

Supplemental Digital Content is available in the text.
Keywords: cell death, lncRNA, neuroinflammation, stroke, TAK1
Abstract
Background and Purpose—
Ischemic stroke is one of the leading causes of morbidity and mortality worldwide and a major cause of long-term disability. Recently, long noncoding RNAs have been revealed, which are tightly associated with several human diseases. However, the functions of long noncoding RNAs in ischemic stroke still remain largely unknown. In the current study, for the first time, we investigated the role of long noncoding RNA Nespas in ischemic stroke.
Methods—
We used in vivo models of middle cerebral artery occlusion and in vitro models of oxygen-glucose deprivation to illustrate the effect of long noncoding RNA Nespas on ischemic stroke.
Results—
We found expression of Nespas was significantly increased in ischemic cerebral tissues and oxygen-glucose deprivation–treated BV2 cells in a time-dependent manner. Silencing of Nespas aggravated middle cerebral artery occlusion operation–induced IR injury and cell death. In addition, proinflammatory cytokine production and NF-κB (nuclear factor-κB) signaling activation were inhibited by Nespas overexpression. TAK1 (transforming growth factor-β–activated kinase 1) was found to directly interact with Nespas, and TAK1 activation was significantly suppressed by Nespas. At last, we found Nespas-inhibited TRIM8 (tripartite motif 8)-induced K63-linked polyubiquitination of TAK1.
Conclusions—
We showed that Nespas played anti-inflammatory and antiapoptotic roles in cultured microglial cells after oxygen-glucose deprivation stimulation and in mice after ischemic stroke by inhibiting TRIM8-related K63-linked polyubiquitination of TAK1.
Stroke is one of the 3 major causes of human death, and ischemic stroke accounts for 80% to 85% of all strokes.1 Although many clinical trials of stroke treatment have been completed, up to date, thrombolysis by intravenous recombinant tPA (tissue-type plasminogen activator) is the only clinically effective therapeutic strategy for ischemic stroke.2 However, only 20% of patients with stroke receive this treatment because of its narrow therapeutic window (<4.5 hours after onset of stroke symptoms) and its association with an increased risk of symptomatic intracerebral hemorrhage.3 Therefore, development of effective therapies is urgently required.
Ischemic stroke initiates complex processes including neuroinflammation, oxidative stress, excitotoxicity, and apoptosis, therefore, leading to the neuronal death.4 Microglial cells, which are rapidly and time dependently activated after ischemia, are considered as the major cellular contributors to neuroinflammation in ischemic stroke,5 and cytokines such as TNF-α (tumor necrosis factor-α), IL (interleukin)-1β, IL-6, and reactive oxygen species produced by microglial cells have been reported as mainly inflammatory factors in ischemic stroke–induced brain injury.6,7 Numerous studies investigating the underlying mechanisms of microglial cells induced neuroinflammation to demonstrate a critical role of NF-κB (nuclear factor-κB) in the induction of these inflammatory cytokines.8,9
Long noncoding RNAs (lncRNAs) are an important class of noncoding RNA, whose transcription sequence is >200 nucleotides.10 Because they cannot be translated into proteins, lncRNAs were originally viewed as the noise of translational process. But recently, lncRNAs have been reported to participate in chromatin modification, transcriptional regulation, post-transcriptional regulation, X chromatin inactivation, genomic imprinting, splicing, transcription interference, cell cycle regulation, epigenetic regulation, and play an important role in numerous pathological and physiological processes.11,12 However, whether lncRNAs are involved in the neuroinflammation and ischemic stroke still remains largely unknown.
LncRNA-Nespas is known to be mainly expressed in the progress zone, mesenchyme and ectoderm of the limb, and the neural tube, and it has been reported to play essential roles in osteoarthritis progression and serves as a potential new prognostic biomarker.13 In the current study, for the first time, we observed that lncRNA-Nespas alleviated ischemic stroke–induced neuroinflammation, and we found that Nespas directly interacts with TAK1 (transforming growth factor-β–activated kinase 1) to inhibit TRIM8 (tripartite motif 8)-induced K63-linked polyubiquitination of TAK1. These findings suggested Nespas as a potential target for related medical research.
Methods
Ethics Approval
All animal experimental procedures and animal care were approved by the Ethics Committee of Capital Medical University and were conducted in accordance with the guidelines of the National Institutes of Health on the care and use of animals. The data that support the findings of this study are available from the corresponding author on reasonable request.
Cerebral Ischemia/Reperfusion Induced by Surgical Middle Cerebral Artery Occlusion
Focal cerebral ischemia was induced in mice by intraluminal middle cerebral artery occlusion (MCAO) as described previously.14 Briefly, male mice (8–10 weeks, 23–25 g) were anesthetized with 1.5% to 3% isoflurane (Henry Schein Animal Health). A 2-cm length of a 7 to 0 rounded-tip nylon suture was gently advanced from the external carotid artery up to the internal carotid artery until regional cerebral blood flow was reduced to <16% of baseline. After 60 minutes of MCAO, blood flow was restored by removing the suture, and the mice were allowed to recover for 1 to 7 days. Changes in cerebral blood flow, arterial blood gases, mean arterial pressure, and heart rate were monitored in animals 30 minutes before, during, and after MCAO. The rectal temperature was controlled at 37.0±0.5°C during surgery.
Stereotaxic Injection of siRNA in Mice
Mice were anesthetized and fixed to a stereotaxic apparatus (Stoelting, Kiel, WI). A total of 5 μL siRNA-negative control or siRNA-Nespas (100 μmol/L) and lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA) was mixed gently, incubated at room temperature for 20 minutes, and injected (intracerebroventricular) into the right cerebral ventricle of mice. One day post-injection, MCAO operation was established.
Cell Cultures and Oxygen-Glucose Deprivation
Microglial cells from mouse brain were isolated and confirmed by immunofluorescence labeling according to the previous study.15,16 Mouse BV2 microglial cells were purchased from ATCC (Gaithersburg, MD). The cells were cultured in normal culture medium containing DMEM solution mixed with 10% FBS and incubated in a humidified incubator at 37°C in 5% CO2. Cells were grown to 85% to 95% confluency before use. BV2 cells were treated with siRNA-negative control or siRNA-Nespas for 48 hours or treated with control or Nespas plasmid for 48 hours before oxygen-glucose deprivation (OGD) exposure. To mimic ischemia-like conditions in vitro, mouse BV2 cultures were exposed to OGD for 24 hours.
Neurological Deficit Scores
Neurological deficits were evaluated as previously reported3 by using a 9-point scale at 24 and 72 hours after MCAO induction on the basis of the following criteria: (1) absence of neurological deficits (0 points); (2) either left forelimb flexion after suspension by the tail or failure to fully extend the right forepaw (1 point); (3) left shoulder adduction after suspension by the tail (2 points); (4) decreased resistance to a lateral push toward the left (3 points); (5) spontaneous movement in all directions with circling to the left only if pulled by the tail (4 points); (6) spontaneously circling or walking only to the left (5 points); (7) walking only when stimulated (6 points); (8) no response to stimulation (7 points); and (9) stroke-related death (8 points).
Infarct Volume Determination
At the indicated times (24 and 72 hours) after MCAO, the brains were removed and sliced into 71-mm coronal sections, which were immersed in a 2% 2,3,5-triphenyl-2H-tetrazolium chloride solution for 15 minutes at 37°C. Normal brain tissue was stained a red color, whereas infarcted tissue stained a pale gray color. The sections were photographed and analyzed using Image-Pro Plus 6.0 (Media Cybernetics), and the infarct volume (percentage) of the 7 slices was calculated after correcting for edema as described previously.3
Western Blot Analysis and Ubiquitination Assay
Microglial cells were harvested using radio immunoprecipitation assay buffer (MDL Biotech, Beijing, China) after MCAO or OGD treatment. The protein concentration was measured using Bio-Rad quantification assay (Bio-Rad Laboratories, Hercules, CA). Proteins were separated using 12% SDS-PAGE and transferred to a polyvinylidene fluoride membrane (Millipore, Billerica). The membrane was blocked with 3% nonfat dry milk for 2 hours. The antibodies for TAK1 or K63-linked ubiquitin (Abcam, Cambridge), p65, phospho-p65, IKKβ (IκB kinase β), phospho-IKKβ, IκBα (inhibitor of NF-κB α), phospho-IκBα (Cell Signaling Technology, Inc, Beverly), TRIM8, GAPDH, hemagglutinin, Flag (Santa Cruz Biotechnology, Santa Cruz) were added and incubated overnight at 4°C. After incubation with the corresponding horseradish peroxidase–conjugated secondary antibody (Santa Cruz Biotechnology), the target protein was visualized by enhanced chemiluminescence (Thermo Fisher Scientific, Bremen, Germany). The ubiquitination of TAK1 was detected as described.17
RNA Isolation and Quantitative Polymerase Chain Reaction Analysis
Total RNA was extracted with TRIzol reagent according to the manufacturer’s instructions. A LightCycler (ABI PRISM 7000; Applied Biosciences) and the SYBR RT-PCR kit (Takara Biotechnology, Dalian, China) were used for real-time polymerase chain reaction analysis. GAPDH was used as the internal control, and the 2−ΔΔCT method was used to evaluate the relative quantities of each amplified product in the samples. Primer sequences used in quantitative polymerase chain reaction were shown in Table I in the online-only Data Supplement.
RNA-Binding Protein Immunoprecipitation Assays
The RNA-binding protein immunoprecipitation (RIP) assay was performed as described previously.18 After treatment of OGD, BV2 cells were lysed in RIP lysis buffer, after incubation with RIP buffer consisting of 150 mmol/L KCl, 25 mmol/L Tris pH 7.4, 5 mmol/L EDTA, 0.5 mmol/L dithiothreitol, 0.5% NP40, and 100 U/mL RNAase inhibitor SUPERASin. The RIP buffer contained magnetic beads conjugated with anti-TAK1 antibody (Millipore) or negative control IgG. The samples were incubated with proteinase K and then immunoprecipitated RNA was isolated. Purified RNAs subjected to real-time polymerase chain reaction to determine the presence of binding targets using respective primers for mouse Nespas are as follows: forward: 5′-AGATGCTGAACCCTGCACAA-3′; reverse: 5′-AGCTAGGATCACATCGGGGT-3′.
Dual-Luciferase Reporter Gene Assays
Luciferase reporter constructs containing the NF-κB promoter region were cloned into pGL3-based vectors, then temporarily transfected with 1 μg of the promoter reporter plasmid into siRNA-negative control–treated or siRNA-Nespas–treated BV2 cells using jetPEI (polyplus-transfection), and 50 ng of the phRL-TK plasmid was cotransfected into the cells to verify transfection efficiency. After 48 hours of transfection, the luciferase activities were measured on a spectraMax M5 reader (Molecular Devices, CA) using the dual-luciferase reporter assay system (Promega, Madison, WI).
Terminal Deoxynucleotidyl Transferase-Mediated dUTP-Biotin Nick End Labeling Assay (TUNEL) Assay
TUNEL assay was performed using DeadEnd Fluorometric TUNEL system (Promega) according to the manufacturer’s instructions. In brief, microglial cells were fixed with 4% formaldehyde in PBS and then incubated in TUNEL reaction mixture in the dark for 1 hour at 37°C. Labeled samples were visualized with a fluorescence microscope (OLYMPUS IX71; Shinjuku-ku, Tokyo, Japan) and counted using Image J software (National Institutes of Health).
Statistical Analysis
All data are presented as mean±SD of 3 experiments. Statistical significance was determined with the 2-tailed Student t test, P<0.05 considered statistically significant. Differences among ≥3 groups were statistically analyzed by 1-way ANOVA followed by Bonferroni post hoc test.
Results
Expression of Nespas Is Upregulated After Ischemia In Vivo and In Vitro
To investigate the role of lncRNA-Nespas after ischemic stroke, at first, we established the mouse MCAO model and detected Nespas expression in the brain tissue at 6, 12, 24, and 72 hours after ischemia. As shown in Figure 1A, expression of Nespas was greatly increased in the hippocampal region, striatal, cerebral cortical, and plasma in MCAO mice compared with the sham group in a time-dependent manner. We next established OGD model in BV2 mouse microglia cells and HT22 mouse hippocampal cells. Interestingly, we observed that expression of Nespas has significantly increased in BV2 cells in a time-dependent manner but not HT22 cells after OGD treatment (Figure 1B and 1C). These data suggested that Nespas may have a critical role in microglial cells during the progression of ischemic stroke.
Figure 1.
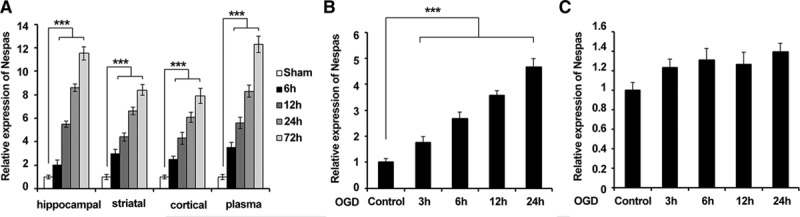
Expression of Nespas is upregulated after ischemia in vivo and in vitro. A, mRNA level of Nespas in hippocampal region, striatal, cerebral cortical, and plasma at 6, 12, 24, and 72 h after ischemia (n=6 per group). B and C, mRNA level of Nespas in BV2 cells (B) or HT22 cells (C) at 3, 6, 12, and 24 h after oxygen-glucose deprivation (OGD) treatment. Data are representative of 3 independent experiments with similar results (mean±SD). ***P<0.001.
Silencing of Nespas Aggravates I/R-Induced Ischemic Brain Damage
To determine whether upregulation of Nespas participates in the pathophysiology of cerebral ischemic stroke, intraventricular injection of Nespas siRNA was used to knock down the expression of Nespas in the brain of MCAO mice. The efficiency of siRNA injection in brain was detected (Figure 2A). As shown in Figure 2B and 2C, silencing of Nespas showed a larger cerebral infarct volume and a significantly more severe neurological deficit (Figure 2D) in response to ischemic insults. These data suggested that Nespas could alleviate I/R-induced ischemic brain damage.
Figure 2.
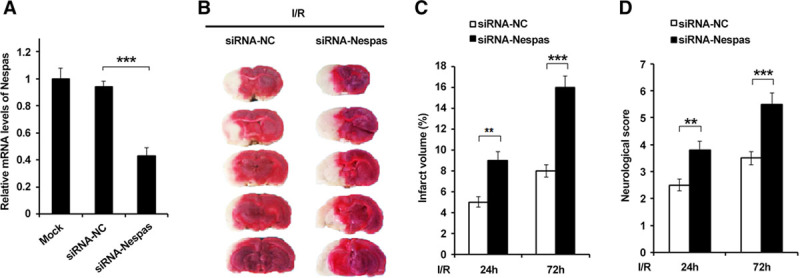
Silencing of Nespas aggravates ischemia/reperfusion (I/R) induced ischemic brain damage. A, Efficiency of siRNA-Nespas in brain tissues of mice. B, TTC-stained sections from siRNA-negative control–treated or siRNA-Nespas–treated mice at 72 h after I/R. C and D, Quantification of the infarct volumes (C) and neurological deficit scores (D) at 24 and 72 h after I/R (n=6 per group). Data are representative of 3 independent experiments with similar results (mean±SD). **P<0.01, ***P<0.001. I/R-induced indicates ischemia/reperfusion induced; siRNA-NC, siRNA-negative control; and TTC, 2,3,5-Triphenyltetrazolium chloride.
Silencing of Nespas Exacerbates I/R-Induced Microglial Cell Death
Because of the high expression of Nespas in microglial cells, we speculated that Nespas might impact the viability of microglia and then affect cerebral ischemia injury. Therefore, we next examined whether Nespas could affect I/R-induced microglial cell death. The isolated microglial cells from mouse brain were confirmed by immunofluorescence labeling of CD11b, which is a widely used microglial marker (Figure IA in the online-only Data Supplement).We found in an MCAO model that silencing of Nespas markedly increased the number of apoptotic microglial cells (Figure 3A). Consistently, we observed that the expression of proapoptotic protein cleaved caspase3 and Bax (BCL2-associated X) was significantly enhanced, but antiapoptotic protein Bcl-2 (B-cell lymphoma-2) expression was decreased in microglial cells of siRNA-Nespas–treated mice after I/R (Figure 3B; Figure IB in the online-only Data Supplement). To further confirm this phenomenon, we evaluated the antiapoptotic effect of Nespas in cultured BV2 cells in vitro after OGD stimulation. As shown in Figure 3C, we found cell viability was decreased and LDH (lactate dehydrogenase) release, which is an indicator of cell injury, was markedly increased in siRNA-Nespas–treated BV2 cells after OGD. Consistent with the in vivo results, we observed that after OGD treatment, cleaved caspase3 and Bax expression was increased and Bcl-2 expression was decreased in Nespas silenced BV2 cells (Figure 3D; Figure IC in the online-only Data Supplement). Taken together, these data suggested that Nespas alleviated I/R-induced microglial cell death.
Figure 3.
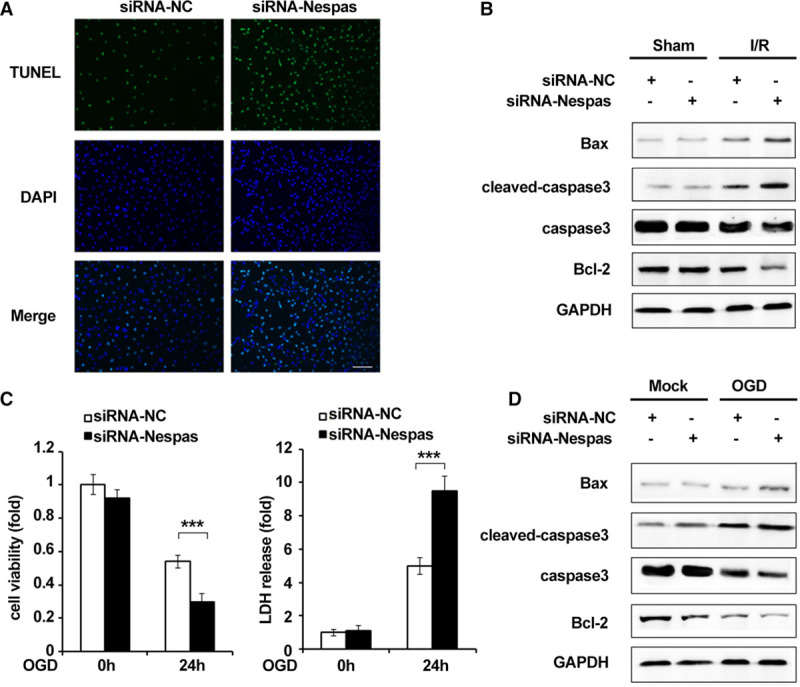
Silencing of Nespas exacerbates I/R-induced microglial cell death. A, TUNEL staining in microglial cells of middle cerebral artery occlusion (MCAO) mice was performed at 24 h after I/R. Scale bars=100 μm. B, Western blot analysis of the protein level of Bax, cleaved caspase3, caspase3, Bcl-2 expression in microglial cells of MCAO model at 24 h after I/R. C, Cell viability (left) and LDH (lactate dehydrogenase) release (right) were assessed and quantified 24 h after oxygen-glucose deprivation (OGD) stimulation. D, Western blot analysis of the protein level of Bax, cleaved caspase3, caspase3, Bcl-2 expression in BV2 microglial cells after OGD treatment for 24 h. Data are representative of 3 independent experiments with similar results (mean±SD). DAPI indicates 4’,6-diamidino-2-phenylin-dole. ***P<0.001. Bax indicates BCL2-associated X; Bcl-2, B-cell lymphoma-2; I/R-induced, ischemia/reperfusion induced; siRNA-NC, siRNA-negative control; and TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP-biotin nick end labeling assay.
Nespas Alleviates I/R-Induced Neuroinflammation Through Suppressing NF-κB Activation
Microglial cells have been considered as the major cellular contributors to neuroinflammation in ischemic stroke, and NF-κB signaling pathway has been proved to be the main regulator of neuroinflammation5,8; therefore, we examined the effect of Nespas on I/R-induced neuroinflammation in microglial cells and the effect of Nespas on NF-κB activation. As shown in Figure 4A, we found that siRNA-Nespas treatment significantly increased the mRNA expression level of proinflammatory cytokines such as TNF-α, IL-1β, IL-6, MCP-1 (monocyte chemoattractant protein-1), VCAM-1 (vascular cell adhesion molecule-1), and ICAM-1 (intercellular cell adhesion molecule-1). Furthermore, we found that after silencing of Nespas, the phosphorylation level of P65, IKKβ, and IκBα were all increased in microglial cells of MCAO mice (Figure 4B; Figure IIA in the online-only Data Supplement). Consistently, we observed that the expression of TNF-α, IL-1β, IL-6, MCP-1, VCAM-1, and ICAM-1 was inhibited in Nespas-overexpressed BV2 cells after OGD treatment (Figure IIB in the online-only Data Supplement; Figure 4C), and the phosphorylation level of P65, IKKβ, IκBα (Figure IIC and IID in the online-only Data Supplement) was all increased in siRNA-Nespas–treated BV2 cells after OGD stimulation. In addition, we used dual-luciferase reporter assay to examine the activation of NF-κB signaling, and we found Nespas markedly suppressed OGD-induced NF-κB activation (Figure 4D; Figure IIE in the online-only Data Supplement). Taken together, these data indicated that Nespas alleviates I/R-induced neuroinflammation through suppressing NF-κB activation.
Figure 4.
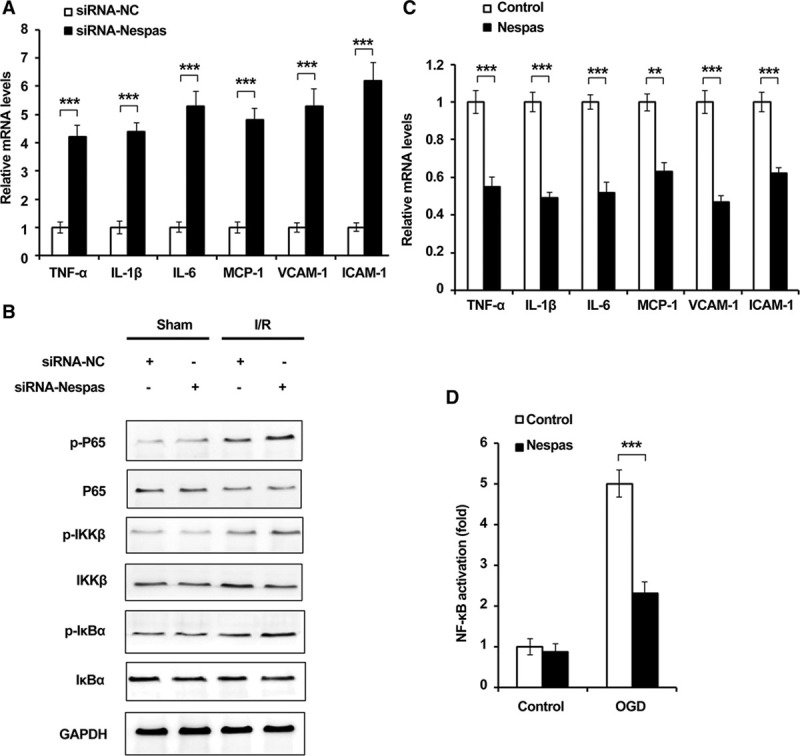
Nespas alleviates I/R-induced neuroinflammation through suppressing NF-κB (nuclear factor-κB) activation. A, mRNA levels of TNF-α (tumor necrosis factor-α), IL (interleukin)-1β, IL-6, MCP-1 (monocyte chemoattractant protein-1), VCAM-1 (vascular cell adhesion molecule-1), and ICAM-1 (intercellular cell adhesion molecule-1) in microglial cells of middle cerebral artery occlusion (MCAO) mice were examined by q-PCR. B, Western blot analysis of phosphorylation level of P65, IKKβ, IκBα in microglial cells of MCAO model at 24 h after I/R. C, mRNA levels of TNF-α, IL-1β, IL-6, MCP-1, VCAM-1, and ICAM-1 in BV2 microglial cells were examined by q-PCR. D, Dual-luciferase reporter gene assay was performed to examine the activation of NF-κB in Nespas-overexpressed BV2 cell after oxygen-glucose deprivation (OGD) treatment for 24 h. Data are the representative of 3 independent experiments with similar results (mean±SD). **P<0.01, ***P<0.001. IKKβ indicates IκB kinase β; IκBα, inhibitor of NF-κB α; I/R-induced, ischemia/reperfusion induced; siRNA-NC, siRNA-negative control; and q-PCR, real-time quantitative PCR.
Nespas Interacts With TAK1
It has been reported that TAK1 played critical roles in the regulation of NF-κB activation; however, the relationship between lncRNA and TAK1 has not been well studied. Therefore, we detected the role of Nespas in TAK1-induced NF-κB activation. As shown in Figure 5A, interestingly, we observed that Nespas obviously suppressed NF-κB activation induced by TAK1. In addition, we performed RIP assay, and we found that Nespas could directly interact with TAK1 (Figure 5B). We also detected the effect of Nespas on TAK1 expression, and the data showed that mRNA level of TAK1 was not affected by Nespas overexpression (Figure 5C). Taken together, these data suggested that lncRNA-Nespas could directly interact with TAK1.
Figure 5.
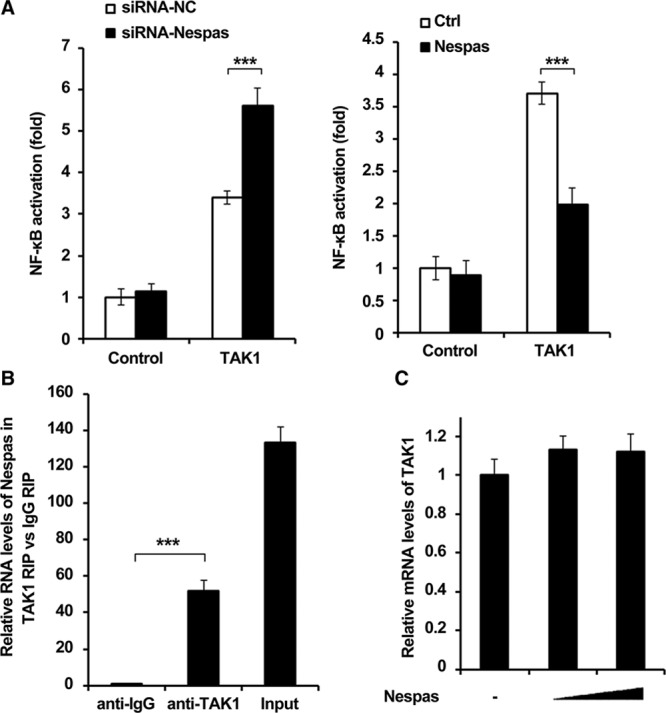
Nespas interacts with TAK1 (transforming growth factor-β–activated kinase 1). A, Dual-luciferase reporter gene assay was performed to examine TAK1-induced NF-κB (nuclear factor-κB) activation in Nespas silenced (left) or Nespas-overexpressed (right) BV2 cell after oxygen-glucose deprivation (OGD) treatment for 24 h. B, RNA-binding protein immunoprecipitation (RIP) assay was used to detect the interaction between Nespas and TAK1 in BV2 cell after OGD treatment for 24 h. C, mRNA level of TAK1 in BV2 microglial cells transfected with increased dose of Nespas. Data are representative of 3 independent experiments with similar results (mean±SD). ***P<0.001. Ctrl indicates control; and siRNA-NC, siRNA-negative control.
Nespas Inhibits TRIM8-Induced K63-Linked Polyubiquitination of TAK1
Because of the invalid effect of Nespas on TAK1 mRNA expression, we suspected whether Nespas could affect the modulation level of TAK1. K63-linked polyubiquitination has been shown to play great roles in TAK1 activation.17 Therefore, we performed the immunoprecipitation experiment to examine the effect of Nespas on K63-linked polyubiquitination of TAK1. We observed that after OGD treatment, the K63-ub level of TAK1 was greatly enhanced in siRNA-Nespas–treated BV2 cells (Figure 6A). Because of the lack of ubiquitin ligase activity, we suspect that this effect may be because of the participation of another TAK1-related E3 ubiquitin ligase or deubiquitinating enzyme. TRIM8 has been reported to directly interact with TAK1 and promote K63-linked polyubiquitination of TAK1. Therefore, we examined the effect of Nespas on the interaction between TRIM8 and TAK1 after OGD stimulation. We found that after OGD treatment, the interaction between TRIM8 and TAK1 was greatly enhanced. However, silencing of Nespas significantly promotes the interaction, and overexpression of Nespas obviously suppressed OGD-induced interaction between TRIM8 and TAK1 (Figure III in the online-only Data Supplement; Figure 6B). We constructed several deletion mutants of TAK1 during immunoprecipitation experiment, and we found that TRIM8 interacts with region 200–291, located in the internal kinase domain of TAK1 (Figure 6C). Most importantly, we observed that the interaction between TRIM8 and 200–291 region of TAK1 was obviously suppressed in Nespas-overexpressed BV2 cells after OGD stimulation (Figure 6D). Taken together, these findings indicated that Nespas inhibits TRIM8-induced K63-linked polyubiquitination of TAK1 through suppressing the interaction between TRIM8 and TAK1.
Figure 6.
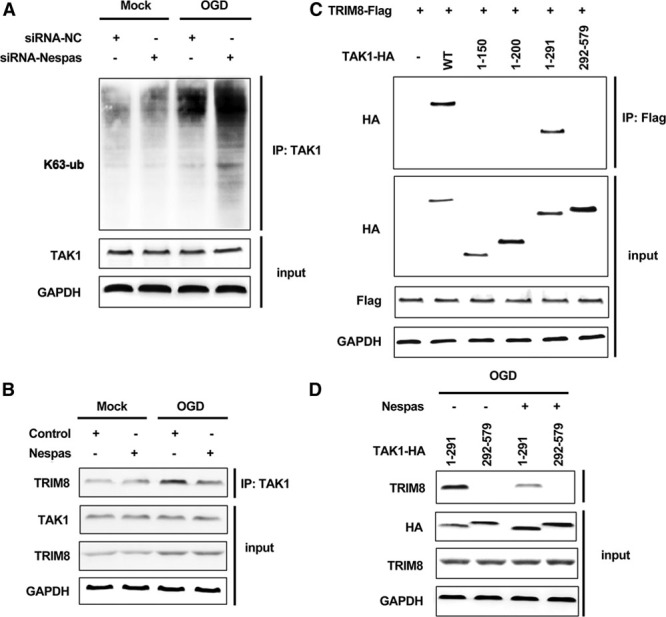
Nespas inhibits TRIM8 (tripartite motif 8)-induced K63-linked polyubiquitination of TAK1 (transforming growth factor-β–activated kinase 1). A, Western blot analysis of K63-linked polyubiquitination level of TAK1 in Nespas silenced BV2 cells after oxygen-glucose deprivation (OGD) treatment for 24 h. B, Immunoprecipitation assay was used to detect the interaction between TRIM8 and TAK1 in Nespas-overexpressed BV2 cells after OGD treatment for 24 h. C, Immunoprecipitation assay was used to detect the interaction between TRIM8 and deletion mutants of TAK1. D, Immunoprecipitation assay was used to detect the interaction between TRIM8 and TAK1 deletion mutants (1–291 and 292–579) in Nespas-overexpressed BV2 cells after OGD treatment for 24 h. HA indicates hemagglutinin; IP, immunoprecipitation; siRNA-NC, siRNA-negative control; and WT, wild type.
Discussion
To the best of our knowledge, the current study is the first article to demonstrate the role of lncRNA-Nespas in ischemic stroke, and it is also the first report to reveal that Nespas could inhibit the interaction between TRIM8 and TAK1, therefore suppressed TRIM8-induced K63-linked polyubiquitinaton of TAK1 and NF-κB activation. These findings suggested Nespas as a potential therapeutic target for the treatment of cerebral ischemia.
Ischemic stroke is one of the major causes of adult morbidity and mortality, representing a large health challenge around the world, and there is an urgent need for better understanding of the molecular pathogenesis during stroke and developing novel strategy against neuronal cerebral ischemia damages.8,19 Recently, more and more researchers revealed that different kinds of proteins or RNAs played great roles in molecular pathogenesis of ischemic stroke.20–23 In the current study, we found that Nespas expression was significantly increased in ischemic cerebral tissues and plasma of acute ischemic stroke as well as in the cultured BV2 microglial cells after OGD stimulation but not HT22 hippocampal cells. In addition, silencing of Nespas showed a larger cerebral infarct volume and a significantly more severe neurological deficit in MCAO mice, which indicated that Nespas could alleviate I/R-induced ischemic brain damage.
LncRNAs have been reported to participate in lots of complicated biological processes including ischemic stroke. LncRNA-H19 has been shown to promote neuroinflammation in ischemic stroke through driving HDAC1 (histone deacetylase 1)-dependent M1 microglial polarization.24 LncRNA-Fos downstream transcript (FosDT) was found to promote ischemic brain injury by interacting with RE-1-silencing transcription factor (REST)-associated chromatin-modifying proteins.25 Zhang et al14 suggested that LncRNA-Malat1 played antiapoptotic and anti-inflammatory roles in ischemic stroke by physically binding to Bim and E-selectin. In the present study, we observed that LncRNA-Nespas significantly suppressed microglial cell death after MCAO or OGD treatment; in addition, the expression of proinflammatory cytokines was also suppressed by Nespas overexpression, which suggested that Nespas played critical protective roles in ischemic stroke through alleviating cell apoptosis and neuroinflammation; in addition, we also found the regulation is not relevant with sexual dimorphism; the expression of Nespas and the effect of neuroinflammation regulation in female or male mice were consistent (female data not shown), which suggested that Nespas is a basic regulator of this process and there is no sexual dimorphism in Nespas-regulated immune responses.
TAK1 is one of the most crucial regulators of the innate immunity and the proinflammatory signaling pathway. Inhibition of TAK1 was shown to be an efficient way to interfere with multiple mechanisms in ischemic stroke, and TAK1 inhibitors were proved as efficient therapeutic method for the treatment of ischemic stroke.26 It has been reported that K63-linked polyubiquitination of TAK1 is necessary for NF-κB activation, and TRIM8 was reported as the main E3 ubiquitin ligase to regulate this process.27–29 In the current study, we found lncRNA-Nespas could negatively regulate NF-κB activation, therefore downregulate the expression of proinflammatory cytokines after MCAO or OGD treatment in microglial cells. Furthermore, we also observed that Nespas could directly interact with TAK1, which has never been reported before. Different from previous studies about the functions of lncRNAs, we found silencing of Nespas aggravates OGD-induced K63-linked polyubiquitination of TAK1, which suggested that Nespas played protective roles in ischemic stroke through modulating the activation of TAK1 by decreasing its K63-linked ubiquitin level. To investigate the specific mechanisms because of the lack of ubiquitin ligase activity, we examined the relationship between TRIM8 and Nespas. Interestingly, we found that overexpression of Nespas markedly suppresses the interaction between TRIM8 and TAK1, and we also observed that region 201–291 was critical for this interaction, which suggested Nespas mainly prevent TRIM8 from binding to and ubiquitinating TAK1, leading to its inactivation.
Conclusions
Collectively, our results demonstrated that Nespas prevents the interaction between TRIM8 and TAK1, leading to the inactivation of TAK1 and NF-κB, therefore alleviated MCAO- or OGD-induced cell death and neuroinflammation in microglial cells and finally protected from cerebral ischemia damages.
Acknowledgments
Drs Deng and Chen performed the experiments and drafted the manuscript. Dr Wang analyzed the data. Drs Gao and Jin performed the neurological evaluation and contributed to the data interpretation. Drs Lv and Zhang performed the immunofluorescence image analysis. Drs Sun and Liu assisted with the cell culture and Western blot. Drs Mo, Ma, and Song performed the experiments and collected the data. Dr Huo assisted with the experiment design. Drs Yan and Miao conceived and designed the study, analyzed and interpreted the data, and critically revised the manuscript.
Sources of Funding
This work was supported by the National Key Research and Development Program of China (Grant No. 2018YFC0115400), the National Natural Science Foundation of China (Grant No. 81671776, 61727807, 61633018), the Beijing Municipal Science & Technology Commission (Grant No. Z161100002616020, Z181100003118007, Z191100010618004), the Beijing Nova Program (Grant No. Z171100001117057), the Beijing Municipal Administration of Hospitals’ Youth Program (Grant No. QML20180506), Capital Medical University (PYZ2018080), the Natural Science Foundation of Beijing (Grant No. 7172070).
Disclosures
None.
Supplementary Material
{kind=link}
Footnotes
Drs Deng and Chen contributed equally.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/STROKEAHA.118.023376.
References
- 1.Zhang H, Sun X, Xie Y, Zan J, Tan W. Isosteviol sodium protects against permanent cerebral ischemia injury in mice via inhibition of NF-κB-mediated inflammatory and apoptotic responses. J Stroke Cerebrovasc Dis. 2017;26:2603–2614. doi: 10.1016/j.jstrokecerebrovasdis.2017.06.023. doi: 10.1016/j.jstrokecerebrovasdis.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 2.Blakeley JO, Llinas RH. Thrombolytic therapy for acute ischemic stroke. J Neurol Sci. 2007;261:55–62. doi: 10.1016/j.jns.2007.04.031. doi: 10.1016/j.jns.2007.04.031. [DOI] [PubMed] [Google Scholar]
- 3.Li T, Qin JJ, Yang X, Ji YX, Guo F, Cheng WL, et al. The ubiquitin E3 ligase TRAF6 exacerbates ischemic stroke by ubiquitinating and activating Rac1. J Neurosci. 2017;37:12123–12140. doi: 10.1523/JNEUROSCI.1751-17.2017. doi: 10.1523/JNEUROSCI.1751-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khoshnam SE, Winlow W, Farzaneh M, Farbood Y, Moghaddam HF. Pathogenic mechanisms following ischemic stroke. Neurol Sci. 2017;38:1167–1186. doi: 10.1007/s10072-017-2938-1. doi: 10.1007/s10072-017-2938-1. [DOI] [PubMed] [Google Scholar]
- 5.Chen S, Dong Z, Cheng M, Zhao Y, Wang M, Sai N, et al. Homocysteine exaggerates microglia activation and neuroinflammation through microglia localized STAT3 overactivation following ischemic stroke. J Neuroinflammation. 2017;14:187. doi: 10.1186/s12974-017-0963-x. doi: 10.1186/s12974-017-0963-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shin JA, Lim SM, Jeong SI, Kang JL, Park EM. Noggin improves ischemic brain tissue repair and promotes alternative activation of microglia in mice. Brain Behav Immun. 2014;40:143–154. doi: 10.1016/j.bbi.2014.03.013. doi: 10.1016/j.bbi.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 7.Lucas SM, Rothwell NJ, Gibson RM. The role of inflammation in CNS injury and disease. Br J Pharmacol. 2006;147(suppl 1):S232–S240. doi: 10.1038/sj.bjp.0706400. doi: 10.1038/sj.bjp.0706400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wen Y, Yu Y, Fu X. Lncrna gm4419 contributes to OGD/R injury of cerebral microglial cells via ikappab phosphorylation and nf-kappab activation. Biochem Biophys Res Commun. 2017;487:923–929. doi: 10.1016/j.bbrc.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 9.Yang L, Liu CC, Zheng H, Kanekiyo T, Atagi Y, Jia L, et al. LRP1 modulates the microglial immune response via regulation of JNK and NF-κB signaling pathways. J Neuroinflammation. 2016;13:304. doi: 10.1186/s12974-016-0772-7. doi: 10.1186/s12974-016-0772-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ponting CP, Belgard TG. Transcribed dark matter: meaning or myth? Hum Mol Genet. 2010;19:R162–168. doi: 10.1093/hmg/ddq362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shi X, Sun M, Liu H, Yao Y, Song Y. Long non-coding RNAs: a new frontier in the study of human diseases. Cancer Lett. 2013;339:159–166. doi: 10.1016/j.canlet.2013.06.013. doi: 10.1016/j.canlet.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 12.Kung JT, Lee JT. RNA in the loop. Dev Cell. 2013;24:565–567. doi: 10.1016/j.devcel.2013.03.009. doi: 10.1016/j.devcel.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park S, Lee M, Chun CH, Jin EJ. The lncRNA, nespas, is associated with osteoarthritis progression and serves as a potential new prognostic biomarker. Cartilage. 2019;10:148–156. doi: 10.1177/1947603517725566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang X, Tang X, Liu K, Hamblin MH, Yin KJ. Long noncoding RNA malat1 regulates cerebrovascular pathologies in ischemic stroke. J Neurosci. 2017;37:1797–1806. doi: 10.1523/JNEUROSCI.3389-16.2017. doi: 10.1523/JNEUROSCI.3389-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee JK, Tansey MG. Microglia isolation from adult mouse brain. Methods Mol Biol. 2013;1041:17–23. doi: 10.1007/978-1-62703-520-0_3. doi: 10.1007/978-1-62703-520-0_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kurland DB, Gerzanich V, Karimy JK, Woo SK, Vennekens R, Freichel M, et al. The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. J Neuroinflammation. 2016;13:130. doi: 10.1186/s12974-016-0599-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo L, Dong W, Fu X, Lin J, Dong Z, Tan X, et al. Tripartite Motif 8 (TRIM8) positively regulates pro-inflammatory responses in pseudomonas aeruginosa-induced keratitis through promoting K63-linked polyubiquitination of TAK1 protein. Inflammation. 2017;40:454–463. doi: 10.1007/s10753-016-0491-3. doi: 10.1007/s10753-016-0491-3. [DOI] [PubMed] [Google Scholar]
- 18.Shao M, Chen G, Lv F, Liu Y, Tian H, Tao R, et al. LncRNA TINCR attenuates cardiac hypertrophy by epigenetically silencing CaMKII. Oncotarget. 2017;8:47565–47573. doi: 10.18632/oncotarget.17735. doi: 10.18632/oncotarget.17735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Winklewski PJ, Radkowski M, Demkow U. Cross-talk between the inflammatory response, sympathetic activation and pulmonary infection in the ischemic stroke. J Neuroinflammation. 2014;11:213. doi: 10.1186/s12974-014-0213-4. doi: 10.1186/s12974-014-0213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tang Z, Gan Y, Liu Q, Yin JX, Liu Q, Shi J, et al. CX3CR1 deficiency suppresses activation and neurotoxicity of microglia/macrophage in experimental ischemic stroke. J Neuroinflammation. 2014;11:26. doi: 10.1186/1742-2094-11-26. doi: 10.1186/1742-2094-11-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Wei R, Zhang L, Tan Y, Qian C. Sirtuin 6 protects the brain from cerebral ischemia/reperfusion injury through NRF2 activation. Neuroscience. 2017;366:95–104. doi: 10.1016/j.neuroscience.2017.09.035. doi: 10.1016/j.neuroscience.2017.09.035. [DOI] [PubMed] [Google Scholar]
- 22.Li XQ, Chen FS, Tan WF, Fang B, Zhang ZL, Ma H. Elevated microRNA-129-5p level ameliorates neuroinflammation and blood-spinal cord barrier damage after ischemia-reperfusion by inhibiting HMGB1 and the TLR3-cytokine pathway. J Neuroinflammation. 2017;14:205. doi: 10.1186/s12974-017-0977-4. doi: 10.1186/s12974-017-0977-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi H, Huang Y, Yang F, Liu W, He S, Hu X. MicroRNA-182 aggravates cerebral ischemia injury by targeting inhibitory member of the ASPP family (iASPP). Arch Biochem Biophys. 2017;620:52–58. doi: 10.1016/j.abb.2016.05.002. doi: 10.1016/j.abb.2016.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Wang J, Zhao H, Fan Z, Li G, Ma Q, Tao Z, et al. Long noncoding RNA H19 promotes neuroinflammation in ischemic stroke by driving histone deacetylase 1-dependent M1 microglial polarization. Stroke. 2017;48:2211–2221. doi: 10.1161/STROKEAHA.117.017387. doi: 10.1161/STROKEAHA.117.017387. [DOI] [PubMed] [Google Scholar]
- 25.Mehta SL, Kim T, Vemuganti R. Long noncoding RNA FosDT promotes ischemic brain injury by interacting with REST-associated chromatin-modifying proteins. J Neurosci. 2015;35:16443–16449. doi: 10.1523/JNEUROSCI.2943-15.2015. doi: 10.1523/JNEUROSCI.2943-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ridder DA, Schwaninger M. TAK1 inhibition for treatment of cerebral ischemia. Exp Neurol. 2013;239:68–72. doi: 10.1016/j.expneurol.2012.09.010. doi: 10.1016/j.expneurol.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 27.Fan Y, Yu Y, Mao R, Zhang H, Yang J. Tak1 lys-158 but not lys-209 is required for il-1beta-induced lys63-linked tak1 polyubiquitination and ikk/nf-kappab activation. Cell Signal. 2011;23:660–665. doi: 10.1016/j.cellsig.2010.11.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fan Y, Yu Y, Shi Y, Sun W, Xie M, Ge N, et al. Lysine 63-linked polyubiquitination of TAK1 at lysine 158 is required for tumor necrosis factor alpha- and interleukin-1beta-induced IKK/NF-kappaB and JNK/AP-1 activation. J Biol Chem. 2010;285:5347–5360. doi: 10.1074/jbc.M109.076976. doi: 10.1074/jbc.M109.076976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li Q, Yan J, Mao AP, Li C, Ran Y, Shu HB, et al. Tripartite motif 8 (trim8) modulates tnfalpha- and il-1beta-triggered nf-kappab activation by targeting tak1 for k63-linked polyubiquitination. Proc Natl Acad Sci USA. 2011;108:19341–19346. doi: 10.1073/pnas.1110946108. [DOI] [PMC free article] [PubMed] [Google Scholar]