

Abstract
Developing in vitro cell culture models that accurately mimic in vivo processes in a manner that also enables near real-time analysis of neurotransmitters is an important research area. New technologies being developed such as 3D scaffolds for cell culture and 3D printed microfluidics provide an opportunity for such advancements. In this work, PC12 cells were used as a model system and they were immobilized onto a 3D scaffold of polystyrene (PS) fibers. These fibers were created by electrospinning onto PS sheets, which were laser cut and, after cell seeding, inserted into a 3D printed microfluidic device. The 3D printed device was designed with threads for connecting commercial fittings (to integrate automated pumps and a 4-port injection system) and a steel pin for simple coupling with PDMS/polystyrene analytical devices. A straight PDMS channel was used for simple (and continuous) flow-based detection by sealing onto a PS base containing an embedded gold array working electrode and a platinum pseudo-reference. Electrochemical detection of stimulated catecholamine release was demonstrated. The insert-based system was then integrated with a bilayer valving PDMS device (for microchip electrophoresis) sealed onto a PS base (with electrodes for electrochemical detection). This base was embedded with a Pd decoupler (for grounding the separation voltage and adsorbing hydrogen) and a 33 µm carbon fiber working electrode for in-channel detection. PC12 cells were stimulated in the 3D cell culture device, and the valving/electrophoresis microchip was able to separate and detect dopamine and norepinephrine release. This work demonstrates the ability to integrate 3D cell scaffolds with microchip-based analysis for detection of multiple analytes released from cells.
Introduction
Microchip devices have been used for the study of various biological systems for several decades.1–4 Fabrication methods such as lithography5 and chemical etching6 can prove to be impractical for commercial applications or for easily transferring technology to other labs. Use of 3D printing technologies is becoming popular for both public interest reasons and for those doing microfluidics.7–11 The development of commercially available 3D printed plastics and metals has helped bring the cost of 3D printing down to a price point that has made it useful for microfluidic researchers. Examples of this work includes work done by Chen et. al. which used 3D printing for the design of a flow device using membrane inserts to analyze components in stored blood.12 Other work used the ability of printers to print flexible materials that allowed them to design a 3D printed O-ring for an ultrafiltration system for the measurement of binding constants.13 3D printers have been used to fabricate many other microfluidic tools including valves,14,15 immunoassay devices,16 and bioreactors17. The build resolution is approaching the limits of fully realized 3D printed microfluidic devices such as electrophoresis chips.18
Another active research area in microfluidics is on-chip cell culture. The majority of cell culture in research today is done using 2-dimensional (2D) cell culture modes.19,20 In vitro cell culture eliminates the obstacles that come with performing experiments on live specimens. However, 2D culture does not accurately mimic the environment experienced in the body. Three-dimensional (3D) culture modes are now used by many researchers to overcome this with substrates such as hydrogels being commonly used.21 Work done by Derda et. al used gels in a paper scaffold to analyze tumor cell migration.22 Lamichhane et. al. analyzed the effects of using polytetrafluoroethylene (PTFE) fiber implants with a static culture method, showing reduced adhesion, activation, and foreign giant body cell formation of macrophages compared to other culture methods.23 Montanez-Sauri et. al. used an automated high-throughput system to seed and co-culture human mammary fibroblasts and human breast carcinoma cells for analysis.24 Another method for fabricating 3D scaffolds for cells is electrospinning. Electrospinning is based on the same principles as electrospray, but the substrate is a high molecular weight polymer which forms long strands of fibers instead of droplets. An advantage of this technique is that a wide variety of substrates can be used and the fiber size and scaffold thickness can be tuned.25 Research, thus far, has mostly focused on characterization and imaging of cells on fibers.26–29 Very little work has been done to incorporate electrospun fibers with flow-based models or incorporate them with other methods of analysis besides optical detection.
A variety of techniques can be used to analyze (detect) molecules in microfluidic devices, such as molecules released from cells.30 Microchip electrophoresis is a commonly used analysis method, however it can have limits, specifically when looking at the release of analytes from cell cultures and active biological systems. The effectiveness of the technique can suffer due to the combination of high-ionic strength buffers with the voltage used for separations.31,32 Combining microchip electrophoresis with electrochemical detection schemes becomes difficult due to the need to decouple the separation voltage from the electrochemical detector.33 One way to address this challenge is to separate the separation buffers from the sample buffers through the use of a “Quake” valve and use of a Pd decoupler to ground the separation and provide a field free region downstream for integrated electrodes.34–36 Polydimethylsiloxane (PDMS)-based pneumatic microvalves in bilayer devices have been used to maintain the isolation of the flow of cell release from electrophoresis on chip, and integrated micropumps can be used to form discrete injection plugs.37–39 This design has been combined with cells using varying methods including capillary sampling from a petri dish,38 sample collection in reservoir,40 and microdialysis sampling.41
In this work, we describe a microfluidic system that utilizes 3D printing and 3D culture scaffolds that can also be integrated with microchip-based analysis technologies. 3D substrates were fabricated using electrospun PS fibers. These fibers were spun onto sheets of PS that were laser cut to secure the fibers into inserts that can be handled as needed. 3D printed housings were designed to fit the inserts providing a continuous perfusion flow device. Automated micropumps and a 4-port valve was used to automate the process of cell buffer and stimulant introduction through the cell device. The cell culture devices were easily interfaced with a microchip-based analysis by dripping the perfusate into a reservoir and applying negative pressure to the microfluidic device. A continuous flow analysis device consisted of a simple straight channel PDMS device sealed onto a PS base with a gold array working electrode and a Pt pseudo-reference embedded in a PS base for the detection of catecholamine release from stimulated PC12 cells. In order to separate and detect different catecholamines released, a bilayer valving device and electrophoresis with electrochemical detection was used. The valving device is sealed onto a base embedded with a carbon fiber working electrode and a Pd decoupler. PC12 cells were stimulated with elevated levels of K+ and catecholamines (dopamine and norepinephrine) and quantitated with the separation/detection device. This approach affords facile methods to integrate 3D cell culture with close-to-real-time analysis of multiple molecules released from cells.
Experimental
Chemicals and materials
The following chemicals and materials were used for the reported studies: Sylgard 184 (Ellsworth Adhesives, Germantown, WI, USA); Nano SU-8 developer, SU-8 50 photoresist (Microchem, Newton, MA, USA); AZ 4620 positive resist and AZ 400 developer (AZ Resist, Somerville, NJ, USA); 5 minute epoxy (Permatex, Harford, CT, USA); dopamine, norepinephrine, potassium chloride, sodium chloride, magnesium chloride, sodium phosphate, TES sodium salt, HEPES, collagen, fluorescein, hexamethyldisilazane, 50% glutaraldehyde solution, DAPI (4′,6-diamidino-2-phenylindole) (Sigma Aldrich, St. Louis, MO, USA), ActinRed 555 ReadyProbes reagent (Thermo Fisher); Medium (Kaighn’s Modification of Ham’s F-12 Medium), horse serum (ATCC, Manassas, VA, USA); Penicillin-Streptomycin (Lonza, Walkersville, MD, USA); Fetal Bovine Serum (Atlanta Biologicals, Atlanta, GA, USA); Hoechst 33258, Pentahydrate (bis-Benzimide) (Thermo Fisher Scientific, Waltham, MA, USA); 0.500 mm Pt wire, and 1 mm palladium wire (Alfa Aesar, Ward Hill, MA, USA); soldering wire and heat shrink tubes (Radioshack); EL30P1.5 High Voltage Power Supply (Glassman High Voltage, Inc., High Bridge, NJ, USA); MJ30P400 High Voltage Power Supply (Glassman High Voltage, Inc., High Bridge, NJ, USA); isopropanol (Fisher Scientific, Springfield, NJ, USA); colloidal silver (Ted Pella, Redding, CA, USA); 15 gauge bent 12.7 mm probe (Virtual Industries, Colorado Springs, CO, USA); a 0.150 mm i.d. capillary (Polymicro Technologies, Phoenix, AZ, USA); 0.033 mm carbon fiber (Avco Specialty Materials, Textron, Lowell, MA, USA); PS powder (0.250 mm particle size, Goodfellow, Oakdale, PA, USA); Stratasys Idea Series Mojo 3D Printer and Stratasys Polyjet Eden 260V Polyjet Printer (Eden Prairie, MN, USA); with ivory P430 ABS model material and SR-30 soluble support material (Stratasys, Ltd., Edina, MN, USA); Autodesk Inventor Professional 2015 (San Rafael, CA, USA); MagicPlot Student 2016 (Saint Petersburg, Russia); a 300 µm ID steel pin (New England Small Tube, Litchfield, NH, USA).
Fabrication of PDMS chips
The molds for the straight channel PDMS devices were fabricated with negative photoresists, and bilayer PDMS microchips were fabricated as described previously using both negative and positive resists.5,37 The straight channel (100 × 100 µm) chips were fabricated using only a negative resist to form the PDMS microchannels. PDMS (20:1 base elastomer : initiator) was poured over master to make a monolayer PDMS microchip. The straight channel is reversibly sealed over the pseudo-reference and the working electrode prior to use. For the bilayer device, negative resist was used for replicant molding of the valving and pump layer, while the positive resist was used for molding the flow layer. The valving layer (5:1 base elastomer:initiator) was partially cured at 75 °C for 8 minutes, after which PDMS (20:1 base elastomer:initiator) was spin coated onto the flow layer master for 1 minute at 2000 rpm. Both layers were aligned and bonded together after an additional 19 minutes of cure time at 75 °C. Channel dimensions of the photoresist structures were measured using a profilometer (Dektak3 ST, Veeco Instruments, Woodbury, NY, USA).
Fabrication of polystyrene bases
The PS devices were made using methods that have been previously described.38,42 An aluminum dish (7 cm diameter) was used to mold the PS powder (0.250 mm particle size, Goodfellow, Oakdale, PA, USA) or milled petri dishes. A 23-gauge needle was used to punch the holes in the aluminum dish for threading the electrodes. The distance between the decoupler and electrode (1 mm) was measured using a caliper. The electrode and decoupler (pseudo-reference for the flow device) were each attached to a piece of copper connecting wire and 5-minute epoxy was used to adhere them together in order to make threading them through the dish easier. Polystyrene powder was poured into the dish and heated on a hot plate (285 °C) for 1 hour. The base was allowed to cool, and a layer of 5-minute epoxy was coated over the capillary for protection. PS was removed from the aluminum dish and shaped by wet polishing using a range (200–1200) of grits (Buehler, Lake Bluff, IL, USA) to achieve a fine polish. As previously described,40 a 3D printed clip was used to maintain a consistent seal of the PDMS chip. The clip was printed and designed to fit on the base where the PDMS chip would be sealed putting pressure on the chip at the valve and pump interface.
Fabrication of Inserts and 3D printed housings
The 3D printed devices used in these studies are shown in Figure 1. Inserts were fabricated by electrospinning polystyrene onto polystyrene sheets. Polystyrene beads (192 kDa) were dissolved in a mixture of THF and DMF (40%/60%) and placed on a shaker for 24 hours. The solution was pumped through a syringe pump at 13.3 µL/min. The syringe was inserted into a hole in a plexiglass box. A high voltage of +25 kV was applied to the needle tip during flow. The voltage in the box was grounded to a piece of aluminum foil placed behind a polystyrene sheet. The foil was secured between the sheet and a piece of cardboard with scotch tape. Fibers were generated and collected on the PS sheet for ~45 mins. The sheet was removed from the foil and laser cut (Glowforge, Seattle, WA, USA) based on a CAD design into 1.8 mm x 26 mm inserts.
Figure 1.

A.) CAD design of the 3D printed culture device. The device is printed together as an assembly of 2 parts consisting of the bottom portion with slots for the inserts tapered to a funnel and the top portion with threads to accommodate commercial fittings. B.) Picture showing the printed device. A pin is epoxied to the end of the device for simple integration with analysis devices. C.) The inserts made up of PS fibers electrospun onto PS sheets. The sheets are then laser cut into individual inserts securing the fibers to the sheet. D.) Cross-sectional view of the slots for the inserts to fit in. E.) Cross-sectional view with the inserts inside the housing.
3D printed housings were printed using a Polyjet printer (Objet Eden 260 V 3D Printer) with Full Cure 720 and 705. The piece was design with 2 slots to accommodate the PS inserts with a 350 µm gap. The 350 µm gap was set due to printer limitations. The housing is designed with female threads to fit commercial IDEX F-120x fittings. The outlet of the housing had a steel pin attached and sealed with epoxy for simple integration with the PDMS chip. Cell counts were done using a Hoechst reagent and read in a 96-well plate using FlexStation 3 Multiwell Plate Reader (Molecular Devices, San Jose, CA, USA).43
PC12 Cell Culture
PC 12 cells (ATCC, Manassas, VA, USA) were cultured at 37° C and 5% CO2 in PS T-25 flasks (Fisher Scientific, Springfield, NJ, USA). The T-25 flasks and all surfaces including fibers for cell adhesion were pretreated with 0.435 mg/mL collagen. The PC 12 cells were grown with media (F-12K supplemented with 1% penicillin–streptomycin solution, 2.5% fetal bovine serum, and 15% horse serum, all from ATCC) being replaced every 1–2 days. Cells were grown to 90% confluency (as determined by optical microscopy) and either passage into new T-25 flasks or used for experiments. Culturing cells on fibers was done in a 3D printed well plate (ABS printed on a FDM printer, MOJO Desktop 3D Printer, Stratsys) designed to fit the inserts in order to hold them at the bottom of the well during treatment and immobilization. Fibers were treated with the collagen solution for 1 hour prior to immobilizing on them. The cells were allowed to immobilize overnight prior to use. A 90% confluent T-25 flask of PC12 cells was scraped and spun down at 2000 rpm for 5 minutes. The cell pellet was suspended in 2.4 mL, and 300 µL of the cell suspension was placed in each of the wells. The cells were immobilized overnight prior to use. As in a previous study with different cell lines, prior to analysis the scaffolds were removed from the well and inserted into the flow device. The previous study also characterized (with microscopy) the flow areas around the insert once in the device.44
For pre-loaded cell experiments cells were placed in solution with 1 mM dopamine or 1 mM dopamine and norepinephrine for 1 hour in physiological buffer prior to stimulation.45 Cells were stimulated with a buffer containing elevated K+ made up of the following: 80 mM KCl, 50 mM NaCl, 0.7 mM MgCl2, 1 mM NaH2PO4, 2 mM CaCl2 and 10 mM HEPES and rinsed with physiological saline: 4.2 mM KCl, 150 mM NaCl, 0.7 mM MgCl2, 1 mM NaH2PO4, 2 mM CaCl2 and 10 mM HEPES. Cell counts were done by immersing inserts in 1 mL of deionized (DI) water to lyse the cells for 1 h. The lysate was used with the Hoechst fluorescence assay as a measure of cell count. The Hoechst solution (10 mg/mL, Sigma-Aldrich, MO, USA) was diluted in TNE buffer (50 mM Tris-HCl, 100 mM NaCl, 0.1 mM EDTA, pH = 7.4) to 20 μg/mL. 200 μL of the cell lysate was pipetted into a 96-well pate, followed by addition of 50 μL of the prepared Hoechst assay. Fluorescence was detected at 460 nm (with 350 nm excitation) by a plate reader (MolecularDevices, CA, USA). A hemocytometer was used for cell counts for standards with 10,000–100,000 cells being used.
Cell Imaging
Fluorescent images of cells on fibers and flat sheets were done by treating the cells with acridine orange (100 µg/mL) (Figure 2A). Cells were fixed for 30 mins in a 2.5% glutaraldehyde solution in PBS and Scanning electron microscopy (SEM) images were taken (Figure 2C–E). For these studies, cells were treated with a glutaraldehyde solution (2.5% in PBS) for 30 minutes to fix the cells on the fibers. For the confocal imaging (Figure 2B), cells were again treated with a 2.5% glutaraldehyde solution to fix the cells in place for 30 mins. Cells were then treated with a staining solution of DAPI (4′,6-diamidino-2-phenylindole) (300 nM in PBS) containing 2 drops of ActinRed555 for 30 minutes after which time the cells were suspended in PBS buffer until imaging. Confocal images where taken at the Saint Louis University Research Microscopy and Histology Core Facility.
Figure 2.
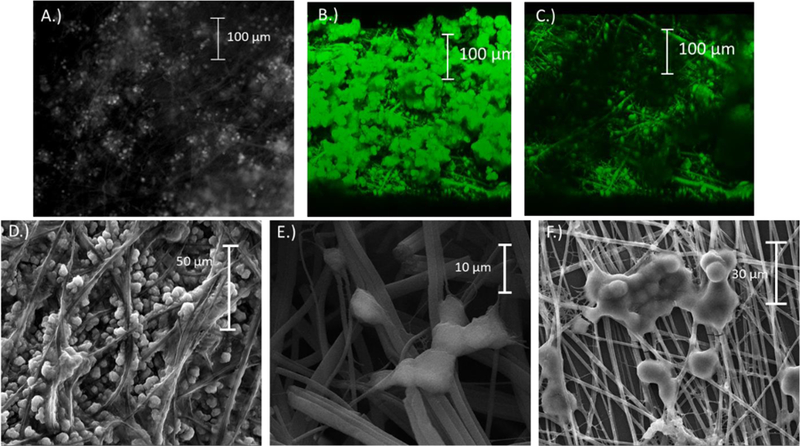
A.) Micrograph showing an insert with PC12 cells immobilized onto the fibers (cells stained with acridine orange); B-C.) Confocal images taken of a section of the collage-coated PS inserts. B.) Micrograph showing clumped cells on top with cells in fibers below. C.) Same view of the cells with the clumped cells on top removed. D.-F) Scanning electron microscopy images showing PC12 fixed onto PS fibers. D.) Well split groups of cells permeate the fibers throughout. E.) Clumps of cells branching out to hold the substrate. F.) Cells bunching together on the surface of a thin sheet of fibers.
Flow studies with electrochemical detection
Automated syringe pumps and a 4-port valve (LabSmith, Livermore, CA, USA) were used for flow and sample introduction into the 3D printed cell device. The system consisted of 2–100 µL micro-syringes with one filled with cell buffer and one filled with either standard or K+ stimulant. The pumps syringes were connected to an automated 4-port with 150 µm ID fused silica capillary. The pumps and valve were controlled with µProcess Software (LabSmith, Livermore, CA, USA). Buffer was flown through the 3D printed insert device at 5.00 µL/min. A PDMS straight channel chip was sealed onto a polystyrene base across a gold array working electrode made from gold gauze (Gold gauze, 100 mesh woven from 0.064 mm diameter wire, Alfa-Aesar, Tewksbury, MA, USA) and a 500 µm diameter Pt wire (Alfa-Aesar, Tewksbury, MA, USA) pseudo-reference. A withdraw pump was used to pull at −5.00 µL/min. Prior to every run, the flow device was primed with cell buffer to ensure minimal air bubbles in the device. For integrating the two different fluidic systems, the steel pin on the 3D printed culture device was placed just above a reservoir on the inlet of the PDMS chip. The reservoir had an overall volume of 1 μL. The reservoir was filled with 2 μL of buffer so that a small, 1 μL droplet sat on top of the reservoir. Flow from the steel pin directly interfaced with this droplet. The pumps were run at the same rates to maintain a constant volume between the cell device and the analysis device. Amperometric detection was performed at +0.9 V (vs a Pt pseudo-reference) with a CH Instruments potentiostat (Austin, TX, USA).
Microchip Electrophoresis with electrochemical detection
The layout of the electrophoresis chip is the same design as previous valving devices.38,40 For these experiments, the field strength was 330 V cm−1, and amperometric detection (at a 33 μm carbon fiber working electrode) was done at +0.9 V (vs a Pt pseudo-reference). For presentation purposes, the CH Instruments potentiostat software (least squares smoothing function) was used to treat the data shown in Figure 5 (to smooth the noise). Electrophoresis was performed using a 1 mm Pd decoupler for grounding of voltages and adsorption of H2 produced during hydrolysis. The sample was continuously withdrawn through the sample portion of the chip before injecting the plug and separating the analytes. 100 µM fluorescein was added to the stimulant for visualization of the injection plug. For buffer flow and sample injection, the system was run in the same manner as the flow experiments with the flow going through the valving and pump setup. For injections, the normally open valve in the flow channel was closed, and the normally closed valve was opened. The peristaltic pumps were run for 5 second injections, and then the valves were returned to their normal state. The run voltages were then initiated and dopamine and norepinephrine were electrophoretically separated and amperometrically detected.
Figure 5.
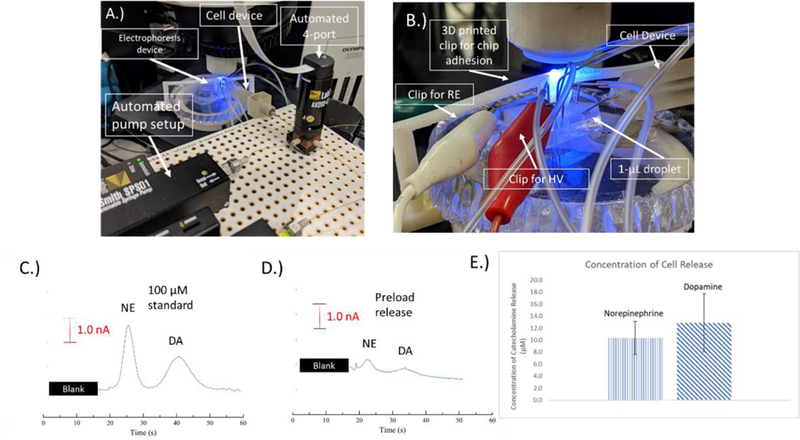
A.) Picture of the electrophoresis setup. The cell device can be seen connected with a capillary to the automated syringe and valve system. The device is connected to the bilayer valving chip. B.) Picture showing the integration of the cell device with the electrophoresis chip. The steel pin can be seen integrated with the 1-µL droplet. C-E.) Electropherograms of C.) 100 µM norepinephrine and dopamine D.) Detected release of norepinephrine and dopamine from pre-loaded PC12 cells E.) The release of norepinephrine and dopamine from cells pre-loaded with 1 mM of both for 1 hr prior to the experiments. E.) A bar graph showing a comparison of the norepinephrine and dopamine detected from the pre-loaded PC12 cells (n=4).
Results/Discussion
Fabrication of Cell Module
In order to improve sample integration of cell release with PDMS-PS devices utilizing electrochemical detection, a 3D printed flow device was fabricated. This device can be fitted with commercial fittings for integration with pumping methods (tubing, connectors etc). The designs for these devices was done using CAD (Figure 1A) and printed with a Stratasys polyjet printer as shown in Figure 1B. In order to better mimic the in vivo environment that cells experience, it is important to use substrates that can form 3-dimensional structures for cells to immobilize. One method for doing this is electrospinning. Electrospinning forms fibrous strands of polymer utilizing the same principles as electrospray. In this study, high molecular weight polystyrene was dissolved in a DMF/THF (60%/40%) mixture and stirred overnight. Fibers were sprayed onto PS sheets and then laser cut to a size that would fit into the slots designed into the 3D printed housing. The laser cutting forms a ridge on the side of the insert that makes a tight fit into the 3D printed piece and sinters the fibers melding them to the sheet.44 This makes the fibers stable on the PS and allows for manipulation methods required to insert and remove them from the housing. An example of laser cut inserts is shown in Figure 1C. Immobilization of cells onto the fibers was done using a 3D printed well plate. The well plate is an ABS print (with an FDM printer). The fibers were coated with collagen in a well plate for 1 hr prior to immobilizing cells, and the cells were immobilized overnight.
To perform flow experiments on the 3D scaffolds, 2 inserts were fitted facing each other into printed slots (Figure 1D–E) and solution introduced. An automated micropump and 4-port valve system was utilized to make reproducible injections of standard and K+ stimulant. The integration of the entire cell culture setup with the analysis devices was done using a droplet reservoir. A steel pin was fixed to the outlet of the 3D printed device and when flow was introduced droplet formed at the end of the pin. The device was placed just over a 1 μL volume reservoir at the microchannel inlet. This reservoir was filled with 2 μL of buffer so that a small, 1 μL droplet sat on top of the reservoir. Flow from the steel pin directly interfaced with this droplet and a negative pressure was used to continuously withdraw the perfusate through the microfluidic device. By matching the positive and negative pressures, the reservoir volume remained constant. Directly inserting the steel pin or tubing from the 3D printed device into the PDMS microchip led to issues with bubble trapping or led to constant bubble generation. This droplet approach resulted in a more robust method for interfacing flow from the 3D culture device to the analysis chip.
For electrochemical detection of analytes, PDMS-PS hybrid devices that have been previously characterized were used.38,46 For flow studies, a straight channel (100 × 100 µm) PDMS device was used with holes punched for the inlet and outlet of the device. A bilayer valving device was used for the electrophoresis studies. Both chips were sealed onto PS bases containing embedded electrodes. For the flow devices, the bases contained a gold array working electrode and a Pt pseudo-reference. For the electrophoresis studies, a carbon fiber working electrode was used in conjunction with a Pd decoupler to allow for in-channel detection.38 The pseudo-reference, in that case, is placed in the waste reservoir.
The 3D printed cell device was integrated with an automated micro syringe pump and 4-port valve system by connecting them with a commercially available fitting. The device can be integrated with any 4- or 6- port system in this method. The system used in this study was chosen due to the ease of automation of the runs utilizing a 4-port timing-based injection. The software was programmed to make 5 µL injections of the stimulant through the cell device. In order to maintain a consistent volume at the outlet of the cell device and the inlet of the analysis device, the withdraw pump pulling through the chip and infusion pumps pushing through the cell device were ran at the same rate.
PC12 Cell Culture on Fibers
PC12 cells have been used as a model for neuroscience for several decades.47–50 They provide an in vitro method to study neurochemistry. They were chosen for this study due to their being well-established and the fact that they can release multiple analytes upon stimulation (dopamine and norepinephrine); therefore, they are a good model system for characterizing the ability to detect multiple analytes in a microfluidic device. Typical cell culture today involves two-dimensional culture which has been shown not to accurately represent the environment cells experience in vivo.51 In order to better mimic the extracellular structure in the body, 3-D cell culture methods can be used. In this study, PS fibers were generated through electrospinning. The fibers were sprayed onto PS sheets which resulted in fiber sizes of 1.9 ± 0.4 µm (n=15) and a wide range of pore sizes with an average of 85 ± 36 µm2 (n=12). The fiber layer (thickness of scaffold) was measured with imaging and found to be 113 ± 12 µm (n=5). Fibers were treated with a collagen solution prior to immobilization. Figure 2A shows a micrograph of acridine orange stained PC12 cells on PS fibers.
In order to get a grasp on how well the cells permeate into the fibers, confocal (Figure 2B and C) and SEM imaging (Figure 2D–F) was done. PC12 cells are notorious for settling in clumps of cells. The cells settled onto the fibers in a couple different scenarios. As shown in Figure 2F and the top portion of 2B, cells would bunch up on top of the fibers forming a layer of cells on top. Other groups of cells would permeate into the fibers and attach as shown in Figure 2D, E, and the lower layer of B and C. Both of these cell layers were present on the inserts used. In order to ensure cells would permeate the fibers, cells were run through a 23-gauge needle using a syringe prior to immobilization. Breaking up the cells helped to prevent clumping and achieve more of the cell dispersion in fibers as shown in Figure 2D.
For comparison purposes, cells were immobilized onto flat polystyrene sheets without any fibers. Figure S-1A and B in the supplemental information shows micrographs of PC12 cells stained with acridine orange along with SEM images of cells cultured onto flat sheets in Figure S-1C. The cells form a monolayer appearing to clump up more than those on the fibers. The cells are relatively stable on the flat sheets, but due to the clumping and the small surface area of the inserts there was vacancies on the flat sheets without cells with the cell suspensions that were used.
PC12 Flow Studies
In order to test the effectiveness of the system for detecting cell release, a flow system was designed to detect the flow through the 3D printed housing containing cell inserts. In conjunction with a commercial automated syringe pump and 4-port system, a hybrid PDMS-PS device was used for the detection of catecholamine release. As previously stated, a steel pin is secured at the outlet of the 3D printed device. The pin is placed in the droplet protruding from the inlet of the PDMS microchannel and the withdraw and infusion pump are run simultaneously at −5.00 µL/min and 5.00 µL/min respectively (to keep a steady state reservoir level). A diagram of the setup is shown in Figure 3A along with a picture of the setup in Figure 3B showing how the cell device was integrated with the PDMS chip. A gold array was used as the detection electrode followed by a Pt pseudo-reference electrode approximately 150 µm downstream. Automated injections were made by programming the software to actuate the valve and do a 5 µL injection of the sample. Standards were ran by doing 5 µL injections of varying concentrations. Figure S-2 shows representation amperograms that are obtained with this setup. A 50 µM standard is shown in Figure S-2A with a blank injection of K+ stimulant without cells shown in Figure S-2B. Samples under 600 nM were indistinguishable from noise, therefore the system had an effective LOD of 600 nM (calibration curve for dopamine in Figure S-3)
Figure 3.
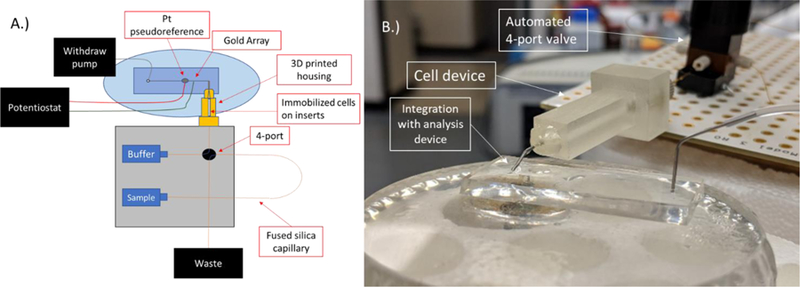
A.) Schematic of the straight channel setup for the flow based electrochemical detection of PC12 catecholamine release. B.) Picture showing the integration of the flow device containing cells with the automated syringe pump and 4-port valve system and the analysis device.
PC12 cells were monitored for cell release by immobilizing cells on inserts and doing injections of stimulant over them through the 3D printed housing. Cell counts were done on the number of cells using a Hoechst assay. Cell counts for each device (which contains 2 inserts) varied but had an average of 150,000 ± 20,000 cells/device. Inserts were kept in physiological buffer for 1 hour prior to running. The inserts were placed into their respective slots, and the device was primed with buffer to remove any air pockets. Analysis time took approximately 10 minutes after filling, priming the device, and stimulated release. For PC12 stimulated on fibers, approximately 1.6 ± 0.4 µM of catecholamine release was detected (n=5) with an example shown in Figure S-2C. In order to get a more significant concentration of release from the PC12 cells, they were preloaded with 1 mM dopamine for 1 hour prior to analysis.45 The cells were rinsed and primed in the device with physiological buffer prior to analysis. When stimulated, 11.0 ± 2 µM of catecholamine release was detected (n=5) from the PC12 cells as shown in Figure S-2D. Both PC12 cells and pre-loaded PC12 cells were treated with reserpine for inhibition of release,52 stimulation of those inserts with K+ did not lead to any detectable release (below the limit of detection for the system). Figure 5A–C shows comparisons of the amount of catecholamine release detected for both sets of cells. Based on cell counts with a Hoechst Assay, the cells were found to release 28 ± 8 pM/cell compared to 72 ± 10 pM/cell for the pre-loaded cells.
Integration of 3D Scaffolds with Microchip-based Electrophoresis with Electrochemical Detection
The system that was used with the flow-based detection was combined with a separation step to allow for the detection of multiple catecholamines released from PC12 cells. This is important for neurochemical studies since dopamine and norepinephrine have similar voltammograms53 and are difficult to distinguish without a separation. A picture of the setup for electrophoresis is shown in Figure 5A, and a close-up image of the integration of the cell device with the electrophoresis chip is shown in Figure 5B. Figure S-4 is a schematic that shows more details on microchip channels and the valve integration/operation. As with the simple channel device, flow from the cell culture system is directed through the steel pin into a reservoir on the valving device. The same withdraw and infusion flow rates are used to continuously introduce sample to the injection portion of the device (valves). The normally closed valve separating the sample and electrophoresis portion of the chip is opened and the normally open valve is closed to allow a series of pneumatic pumps to inject the sample. The valves are then returned to their normal position, and the sample is electrophoretically separated. For in-channel detection, a 1 mm Pd decoupler is used to ground the electrophoresis voltages and adsorb H2. A 33 µm carbon fiber electrode was used as the detection electrode because of optimization done in prior work.38 The buffer used for all separations was 25 mM TES and 25 mM SDS (pH=7.4), and the field strength was 330 V cm−1. Figure 5C shows a 100 µM injection of dopamine and norepinephrine in K+ stimulant. The norepinephrine and dopamine are clearly baseline resolved with the separation parameters.
In order to show that the cells can be integrated with the PDMS-PS bilayer valving device, cells were preloaded as in the previous flow studies38,40 with 1 mM dopamine and norepinephrine for 1 hr prior to the analysis to have a significant detectable amount of both. Cells were then stimulated as done in the flow studies described above. A small amount of fluorescein was used in the stimulant to indicate when the release would be coming through the device and to also monitor plug size. Figure 5D shows an example of detected pre-loaded cell release, and Figure 5E shows a comparison of the amounts of norepinephrine and dopamine released. A blank injection with no cells present can be seen in Figure S-5 (no peaks detected). There was 13 ± 5 µM of dopamine and 10 ± 3 µM of norepinephrine detected (n=4). On a per cell basis there was 77 ± 22 pM/cell for dopamine and 64 ± 11 pM/cell of norepinephrine release detected for this system. This compares favorably to previous studies sampling from cell flasks of pre-loaded PC12 cells (reported concentrations between 25–50 pM/cell).38,40
Conclusion
In this study, a 3D cell culture model utilizing fibers spun onto PS sheets was combined with 3D printed flow housings for 3D culture of PC 12 cells. The cell culture system was integrated with an automated micropump injection system for introduction of buffer and K+ stimulant for the release of catecholamines from PC12 cells. Cell buffer and cell release through the cell housing was flowed into either a PDMS straight channel device or bilayer valving/electrophoresis device sealed onto PS bases containing embedded electrodes for electrochemical detection. PC12 cells and dopamine pre-loaded PC12 cells cultured on fibers were combined with the flow device for catecholamine detection. The bilayer PDMS valving chip was then used in combination with pre-loaded PC12 cells for the separation and detection of dopamine and norepinephrine release. This study shows that this insert based method of cell culture and analysis can be integrated with PDMS analysis devices. Future work can be done to integrate other cell systems and also utilize co-culture models with this setup; however, here we have shown this approach affords facile methods to integrate 3D cell culture with close-to-real-time analysis of multiple molecules released from cells.
Supplementary Material
Figure 4.
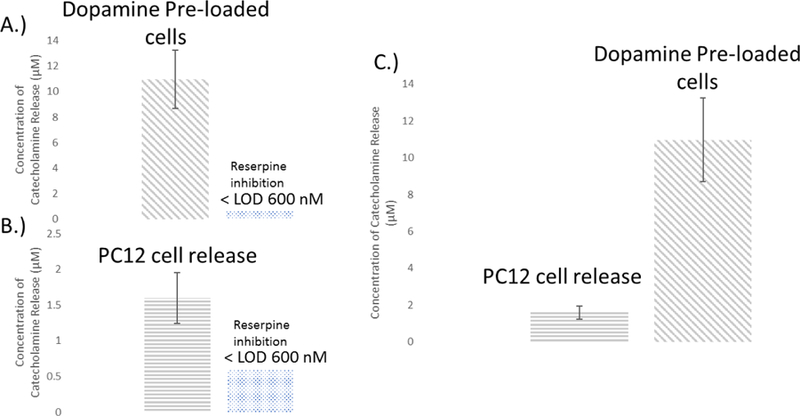
A and B.) Bar graphs comparing the detected release of catecholamines from both PC12 and PC12 cells pre-loaded with dopamine. The concentration of detected catecholamine release was 4 ± 1 µM and 11 ± 2 µM or PC12 and preloaded PC12 cells respectively (n=5). Reserpine inhibition resulted in catecholamine levels released below the lowest detectable concentration (n=3). C.) Shows a comparison of the concentration released between both sets of cells and per cell respectively.
Acknowledgement
Support from the National Institute of General Medical Sciences (Award Number R15GM084470–04) is acknowledged.
Footnotes
Conflicts of interest
There are no conflicts of interest to declare.
References
- 1.Manz A, Harrison DJ, Verpoorte EMJ, Fettinger JC, Paulus A, Ludi H and Widmer HM, J Chromatogr A, 1992, 593, 253–258. [Google Scholar]
- 2.Manz A, Graber N and Widmer HM, Sens Actuators, B, 1990, 1, 244–248. [Google Scholar]
- 3.Harrison DJ, Manz A, Fan Z, Luedi H and Widmer HM, Anal Chem, 1992, 64, 1926–1932. [Google Scholar]
- 4.Woolley AT and Mathies RA, Proc Natl Acad Sci USA, 1994, 91, 11348–11352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duffy DC, McDonald JC, Schueller OJA and Whitesides GM, Anal. Chem, 1998, 70, 4974–4984. [DOI] [PubMed] [Google Scholar]
- 6.Bustillo JM, Howe RT and Muller RS, Proceedings IEEE, 1998, 86, 1552–1574. [Google Scholar]
- 7.Ho CM, Ng SH, Li KH and Yoon YJ, Lab Chip, 2015, 15, 3627–3637. [DOI] [PubMed] [Google Scholar]
- 8.Waheed S, Cabot JM, Macdonald NP, Lewis T, Guijt RM, Paull B and Breadmore MC, Lab Chip, 2016, 16, 1993–2013. [DOI] [PubMed] [Google Scholar]
- 9.Brimmo A, Goyette PA, Alnemari R, Gervais T and Qasaimeh MA, Sci Rep, 2018, 8, 10995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen C, Mehl BT, Munshi AS, Townsend AD, Spence DM and Martin RS, Anal. Methods, 2016, 8, 6005–6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gross B, Lockwood SY and Spence DM, Anal Chem, 2017, 89, 57–70. [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Wang Y, Lockwood SY and Spence DM, Analyst, 2014, 139, 3219–3226. [DOI] [PubMed] [Google Scholar]
- 13.Castiaux AD, Pinger CW and Spence DM, Anal Bioanal Chem, 2018, 410, 7565–7573. [DOI] [PubMed] [Google Scholar]
- 14.Han N, Shin JH and Han K-H, RSC Adv, 2014, 4, 9160. [Google Scholar]
- 15.Rogers CI, Qaderi K, Woolley AT and Nordin GP, Biomicrofluidics, 2015, 9, 016501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kadimisetty K, Mosa IM, Malla S, Satterwhite-Warden JE, Kuhns TM, Faria RC, Lee NH and Rusling JF, Biosens Bioelectron, 2016, 77, 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Su CK, Yen SC, Li TW and Sun YC, Anal Chem, 2016, 88, 6265–6273. [DOI] [PubMed] [Google Scholar]
- 18.Gong H, Bickham BP, Woolley AT and Nordin GP, Lab Chip, 2017, 17, 2899–2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Young EWK and Beebe DJ, Chem. Soc. Rev, 2010, 39, 1036–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu M-H, Huang S-B and Lee G-B, Lab Chip, 2010, 10, 939–956. [DOI] [PubMed] [Google Scholar]
- 21.van Duinen V, Trietsch SJ, Joore J, Vulto P and Hankemeier T, Curr. Opin. Biotechnol, 2015, 35, 118–126. [DOI] [PubMed] [Google Scholar]
- 22.Derda R, Tang SK, Laromaine A, Mosadegh B, Hong E, Mwangi M, Mammoto A, Ingber DE and Whitesides GM, PLoS One, 2011, 6, e18940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lamichhane S, Anderson JA, Vierhout T, Remund T, Sun H and Kelly P, J Biomed Mater Res A, 2017, 105, 2441–2450. [DOI] [PubMed] [Google Scholar]
- 24.Montanez-Sauri S, Sung KE, Puccinelli JP, Pehlke C and Beebe DJ, JALA, 2011, 171–185. [DOI] [PMC free article] [PubMed]
- 25.Xue J, Xie J, Liu W and Xia Y, Acc. Chem. Res, 2017, 50, 1976–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wolun-Cholewa M, Langer K, Szymanowski K, Glodek A, Jankowska A, Warchol W and Langer J, PLoS One, 2013, 8, e72936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.He F-L, Li D-W, He J, Liu Y-Y, Ahmad F, Liu Y-L, Deng X, Ye Y-J and Yin D-C, Mater. Sci. Eng. C, 2018, 86, 18–27. [DOI] [PubMed] [Google Scholar]
- 28.Badami AS, Kreke MR, Thompson MS, Riffle JS and Goldstein AS, Biomaterials, 2006, 27, 596–606. [DOI] [PubMed] [Google Scholar]
- 29.Park J-S, Choi J-B, Jo S-Y, Lim Y-M, Gwon H-J, Khil MS and Nho Y-C, J. Appl. Polym. Sci, 2012, 125, E595–E603. [Google Scholar]
- 30.Pasas SA, Fogarty BA, Huynh BH, Lacher NA, Carlson B, Martin RS, Vandaveer WRI and Lunte SM, in Separation Methods in Microanalytical Systems, ed. Kutter JP, Fintschenko Y, CRC Press LLC, Boca Raton, Fl, Editon edn., 2006, pp. 433–497. [Google Scholar]
- 31.Gillogly JA and Lunte CE, Electrophoresis, 2005, 26, 633–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arnett SD and Lunte CE, Electrophoresis, 2003, 24, 1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin RS, in Methods in Molecular Biology, vol. 339: Microchip Capillary Electrophoresis: Methods and Protocols, ed. Henry CS, Humana Press, Towtowa, NJ, Editon edn., 2006. [Google Scholar]
- 34.Bowen AL and Martin RS, Electrophoresis, 2009, 30, 3347–3354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Johnson AS, Selimovic A and Martin RS, Electrophoresis, 2011, 32, 3121–3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mecker LC and Martin RS, Electrophoresis, 2006, 27, 5032–5042. [DOI] [PubMed] [Google Scholar]
- 37.Bowen AL and Martin RS, Electrophoresis, 2010, 31, 2534–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johnson AS, Mehl BT and Martin RS, Anal. Methods, 2015, 7, 884–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li MW and Martin RS, Electrophoresis, 2007, 28, 2478–2488. [DOI] [PubMed] [Google Scholar]
- 40.Mehl BT and Martin RS, Anal. Methods, 2018, 10, 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mecker LC and Martin RS, Anal. Chem, 2008, 80, 9257–9264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Anderson KB, Halpin ST, Johnson AS, Martin RS and Spence DM, Analyst, 2013, 138, 137–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rage R, Mitchen J and Wilding G, Anal. Biochem, 1990, 191, 31–34. [DOI] [PubMed] [Google Scholar]
- 44.Chen C, Townsend AD, Hayter EA, Birk HM, Sell SA and Martin RS, Anal Bioanal Chem, 2018, 410, 3025–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Moore JM, Papke JB, Cahill AL and Harkins AB, Am. J. Physiol. Cell Physiol, 2006, 291, C270–281. [DOI] [PubMed] [Google Scholar]
- 46.Johnson AS, Anderson KB, Halpin ST, Kirkpatrick DC, Spence DM and Martin RS, Analyst, 2013, 138, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Greene LA and Rein G, Brain Res, 1977, 138, 521–528. [DOI] [PubMed] [Google Scholar]
- 48.Chen TK, Luo G and Ewing AG, Anal. Chem, 1994, 66, 3031–3035. [DOI] [PubMed] [Google Scholar]
- 49.Kozminski KD, Gutman DA, Davila V, Sulzer D and Ewing AG, Anal Chem, 1998, 70, 3123–3130. [DOI] [PubMed] [Google Scholar]
- 50.Westerink RH and Ewing AG, Acta Physiol, 2008, 192, 273–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Antoni D, Burckel H, Josset E and Noel G, Int J Mol Sci, 2015, 16, 5517–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kozminski KD, Gutman DA, Davila V, Sulzer D, Ewing AG, Anal. Chem, 1998, 70, 3123–3130. [DOI] [PubMed] [Google Scholar]
- 53.Heien ML, Phillips PE, Stuber GD, Seipel AT and Wightman RM, Analyst, 2003, 128, 1413–1419. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.