

A genomic assembly of the goldfish (Carrassius auratus) increases the utility of goldfish in research and agriculture.
Abstract
For over a thousand years, the common goldfish (Carassius auratus) was raised throughout Asia for food and as an ornamental pet. As a very close relative of the common carp (Cyprinus carpio), goldfish share the recent genome duplication that occurred approximately 14 million years ago in their common ancestor. The combination of centuries of breeding and a wide array of interesting body morphologies provides an exciting opportunity to link genotype to phenotype and to understand the dynamics of genome evolution and speciation. We generated a high-quality draft sequence and gene annotations of a “Wakin” goldfish using 71X PacBio long reads. The two subgenomes in goldfish retained extensive synteny and collinearity between goldfish and zebrafish. However, genes were lost quickly after the carp whole-genome duplication, and the expression of 30% of the retained duplicated gene diverged substantially across seven tissues sampled. Loss of sequence identity and/or exons determined the divergence of the expression levels across all tissues, while loss of conserved noncoding elements determined expression variance between different tissues. This assembly provides an important resource for comparative genomics and understanding the causes of goldfish variants.
INTRODUCTION
Over a thousand years of ornamental breeding has generated more than 300 goldfish variants in body shape, fin configuration, eye style, and coloration (1), which make goldfish an excellent genetic model system for understanding the evolution of body shape (1). In addition, goldfish have long been used in research to study a wide array of biological processes such as pigmentation (2, 3), disease and environment interactions (4, 5), behavior (6), physiology (7), neurobiology (8, 9), reproduction and growth (10), and neuroendocrine signaling (11). The best estimate, based on mitochondrial DNA analysis from domesticated and wild-caught goldfish, is that domesticated goldfish were derived from fish in southern Asia, possibly from the lower Yangtze River (12).
Like the closely related common carp, goldfish share the same whole-genome duplication (WGD) event that occurred ≈8 to 12 million years (Ma) ago, which is believed to have been an allotetraploidy event (i.e., a hybrid of two closely related species created with both chromosome sets of each parents being present in gametes) (13). This fusion occurred after the divergence from grass carp (Ctenopharyngodon idella) but before goldfish diverged from the common carp. This event is quite recent compared to other animal WGD events such as the one that occurred in teleosts (320 to 350 Ma ago) (14), in the Salmoniformes including salmon (50 to 80 Ma ago) (15), and the allotetraploid event of Xenopus laevis (17 to 18 Ma ago) (16), and we now have two closely related but different species that resulted from the same genome duplication event with near-complete genome sequences. Thus, comparing how the goldfish genome has diverged from the common carp genome provides an excellent opportunity to study how genes change during the course of speciation. In addition, the relative evolutionary proximity of goldfish and carp to the commonly used model organism zebrafish provides additional reference sequences for identifying conserved elements involved in gene regulation [conserved noncoding elements (CNEs)] (17, 18), at sensitivities not available from comparing much more distantly related vertebrate genomes, including most other sequenced fish species.
Here, we report a contiguous, accurate, and proximately complete genome assembly of a common goldfish line, Wakin, and shed light on how the genome and gene expression evolved after the carp WGD. The genome represents an essential resource for the study of the greater than 300 goldfish variants and for the understanding of genome evolution in related fish species.
RESULTS
Genomic assembly and annotation
The estimated size of the goldfish genome ranges from 1.6 to 2.08 pg according to the Animal Genome Size Database (19), similar to that of the common carp (1.8 pg). Using a Wakin goldfish generated by heat-shock gynogenesis (Fig. 1A) (20), we generated ~16.4 million reads (71X coverage) from PacBio Single Molecule, Real-Time (SMRT) cells, which were corrected and assembled into 9415 contigs by the Canu assembler (21). The Canu assembly was ~1849 megabase pairs (Mbp) with an N50 of 817 kbp. There were 6937 contigs (497 Mbp) with a relative read coverage of <0.6, which indicated that our sample was not fully homozygous with ~249 Mbp being heterozygous. This was consistent with the 25-nucleotide oligomer spectrum from Illumina short-read sequencing (fig. S1 and table S2). We then made linkage groups (LGs) using a published genetic map for the goldfish (22) in combination with the OneMap program (23). This chromosome-sized, final assembly (cauAur01) contained 50 large LGs, with a total length of 1246 Mbp linked and approximately 500 Mbp in unplaced contigs or scaffolds (for summary, see Table 1). By mapping the Illumina short reads to the carAur01 assembly, we estimated that the assembly has an error rate of <1 per 50,000 bases, and 98.5% reads were mappable (96% properly paired), indicating a highly accurate assembly.
Fig. 1. Basic statistics for the goldfish genome in comparison to grass carp, common carp, and zebrafish.
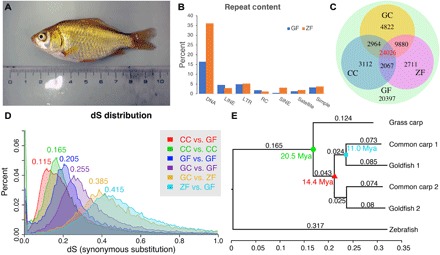
(A) The gynogenetic goldfish used for sequencing before sacrifice. (B) Transposable elements distribution for goldfish (GF) and zebrafish (ZF). (C) Distribution of orthologous/ohnologous gene pairs by synonymous substitution among four species: zebrafish, grass carp (GC), common carp (CC), and goldfish. Numbers are a count of the homologous genes shared among zebrafish, common carp, and goldfish. (D) Rate of synonymous base changes (dS) for various species comparisons. (E) The phylogenetic tree shows the time of divergence of grass carp from goldfish and common carp (green circle), the WGD (red triangle), and divergence common carp and goldfish (cyan square). Each genome from the duplication was analyzed separately (chromosomes randomly assigned) and are denoted with _1 or _2 for both common carp and goldfish. (Photo credit: Yoshihiro Omori, Osaka University).
Table 1. Assembly statistics.
| CarAur01 (Canu + genetic map) | |
| Longest scaffold | 37,185 kbp |
| N10 | 30,202 kbp (n = 10) |
| N50 | 22,763 kbp (n = 14) |
| N90 | 86.8 kbp (n = 1506) |
| Total length | 1,820,635,051 bp |
| No. of LGs | 50 |
| Total length of LGs | 1,246,641,604 bp |
We sequenced one additional gynogenetic and one “wild-type” Wakin fish to ~70X coverage using Illumina short-read sequencing. In aggregate, we identified 12,163,467 unique single nucleotide variants (SNVs) and 2,316,524 deletion/insertion variants (DIVs) from these fish and estimated the general polymorphism rate in goldfish to be approximately 1%. The number of SNVs and DIVs was comparable to those of the common carp (18,949,596 SNVs and 1,694,102 DIVs) (24).
The goldfish genome showed an overall repeat content of 39.6%, which was similar to the 39.2% for common carp (24), higher than that for many of the sequenced teleost genomes [33% in cave fish (25) and 30.68% in Oryzias latipes (26)], but much lower than that of the zebrafish (54.3%) (27) and Atlantic salmon (58%) (15). The most enriched repeat classes were DNA transposons, of which hAT (3.87%), DNA (3.08%), TC1/mariner (2.28%), and CMC (2.05%) were the top enriched superfamiles. The long interspersed nuclear element (LINE)/L2 (2.67%), long terminal repeat (LTR)/Gypsy (2.14%), rolling circle transposition (RC)/Helitron (1.89%), and LTR/DIRS (1.18%) superfamilies were also somewhat enriched (>1%). Goldfish contained more LINEs but fewer short interspersed nuclear elements (SINEs) and DNA transposons than zebrafish (Fig. 1B and table S3).
We sequenced and assembled total RNA from seven adult tissues (brain, gill, bone, eye heart, skeletal muscle, and tail fin). Using MAKER (28), we identified 80,062 protein-coding genes, of which 9738 genes were masked because they were counted twice in the genomic regions that remained heterozygous. The final assembly, carAur01, contained 70,324 unmasked gene models and 479,594 exons. The gene completeness was assessed by Benchmarking Universal Single-Copy Orthologs (BUSCOs) (29) using the vertebrate core gene sets, resulting in 2710 complete (90%), 157 fragmented (5%), and 156 (5%) missing BUSCOs of 3023 total BUSCOs (see Table 2 and table S4). Fifty-eight percent of the BUSCO genes could be found in two complete copies; 83.11 to 96.93% of the RNA sequencing (RNA-seq) reads from seven goldfish tissues could be mapped to the assembly. These assessments indicated that our gene models were of very good quality and significantly more complete than that of the published common carp assembly. On the basis of alignment evidence from Ensembl noncoding RNA sequences (release 85), we predicted 11,820 noncoding RNA transcripts, including 574 microRNAs (miRNAs). miRBase (http://www.mirbase.org) hairpin sequence alignment identified 1037 miRNA loci.
Table 2. Annotations statistics.
CNE, conserved noncoding element (i.e., potential enhancers/promoters); GO, gene ontology.
| Goldfish | Common carp |
Zebrafish (danRer10) |
|
|
Assembly size (bp) |
1,820,635,051 | 1,713,641,436 | 1,371,719,383 |
| GC content | 37.48% | 36.99% | 36.64% |
| Repeats (bp) | 721,087,053 (39.6%) | 672,246,354 (39.2%) | 745,150,642 (54.3%) |
|
Protein-coding genes |
70,324 | 66,999 | 25,600 (Ensembl release 85) |
| Genes with GO | 49,272 | – | 18,779* |
| Exons | 556,731 | 547,164 | 276,021 |
|
Genes with InterPro |
49,272 | 44,845* | 24,204* |
| miRNA | 1,037 | – | 769 |
|
ncRNA (noncoding RNA) |
11,820 | – | – |
|
4-way CNE counts |
486,767 | 484,139* | 237,891* |
| 4-way CNE bp | 95,815,233 | 97,818,440* | 44,090,004* |
|
Missing BUSCOs (of 3023) |
167 | 330* | 0 (used for original BUSCO set) |
*Data generated from this study.
Approximately 50,000 coding genes had a reciprocal best hit (RBH) or second best hit to genes in zebrafish, grass carp, or common carp out of which 24,026 genes aligned to all three species (Fig. 1C). The spectrum of synonymous substitutions (dS) between RBH pairs showed peaks at 0.115, 0.205, and 0.415 for common carp–goldfish (Fig. 1D, CC versus GF), between goldfish WGD paralogs (Fig. 1D, GF versus GF), and zebrafish-goldfish (Fig. 1D, ZF versus GF) comparisons, respectively. As expected, this indicated that the WGD happened before the divergence of goldfish and common carp. On the basis of the maximum likelihood phylogenetic tree and using 20.5 Ma ago (30) as the grass carp–common carp divergence point, we deduced that the speciation time for common carp and goldfish was ~11.0 Ma ago and that the WGD time was ~14.4 Ma ago (Fig. 1E), which was consistent with Larhammer and Risinger’s estimate (31) but slightly longer ago than other more recent publications’ predictions (13, 24). A fully implemented University of California, Santa Cruz-style genome browser for carAur01 is available at https://research.nhgri.nih.gov/goldfish/ (fig. S2).
Extensive retention of synteny and collinearity after WGD
Although goldfish diverged from zebrafish ~60 Ma ago, the genome of goldfish retained extensive collinearity/synteny with that of zebrafish (or grass carp). About 97.4% of RBH or second best ortholog gene pairs between goldfish and zebrafish were located in the 25 synteny triples, including one zebrafish chromosome and two corresponding goldfish LGs (figs. S4 to S7). No large interchromosomal translocations were found between the 25 zebrafish chromosomes and the 50 goldfish LGs (Fig. 2, A and B). The 1:2 relationship and extensive retained collinearity/synteny between zebrafish and goldfish chromosome were consistent with the WGD hypothesis (13). Alignment between zebrafish chromosomes and two WGD descended goldfish LGs showed large collinear blocks, although there were large intrachromosomal rearrangements (Fig. 2C and fig. S7), which indicated that the overall gene order in the goldfish genome remained very stable after divergence from zebrafish.
Fig. 2. Chromosome collinearity is stable from zebrafish to goldfish.
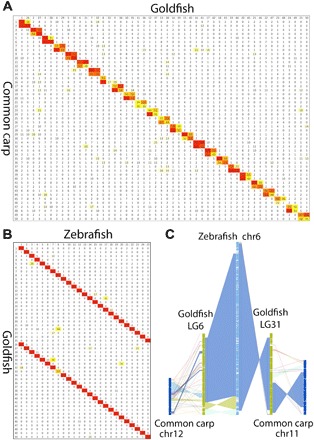
(A) Reciprocal BLAST best gene pair counts for each pair of chromosomes between common carp and goldfish. Color from yellow to red indicates low to high counts, respectively. (B) Reciprocal BLAST best gene pair counts for each pair of chromosomes between goldfish and zebrafish. Color from yellow to red indicates low to high counts, respectively. Goldfish to common carp results in 50 bivalents, and goldfish to zebrafish shows a clear 1:2 relationship. (C) Chain alignment along zebrafish chromosome six and the two duplicated chromosomes from goldfish and common carp. Very large stretches of collinearity are readily visible between zebrafish and goldfish, as are simple intrachromosomal inversions. The more fragmented relationship with common carp (e.g., chr12) may be the result of a more fragmented common carp assembly.
Only 55.3% of RBH orthologous pairs were located in the 25 LG quadruplets (two goldfish paralog LGs and two common carp paralog LGs derived from the same WGD ancestral chromosome), and there were also plenty of interchromosomal translocations between the paralog LGs, suggesting intensive interchromosomal translocations between common carp LGs after the WGD, especially after speciation from goldfish (Fig. 2). Comparisons between common carp and goldfish orthologous LGs suggested that there were some small, interchromosomal translocations, although they maintained very strong colinearity (Fig. 2 and fig. S7).
Evolution after WGD
Several fish genomes in the Cyprinidae family have been sequenced now, including zebrafish (27), grass carp (32), common carp (24), fathead minnow (33), Amur ide (34), and Sinocyclocheilus (35). We used four of them: zebrafish, grass carp, common carp, and now goldfish, which have a very useful evolutionary relationship for directly examining the processes of gene nonfunctionalization (non-F), subfunctionalization (sub-F), and neofunctionalization (neo-F) (36) over a relatively short time (10 ~ 20 Ma) after WGD. Zebrafish is distantly and equally related to all three carps (common ancestor was ~60 Ma ago, roughly similar to a human-to-mouse genomic comparison), such that the conserved sequences from zebrafish to carp are limited to exonic sequences and CNEs (17, 18) that are strongly enriched for enhancers and promoters. Common carp and goldfish speciated from grass carp ~20 Ma ago (30), the genome duplication occurred ~14 Ma ago, and then goldfish and common carp speciated roughly 11 Ma ago (Fig. 1E). This timeline allows us to watch as duplicated genes naturally decay from the tetraploid state, as was done for the common carp (37), and the common carp–goldfish separation allows us to watch this occur twice in parallel.
Gene loss
We should be able to map one grass carp or zebrafish gene to two goldfish or common carp “ohnologous” genes. We identified 19,040 ortholog-paralog gene clusters with at least one zebrafish gene in each cluster. There were 16,455 (15,202) clusters with both paralogs retained and 2341 (3462) singletons in goldfish (common carp). Therefore, 12.4% of the duplicated gene pairs had lost one copy in goldfish, while the common carp appeared to have had a higher rate of gene loss (18.5%) (Fig. 3). The higher loss rates in common carp may reflect the more fragmented assembly of that genome or differences in gene identification methodology and not an actual increase in gene loss, as suggested by the lower completeness of the BUSCO genes in the common carp assembly (Table 2). In total, genes were lost at a rate of 0.43% per million years (equal to 12.4% in 14.4 Ma) in goldfish after the WGD, compared to 0.25% per Ma (see the Supplementary Materials for computational details) in salmon during the 80 Ma following the Salmoniformes WGD (15) and the 0.06% per Ma (approximately 7.4% gene clusters without zebrafish or grass carp orthologs, i.e., 7.4% of gene loss occurred between zebrafish and grass carp over 120 Ma), suggesting that gene loss rate increased after the WGD event, which is supported by the observed faster loss [22% = (1 to 56%)/2 in 18 Ma or 1.2% per Ma] in X. laevis after the frog allotetraploid event (16). We then went on to ask whether there were specific classes of genes that were either more or less likely than average to be lost. We examined the percentage of genes in a Gene Ontology (GO) term category that were lost compared to the total percentage the category represented. Oxidoreductase activity, nuclease activity, and methyltransferase activity were much more likely than average to be lost, while protein binding and transcription factors were retained at a higher than average rate (see fig. S8).
Fig. 3. The evolutionary relationships between zebrafish, grass carp, common carp, and goldfish can be used to study the dynamics of gene loss after WGD events.
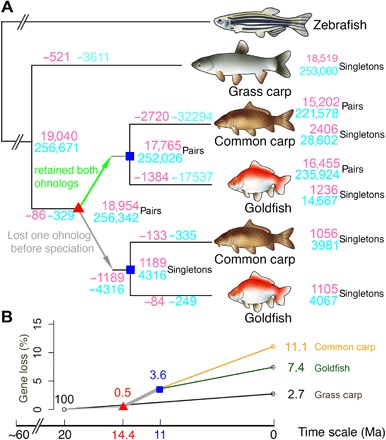
(A) Using zebrafish as the reference, the tree tracks gene and CNE loss at different evolutionary branch points. Numbers on nodes or leaves indicate retained genes (pink) or CNEs (skyblue). Negative number on the branches indicates the number of lost genes (pink) or CNEs (skyblue) on the corresponding branch. The red triangle represents the carp WGD event at 14.4 Ma ago. The blue square marks the speciation of common carp and goldfish at 11.0 Ma ago. A maximum-likelihood phylogenetic tree was constructed by using the third position of all codons of ohnologous genes. (B) Decay curve of gene loss. The rates of gene loss accelerated after the genome duplication event (i.e., thick gray line between the red triangle and blue square). We assume that most cases where both copies of a gene were lost in either goldfish or carp occurred after separation from grass carp but before the WGD.
CNE loss
We were able to analyze enhancer/promoter loss rates in a four-way comparison using CNE loss as the proxy for altered regulatory function. When we directly compared zebrafish and grass carp (using common carp or goldfish as the reference), 15,745 CNEs were not shared between them. Assuming that they were lost or gained either in zebrafish or grass carp, we estimated that the lost rate was less than 131 CNEs per Ma. Using zebrafish as the reference, 3611 CNEs were lost during the 40 Ma (or 90 CNEs per Ma) from grass carp. There were 329 CNEs (54 CNEs per Ma), where the two duplicated copies were missing in both goldfish and common carp. These were CNE losses that presumably happened after the split from grass carp but before the WGD. Goldfish and common carp shared 4316 one-copy CNE losses, presumably all or most of those occurred in the 3 Ma between the genome duplication and speciation events, resulting in a rate of 1439 per Ma. In the ~11 Ma since the common carp–goldfish split, 16,102 and 28,937 CNE paralog pairs became singleton or totally lost in goldfish and common carp, respectively, or 1463 and 2631 CNEs per Ma (Fig. 3). The above scenario indicated an accelerated CNE loss after the WGD, and the effect persisted after the speciation of goldfish and common carp. A full history of exon/CNE loss is shown in Fig. 3.
Divergence of gene expression
It is logical to assume that as a genome goes through the evolutionary process of rediploidization, genes that were once duplicates of each other will begin to diverge in location of expression or in specific function from each other. The goldfish–carp duplication event was relatively recent, which makes it possible to illuminate how sequence divergence, exon loss, and CNE loss shaped the expression pattern of ohnolog genes in the ~14 Ma after the WGD. We identified 2481 colinear ohnolog blocks covering 1004 Mbp of the carAur01 assembly, including 44,650 protein-coding genes (6385 singleton), 14,527 singleton exons, and 8617 singleton CNEs.
We compared the RNA expression level between 10,399 ohnolog gene pairs (20,798 genes) in the ohnolog blocks across seven tissues. Six hundred forty-nine (6.2%) of these gene pairs contained one silenced gene [i.e., TPM (transcripts per million) < 1 in all tissues], which may be genes that had become nonfunctionalized or simply not expressed in the tissues profiled. The silenced genes showed a significantly higher rate of exon loss compared to the other genes (Fisher’s exact test); 2895 (29.7%) of the remaining ohnolog pairs showed divergent expression (i.e., a Pearson’s correlation coefficient of <0.6 or a Euclidean distance of ≥5; Fig. 4A), 7149 (68.7%) pairs coexpressed (TPM ≥ 1) in at least three tissues compared to 75% in common carp (37), and 1273 (13%) ohnolog pairs contained at least one tissue in which only one ohnolog exhibited expression.
Fig. 4. Gene expression is affected by changes in sequence, exon loss, and CNE loss.
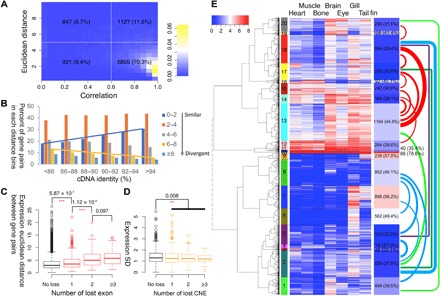
(A) Histogram of expression correlation (x axis) and expression Euclidean distance (y axis) between WGD ohnolog gene pairs. Each box lists the number of ohnolog pairs (×2 for total genes) and the percentage of the total number of pairs this group represents. Most of the genes (70.3%) had a correlation of 0.6 or better. (B) Expression distance distribution in different cDNA identity groups. The more closely related the cDNA sequence, the more closely correlated gene expression was. (C) Boxplot of expression distance in gene groups with different numbers of lost exons. The more exons lost, the less related gene expression becomes. Asterisks mark statistically significant differences. (D) Boxplot of tissue expression SD in gene groups with different numbers of CNEs lost. Similar to exons, loss of CNEs correlates with loss of concordant expression, but the effect size is smaller. Asterisks denote significant differences. (E) Gene expression clustered into 20 groups for the 19,500 ohnologous genes. Heatmap and the keys indicate the value of log2(TPM + 1). Left color bar indicates different clusters. Right bars show the number and percentage of the gene pairs in the same cluster. Colored links indicate the number of gene pairs split between different clusters, only numbers larger than 100 were plotted, and thicker links indicate larger counts.
To illuminate which type of mutations correlate with divergence of the expression between ohnolog gene pairs, we divided these gene pairs into different groups according to their cDNA sequence identity, number of exons lost, or number of CNE lost and looked for correlations between group assignment and expression divergence. We found that in the low–sequence identity groups, there was a greater percentage of diverged gene pairs and a lower percentage of diverged gene pairs in the high–sequence identity groups (Fig. 4B, yellow line), while the trend was reversed for less-diverged gene pairs (Fig. 4B, blue line), indicating that expression distance increased as the sequence identity decreased. Function enrichment analysis revealed that low identity gene pairs were enriched in functions involving the immune system, while “regulation of metabolic process,” “development,” “regulation of gene expression,” “regulation of biosynthetic process,” “cell surface receptor signaling pathway,” and “ion transport” were enriched in ohnolog pairs with high sequence identity (Fisher’s exact test, P < 0.05 and false discovery rate < 0.1). There was a significant increase in expression distance between the no-exon-lost (gene without exon loss) group and the one-exon-lost (gene pairs that lost exactly one exon) groups (one-sided Fisher’s exact test, P = 5.87 × 10−07). The more exons a gene pair lost, the more the expression diverged (Fig. 4C). We did not find a significant relationship between the number of nearby CNEs lost and the expression distance or correlation. However, in the ohnolog gene pairs with CNE loss but no exon loss, the tissue expression SD decreased in the genes that lost CNEs (one-sided Fisher’s exact test, P = 0.008), which indicated that the loss of CNEs reduced the expression variance among different tissues (dynamic range) rather than affected the expression divergence between ohnolog gene pairs, i.e., CNE loss reduced tissue-specific expression variation (Fig. 4D and example in fig. S10) (38).
Nineteen thousand five hundred genes (or 9750 gene pairs, not including the silenced singletons) were classified into 20 clusters according to a plateau in their expression Euclidean distance (Fig. 4E and figs. S11 to S13). Ohnologs were classified into different clusters in 62.4% of gene pairs, which decrease to 46.9% when classified into eight clusters (another local plateau), suggesting either a rapid expression divergence between ohnolog gene pairs in the first ~14 Ma since the WGD event or some significant differences in gene expression that existed among parental species before the allotetraploid genome fusion event. Most of the shared gene pairs fell within two super clusters: clusters 1 to 9 (Fig. 4E, blue curve bundles) and clusters 12 to 20 (Fig. 4E, red curve bundles). However, there were 2508 gene pairs that were not in the same cluster within the two different super clusters. We found that there were fewer numbers of genes with lost exons or CNEs in the four most highly expressed clusters (10, 11, 12, and 15), especially in the highest expression cluster 10, in which there were no exon or CNE losses between the pairs. Similar to gene loss, genes that were more likely to maintain concordant expression were often involved in cell signaling and gene regulation (signaling molecules and transcription factors) (fig. S13).
The four major evolutionary mechanisms of retained duplicated ohnologs are as follows: conserved coexpression of the two ohnologs, non-F of one copy (39), sub-F (36, 38), and neo-F (36, 38, 40, 41). To identify genes representing each mechanism, we analyzed expression [(FPKM) fragment per kilo base pairs per million reads] of 8483 gene triplets (one zebrafish ortholog and both duplicated goldfish ohnologs, zebrafish orthologs with an FPKM of ≥1 in at least one tissue, and 177 triplets silenced in all three genes were removed and not included), across six common tissues between zebrafish and goldfish (Fig. 5A). A triplet was denoted as double correlated if the expression pattern of the zebrafish gene was correlated with both goldfish ohnologs and single correlated if it correlated with exactly only one goldfish ohnolog. We denoted FPKM of an “ohno-pair” as the sum of FPKM of both goldfish ohnologs. A triplet was defined as dosage correlated if the expression pattern of zebrafish ortholog and ohno-pair was correlated.
Fig. 5. Systematic analysis of gene expression changes between duplicated genes can detect gene extinction, sub-F, and neo-F events.
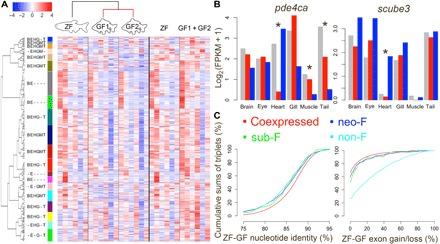
(A) Genes clustered into 20 groups for the 8483 zebrafish-goldfish gene triplets. Heatmap and the keys indicate the normalized value (z score) of log2(FPKM + 1). The left color bar indicates different clusters, the text next to the cluster color bar indicates major zebrafish-expressed tissue in each cluster, and unlabeled ones are expressed in all zebrafish tissues. B, brain; E, eye; H, heart; G, gill; M, muscle; T, tail fin. (B) Example of expression of subfunctionalized (left) and neofunctionalized (right) genes. Gray bar, zebrafish; red and blue bar, two goldfish orthologs. Asterisks indicate tissue(s) associated with sub-F or neo-F. (C) Cumulative sum of triplets in different zebrafish-goldfish nucleotide identity groups (left) and exon gain/loss groups. Genes in non-F, neo-F, and sub-F triplets have low nucleotide identity and higher exon gain/loss than the coexpressed group. Genes in sub-F and neo-F triplets have medial exon gain/loss.
We found that goldfish usually showed higher sequence similarity between one ohnolog and the corresponding zebrafish ortholog, while the other ohnolog showed much higher divergence (fig. S14). About 6% of ancestral exons/CNEs with zebrafish orthologs lost exactly one copy in goldfish, and there were 547 ohnolog pairs with reciprocal exon loss (tables S5 and S6).
Goldfish ohnologs showed a high (or medial; see Methods for definitions) expression correlation to their zebrafish ortholog in 58.8% (73.7%) of triplets in one of three configurations: double correlated, single correlated, or dosage correlated. Expression was significantly more correlated between goldfish ohnologs than between zebrafish and the sum of goldfish pairs (two-sided paired t test between correlations coefficients, P < 1 × 10−6), which was still more conserved than between zebrafish and each individual goldfish ohnolog (two-sided paired t test, P < 1 × 10−6; fig. S15). This trend can also be visualized in the expression patterns in Fig. 5A. Preservation of expression patterns for one or both ohnologs after WGD was also observed in Atlantic salmon that underwent the salmonid-specific WGD (Ss4R) (15), as well as for the teleost WGD ohnologs in zebrafish compared to the gar ortholog (42). All these correlations indicated a strong purifying selection maintaining expression of ohnolog(s) after a WGD. Of the 2084 triplets with differentially expressed genes between zebrafish and goldfish ohnologs, only 372 were differentially expressed between the zebrafish gene and the summed expression of the goldfish “ohno-pairs.” In addition, more goldfish ohno-pairs coexpressed in all six tissues compared to zebrafish than did individual goldfish ohnologs (table S7), suggesting that most of these genes were subject to strong selection to maintain dosage balance, possibly stabilizing duplicate gene retention (43).
We found 5345 (63.0%) coexpressed, 672 (7.92%) non-F, 39 (0.46%) sub-F, and 321 (3.78%) neo-F triplets using an “on-off” identification method (table S8; see Methods), and that 3506 (41.3% of total) of the coexpressed ohnologs also coexpressed with the zebrafish ortholog. Examples of expression from sub-F and neo-F triplets are shown in Fig. 5B; gene pde4ca was subfunctionalized, gene expression in heart was shifted to the ohnolog on LG27, while expression in tail (or muscle) shifted to the one on LG2. One ohnolog of the gene scube3 gained new expression in heart, while the other scube3 copy maintained the same expression pattern as that in zebrafish, i.e., neo-F (Fig. 5B). We found 1169 (13.78%) partial non-F (non-F in at least one tissue in exactly one ohnolog while coexpressed in all other tissues), 6 (0.07%) partial sub-F, and 286 (3.37%) partial neo-F triplets (see Methods and fig. S16 for definition), suggesting that coexpression and non-F were the dominant fates of retained WGD genes, and the major mechanism to escape non-F was neo-F instead of sub-F, at least in the first ~10 Ma since the carp WGD.
The non-F, sub-F, and neo-F groups had more genes with low zebrafish-goldfish nucleotide identity compared to those in the coexpressed triplets (Wilcox rank test, P < 1 × 10−6; Fig. 5C, left). In non-F triplets, the expressed ohnologs had more zebrafish exon coverage (and less exon gain/loss) than the silent ones (Wilcoxon rank test, P < 1 × 10−6), and more, but not to the point of significance, nucleotide identity than the silenced gene, suggesting that exon loss was a major mutation correlated with non-F (table S9). Zebrafish-goldfish exon gain/loss of sub-F and neo-F triplets was lower than for non-F (Wilcoxon rank test, P < 1 × 10−6) but higher than for the coexpressed group (Wilcoxon rank test, P < 1 × 10−6), suggesting that exon gain/loss also played an important role in sub-F and neo-F expression of ohnolog pairs (Fig. 5C, right). The neo-F and non-F triplets also came with higher CNE gain/loss (between zebrafish and goldfish) than the coexpressed triplets, and the expression intensity and number of tissues were more similar for coexpressed triplets than for either the neo-F or non-F triplets (fig. S17 and table S10), suggesting that high-expressed and multiple-tissue–expressed genes were more likely to subfunctionalize after WGD.
GO analysis of the coexpressed, non-F, sub-F, and neo-F triplets revealed that each functionalization category was associated with different GO functional classes. The coexpressed triplets were enriched in functions involved in development, ion transport, “macromolecule biosynthetic process,” “kinase activity,” “transcription factor activity,” and “metabolic process.” The non-F triplets were enriched in “oxidoreductase activity,” “cellular nitrogen compound metabolic process,” and “serine hydrolase activity.” Neo-F triplets were enriched in functions involved in “cell-cell adhesion” and “signaling receptor and transductor activity.” Sub-F triplets were enriched in “signal transduction and dioxygenase activity” (figs. S18 to S21).
DISCUSSION
Steady advances in sequencing technology and reductions in cost are improving our ability to generate high-quality genomic sequences, even in cases such as the goldfish, where the efforts are complicated by a recent WGD. Interest in the goldfish has a long history, and goldfish still maintain a special position in both the scientific and ornamental fish communities. We have generated and made publicly available a high-quality annotated assembly of the goldfish genome. Our genomic assembly and gene annotations represent an important resource to these communities as they continue efforts to link phenotypes to genotypes. In addition, the cluster of sequenced fish genomes that includes zebrafish, grass carp, common carp, and goldfish is nicely situated in their evolutionary relationship to provide further insights into the process of rediploidization after a WGD.Comparing gene loss rates between that of zebrafish to grass carp and zebrafish to goldfish, grass carp shows half as many gene losses as goldfish, consistent with a hypothesis of accelerated gene copy loss after the WGD. However, specific functional classes of genes such as transcription factors were more likely to be preserved in two copies. It is known that not only binary expression but also the actual level of transcription factor expression can determine cell fate outcomes [e.g., (44)], so one possible explanation for the specific retention of transcription factors is that a change in dose balance for developmentally key transcription factors could result in disrupted or altered development of the larvae.
In general, the retained duplicate genes maintained an overall expression that correlates closely with zebrafish and to each other. However, accelerated expression divergence of goldfish genes began at the carp WGD, which was also observed in the common carp (37), in zebrafish after the teleost WGD (42), and in Atlantic salmon and rainbow trout after the salmonid-specific WGD (15, 45). Dosage compensation appears to be a major driver of goldfish duplicate gene retention after the carp WGD, which introduced a strong negative selection against loss of either duplicated gene, particularly genes involved in metabolic processes and protein complex formation (46, 47). Gene expression divergence after carp WGD follows the usual paths of non-F and neo-F (either partially or totally) and, to a lesser extent, sub-F. Those genes under partial non-F may become completely inactive (specialization) and finally lost such as in the vertebrate 2R WGD and teleost 3R WGD (48). Goldfish ohnologs escaped from non-F more often through neo-F rather than sub-F, which was also observed in mouse-zebrafish comparisons (49) and salmon but not in X. laevis (50). Neo-F favored the retention of GO terms “kinase” and “G protein–coupled receptor.” It is easy to imagine how genes directly involved in cell-to-cell communication could create interesting evolutionary changes in body form by altering where and when the signaling molecule is expressed. Although sub-F may not be a dominant outcome of goldfish genes, at least in the short time after the carp WGD, we found a few ohnologs adopting an obvious sub-F, e.g., pde4ca and ogn. ogn had also subfunctionalized after the teleost WGD (51), suggesting that there may be evolutionary hotspots for particular genes to neo- or subfunctionalize.
Several other features of genome sequence evolution affect how gene pairs diverge in expression over time. Key factors include divergence of the primary genomic sequence through base substitution, gain/loss of exons, and gain/loss of CNEs, all of which affect gene expression in different ways. Gain/loss of exons is the most important mutation correlated with non-F, neo-F, and sub-F. This process is one that has been proposed to be a critical evolutionary phenomenon that drives vertebrate diversity, and the goldfish–carp speciation is a useful case to explore this evolutionary process.
METHODS
Additional methods and assembly information are included in the Supplementary Materials.
Preparation of genomic DNA and total RNA from goldfish
Gynogenic offspring were generated, as previously described with some modifications (20). The Wakin goldfish eggs were treated with common carp sperm irradiated by ultraviolet light (8000 erg/mm2). After a 34-min incubation at 20°C, the eggs were subjected to a post-fertilization heat-shock treatment at 40°C for 40 s. After a 1-min incubation at 20°C, the eggs were subjected to a second heat-shock treatment at 40°C for 40 s. After the heat-shock treatment, the fertilized eggs were incubated at 20°C. The muscle tissue was dissected from gynogenic goldfish at 3 months of age, and high–molecular weight genomic DNAs were purified using TissueLyser II (Qiagen) and Blood & Cell Culture DNA Maxi Kit (Qiagen). The molecular size of genomic DNA at the peak of 40 to 50 kb was confirmed using the Pippin pulse electroporation system (NIPPON Genetics). Tissues for RNA-seq were dissected from Wakin goldfish at 2 years of age and were stored in RNAlater (Sigma-Aldrich) at −80°C. Total RNA from these tissues was purified using TRIzol reagent (Invitrogen). All procedures were approved by the Animal Experimental Committees of the Institute for Protein Research at Osaka University and were performed in compliance with the institutional guidelines.
Genome assembly
Genomic DNAs from the samples described above were used to perform whole-genome shotgun sequencing on a PacBio RS II sequencer. About 16.4 million PacBio subreads (~71X) with a peak length of ~8 kbp were corrected and assembled into 9415 contigs using the Canu assembler and improved the accuracy using Arrow (21). The total length of the assembly was 1848 Mbp, and N50 reached 816.8 kbp. The longest contig was 12.8 Mbp. We remapped all PacBio subreads to this assembly and found that 6607 contigs had a read coverage of less than 0.6 with a total length of 596 Mbp. The reason for this appears to be that the heat-shock gynogenesis resulted in a meiosis II block creating heterogeneous diploid regions in approximately 22% of our fish genome, as opposed to the expected mitosis I block that would have resulted in a fully homozygous animal. It was possible that the fully homozygous fish in the heat-shocked samples were not viable because of too many potentially harmful mutations in the background. The homozygous regions had 2667 contigs (total length, ~1247 Mbp) with a read coverage in a range of 0.6 to 1.8. This was consistent with the results from our Illumina short-read sequencing, which indicated that about one-fourth of the genome was heterogeneous. By summing all contig lengths normalized by read coverage, we obtained an actual genome size of at least 1.6 Gbp. To remove the alternate alleles from the primary assembly, all contigs were aligned to one another using nucmer (52), and 928 contigs fully contained in other contigs were removed (when the relative read coverage was <0.6 and the identity was >97% to prevent WGD ohnolog removal), which was 27.3 Mbp in total.
LG construction
RNA-seq data from two goldfish parents and their family were downloaded from the National Center for Biotechnology Information (NCBI) (bioproject: PRJEB12518) (22). All reads were trimmed using Trimmomatic (same configuration as in the “Genome annotation” section) and aligned to the Canu assembly using the Hisat2 (53). Variant calling was performed via SAMtools mpileup and bcftools call (parameter “-m”) (54). We identified ~5.6 million variants in total. SNPs without a matching genotype or low read depth (<4) in more than 25% of the samples or with a missing genotype from one of two parents were removed (other filter: bcftools filter “-g 10 -Ov -i 'TYPE="snp" && QUAL>=10 && INFO/DP>=50”). SNPs that were homozygous in both parents or failed a Mendelian test were also removed. We also made sure that two SNPs on the same contig were separated by at least 10 kbp. SNPs (14,022) were kept after filtering and used for constructing the genetic maps.
SNPs from the same contigs were grouped and ordered using “group” and “seq.order” from the R package “onemap” (23), with an LOD (log of odds) threshold of 5.5. Contigs with two or more groups (with ≥3 markers each) were broken at the position where read depth valley and depth were <20 and depth was in the <20% quantile. In total, 16 contigs were broken. Contigs were placed in each LG according to the ordered SNPs using Chromonomer (v1.06). After manual corrections, 50 long LGs were retained and named according to their alignment to the zebrafish genome (e.g., LG1 and LG26 map to zebrafish chr1, LG2 and LG27 map to zebrafish chr2, etc.).
Genome annotation
A custom repeat library for goldfish was built using RepeatModeler (55) (http://repeatmasker.org/) based on the Canu assembly. A zebrafish and the custom repeat library were used to mask the genome by RepeatMasker [http://repeatmasker.org/; performed in MAKER 3.0 (28)].
RNA-seq from seven goldfish tissues was performed to aid with gene annotation, including bone, brain (three samples), eye, gill (two samples), heart, muscle, and tail fin. Reads were assembled via the Trinity assembler (56) without a genome guide for each sample. All assemblies were clustered via CD-HIT (-c 0.95 -aS 0.95 -uS 0.05), as EST (expressed sequence tag) evidence for MAKER 3.0. cDNA sequences from the Ensembl database (version 85; 69 species), NCBI vertebrate RefSeq, and common carp (www.carpbase.org/gbrowse.php) were used as additional RNA evidence. Proteins from the Ensembl database, common carp, and UniProt database (UniRef90) were used as protein evidence. To annotate gene structure, we performed MAKER 3.0 on the Canu assembly with Augustus prediction and the EST, RNA, and protein evidence. Gene structures were lifted over to the carAur01 assembly using liftover (57) or crossmap (https://sourceforge.net/projects/crossmap/files/). InterProScan5 (58) was used to annotate the InterPro/GO/Pathway function for all protein-coding genes.
CNE annotation
All-to-all pairwise genomic alignment was performed using lastz (--gapped --ambiguous=n --step=10 --strand=both --masking=10 --maxwordcount=500 --identity=70..100 --format=axt) and axtToChain for four species (goldfish, common carp, grass carp, and zebrafish). Alignments in repeat regions were subtracted and transformed to multiple alignment format (MAF) format, splitting at gaps longer than 30 bp (chainToAxt –maxGap=30, then axtToMaf -score). All the pairwise MAF files were transformed to multiple alignment MAF files using roast (P = multic). Phylogenetic model was fit for each chromosome, LG or scaffold using phyloFit [--tree '(ZF,(GC,(GF,CC)))' --subst-mod REV --nrate 4], which was used by PhastCons for computing conserve score and regions. The conserved regions out of exons (of coding or noncoding genes) were defined as CNEs for each of the four species. DNA sequences were also extracted from these elements.
Computation of gene (CNE) loss on each branch in the species tree
Ohnolog gene clusters that contained at least one zebrafish ortholog were used to count the number of genes lost on each branch as follows [N(.), number of ohnolog clusters; GC, grass carp; GF, goldfish; CC, common carp]:
∙ Carp ancestry-grass carp: N(GC = 0) (i.e., number of clusters contained none of grass carp gene)
∙ Carp ancestry-WGD: N(GF = 0 AND CC = 0)
∙ WGD speciation of goldfish and common carp: N((GF = 1 AND CC ≤ 1) OR (CC = 1 AND GF ≤ 1)) (according to parsimony)
∙ Speciation – goldfish, single-copy loss: N(GF = 1 AND CC = 2 OR GF = 0 AND CC = 1)
∙ Speciation – goldfish, double-copies loss: 2*N(GF = 0 AND CC = 2)
∙ Speciation – goldfish, total loss: single-copy loss + double-copies loss on the branch
∙ Speciation – common carp, single-copy loss: N(CC = 1 AND GF = 2 OR CC = 0 AND GF = 1)
∙ Speciation – common carp, double-copies loss: 2*N(CC = 0 AND GF = 2)
∙ Speciation – common carp, total loss: single-copy loss + double-copies loss on the branch
Calculation for CNE loss was the same as gene loss. Retention at each node was derived from retention at the direct parent node minus loss at the branch leading to the current node.
Exon/CNE gain/loss between orthologs/ohnologs
Goldfish-zebrafish chain-net alignment (>20 kbp) was divided into two different sets, each representing the alignment between the zebrafish and one goldfish ohnolog. Exons/CNEs from goldfish were liftover to zebrafish based on the two chain-net alignments and annotated using Exon/CNE information of zebrafish, and an exon/CNE was considered as a loss in goldfish (or a gain in zebrafish) if less than 0.5 of the Exon/CNE was mapped to the genome of zebrafish. CNE liftovered to exons was considered as an exon instead of a CNE. The same process was applied for zebrafish exons/CNEs. Exon/CNE triplets with one zebrafish ortholog and two goldfish ohnologs were identified and mapped to gene pairs, and only unique one to two gene pairs were retained for further analysis. CNE was assigned to its nearest gene within 5 kbp (this window cover most of CNEs). The number and length of exons/CNEs in seven configurations were counted for each gene triplet: (ZF,GF1,GF2), (ZF,GF1), (ZF,GF2), (GF1,GF2), (ZF), (GF1), and (GF2), where (.) means that the exon/CNE exists in the corresponding genes. Length was calculated according to ZF exon/CNE if the ZF exon/CNE exists, otherwise according to GF1. Percentage for each configuration was computed as the length of the configurations divided by the total length of all configurations of the gene. Exon gain/loss (difference) between any gene pairs in each gene triplet was computed from the seven configurations, e.g., Dpercent(ZF,GF1) = Percent(ZF,GF2) + Percent(ZF) + Percent(GF1) + Percent(GF1,GF2), where Dpercent(ZF,GF1) is the exon gain/loss between ZF and GF1, Percent(.) is the percentage of the configuration. We did not distinguish between gain and loss.
Expression comparison between zebrafish orthologs and goldfish ohnologs
RNA-seq reads from six shared tissues (brain, eye, heart, gill, muscle, and tail fin) were fetched from NCBI SRA (SRP044781, eye from ERP012920). Reads were mapped to zebrafish genome (danRer10/GRC_z10/Ensembl release 85) using STAR (59). Expression levels (FPKM) were estimated using RSEM (60). A gene was said to be expressed if FPKM ≥ 1 in at least one tissue. Triplets without expressed genes were removed. Triplets with very high identity (≥98%) between any gene pairs were also removed to not collapse the expression of similar genes. We also computed the sum of FPKM from both goldfish ohnologs (i.e., ohno-pairs). The Pearson’s correlation coefficient of expression patterns between zebrafish and individual goldfish ohnologs and between zebrafish and ohno-pair was used to detect expression correlation. Two genes were denoted as highly correlated if the Pearson’s correlation coefficient between their log2(FPKM + 1) was greater than 0.75 and with correlation test (cor.test in R) P < 0.1, medially correlated if their correlation coefficient was greater than 0.6, and differentially expressed if the t test between their log2(FPKM + 1) was less than 0.01. We defined that gene A is “on” relative to gene B if FPKM(A) ≥ 2 and FPKM(B) < 1 and identified coexpressed, nonfunctionalized, subfunctionalized, and neofunctionalized triplets following on-off conditions (fig. S16).
The condition for coexpressed gene A and gene B:
1) A is “on” relative to B in none of the tissues, AND
2) B is “on” relative to A in none of the tissues.
The condition for nonfunctionalized triplet:
1) One goldfish ohnolog does not express (FPKM < 1), AND
2) the zebrafish ortholog expresses with FPKM ≥ 2 in at least one tissue, AND
3) the other goldfish ohnolog expresses with FPKM ≥ 2 in at least one tissue.
The condition for sub-F (where ZF is the zebrafish ortholog and GF1,2 are the two goldfish orthologs):
1) There is at least one tissue T1 in which ZF is “on” relative to GF1 and GF2 is “on” relative to GF1; AND
2) there is at least one other tissue T2 different from T1, in which ZF is “on” relative to GF2 and GF1 is “on” relative to GF2; AND
3) GF1 is ‘on’ relative to ZF in none of the tissues; AND
4) GF2 is ‘on’ relative to ZF in none of the tissues.
The condition for a neo-F triplet (assume that GF1 is the newly expressed gene):
1) There is at least one tissue T1 in which GF1 is “on” relative to ZF and GF1 is “on” relative to GF2; AND.
2) GF2 is “on” relative to GF1 in none of the tissues; AND
3) ZF is “on” relative to GF1 in none of the tissues; AND
4) ZF is “on” relative to GF2 in none of the tissues.
The condition for a partial non-F triplet (assume GF1 is nonfunctionalized):
1) ZF and GF2 are “on” relative to GF1 in some but not all tissues; AND
2) GF1 and GF2 are not “on” relative to ZF in all the other tissues; AND
3) ZF is not “on” relative to GF2 in all the other tissues; AND
4) is not non-F.
The condition for a partial (or mixed) subfunctionalized triplet:
1) There is at least one tissue T1 in which ZF is “on” relative to GF1 and GF2 is “on” relative to GF1; AND
2) there is at least one other tissue T2 different from T1, in which ZF is “on” relative to GF2 and GF1 is “on” relative to GF2; AND
3) is neither non-F nor sub-F.
The condition for a partial (or mixed) non-F triplet (assume GF1 is newly expressed gene):
1) There is at least one tissue T1 in which GF1 is “on” relative to ZF and GF1 is “on” relative to GF2; AND
2) is not non-F or neo-F.
Supplementary Material
Acknowledgments
Funding: This work was supported by Grant-in-Aid for Scientific Research (C) (16 K08583 to Y.O.) from the Japan Society for the Promotion of Science (JSPS) and NIG Collaborative Research Program (2016B5 to Y.O). This research was also funded by the Intramural Research Program of the National Human Genome Research Institute, National Institutes of Health (1ZIAHG000183 to S.M.B., 1ZIBHG000196 to J.C.M., and 1ZIAHG200398 to A.M.P.). Author contributions: Y.O. produced goldfish samples, performed the analysis, and assisted in editing the manuscript. A.M.P. and S.K. assisted in assembly analysis. K.K., T.S., T.K., A.M., H.W., A.F., and A.T. generated goldfish samples. S.Z. and T.G.W. developed the genome browser website. The NISC Comparative Sequencing Program and J.C.M. performed the sequencing and assisted in the assembly and analysis. S.M.B. assisted in the analysis and in the writing and editing of the manuscript. Z.C. analyzed the data and wrote the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: PacBio raw reads have been deposited in the SRA (project ID: PRJNA481500). The BioSample accession number is SAMN09670328. The Canu assembly was deposited in GenBank under accession number QPKE00000000 (data release date: 1 August 2018). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav0547/DC1
Supplementary Methods and Analysis
Table S1. PacBio read statistics.
Table S2. Assembly statistics for different coverage groups.
Table S3. Repeated DNA statistics.
Table S4. Core eukaryotic genes using BUSCOs.
Table S5. Statistics of exon gain/loss.
Table S6. Statistics of CNE gain/loss.
Table S7. Triplets with different number of coexpressed tissues.
Table S8. Number and percentage of ohnolog clusters in evolutionary fate categories.
Table S9. Comparison of features between ZF-GF1 and ZF-GF2, where “Mean1” and “Mean2” are the mean of features between ZF-GF1 and ZF-GF2, respectively.
Table S10. Comparison of features between different gene evolutionary fate.
Fig. S1. Twenty-five–nucleotide oligomer occurrence distribution from 2 × 125 bp Illumina paired-end reads.
Fig. S2. Screenshot of the UCSC Genome Browser implementation of the carAur01 assembly.
Fig. S3. Distribution of exon and intron lengths.
Fig. S4. RBH gene counts between zebrafish and common carp chromosomes.
Fig. S5. RBH gene counts between grass carp and goldfish chromosomes.
Fig. S6. RBH gene counts between goldfish whole-genome duplicated chromosomes.
Fig. S7. Chain-net alignment between each zebrafish chromosome (middle light blue bars) and two corresponding whole-genome duplicated goldfish chromosomes (green bars), and goldfish to common carp (blue bars).
Fig. S8. GO terms prone to retaining both gene copies (blue rectangle) or losing one copy (blue rectangle) after WGD in goldfish.
Fig. S9. GO molecular function comparison among zebrafish (ZF), grass carp (GC), common carp (CC), goldfish (GF).
Fig. S10. Example of neo-F.
Fig. S11. Expression of ohnolog gene pairs in seven tissues.
Fig. S12. Number of ohnolog gene pairs in the same cluster (diagonal) or between each of the 20 clusters (top triangle).
Fig. S13. Function enrichment and reduction in divergent expressed gene pairs.
Fig. S14. Sequence divergence among zebrafish-goldfish triplets.
Fig. S15. Pearson’s correlation coefficient between zebrafish ortholog (ZF)–goldfish ohnolog (GF) and goldfish ohnolog-ohnolog (GF1-GF2).
Fig. S16. Definition of neo-F, sub-F, and neo-F.
Fig. S17. Correlation between different classes of gene expression changes and gain/loss of CNEs.
Fig. S18. Function enrichment (red) or reduction (blue) of genes in coexpressed groups.
Fig. S19. Function enrichment (red) or reduction (blue) of genes in nonfunctionalized groups.
Fig. S20. Function enrichment (red) or reduction (blue) of genes in subfunctionalized groups.
Fig. S21. Function enrichment (red) or reduction (blue) of genes in neofunctionalized groups.
List of members of the NISC Comparative Sequencing Program.
REFERENCES AND NOTES
- 1.Ota K. G., Abe G., Goldfish morphology as a model for evolutionary developmental biology. Wiley Interdiscip. Rev. Dev. Biol. 5, 272–295 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen S. C., Transparency and mottling, a case of Mendelian inheritance in the goldfish Carassius auratus. Genetics 13, 434–452 (1928). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cerdá-Reverter J. M., Haitina T., Schiöth H. B., Peter R. E., Gene structure of the goldfish agouti-signaling protein: A putative role in the dorsal-ventral pigment pattern of fish. Endocrinology 146, 1597–1610 (2005). [DOI] [PubMed] [Google Scholar]
- 4.Munkittrick K. R., Moccia R. D., Leatherland J. F., Polycystic kidney disease in goldfish (Carassius auratus) from Hamilton Harbour, Lake Ontario, Canada. Vet. Pathol. 22, 232–237 (1985). [DOI] [PubMed] [Google Scholar]
- 5.Sahoo P. K., Swaminathan T. R., Abraham T. J., Kumar R., Pattanayak S., Mohapatra A., Rath S. S., Patra A., Adikesavalu H., Sood N., Pradhan P. K., Das B. K., Jayasankar P., Jena J. K., Detection of goldfish haematopoietic necrosis herpes virus (Cyprinid herpesvirus-2) with multi-drug resistant Aeromonas hydrophila infection in goldfish: First evidence of any viral disease outbreak in ornamental freshwater aquaculture farms in India. Acta Trop. 161, 8–17 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Geller I., Conditioned "anxiety" and punishment effects on operant behavior of goldfish (Carassius auratus). Science 141, 351–353 (1963). [DOI] [PubMed] [Google Scholar]
- 7.Osborne W. A., Muntz E., The action of carbon di-oxide on the respiration of the goldfish. Biochem. J. 1, 377–382 (1906). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wullimann M. F., Northcutt R. G., Afferent connections of the valvula cerebelli in two teleosts, the common goldfish and the green sunfish. J. Comp. Neurol. 289, 554–567 (1989). [DOI] [PubMed] [Google Scholar]
- 9.Yazulla S., Zucker C. L., Synaptic organization of dopaminergic interplexiform cells in the goldfish retina. Vis. Neurosci. 1, 13–29 (1988). [DOI] [PubMed] [Google Scholar]
- 10.Blázquez M., Bosma P. T., Chang J. P., Docherty K., Trudeau V. L., γ-Aminobutyric acid up-regulates the expression of a novel secretogranin-II messenger ribonucleic acid in the goldfish pituitary. Endocrinology 139, 4870–4880 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Popesku J. T., Martyniuk C. J., Mennigen J., Xiong H., Zhang D., Xia X., Cossins A. R., Trudeau V. L., The goldfish (Carassius auratus) as a model for neuroendocrine signaling. Mol. Cell. Endocrinol. 293, 43–56 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Wang S.-Y., Luo J., Murphy R. W., Wu S.-F., Zhu C.-L., Gao Y., Zhang Y.-P., Origin of Chinese goldfish and sequential loss of genetic diversity accompanies new breeds. PLOS ONE 8, e59571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ma W., Zhu Z.-H., Bi X.-Y., Murphy R. W., Wang S.-Y., Gao Y., Xiao H., Zhang Y.-P., Luo J., Allopolyploidization is not so simple: Evidence from the origin of the tribe Cyprinini (Teleostei: Cypriniformes). Curr. Mol. Med. 14, 1331–1338 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Glasauer S. M. K., Neuhauss S. C. F., Whole-genome duplication in teleost fishes and its evolutionary consequences. Mol. Genet. Genomics 289, 1045–1060 (2014). [DOI] [PubMed] [Google Scholar]
- 15.Lien S., Koop B. F., Sandve S. R., Miller J. R., Kent M. P., Nome T., Hvidsten T. R., Leong J. S., Minkley D. R., Zimin A., Grammes F., Grove H., Gjuvsland A., Walenz B., Hermansen R. A., von Schalburg K., Rondeau E. B., di Genova A., Samy J. K. A., Olav Vik J., Vigeland M. D., Caler L., Grimholt U., Jentoft S., Inge Våge D., de Jong P., Moen T., Baranski M., Palti Y., Smith D. R., Yorke J. A., Nederbragt A. J., Tooming-Klunderud A., Jakobsen K. S., Jiang X., Fan D., Hu Y., Liberles D. A., Vidal R., Iturra P., Jones S. J. M., Jonassen I., Maass A., Omholt S. W., Davidson W. S., The Atlantic salmon genome provides insights into rediploidization. Nature 533, 200–205 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Session A. M., Uno Y., Kwon T., Chapman J. A., Toyoda A., Takahashi S., Fukui A., Hikosaka A., Suzuki A., Kondo M., van Heeringen S. J., Quigley I., Heinz S., Ogino H., Ochi H., Hellsten U., Lyons J. B., Simakov O., Putnam N., Stites J., Kuroki Y., Tanaka T., Michiue T., Watanabe M., Bogdanovic O., Lister R., Georgiou G., Paranjpe S. S., van Kruijsbergen I., Shu S., Carlson J., Kinoshita T., Ohta Y., Mawaribuchi S., Jenkins J., Grimwood J., Schmutz J., Mitros T., Mozaffari S. V., Suzuki Y., Haramoto Y., Yamamoto T. S., Takagi C., Heald R., Miller K., Haudenschild C., Kitzman J., Nakayama T., Izutsu Y., Robert J., Fortriede J., Burns K., Lotay V., Karimi K., Yasuoka Y., Dichmann D. S., Flajnik M. F., Houston D. W., Shendure J., DuPasquier L., Vize P. D., Zorn A. M., Ito M., Marcotte E. M., Wallingford J. B., Ito Y., Asashima M., Ueno N., Matsuda Y., Veenstra G. J. C., Fujiyama A., Harland R. M., Taira M., Rokhsar D. S., Genome evolution in the allotetraploid frog Xenopus laevis. Nature 538, 336–343 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Margulies E. H., Blanchette M.; NISC Comparative Sequencing Program, Haussler D., Green E. D., Identification and characterization of multi-species conserved sequences. Genome Res. 13, 2507–2518 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Woolfe A., Goodson M., Goode D. K., Snell P., McEwen G. K., Vavouri T., Smith S. F., North P., Callaway H., Kelly K., Walter K., Abnizova I., Gilks W., Edwards Y. J. K., Cooke J. E., Elgar G., Highly conserved non-coding sequences are associated with vertebrate development. PLOS Biol. 3, e7 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.T. R. Gregory, Animal Genome Size Database (2018).
- 20.Koie H., Tsuzuki M., Mizuno M., Appearance of recessive globe-eye character by gynogenesis with suppression of first cleavage in goldfish (Carassius auratus). Bull. Aichi Fish. Res. Inst. 7, 13–16 (2000). [Google Scholar]
- 21.Koren S., Walenz B. P., Berlin K., Miller J. R., Bergman N. H., Phillippy A. M., Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuang Y.-Y., Zheng X.-H., Li C.-Y., Li X.-M., Cao D.-C., Tong G.-X., Lv W.-H., Xu W., Zhou Y., Zhang X.-F., Sun Z.-P., Mahboob S., Al-Ghanim K. A., Li J.-T., Sun X.-W., The genetic map of goldfish (Carassius auratus) provided insights to the divergent genome evolutions in the Cyprinidae family. Sci. Rep. 6, 34849 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Margarido G. R. A., Souza A. P., Garcia A. A. F., OneMap: Software for genetic mapping in outcrossing species. Hereditas 144, 78–79 (2007). [DOI] [PubMed] [Google Scholar]
- 24.Xu P., Zhang X., Wang X., Li J., Liu G., Kuang Y., Xu J., Zheng X., Ren L., Wang G., Zhang Y., Huo L., Zhao Z., Cao D., Lu C., Li C., Zhou Y., Liu Z., Fan Z., Shan G., Li X., Wu S., Song L., Hou G., Jiang Y., Jeney Z., Yu D., Wang L., Shao C., Song L., Sun J., Ji P., Wang J., Li Q., Xu L., Sun F., Feng J., Wang C., Wang S., Wang B., Li Y., Zhu Y., Xue W., Zhao L., Wang J., Gu Y., Lv W., Wu K., Xiao J., Wu J., Zhang Z., Yu J., Sun X., Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nat. Genet. 46, 1212–1219 (2014). [DOI] [PubMed] [Google Scholar]
- 25.McGaugh S. E., Gross J. B., Aken B., Blin M., Borowsky R., Chalopin D., Hinaux H., Jeffery W. R., Keene A., Ma L., Minx P., Murphy D., O’Quin K. E., Rétaux S., Rohner N., Searle S. M. J., Stahl B. A., Tabin C., Volff J.-N., Yoshizawa M., Warren W. C., The cavefish genome reveals candidate genes for eye loss. Nat. Commun. 5, 5307 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kasahara M., Naruse K., Sasaki S., Nakatani Y., Qu W., Ahsan B., Yamada T., Nagayasu Y., Doi K., Kasai Y., Jindo T., Kobayashi D., Shimada A., Toyoda A., Kuroki Y., Fujiyama A., Sasaki T., Shimizu A., Asakawa S., Shimizu N., Hashimoto S.-I., Yang J., Lee Y., Matsushima K., Sugano S., Sakaizumi M., Narita T., Ohishi K., Haga S., Ohta F., Nomoto H., Nogata K., Morishita T., Endo T., Shin-I T., Takeda H., Morishita S., Kohara Y., The medaka draft genome and insights into vertebrate genome evolution. Nature 447, 714–719 (2007). [DOI] [PubMed] [Google Scholar]
- 27.Howe K., Clark M. D., Torroja C. F., Torrance J., Berthelot C., Muffato M., Collins J. E., Humphray S., McLaren K., Matthews L., McLaren S., Sealy I., Caccamo M., Churcher C., Scott C., Barrett J. C., Koch R., Rauch G.-J., White S., Chow W., Kilian B., Quintais L. T., Guerra-Assunção J. A., Zhou Y., Gu Y., Yen J., Vogel J.-H., Eyre T., Redmond S., Banerjee R., Chi J., Fu B., Langley E., Maguire S. F., Laird G. K., Lloyd D., Kenyon E., Donaldson S., Sehra H., Almeida-King J., Loveland J., Trevanion S., Jones M., Quail M., Willey D., Hunt A., Burton J., Sims S., McLay K., Plumb B., Davis J., Clee C., Oliver K., Clark R., Riddle C., Elliot D., Threadgold G., Harden G., Ware D., Begum S., Mortimore B., Kerry G., Heath P., Phillimore B., Tracey A., Corby N., Dunn M., Johnson C., Wood J., Clark S., Pelan S., Griffiths G., Smith M., Glithero R., Howden P., Barker N., Lloyd C., Stevens C., Harley J., Holt K., Panagiotidis G., Lovell J., Beasley H., Henderson C., Gordon D., Auger K., Wright D., Collins J., Raisen C., Dyer L., Leung K., Robertson L., Ambridge K., Leongamornlert D., McGuire S., Gilderthorp R., Griffiths C., Manthravadi D., Nichol S., Barker G., Whitehead S., Kay M., Brown J., Murnane C., Gray E., Humphries M., Sycamore N., Barker D., Saunders D., Wallis J., Babbage A., Hammond S., Mashreghi-Mohammadi M., Barr L., Martin S., Wray P., Ellington A., Matthews N., Ellwood M., Woodmansey R., Clark G., Cooper J., Tromans A., Grafham D., Skuce C., Pandian R., Andrews R., Harrison E., Kimberley A., Garnett J., Fosker N., Hall R., Garner P., Kelly D., Bird C., Palmer S., Gehring I., Berger A., Dooley C. M., Ersan-Ürün Z., Eser C., Geiger H., Geisler M., Karotki L., Kirn A., Konantz J., Konantz M., Oberländer M., Rudolph-Geiger S., Teucke M., Lanz C., Raddatz G., Osoegawa K., Zhu B., Rapp A., Widaa S., Langford C., Yang F., Schuster S. C., Carter N. P., Harrow J., Ning Z., Herrero J., Searle S. M., Enright A., Geisler R., Plasterk R. H., Lee C., Westerfield M., de Jong P. J., Zon L. I., Postlethwait J. H., Nüsslein-Volhard C., Hubbard T. J., Roest Crollius H., Rogers J., Stemple D. L., The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cantarel B. L., Korf I., Robb S. M. C., Parra G., Ross E., Moore B., Holt C., Sánchez Alvarado A., Yandell M., MAKER: An easy-to-use annotation pipeline designed for emerging model organism genomes. Genome Res. 18, 188–196 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waterhouse R. M., Seppey M., Simão F. A., Manni M., Ioannidis P., Klioutchnikov G., Kriventseva E. V., Zdobnov E. M., BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 35, 543–548 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.David L., Blum S., Feldman M. W., Lavi U., Hillel J., Recent duplication of the common carp (Cyprinus carpio L.) genome as revealed by analyses of microsatellite loci. Mol. Biol. Evol. 20, 1425–1434 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Larhammar D., Risinger C., Molecular genetic aspects of tetraploidy in the common carp Cyprinus carpio. Mol. Phylogenet. Evol. 3, 59–68 (1994). [DOI] [PubMed] [Google Scholar]
- 32.Wang Y., Lu Y., Zhang Y., Ning Z., Li Y., Zhao Q., Lu H., Huang R., Xia X., Feng Q., Liang X., Liu K., Zhang L., Lu T., Huang T., Fan D., Weng Q., Zhu C., Lu Y., Li W., Wen Z., Zhou C., Tian Q., Kang X., Shi M., Zhang W., Jang S., du F., He S., Liao L., Li Y., Gui B., He H., Ning Z., Yang C., He L., Luo L., Yang R., Luo Q., Liu X., Li S., Huang W., Xiao L., Lin H., Han B., Zhu Z., The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nat. Genet. 47, 625–631 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Burns F. R., Cogburn A. L., Ankley G. T., Villeneuve D. L., Waits E., Chang Y.-J., Llaca V., Deschamps S. D., Jackson R. E., Hoke R. A., Sequencing and de novo draft assemblies of a fathead minnow (Pimephales promelas) reference genome. Environ. Toxicol. Chem. 35, 212–217 (2016). [DOI] [PubMed] [Google Scholar]
- 34.Xu J., Li J.-T., Jiang Y., Peng W., Yao Z., Chen B., Jiang L., Feng J., Ji P., Liu G., Liu Z., Tai R., Dong C., Sun X., Zhao Z.-X., Zhang Y., Wang J., Li S., Zhao Y., Yang J., Sun X., Xu P., Genomic basis of adaptive evolution: The survival of Amur Ide (Leuciscus waleckii) in an extremely alkaline environment. Mol. Biol. Evol. 34, 145–159 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang J., Chen X., Bai J., Fang D., Qiu Y., Jiang W., Yuan H., Bian C., Lu J., He S., Pan X., Zhang Y., Wang X., You X., Wang Y., Sun Y., Mao D., Liu Y., Fan G., Zhang H., Chen X., Zhang X., Zheng L., Wang J., Cheng L., Chen J., Ruan Z., Li J., Yu H., Peng C., Ma X., Xu J., He Y., Xu Z., Xu P., Wang J., Yang H., Wang J., Whitten T., Xu X., Shi Q., The Sinocyclocheilus cavefish genome provides insights into cave adaptation. BMC Biol. 14, 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.S. Ohno, Evolution by Gene Duplication (Springer-Verlag, New York, 1970), pp. 160. [Google Scholar]
- 37.Li J.-T., Hou G.-Y., Kong X.-F., Li C.-Y., Zeng J.-M., Li H.-D., Xiao G.-B., Li X.-M., Sun X.-W., The fate of recent duplicated genes following a fourth-round whole genome duplication in a tetraploid fish, common carp (Cyprinus carpio). Sci. Rep. 5, 8199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Force A., Lynch M., Pickett F. B., Amores A., Yan Y.-l., Postlethwait J., Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151, 1531–1545 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lynch M., Conery J. S., The evolutionary fate and consequences of duplicate genes. Science 290, 1151–1155 (2000). [DOI] [PubMed] [Google Scholar]
- 40.Rastogi S., Liberles D. A., Subfunctionalization of duplicated genes as a transition state to neofunctionalization. BMC Evol. Biol. 5, 28 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vonk F. J., Casewell N. R., Henkel C. V., Heimberg A. M., Jansen H. J., McCleary R. J. R., Kerkkamp H. M. E., Vos R. A., Guerreiro I., Calvete J. J., Wüster W., Woods A. E., Logan J. M., Harrison R. A., Castoe T. A., de Koning A. P. J., Pollock D. D., Yandell M., Calderon D., Renjifo C., Currier R. B., Salgado D., Pla D., Sanz L., Hyder A. S., Ribeiro J. M. C., Arntzen J. W., van den Thillart G. E. E. J. M., Boetzer M., Pirovano W., Dirks R. P., Spaink H. P., Duboule D., McGlinn E., Kini R. M., Richardson M. K., The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. U.S.A. 110, 20651–20656 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Braasch I., Gehrke A. R., Smith J. J., Kawasaki K., Manousaki T., Pasquier J., Amores A., Desvignes T., Batzel P., Catchen J., Berlin A. M., Campbell M. S., Barrell D., Martin K. J., Mulley J. F., Ravi V., Lee A. P., Nakamura T., Chalopin D., Fan S., Wcisel D., Cañestro C., Sydes J., Beaudry F. E. G., Sun Y., Hertel J., Beam M. J., Fasold M., Ishiyama M., Johnson J., Kehr S., Lara M., Letaw J. H., Litman G. W., Litman R. T., Mikami M., Ota T., Saha N. R., Williams L., Stadler P. F., Wang H., Taylor J. S., Fontenot Q., Ferrara A., Searle S. M. J., Aken B., Yandell M., Schneider I., Yoder J. A., Volff J.-N., Meyer A., Amemiya C. T., Venkatesh B., Holland P. W. H., Guiguen Y., Bobe J., Shubin N. H., di Palma F., Alföldi J., Lindblad-Toh K., Postlethwait J. H., The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 48, 427–437 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hughes T., Ekman D., Ardawatia H., Elofsson A., Liberles D. A., Evaluating dosage compensation as a cause of duplicate gene retention in Paramecium tetraurelia. Genome Biol. 8, 213 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Niwa H., Miyazaki J.-i., Smith A. G., Quantitative expression of Oct-3/4 defines differentiation, dedifferentiation or self-renewal of ES cells. Nat. Genet. 24, 372–376 (2000). [DOI] [PubMed] [Google Scholar]
- 45.Berthelot C., Brunet F., Chalopin D., Juanchich A., Bernard M., Noël B., Bento P., da Silva C., Labadie K., Alberti A., Aury J.-M., Louis A., Dehais P., Bardou P., Montfort J., Klopp C., Cabau C., Gaspin C., Thorgaard G. H., Boussaha M., Quillet E., Guyomard R., Galiana D., Bobe J., Volff J.-N., Genêt C., Wincker P., Jaillon O., Crollius H. R., Guiguen Y., The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 5, 3657 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Edger P. P., Pires J. C., Gene and genome duplications: The impact of dosage-sensitivity on the fate of nuclear genes. Chromosome Res. 17, 699–717 (2009). [DOI] [PubMed] [Google Scholar]
- 47.Jiang W.-k., Liu Y.-l., Xia E.-h., Gao L.-z., Prevalent role of gene features in determining evolutionary fates of whole-genome duplication duplicated genes in flowering plants. Plant Physiol. 161, 1844–1861 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marletaz F., Firbas P. N., Maeso I., Tena J. J., Bogdanovic O., Perry M., Wyatt C. D. R., de la Calle-Mustienes E., Bertrand S., Burguera D., Acemel R. D., van Heeringen S. J., Naranjo S., Herrera-Ubeda C., Skvortsova K., Jimenez-Gancedo S., Aldea D., Marquez Y., Buono L., Kozmikova I., Permanyer J., Louis A., Albuixech-Crespo B., Le Petillon Y., Leon A., Subirana L., Balwierz P. J., Duckett P. E., Farahani E., Aury J.-M., Mangenot S., Wincker P., Albalat R., Benito-Gutiérrez È., Cañestro C., Castro F., D’Aniello S., Ferrier D. E. K., Huang S., Laudet V., Marais G. A. B., Pontarotti P., Schubert M., Seitz H., Somorjai I., Takahashi T., Mirabeau O., Xu A., Yu J.-K., Carninci P., Martinez-Morales J. R., Crollius H. R., Kozmik Z., Weirauch M. T., Garcia-Fernàndez J., Lister R., Lenhard B., Holland P. W. H., Escriva H., Gómez-Skarmeta J. L., Irimia M., Amphioxus functional genomics and the origins of vertebrate gene regulation. Nature 564, 64–70 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kassahn K. S., Dang V. T., Wilkins S. J., Perkins A. C., Ragan M. A., Evolution of gene function and regulatory control after whole-genome duplication: Comparative analyses in vertebrates. Genome Res. 19, 1404–1418 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sémon M., Wolfe K. H., Preferential subfunctionalization of slow-evolving genes after allopolyploidization in Xenopus laevis. Proc. Natl. Acad. Sci. U.S.A. 105, 8333–8338 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Costa R. A., Martins R. S. T., Capilla E., Anjos L., Power D. M., Vertebrate SLRP family evolution and the subfunctionalization of osteoglycin gene duplicates in teleost fish. BMC Evol. Biol. 18, 191 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kurtz S., Phillippy A., Delcher A. L., Smoot M., Shumway M., Antonescu C., Salzberg S. L., Versatile and open software for comparing large genomes. Genome Biol. 5, R12 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim D., Langmead B., Salzberg S. L., HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R.; Genome Project Data Processing Subgroup , The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huda A., Jordan I. K., Analysis of transposable element sequences using CENSOR and RepeatMasker. Methods Mol. Biol. 537, 323–336 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Grabherr M. G., Haas B. J., Yassour M., Levin J. Z., Thompson D. A., Amit I., Adiconis X., Fan L., Raychowdhury R., Zeng Q., Chen Z., Mauceli E., Hacohen N., Gnirke A., Rhind N., di Palma F., Birren B. W., Nusbaum C., Lindblad-Toh K., Friedman N., Regev A., Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kuhn R. M., Haussler D., Kent W. J., The UCSC genome browser and associated tools. Brief. Bioinformatics 14, 144–161 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones P., Binns D., Chang H.-Y., Fraser M., Li W., McAnulla C., McWilliam H., Maslen J., Mitchell A., Nuka G., Pesseat S., Quinn A. F., Sangrador-Vegas A., Scheremetjew M., Yong S.-Y., Lopez R., Hunter S., InterProScan 5: Genome-scale protein function classification. Bioinformatics 30, 1236–1240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li B., Dewey C. N., RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delcher A. L., Salzberg S. L., Phillippy A. M., Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinformatics Chapter 10, Unit 10.3 (2003). [DOI] [PubMed] [Google Scholar]
- 62.Bolger A. M., Lohse M., Usadel B., Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D., The human genome browser at UCSC. Genome Res. 12, 996–1006 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu C., Bai B., Skogerbø G., Cai L., Deng W., Zhang Y., Bu D., Zhao Y., Chen R., NONCODE: An integrated knowledge database of non-coding RNAs. Nucleic Acids Res. 33, D112–D115 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.The RNAcentral Consortium , RNAcentral: A comprehensive database of non-coding RNA sequences. Nucleic Acids Res. 45, D128–D134 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hubbard T. J. P., Aken B. L., Ayling S., Ballester B., Beal K., Bragin E., Brent S., Chen Y., Clapham P., Clarke L., Coates G., Fairley S., Fitzgerald S., Fernandez-Banet J., Gordon L., Graf S., Haider S., Hammond M., Holland R., Howe K., Jenkinson A., Johnson N., Kahari A., Keefe D., Keenan S., Kinsella R., Kokocinski F., Kulesha E., Lawson D., Longden I., Megy K., Meidl P., Overduin B., Parker A., Pritchard B., Rios D., Schuster M., Slater G., Smedley D., Spooner W., Spudich G., Trevanion S., Vilella A., Vogel J., White S., Wilder S., Zadissa A., Birney E., Cunningham F., Curwen V., Durbin R., Fernandez-Suarez X. M., Herrero J., Kasprzyk A., Proctor G., Smith J., Searle S., Flicek P., Ensembl 2009. Nucleic Acids Res. 37, D690–D697 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Camacho C., Coulouris G., Avagyan V., Ma N., Papadopoulos J., Bealer K., Madden T. L., BLAST+: Architecture and applications. BMC Bioinformatics 10, 421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Slater G. S., Birney E., Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics 6, 31 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Finn R. D., Bateman A., Clements J., Coggill P., Eberhardt R. Y., Eddy S. R., Heger A., Hetherington K., Holm L., Mistry J., Sonnhammer E. L. L., Tate J., Punta M., Pfam: The protein families database. Nucleic Acids Res. 42, D222–D230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kong L., Zhang Y., Ye Z.-Q., Liu X.-Q., Zhao S.-Q., Wei L., Gao G., CPC: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nawrocki E. P., Eddy S. R., Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kalvari I., Argasinska J., Quinones-Olvera N., Nawrocki E. P., Rivas E., Eddy S. R., Bateman A., Finn R. D., Petrov A. I., Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 46, D335–D342 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Teer J. K., Bonnycastle L. L., Chines P. S., Hansen N. F., Aoyama N., Swift A. J., Abaan H. O., Albert T. J.; NISC Comparative Sequencing Program, Margulies E. H., Green E. D., Collins F. S., Mullikin J. C., Biesecker L. G., Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 20, 1420–1431 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chen Y., Shi M., Zhang W., Cheng Y., Wang Y., Xia X.-Q., The Grass Carp Genome Database (GCGD): An online platform for genome features and annotations. Database (Oxford) 2017, bax051 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Katoh K., Standley D. M., MAFFT: Iterative refinement and additional methods. Methods Mol. Biol. 1079, 131–146 (2014). [DOI] [PubMed] [Google Scholar]
- 76.Rice P., Longden I., Bleasby A., EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 16, 276–277 (2000). [DOI] [PubMed] [Google Scholar]
- 77.Castresana J., Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552 (2000). [DOI] [PubMed] [Google Scholar]
- 78.Stamatakis A., RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yang Z., PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav0547/DC1
Supplementary Methods and Analysis
Table S1. PacBio read statistics.
Table S2. Assembly statistics for different coverage groups.
Table S3. Repeated DNA statistics.
Table S4. Core eukaryotic genes using BUSCOs.
Table S5. Statistics of exon gain/loss.
Table S6. Statistics of CNE gain/loss.
Table S7. Triplets with different number of coexpressed tissues.
Table S8. Number and percentage of ohnolog clusters in evolutionary fate categories.
Table S9. Comparison of features between ZF-GF1 and ZF-GF2, where “Mean1” and “Mean2” are the mean of features between ZF-GF1 and ZF-GF2, respectively.
Table S10. Comparison of features between different gene evolutionary fate.
Fig. S1. Twenty-five–nucleotide oligomer occurrence distribution from 2 × 125 bp Illumina paired-end reads.
Fig. S2. Screenshot of the UCSC Genome Browser implementation of the carAur01 assembly.
Fig. S3. Distribution of exon and intron lengths.
Fig. S4. RBH gene counts between zebrafish and common carp chromosomes.
Fig. S5. RBH gene counts between grass carp and goldfish chromosomes.
Fig. S6. RBH gene counts between goldfish whole-genome duplicated chromosomes.
Fig. S7. Chain-net alignment between each zebrafish chromosome (middle light blue bars) and two corresponding whole-genome duplicated goldfish chromosomes (green bars), and goldfish to common carp (blue bars).
Fig. S8. GO terms prone to retaining both gene copies (blue rectangle) or losing one copy (blue rectangle) after WGD in goldfish.
Fig. S9. GO molecular function comparison among zebrafish (ZF), grass carp (GC), common carp (CC), goldfish (GF).
Fig. S10. Example of neo-F.
Fig. S11. Expression of ohnolog gene pairs in seven tissues.
Fig. S12. Number of ohnolog gene pairs in the same cluster (diagonal) or between each of the 20 clusters (top triangle).
Fig. S13. Function enrichment and reduction in divergent expressed gene pairs.
Fig. S14. Sequence divergence among zebrafish-goldfish triplets.
Fig. S15. Pearson’s correlation coefficient between zebrafish ortholog (ZF)–goldfish ohnolog (GF) and goldfish ohnolog-ohnolog (GF1-GF2).
Fig. S16. Definition of neo-F, sub-F, and neo-F.
Fig. S17. Correlation between different classes of gene expression changes and gain/loss of CNEs.
Fig. S18. Function enrichment (red) or reduction (blue) of genes in coexpressed groups.
Fig. S19. Function enrichment (red) or reduction (blue) of genes in nonfunctionalized groups.
Fig. S20. Function enrichment (red) or reduction (blue) of genes in subfunctionalized groups.
Fig. S21. Function enrichment (red) or reduction (blue) of genes in neofunctionalized groups.
List of members of the NISC Comparative Sequencing Program.