–45. doi: 10.4088/JCP.14m09320 [DOI] [PubMed] [Google Scholar]
- 10.Schwab SG, Wildenauer DB. Genetics of psychiatric disorders in the GWAS era: an update on schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2013. November;263(Suppl 2):S147–54. doi: 10.1007/s00406-013-0450-z [DOI] [PubMed] [Google Scholar]
- 11.Sullivan PF, Kendler KS, Neale MC. Schizophrenia as a complex trait: evidence from a meta-analysis of twin studies. Arch Gen Psychiatry. 2003. December;60(12):1187–92. doi: 10.1001/archpsyc.60.12.1187 [DOI] [PubMed] [Google Scholar]
- 12.Lichtenstein P, Yip BH, Bjork C, Pawitan Y, Cannon TD, Sullivan PF, et al. Common genetic determinants of schizophrenia and bipolar disorder in Swedish families: a population-based study. Lancet. 2009. January 17;373(9659):234–9. doi: 10.1016/S0140-6736(09)60072-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gottesman II, Laursen TM, Bertelsen A, Mortensen PB. Severe mental disorders in offspring with 2 psychiatrically ill parents. Arch Gen Psychiatry. 2010. March;67(3):252–7. doi: 10.1001/archgenpsychiatry.2010.1 [DOI] [PubMed] [Google Scholar]
- 14.Walder DJ, Faraone SV, Glatt SJ, Tsuang MT, Seidman LJ. Genetic liability, prenatal health, stress and family environment: risk factors in the Harvard Adolescent Family High Risk for schizophrenia study. Schizophr Res. 2014. August;157(1–3):142–8. doi: 10.1016/j.schres.2014.04.015 [DOI] [PubMed] [Google Scholar]
- 15.Baron M Genetics of schizophrenia and the new millennium: progress and pitfalls. Am J Hum Genet. 2001. February;68(2):299–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lewis CM, Levinson DF, Wise LH, DeLisi LE, Straub RE, Hovatta I, et al. Genome Scan Meta-Analysis of Schizophrenia and Bipolar Disorder, Part II: Schizophrenia. Am J Hum Genet. 2003. July;73(1):34–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Owen MJ, Williams NM, O’Donovan MC. The molecular genetics of schizophrenia: new findings promise new insights. Mol Psychiatry. 2004. January;9(1):14–27. [DOI] [PubMed] [Google Scholar]
- 18.Gogos JA, Gerber DJ. Schizophrenia susceptibility genes: emergence of positional candidates and future directions. Trends Pharmacol Sci. 2006. April;27(4):226–33. [DOI] [PubMed] [Google Scholar]
- 19.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005. January;10(1):40–68; image 5. [DOI] [PubMed] [Google Scholar]
- 20.Schork NJ, Greenwood TA, Braff DL. Statistical genetics concepts and approaches in schizophrenia and related neuropsychiatric research. Schizophr Bull. 2007. January;33(1):95–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008. September;40(9):1053–5. doi: 10.1038/ng.201 [DOI] [PubMed] [Google Scholar]
- 22.Shi J, Levinson DF, Duan J, Sanders AR, Zheng Y, Pe’er I, et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature. 2009. August 6;460(7256):753–7. doi: 10.1038/nature08192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stefansson H, Ophoff RA, Steinberg S, Andreassen OA, Cichon S, Rujescu D, et al. Common variants conferring risk of schizophrenia. Nature. 2009. August 6;460(7256):744–7. doi: 10.1038/nature08186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009. August 6;460(7256):748–52. doi: 10.1038/nature08185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013. October;45(10):1150–9. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Altshuler D, Daly MJ, Lander ES. Genetic mapping in human disease. Science. 2008. November 7;322(5903):881–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009. October 8;461(7265):747–53. doi: 10.1038/nature08494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rutkowski TP, Schroeder JP, Gafford GM, Warren ST, Weinshenker D, Caspary T, et al. Unraveling the genetic architecture of copy number variants associated with schizophrenia and other neuropsychiatric disorders. J Neurosci Res. 2017. May;95(5):1144–60. doi: 10.1002/jnr.23970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, Cooper GM, et al. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008. April 25;320(5875):539–43. doi: 10.1126/science.1155174 [DOI] [PubMed] [Google Scholar]
- 30.Stefansson H, Rujescu D, Cichon S, Pietilainen OP, Ingason A, Steinberg S, et al. Large recurrent microdeletions associated with schizophrenia. Nature. 2008. September 11;455(7210):232–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.International Schizophrenia Consortium. Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008. September 11;455(7210):237–41. doi: 10.1038/nature07239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012. February;17(2):142–53. doi: 10.1038/mp.2011.154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Marshall CR, Howrigan DP, Merico D, Thiruvahindrapuram B, Wu W, Greer DS, et al. Contribution of copy number variants to schizophrenia from a genome-wide study of 41,321 subjects. Nat Genet. 2017. January;49(1):27–35. doi: 10.1038/ng.3725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Need AC, McEvoy JP, Gennarelli M, Heinzen EL, Ge D, Maia JM, et al. Exome sequencing followed by large-scale genotyping suggests a limited role for moderately rare risk factors of strong effect in schizophrenia. Am J Hum Genet. 2012. August 10;91(2):303–12. doi: 10.1016/j.ajhg.2012.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Purcell SM, Moran JL, Fromer M, Ruderfer D, Solovieff N, Roussos P, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014. February 13;506(7487):185–90. doi: 10.1038/nature12975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gulsuner S, Walsh T, Watts AC, Lee MK, Thornton AM, Casadei S, et al. Spatial and temporal mapping of de novo mutations in schizophrenia to a fetal prefrontal cortical network. Cell. 2013. August 1;154(3):518–29. doi: 10.1016/j.cell.2013.06.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang Z, Li L, Li M. De novo mutations as causes of schizophrenia. Psychiatry Res. 2018. December;270:1168–9. doi: 10.1016/j.psychres.2018.05.037. [DOI] [PubMed] [Google Scholar]
- 38.Glatt SJ, Stone WS, Nossova N, Liew CC, Seidman LJ, Tsuang MT. Similarities and differences in peripheral blood gene-expression signatures of individuals with schizophrenia and their first-degree biological relatives. Am J Med Genet B Neuropsychiatr Genet. 2011. December;156B(8):869–87. doi: 10.1002/ajmg.b.31239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Avramopoulos D Recent Advances in the Genetics of Schizophrenia. Mol Neuropsychiatry. 2018. June;4(1):35–51. doi: 10.1159/000488679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Guidotti A, Auta J, Davis JM, Dong E, Gavin DP, Grayson DR, et al. Toward the identification of peripheral epigenetic biomarkers of schizophrenia. J Neurogenet. 2014. Mar-Jun;28(1–2):41–52. doi: 10.3109/01677063.2014.892485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Montano C, Taub MA, Jaffe A, Briem E, Feinberg JI, Trygvadottir R, et al. Association of DNA Methylation Differences With Schizophrenia in an Epigenome-Wide Association Study. JAMA Psychiatry. 2016. May 1;73(5):506–14. doi: 10.1001/jamapsychiatry.2016.0144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Girdhar K, Hoffman GE, Jiang Y, Brown L, Kundakovic M, Hauberg ME, et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat Neurosci. 2018. August;21(8):1126–36. doi: 10.1038/s41593-018-0187-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Javidfar B, Park R, Kassim BS, Bicks LK, Akbarian S. The epigenomics of schizophrenia, in the mouse. Am J Med Genet B Neuropsychiatr Genet. 2017. September;174(6):631–40. doi: 10.1002/ajmg.b.32566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gottesman I, Shields J. Schizophrenia and Genetics: A Twin Study Vantage Point. New York(US): Academic Press; 1972. [Google Scholar]
- 45.Braff DL, Freedman R. The importance of endophenotypes in studies of the genetics of schizophrenia In: Davis KL, Charney D, Coyle JT, Nemeroff C, editors. Neuropsychopharmacology: The Fifth Generation of Progress. Baltimore(US): Lippincott, Williams & Wilkins; 2002. p. 703–16. [Google Scholar]
- 46.Braff DL, Greenwood TA, Swerdlow NR, Light GA, Schork NJ, Investigators of the Consortium on the Genetics of S. Advances in endophenotyping schizophrenia. World Psychiatry. 2008. February;7(1):11–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cannon TD, Keller MC. Endophenotypes in the genetic analyses of mental disorders. Annu Rev Clin Psychol. 2006;2:267–90. doi: 10.1146/annurev.clinpsy.2.022305.095232 [DOI] [PubMed] [Google Scholar]
- 48.Thaker GK. Schizophrenia endophenotypes as treatment targets. Expert Opin Ther Targets. 2007. September;11(9):1189–206. doi: 10.1517/14728222.11.9.1189. [DOI] [PubMed] [Google Scholar]
- 49.Wadhera RK, Steen DL, Khan I, Giugliano RP, Foody JM. A review of low-density lipoprotein cholesterol, treatment strategies, and its impact on cardiovascular disease morbidity and mortality. J Clin Lipidol. 2016. May-Jun;10(3):472–89. doi: 10.1016/j.jacl.2015.11.010 [DOI] [PubMed] [Google Scholar]
- 50.Gottesman II, Shields J. A polygenic theory of schizophrenia. Proc Natl Acad Sci U S A. 1967. July;58(1):199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Neale BM, Sklar P. Genetic analysis of schizophrenia and bipolar disorder reveals polygenicity but also suggests new directions for molecular interrogation. Curr Opin Neurobiol. 2015. February;30:131–8. doi: 10.1016/j.conb.2014.12.001 [DOI] [PubMed] [Google Scholar]
- 52.Gould TD. Gottesman Irving I. (1930-2016): the multifactorial threshold model of complex phenotypes mediated by endophenotype strategies. Genes Brain Behav. 2016. November;15(8):775–6. doi: 10.1111/gbb.12345 [DOI] [PubMed] [Google Scholar]
- 53.Blangero J, Williams JT, Almasy L. Novel family-based approaches to genetic risk in thrombosis. J Thromb Haemost. 2003. July;1(7):1391–7. [DOI] [PubMed] [Google Scholar]
- 54.Lee SH, Wray NR. Novel genetic analysis for case-control genome-wide association studies: quantification of power and genomic prediction accuracy. PLoS One. 2013;8(8):e71494. doi: 10.1371/journal.pone.0071494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abecasis GR, Cookson WO, Cardon LR. The power to detect linkage disequilibrium with quantitative traits in selected samples. Am J Hum Genet. 2001. June;68(6):1463–74. doi: 10.1086/320590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Saykin AJ, Gur RC, Gur RE, Mozley PD, Mozley LH, Resnick SM, et al. Neuropsychological function in schizophrenia. Selective impairment in memory and learning. Arch Gen Psychiatry. 1991. July;48(7):618–24. [DOI] [PubMed] [Google Scholar]
- 57.Green MF, Kern RS, Braff DL, Mintz J. Neurocognitive deficits and functional outcome in schizophrenia: are we measuring the “right stuff”? Schizophr Bull. 2000;26(1):119–36. [DOI] [PubMed] [Google Scholar]
- 58.Elvevag B, Goldberg TE. Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol. 2000;14(1):1–21. [PubMed] [Google Scholar]
- 59.Nuechterlein KH, Green MF, Kern RS, Baade LE, Barch DM, Cohen JD, et al. The MATRICS Consensus Cognitive Battery, part 1: test selection, reliability, and validity. Am J Psychiatry. 2008. February;165(2):203–13. doi: 10.1176/appi.ajp.2007.07010042 [DOI] [PubMed] [Google Scholar]
- 60.Kern RS, Nuechterlein KH, Green MF, Baade LE, Fenton WS, Gold JM, et al. The MATRICS Consensus Cognitive Battery, part 2: co-norming and standardization. Am J Psychiatry. 2008. February;165(2):214–20. doi: 10.1176/appi.ajp.2007.07010043 [DOI] [PubMed] [Google Scholar]
- 61.Seidman LJ, Shapiro DI, Stone WS, Woodberry KA, Ronzio A, Cornblatt BA, et al. Association of Neurocognition With Transition to Psychosis: Baseline Functioning in the Second Phase of the North American Prodrome Longitudinal Study. JAMA Psychiatry. 2016. December 1;73(12):1239–48. doi: 10.1001/jamapsychiatry.2016.2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reichenberg A, Mollon J. Challenges and Opportunities in Studies of Cognition in the Prodrome to Psychosis: No Detail Is Too Small. JAMA Psychiatry. 2016. December 1;73(12):1249–50. doi: 10.1001/jamapsychiatry.2016.2655 [DOI] [PubMed] [Google Scholar]
- 63.Braff DL, Light GA. The use of neurophysiological endophenotypes to understand the genetic basis of schizophrenia. Dialogues Clin Neurosci. 2005;7(2):125–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Light GA, Swerdlow NR, Rissling AJ, Radant A, Sugar CA, Sprock J, et al. Characterization of neurophysiologic and neurocognitive biomarkers for use in genomic and clinical outcome studies of schizophrenia. PLoS One. 2012;7(7):e39434. doi: 10.1371/journal.pone.0039434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gur RE, Calkins ME, Gur RC, Horan WP, Nuechterlein KH, Seidman LJ, et al. The Consortium on the Genetics of Schizophrenia: neurocognitive endophenotypes. Schizophr Bull. 2007. January;33(1):49–68. doi: 10.1093/schbul/sbl055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Turetsky BI, Calkins ME, Light GA, Olincy A, Radant AD, Swerdlow NR. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr Bull. 2007. January;33(1):69–94. doi: 10.1093/schbul/sbl060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Owens EM, Bachman P, Glahn DC, Bearden CE. Electrophysiological Endophenotypes for Schizophrenia. Harv Rev Psychiatry. 2016. Mar-Apr;24(2):129–47. doi: 10.1097/HRP.0000000000000110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ivleva EI, Clementz BA, Dutcher AM, Arnold SJM, Jeon-Slaughter H, Aslan S, et al. Brain Structure Biomarkers in the Psychosis Biotypes: Findings From the Bipolar-Schizophrenia Network for Intermediate Phenotypes. Biol Psychiatry. 2017. July 1;82(1):26–39. doi: 10.1016/j.biopsych.2016.08.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cao H, Dixson L, Meyer-Lindenberg A, Tost H. Functional connectivity measures as schizophrenia intermediate phenotypes: advances, limitations, and future directions. Curr Opin Neurobiol. 2016. February;36:7–14. doi: 10.1016/j.conb.2015.07.008 [DOI] [PubMed] [Google Scholar]
- 70.Swerdlow NR, Light GA, Sprock J, Calkins ME, Green MF, Greenwood TA, et al. Deficient prepulse inhibition in schizophrenia detected by the multi-site COGS. Schizophr Res. 2014. February;152(2–3):503–12. doi: 10.1016/j.schres.2013.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Swerdlow NR, Light GA, Thomas ML, Sprock J, Calkins ME, Green MF, et al. Deficient prepulse inhibition in schizophrenia in a multi-site cohort: Internal replication and extension. Schizophr Res. 2017. May 23. doi: 10.1016/j.schres.2017.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kumari V, Soni W, Sharma T. Normalization of information processing deficits in schizophrenia with clozapine. Am J Psychiatry. 1999. July;156(7):1046–51. [DOI] [PubMed] [Google Scholar]
- 73.Weike AI, Bauer U, Hamm AO. Effective neuroleptic medication removes prepulse inhibition deficits in schizophrenia patients. Biol Psychiatry. 2000. January 1;47(1):61–70. [DOI] [PubMed] [Google Scholar]
- 74.Greenwood TA, Braff DL, Light GA, Cadenhead KS, Calkins ME, Dobie DJ, et al. Initial heritability analyses of endophenotypic measures for schizophrenia: the consortium on the genetics of schizophrenia. Arch Gen Psychiatry. 2007. November;64(11):1242–50. doi: 10.1001/archpsyc.64.11.1242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hasenkamp W, Epstein MP, Green A, Wilcox L, Boshoven W, Lewison B, et al. Heritability of acoustic startle magnitude, prepulse inhibition, and startle latency in schizophrenia and control families. Psychiatry Res. 2010. July 30;178(2):236–43. doi: 10.1016/j.psychres.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Aukes MF, Alizadeh BZ, Sitskoorn MM, Selten JP, Sinke RJ, Kemner C, et al. Finding suitable phenotypes for genetic studies of schizophrenia: heritability and segregation analysis. Biol Psychiatry. 2008. July 15;64(2):128–36. [DOI] [PubMed] [Google Scholar]
- 77.Greenwood TA, Light GA, Swerdlow NR, Calkins ME, Green MF, Gur RE, et al. Gating Deficit Heritability and Correlation With Increased Clinical Severity in Schizophrenia Patients With Positive Family History. Am J Psychiatry. 2016. April 1;173(4):385–91. doi: 10.1176/appi.ajp.2015.15050605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Naatanen R, Paavilainen P, Tiitinen H, Jiang D, Alho K. Attention and mismatch negativity. Psychophysiology. 1993. September;30(5):436–50. [DOI] [PubMed] [Google Scholar]
- 79.Naatanen R, Paavilainen P, Rinne T, Alho K. The mismatch negativity (MMN) in basic research of central auditory processing: a review. Clin Neurophysiol. 2007. December;118(12):2544–90. doi: 10.1016/j.clinph.2007.04.026 [DOI] [PubMed] [Google Scholar]
- 80.Lawrie SM, Olabi B, Hall J, McIntosh AM. Do we have any solid evidence of clinical utility about the pathophysiology of schizophrenia? World Psychiatry. 2011. February;10(1):19–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Umbricht D, Krljes S. Mismatch negativity in schizophrenia: a meta-analysis. Schizophr Res. 2005. July 1;76(1):1–23. doi: 10.1016/j.schres.2004.12.002 [DOI] [PubMed] [Google Scholar]
- 82.Shinozaki N, Yabe H, Sato Y, Hiruma T, Sutoh T, Nashida T, et al. The difference in Mismatch negativity between the acute and post-acute phase of schizophrenia. Biol Psychol. 2002. March;59(2):105–19. [DOI] [PubMed] [Google Scholar]
- 83.Korostenskaja M, Dapsys K, Siurkute A, Maciulis V, Ruksenas O, Kahkonen S. Effects of olanzapine on auditory P300 and mismatch negativity (MMN) in schizophrenia spectrum disorders. Prog Neuropsychopharmacol Biol Psychiatry. 2005. May;29(4):543–8. doi: 10.1016/j.pnpbp.2005.01.019 [DOI] [PubMed] [Google Scholar]
- 84.Light GA, Swerdlow NR, Thomas ML, Calkins ME, Green MF, Greenwood TA, et al. Validation of mismatch negativity and P3a for use in multi-site studies of schizophrenia: Characterization of demographic, clinical, cognitive, and functional correlates in COGS-2. Schizophr Res. 2015. April;163(1–3):63–72. doi: 10.1016/j.schres.2014.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hall MH, Schulze K, Rijsdijk F, Picchioni M, Ettinger U, Bramon E, et al. Heritability and reliability of P300, P50 and duration mismatch negativity. Behav Genet. 2006. November;36(6):845–57. doi: 10.1007/s10519-006-9091-6 [DOI] [PubMed] [Google Scholar]
- 86.Jessen F, Fries T, Kucharski C, Nishimura T, Hoenig K, Maier W, et al. Amplitude reduction of the mismatch negativity in first-degree relatives of patients with schizophrenia. Neurosci Lett. 2001. August 31;309(3):185–8. [DOI] [PubMed] [Google Scholar]
- 87.Price GW, Michie PT, Johnston J, Innes-Brown H, Kent A, Clissa P, et al. A multivariate electrophysiological endophenotype, from a unitary cohort, shows greater research utility than any single feature in the Western Australian family study of schizophrenia. Biol Psychiatry. 2006. July 1;60(1):1–10. doi: 10.1016/j.biopsych.2005.09.010 [DOI] [PubMed] [Google Scholar]
- 88.Schreiber H, Stolz-Born G, Kornhuber HH, Born J. Event-related potential correlates of impaired selective attention in children at high risk for schizophrenia. Biol Psychiatry. 1992. October 15;32(8):634–51. [DOI] [PubMed] [Google Scholar]
- 89.Michie PT, Innes-Brown H, Todd J, Jablensky AV. Duration mismatch negativity in biological relatives of patients with schizophrenia spectrum disorders. Biol Psychiatry. 2002. October 1;52(7):749–58. [DOI] [PubMed] [Google Scholar]
- 90.Atkinson RJ, Michie PT, Schall U. Duration mismatch negativity and P3a in first-episode psychosis and individuals at ultra-high risk of psychosis. Biol Psychiatry. 2012. January 15;71(2):98–104. doi: 10.1016/j.biopsych.2011.08.023 [DOI] [PubMed] [Google Scholar]
- 91.Light GA, Swerdlow NR, Braff DL. Preattentive sensory processing as indexed by the MMN and P3a brain responses is associated with cognitive and psychosocial functioning in healthy adults. J Cogn Neurosci. 2007. October;19(10):1624–32. doi: 10.1162/jocn.2007.19.10.1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bodatsch M, Ruhrmann S, Wagner M, Muller R, Schultze-Lutter F, Frommann I, et al. Prediction of psychosis by mismatch negativity. Biol Psychiatry. 2011. May 15;69(10):959–66. doi: 10.1016/j.biopsych.2010.09.057 [DOI] [PubMed] [Google Scholar]
- 93.Light GA, Naatanen R. Mismatch negativity is a breakthrough biomarker for understanding and treating psychotic disorders. Proc Natl Acad Sci U S A. 2013. September 17;110(38):15175–6. doi: 10.1073/pnas.1313287110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Perez VB, Woods SW, Roach BJ, Ford JM, McGlashan TH, Srihari VH, et al. Automatic auditory processing deficits in schizophrenia and clinical high-risk patients: forecasting psychosis risk with mismatch negativity. Biol Psychiatry. 2014. March 15;75(6):459–69. doi: 10.1016/j.biopsych.2013.07.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nagai T, Tada M, Kirihara K, Araki T, Jinde S, Kasai K. Mismatch negativity as a “translatable” brain marker toward early intervention for psychosis: a review. Front Psychiatry. 2013. September 23;4:115. doi: 10.3389/fpsyt.2013.00115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Thomas ML, Green MF, Hellemann G, Sugar CA, Tarasenko M, Calkins ME, et al. Modeling Deficits From Early Auditory Information Processing to Psychosocial Functioning in Schizophrenia. JAMA Psychiatry. 2017. January 1;74(1):37–46. doi: 10.1001/jamapsychiatry.2016.2980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nieman D, Becker H, van de Fliert R, Plat N, Bour L, Koelman H, et al. Antisaccade task performance in patients at ultra high risk for developing psychosis. Schizophr Res. 2007. September;95(1–3):54–60. doi: 10.1016/j.schres.2007.06.022 [DOI] [PubMed] [Google Scholar]
- 98.Radant AD, Dobie DJ, Calkins ME, Olincy A, Braff DL, Cadenhead KS, et al. Antisaccade performance in schizophrenia patients, their first-degree biological relatives, and community comparison subjects: data from the COGS study. Psychophysiology. 2010. September;47(5):846–56. doi: 10.1111/j.1469-8986.2010.01004.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Radant AD, Millard SP, Braff DL, Calkins ME, Dobie DJ, Freedman R, et al. Robust differences in antisaccade performance exist between COGS schizophrenia cases and controls regardless of recruitment strategies. Schizophr Res. 2015. April;163(1–3):47–52. doi: 10.1016/j.schres.2014.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Cutsuridis V, Kumari V, Ettinger U. Antisaccade performance in schizophrenia: a neural model of decision making in the superior colliculus. Front Neurosci. 2014;8:13. doi: 10.3389/fnins.2014.00013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Garcia-Blanco AC, Perea M, Salmeron L. Attention orienting and inhibitory control across the different mood states in bipolar disorder: an emotional antisaccade task. Biol Psychol. 2013. December;94(3):556–61. doi: 10.1016/j.biopsycho.2013.10.005 [DOI] [PubMed] [Google Scholar]
- 102.Malone SM, Iacono WG. Error rate on the antisaccade task: heritability and developmental change in performance among preadolescent and late-adolescent female twin youth. Psychophysiology. 2002. September;39(5):664–73. [PubMed] [Google Scholar]
- 103.Levy DL, O’Driscoll G, Matthysse S, Cook SR, Holzman PS, Mendell NR. Antisaccade performance in biological relatives of schizophrenia patients: a meta-analysis. Schizophr Res. 2004. November 1;71(1):113–25. doi: 10.1016/j.schres.2003.11.006 [DOI] [PubMed] [Google Scholar]
- 104.Calkins ME, Iacono WG, Curtis CE. Smooth pursuit and antisaccade performance evidence trait stability in schizophrenia patients and their relatives. Int J Psychophysiol. 2003. August;49(2):139–46. [DOI] [PubMed] [Google Scholar]
- 105.Baddeley A Working memory. Science. 1992. January 31;255(5044):556–9. [DOI] [PubMed] [Google Scholar]
- 106.Lee J, Park S. Working memory impairments in schizophrenia: a meta-analysis. J Abnorm Psychol. 2005. November;114(4):599–611. doi: 10.1037/0021-843X.114.4.599 [DOI] [PubMed] [Google Scholar]
- 107.Glahn DC, Therman S, Manninen M, Huttunen M, Kaprio J, Lonnqvist J, et al. Spatial working memory as an endophenotype for schizophrenia. Biol Psychiatry. 2003. April 1;53(7):624–6. [DOI] [PubMed] [Google Scholar]
- 108.Goldman-Rakic PS. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 1994. Fall;6(4):348–57. doi: 10.1176/jnp.6.4.348 [DOI] [PubMed] [Google Scholar]
- 109.Wechsler D Wechsler Memory Scale. 3rd ed. San Antonio(US): Psychological Corporation; 1997. [Google Scholar]
- 110.Seidman LJ, Hellemann G, Nuechterlein KH, Greenwood TA, Braff DL, Cadenhead KS, et al. Factor structure and heritability of endophenotypes in schizophrenia: findings from the Consortium on the Genetics of Schizophrenia (COGS-1). Schizophr Res. 2015. April;163(1–3):73–9. doi: 10.1016/j.schres.2015.01.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Nuechterlein KH, Parasuraman R, Jiang Q. Visual sustained attention: image degradation produces rapid sensitivity decrement over time. Science. 1983. April 15;220(4594):327–9. [DOI] [PubMed] [Google Scholar]
- 112.Nuechterlein KH. Signal detection in vigilance tasks and behavioral attributes among offspring of schizophrenic mothers and among hyperactive children. J Abnorm Psychol. 1983. February;92(1):4–28. [DOI] [PubMed] [Google Scholar]
- 113.Cornblatt BA, Risch NJ, Faris G, Friedman D, Erlenmeyer-Kimling L. The Continuous Performance Test, identical pairs version (CPT-IP): I. New findings about sustained attention in normal families. Psychiatry Res. 1988. November;26(2):223–38. [DOI] [PubMed] [Google Scholar]
- 114.Cornblatt BA, Lenzenweger MF, Erlenmeyer-Kimling L. The continuous performance test, identical pairs version: II. Contrasting attentional profiles in schizophrenic and depressed patients. Psychiatry Res. 1989. July;29(1):65–85. [DOI] [PubMed] [Google Scholar]
- 115.Heinrichs RW, Zakzanis KK. Neurocognitive deficit in schizophrenia: a quantitative review of the evidence. Neuropsychology. 1998. July;12(3):426–45. [DOI] [PubMed] [Google Scholar]
- 116.Finkelstein JR, Cannon TD, Gur RE, Gur RC, Moberg P. Attentional dysfunctions in neuroleptic-naive and neuroleptic-withdrawn schizophrenic patients and their siblings. J Abnorm Psychol. 1997. May;106(2):203–12. [DOI] [PubMed] [Google Scholar]
- 117.Wohlberg GW, Kornetsky C. Sustained attention in remitted schizophrenics. Arch Gen Psychiatry. 1973. April;28(4):533–7. [DOI] [PubMed] [Google Scholar]
- 118.Nuechterlein K, Dawson M, Ventura J, Yee-Bradbury C. Longitudinal stability of vigilance and span of apprehension deficits in the early phase of schizophrenia. In Sixth Annual Meeting of the Society for Research in Psychopathology; 1991 Dec; Cambridge, USA. [Google Scholar]
- 119.Chen WJ, Liu SK, Chang CJ, Lien YJ, Chang YH, Hwu HG. Sustained attention deficit and schizotypal personality features in nonpsychotic relatives of schizophrenic patients. Am J Psychiatry. 1998. September;155(9):1214–20. doi: 10.1176/ajp.155.9.1214. [DOI] [PubMed] [Google Scholar]
- 120.Grove WM, Lebow BS, Clementz BA, Cerri A, Medus C, Iacono WG. Familial prevalence and coaggregation of schizotypy indicators: a multitrait family study. J Abnorm Psychol. 1991. May;100(2):115–21. [DOI] [PubMed] [Google Scholar]
- 121.Roussos P, Giakoumaki SG, Zouraraki C, Fullard JF, Karagiorga VE, Tsapakis EM, et al. The Relationship of Common Risk Variants and Polygenic Risk for Schizophrenia to Sensorimotor Gating. Biol Psychiatry. 2016. June 15;79(12):988–96. doi: 10.1016/j.biopsych.2015.06.019 [DOI] [PubMed] [Google Scholar]
- 122.Qin XY, Wu HT, Cao C, Loh YP, Cheng Y. A meta-analysis of peripheral blood nerve growth factor levels in patients with schizophrenia. Mol Psychiatry. 2017. September;22(9):1306–12. doi: 10.1038/mp.2016.235 [DOI] [PubMed] [Google Scholar]
- 123.Quednow BB, Ejebe K, Wagner M, Giakoumaki SG, Bitsios P, Kumari V, et al. Meta-analysis on the association between genetic polymorphisms and prepulse inhibition of the acoustic startle response. Schizophr Res. 2018. August;198:52–9. doi: 10.1016/j.schres.2017.12.011 [DOI] [PubMed] [Google Scholar]
- 124.Lencer R, Mills LJ, Alliey-Rodriguez N, Shafee R, Lee AM, Reilly JL, et al. Genome-wide association studies of smooth pursuit and antisaccade eye movements in psychotic disorders: findings from the B-SNIP study. Transl Psychiatry. 2017. October 24;7(10):e1249. doi: 10.1038/tp.2017.210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hagenaars SP, Harris SE, Davies G, Hill WD, Liewald DC, Ritchie SJ, et al. Shared genetic aetiology between cognitive functions and physical and mental health in UK Biobank (N = 112 151) and 24 GWAS consortia. Mol Psychiatry. 2016. November;21(11):1624–32. doi: 10.1038/mp.2015.225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Hubbard L, Tansey KE, Rai D, Jones P, Ripke S, Chambert KD, et al. Evidence of common genetic overlap between schizophrenia and cognition. Schizophr Bull. 2016. May;42(3):832–42. doi: 10.1093/schbul/sbv168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Trampush JW, Yang ML, Yu J, Knowles E, Davies G, Liewald DC, et al. GWAS meta-analysis reveals novel loci and genetic correlates for general cognitive function: a report from the COGENT consortium. Mol Psychiatry. 2017. March;22(3):336–45. doi: 10.1038/mp.2016.244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Greenwood TA, Lazzeroni LC, Murray SS, Cadenhead KS, Calkins ME, Dobie DJ, et al. Analysis of 94 candidate genes and 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. Am J Psychiatry. 2011. September;168(9):930–46. doi: 10.1176/appi.ajp.2011.10050723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Greenwood TA, Light GA, Swerdlow NR, Radant AD, Braff DL. Association analysis of 94 candidate genes and schizophrenia-related endophenotypes. PLoS One. 2012;7(1):e29630. doi: 10.1371/journal.pone.0029630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Greenwood TA, Lazzeroni LC, Calkins ME, Freedman R, Green MF, Gur RE, et al. Genetic assessment of additional endophenotypes from the Consortium on the Genetics of Schizophrenia Family Study. Schizophr Res. 2016. January;170(1):30–40. doi: 10.1016/j.schres.2015.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Greenwood TA, Swerdlow NR, Gur RE, Cadenhead KS, Calkins ME, Dobie DJ, et al. Genome-wide linkage analyses of 12 endophenotypes for schizophrenia from the Consortium on the Genetics of Schizophrenia. Am J Psychiatry. 2013. May;170(5):521–32. doi: 10.1176/appi.ajp.2012.12020186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7. doi: 10.1038/nature13595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ohi K, Hashimoto R, Ikeda M, Yamamori H, Yasuda Y, Fujimoto M, et al. Glutamate Networks Implicate Cognitive Impairments in Schizophrenia: Genome-Wide Association Studies of 52 Cognitive Phenotypes. Schizophr Bull. 2015. July;41(4):909–18. doi: 10.1093/schbul/sbu171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fiorentino A, Sharp SI, McQuillin A. Association of rare variation in the glutamate receptor gene SLC1A2 with susceptibility to bipolar disorder and schizophrenia. Eur J Hum Genet. 2015. September;23(9):1200–6. doi: 10.1038/ejhg.2014.261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fromer M, Pocklington AJ, Kavanagh DH, Williams HJ, Dwyer S, Gormley P, et al. De novo mutations in schizophrenia implicate synaptic networks. Nature. 2014. February 13;506(7487):179–84. doi: 10.1038/nature12929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Iossifov I, Ronemus M, Levy D, Wang Z, Hakker I, Rosenbaum J, et al. De novo gene disruptions in children on the autistic spectrum. Neuron. 2012. April 26;74(2):285–99. doi: 10.1016/j.neuron.2012.04.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Neale BM, Kou Y, Liu L, Ma’ayan A, Samocha KE, Sabo A, et al. Patterns and rates of exonic de novo mutations in autism spectrum disorders. Nature. 2012. May 10;485(7397):242–5. doi: 10.1038/nature11011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.O’Roak BJ, Vives L, Girirajan S, Karakoc E, Krumm N, Coe BP, et al. Sporadic autism exomes reveal a highly interconnected protein network of de novo mutations. Nature. 2012. May 10;485(7397):246–50. doi: 10.1038/nature10989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Rauch A, Wieczorek D, Graf E, Wieland T, Endele S, Schwarzmayr T, et al. Range of genetic mutations associated with severe non-syndromic sporadic intellectual disability: an exome sequencing study. Lancet. 2012. November 10;380(9854):1674–82. doi: 10.1016/S0140-6736(12)61480-9 [DOI] [PubMed] [Google Scholar]
- 140.Sanders SJ, Murtha MT, Gupta AR, Murdoch JD, Raubeson MJ, Willsey AJ, et al. De novo mutations revealed by whole-exome sequencing are strongly associated with autism. Nature. 2012. May 10;485(7397):237–41. doi: 10.1038/nature10945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.de Ligt J, Willemsen MH, van Bon BW, Kleefstra T, Yntema HG, Kroes T, et al. Diagnostic exome sequencing in persons with severe intellectual disability. N Engl J Med. 2012. November 15;367(20):1921–9. doi: 10.1056/NEJMoa1206524 [DOI] [PubMed] [Google Scholar]
- 142.Li JM, Lu CL, Cheng MC, Luu SU, Hsu SH, Hu TM, et al. Role of the DLGAP2 gene encoding the SAP90/PSD-95-associated protein 2 in schizophrenia. PLoS One. 2014;9(1):e85373. doi: 10.1371/journal.pone.0085373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Guilmatre A, Dubourg C, Mosca AL, Legallic S, Goldenberg A, Drouin-Garraud V, et al. Recurrent rearrangements in synaptic and neurodevelopmental genes and shared biologic pathways in schizophrenia, autism, and mental retardation. Arch Gen Psychiatry. 2009. September;66(9):947–56. doi: 10.1001/archgenpsychiatry.2009.80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006. Jul-Aug;26(4–6):365–84. doi: 10.1007/s10571-006-9062-8 [DOI] [PubMed] [Google Scholar]
- 145.Sodhi M, Wood KH, Meador-Woodruff J. Role of glutamate in schizophrenia: integrating excitatory avenues of research. Expert Rev Neurother. 2008. September;8(9):1389–406. doi: 10.1586/14737175.8.9.1389 [DOI] [PubMed] [Google Scholar]
- 146.Buonanno A The neuregulin signaling pathway and schizophrenia: from genes to synapses and neural circuits. Brain Res Bull. 2010. September 30;83(3–4):122–31. doi: 10.1016/j.brainresbull.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ren H, Li M, Wang Q, Li T. Identification of risk-conferring genes of schizophrenia using endophenotypes. Neuropsychiatry. 2017;7(5):717–26. [Google Scholar]
- 148.Insel T, Cuthbert B, Garvey M, Heinssen R, Pine DS, Quinn K, et al. Research domain criteria (RDoC): toward a new classification framework for research on mental disorders. Am J Psychiatry. 2010. July;167(7):748–51. doi: 10.1176/appi.ajp.2010.09091379 [DOI] [PubMed] [Google Scholar]
- 149.Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology (Berl). 2008. August;199(3):331–88. doi: 10.1007/s00213-008-1072-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Shilling PD, Saint Marie RL, Shoemaker JM, Swerdlow NR. Strain differences in the gating-disruptive effects of apomorphine: relationship to gene expression in nucleus accumbens signaling pathways. Biol Psychiatry. 2008. April 15;63(8):748–58. doi: 10.1016/j.biopsych.2007.10.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Shilling P, Greenwood T, Braff D, Light G, Cadenhead K, Sprock J. Prioritizing candidate genes for schizophrenia: In Convergence of association and gene expression analyses. World Congress on Psychiatric Genetics; 2009 Nov 5–8; San Diego, CA (US). [Google Scholar]
- 152.Young JW, Geyer MA, Rissling AJ, Sharp RF, Eyler LT, Asgaard GL, et al. Reverse translation of the rodent 5C-CPT reveals that the impaired attention of people with schizophrenia is similar to scopolamine-induced deficits in mice. Transl Psychiatry. 2013;3:e324. doi: 10.1038/tp.2013.82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003. April;160(4):636–45. [DOI] [PubMed] [Google Scholar]
- 154.Braff D, Schork NJ, Gottesman, II. Endophenotyping schizophrenia. Am J Psychiatry. 2007. May;164(5):705–7. [DOI] [PubMed] [Google Scholar]
- 155.Insel TR, Cuthbert BN. Endophenotypes: bridging genomic complexity and disorder heterogeneity. Biol Psychiatry. 2009. December 1;66(11):988–9. doi: 10.1016/j.biopsych.2009.10.008 [DOI] [PubMed] [Google Scholar]
- 156.Liang SG, Greenwood TA. The impact of clinical heterogeneity in schizophrenia on genomic analyses. Schizophr Res. 2015. February;161(2–3):490–5. doi: 10.1016/j.schres.2014.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Manchia M, Cullis J, Turecki G, Rouleau GA, Uher R, Alda M. The impact of phenotypic and genetic heterogeneity on results of genome wide association studies of complex diseases. PLoS One. 2013;8(10):e76295. doi: 10.1371/journal.pone.0076295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hart AB, Gamazon ER, Engelhardt BE, Sklar P, Kahler AK, Hultman CM, et al. Genetic variation associated with euphorigenic effects of d-amphetamine is associated with diminished risk for schizophrenia and attention deficit hyperactivity disorder. Proc Natl Acad Sci U S A. 2014. April 22;111(16):5968–73. doi: 10.1073/pnas.1318810111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Clementz BA, Sweeney JA, Hamm JP, Ivleva EI, Ethridge LE, Pearlson GD, et al. Identification of Distinct Psychosis Biotypes Using Brain-Based Biomarkers. Am J Psychiatry. 2016. April 1;173(4):373–84. doi: 10.1176/appi.ajp.2015.14091200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Arnedo J, Svrakic DM, Del Val C, Romero-Zaliz R, Hernandez-Cuervo H, Molecular Genetics of Schizophrenia C, et al. Uncovering the hidden risk architecture of the schizophrenias: confirmation in three independent genome-wide association studies. Am J Psychiatry. 2015. February 1;172(2):139–53. doi: 10.1176/appi.ajp.2014.14040435 [DOI] [PubMed] [Google Scholar]
- 161.Millard SP, Shofer J, Braff D, Calkins M, Cadenhead K, Freedman R, et al. Prioritizing schizophrenia endophenotypes for future genetic studies: An example using data from the COGS-1 family study. Schizophr Res. 2016. July;174(1–3):1–9. doi: 10.1016/j.schres.2016.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]