

A migration-associated chromosome rearrangement in Atlantic cod reveals genetic structure and past signatures of stock collapse.
Abstract
Chromosome structural variation may underpin ecologically important intraspecific diversity by reducing recombination within supergenes containing linked, coadapted alleles. Here, we confirm that an ancient chromosomal rearrangement is strongly associated with migratory phenotype and individual genetic structure in Atlantic cod (Gadus morhua) across the Northwest Atlantic. We reconstruct trends in effective population size over the last century and reveal declines in effective population size matching onset of industrialized harvest (after 1950). We find different demographic trajectories between individuals homozygous for the chromosomal rearrangement relative to heterozygous or homozygous individuals for the noninverted haplotype, suggesting different selective histories across the past 150 years. These results illustrate how chromosomal structural diversity can mediate fine-scale genetic, phenotypic, and demographic variation in a highly connected marine species and show how overfishing may have led to loss of biocomplexity within Northern cod stock.
INTRODUCTION
Consideration of intraspecific variation is central to the effective management of natural resources and ecosystem services (1, 2). Individual phenotypic and genetic variation can play a key role in dictating ecological composition and function, and accordingly, perturbations have been shown to alter ecosystem structure (3). Incorporating intraspecific diversity into management plans requires the identification of heritable phenotypes linked to ecological resilience and biocomplexity (4, 5). Genomic analyses have increasingly revealed chromosome structural rearrangements (e.g., inversions and translocations) underpinning ecologically variable traits, supporting an important role for genomic architecture in promoting intraspecific ecological diversity (6, 7). For example, chromosomal inversions may facilitate the evolution of complex phenotypes in sympatry through the formation of supergenes or clusters of linked adaptive loci in regions of reduced recombination (6). Quantifying the relationship between genomic architectural variation and ecologically relevant phenotypes can provide a deeper understanding of the genomic drivers of biocomplexity, enabling genomics-guided species management and precise measurement of human impacts within species (8). However, the relationship between genomic architecture and key ecological traits remains largely unknown in most exploited species (9).
The Atlantic cod (Gadus morhua) has an extensive history of exploitation and increasing evidence of genomic structural variation associated with ecological adaptation (7, 9, 10). The Northern cod population in Northwest Atlantic waters around Newfoundland and Labrador has undergone multiple population declines, the most recent and drastic of which have been driven largely by overharvesting following adoption of industrial-scale fishing, in tandem with climate shifts (11). In conjunction with recent declines in abundance, size and age at maturity have also declined within the Northern cod stock since the 1970s (12, 13), suggesting that fishery-induced selection has occurred (13).
Individual and regional genomic structural diversity that differentiates Atlantic cod populations by environment and migratory behavior has been identified across the species range in the North Atlantic (10). Recent genomic analyses have revealed large chromosomal rearrangements on four linkage groups (LG1, LG2, LG7, and LG12) (7, 10); two adjacent inversions within LG1 (14) have been shown to consistently differentiate offshore migratory and coastal nonmigratory populations (7, 9, 14, 15). The migratory phenotype exhibited by Northern cod may have imposed increased vulnerability to overfishing in these populations (16). However, the link between these rearrangements, migratory phenotype, and the consequence of overharvest remains largely unexamined. Understanding the relationship between phenotypic variation, population stability, and genomic architectural variation is particularly important in Northern cod, which have undergone one of the most severe population crashes recorded in an exploited marine species and have failed to recover to historical abundance despite a lengthy and continuing fishing moratorium (17). Here, we explore the genomic basis of individual variation within the Northern cod stock around Newfoundland and Labrador (Fig. 1A) using a 12 K single-nucleotide polymorphism (SNP) array. We examine the role of chromosomal rearrangements associated with migration phenotype and temporal trends in effective population size over the last century.
Fig. 1. Map of Atlantic cod (G. morhua) sampling locations in the Northwest Atlantic and genetic structuring across all sampled sites and within Northern cod.
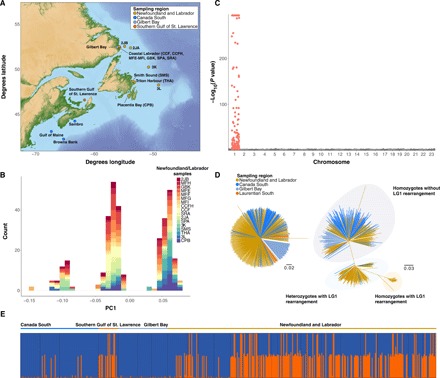
(A) Sampling sites across North America color coded by population. (B) Genetic structure within Northern cod visualized using the first PC calculated from 6030 SNPs in pcadapt. Colors correspond to sample groups taken from sites within the Northwest Atlantic cod population. (C) Genomic sources of individual-level genetic variation within Northern cod assessed by significance of correlation (P value) of each SNP locus with the first PC axis obtained from transformed Mahalanobis distances. SNPs within the LG1 rearrangement are colored in red. (D) Individual neighbor-joining trees for 511 individuals from all sampling locations calculated using 2500 randomly selected SNPs outside of Northern cod rearrangements (left) and 237 SNPs in the LG1 rearranged region (right). (E) Bayesian clustering results from STRUCTURE for 237 SNPs in the LG1 rearranged region calculated among all 511 individuals from all sampling locations for K = 2 genetic clusters. The LG1 inverted genetic background is represented in orange, and the LG1 noninverted genetic background is represented in blue.
RESULTS AND DISCUSSION
Within the Northern cod stock, principal component (PC) analysis (PCA) using 6669 informative SNPs identified three distinct clusters along the first PC axis (Fig. 1B). Outlier identification using the Mahalanobis distance for each SNP and the first PC revealed that individual genetic structuring was greatest among SNPs localized between 9.89 and 27.22 Mbp on LG1, consistent with boundaries of the chromosomal rearrangement consisting of two previously identified adjacent inversions on LG1 (14). Suppressed recombination along the entire rearrangement between different orientations may allow this genomic region to function as a single supergene (7, 18). Recent sequence comparisons indicate that these inversions arose in the Eastern Atlantic 1.6 to 2.0 million years ago, suggesting the presence of this inversion as standing or introduced variation in the Northwest Atlantic (14). To further evaluate our conclusions that the genomic region in question on LG1 represents a chromosomal inversion associated with individual genetic structure, we conducted PCA using previously published whole-genome data from North America (19), including 20,000 SNPs across LG1. Again, we find genetic structuring along LG1 driven by the same rearrangement region consistent with the boundaries of the inversion previously identified using mapping crosses and linkage analysis by Kirubakaran et al. (14) (fig. S1A). Moreover, pairwise linkage disequilibrium (LD) estimates from averaged 100 SNP pairwise r2 revealed elevated LD within previously identified boundaries again consistent with an inversion (fig. S1B).
To evaluate the prevalence of intrapopulation variation associated with the rearrangement on LG1, we geographically expanded the population structure analysis to include samples from throughout the Northwest Atlantic in waters around Newfoundland and Labrador, southern Canada, the Laurentian Gulf, and coastal Labrador. We conducted separate analyses using genome-wide SNPs from regions without known inversions (n = 2500) and SNPs within the rearranged region on LG1 (n = 237). Individual neighbor-joining trees revealed little structuring at neutral loci and again uncovered the presence of three discrete groups using only SNPs within the LG1 rearranged region (Fig. 1D). Bayesian clustering analysis of SNPs within the LG1 rearranged region also demonstrated ancestry proportions consistent with homozygous or heterozygous genotypes from inverted or noninverted LG1 haplotypes (Fig. 1E).
To quantify the relationship between migratory phenotype and chromosomal rearrangements in the Atlantic cod genome, we conducted a range-wide comparison of cod populations in the Northwest Atlantic with assigned migratory phenotypes and used genome-wide association analysis to confirm LG1-migration associations revealed in previous divergence-based studies (7, 10). We assigned regional-level migratory and nonmigratory phenotypes to individuals from each sampled location using previously collected migratory behavior data (table S1) (20) and detected migration-associated SNPs using random forest (RF), partial redundancy analysis (RDA), and a latent factor mixed model (LFMM). We uncovered 24 SNPs (table S2) associated with migratory phenotype across all three association methods, all located within 9.89 and 27.22 Mbp in LG1, corresponding to the position of the rearrangement (Fig. 2, A and B and fig. S2) and exhibiting high LD (r2 = 0.82; SD = 0.09). To detect selective sweeps within LG1 in homozygous inverted or noninverted groups, we used two methods, composite likelihood ratio (CLR) and integrated haplotype score (iHS), which have been previously shown to be robust to variation in recombination rate (21, 22, 23, 24). Both methods detected evidence of a selective sweep within the LG1 rearrangement not identified in the noninverted homozygous individuals, suggesting selection distinct to this structural variant, potentially for migratory phenotype, although alternative sources of selection are also possible (Fig. 2, C and D). A large suite of variable sites within this rearrangement with predicted effects on gene function in greater than 300 genes were previously shown to be differentiated between coastal and migratory ecotypes in Norwegian waters (14). Several of these variants are suggested to affect swim bladder function and muscular efficiency, facilitating vertical movement at variable depths in offshore sites and enhancing muscular capacity for strenuous migration (7, 14).
Fig. 2. Genome-wide association of 6669 SNPs with migration phenotype, across all sampling locations, and selective sweeps within LG1.
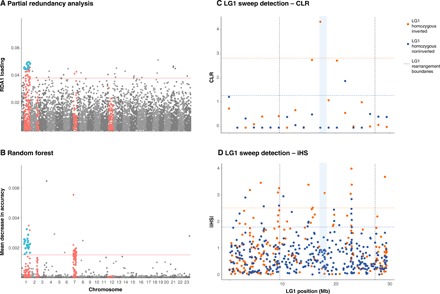
(A) Absolute values of scores for each SNP along the first redundancy canonical axis (RDA1) in partial RDA of migratory phenotype conditioned by geographic location, treating all SNPs in the 99th percentile as significantly associated. SNPs within rearrangements are colored in red, and SNPs identified in association with migratory phenotype across all three methods are identified in blue. (B) Mean decrease in accuracy per SNP in RF prediction of migratory phenotype, categorizing SNPs with a mean decrease in accuracy of >0.015 as significantly associated. (C) Positive selective sweeps in LG1 for LG1 homozygous noninverted and LG1 homozygous inverted individuals detected by calculating CLR per 20 SNP window with regions exceeding the 95th percentile were considered likely candidates for selection. Cutoff values for the 95th percentile in LG1 homozygous inverted and noninverted individuals are represented by dashed lines in orange and blue, respectively. The blue bar represents the region that exhibited a sweep specific to LG1 inverted homozygous individuals across both methods. (D) Positive selective sweeps in LG1 for LG1 homozygous noninverted and LG1 homozygous inverted individuals detected by calculating iHS with regions exceeding the 95th percentile were considered likely candidates for selection.
Northern cod experienced substantial overharvest, reflected in the occurrence of one of the biggest population crashes of any marine vertebrate that resulted in >90% decline in abundance during the 20th century (12, 16, 25). Despite the scale of stock collapse, previous neutral molecular marker–based attempts have failed to detect signatures of this decline across the genome (26–28). Signatures of temporal change in allele frequency at individual loci within LG1 have indicated the importance of this rearrangement to collapse of cod stocks, including Canadian stocks, but the association of this rearrangement with demographic history across the genome and loss of genetic diversity in Northern cod has not been characterized (27, 28). Here, we find that genomic data can detect declines, as well as demographic histories specific to groups with noninverted or inverted copies of the LG1 rearrangement, revealing different patterns of change in effective size between individuals with differing rearrangement genotype. We used PCA clusters to infer inversion genotype and separate individuals into LG1 homozygous inverted, LG1 homozygous noninverted, and heterozygous groups. Using a linkage-based method, we reconstructed trends in recent effective population size (Ne) in each group using genome-wide SNPs outside of LG1 and LG12 rearrangements identified in Northern cod, selecting a time interval to prioritize recent changes in effective size (29). We then compared the proportional change in Ne relative to the maximum observed effective size within each group during the past 150 years to observed and reconstructed stock abundance (11).
We observed different demographic histories between individuals with different LG1 rearrangement haplotypes; we found a similar starting effective population size at the 1860 time point for each group range (Ne range = 1976 – 2629) but observed different patterns of expansion and decline among LG1 homozygous noninverted, LG1 homozygous inverted, and heterozygous groups (Fig. 3, A and B), suggesting different selective histories within the past century. Within LG1 homozygous noninverted and heterozygous groups, we observed an increase in effective size indicated by large Ne estimates with wide confidence intervals, including negative values, which reflect very large to infinite estimated Ne, coinciding with a period of population productivity from 1900 to approximately 1970 (11). In contrast, this pattern was not observed in LG1 homozygous inverted individuals. From 1970 to 2004, we observe a shared pattern of decline resulting in different contemporary effective sizes of each group. These results indicate that different demographic histories may have resulted in distinct genetic consequences from the shared bottleneck imposed by overharvest of Northern cod, although additional factors shaping demographic history such as change in climate have also potentially played a role (11). Although drift and nonindependence of SNPs may affect precision of estimates of Ne (29, 30), we do not expect these effects to produce different demographic histories from the same SNP panel as observed here. Comparing changes in effective size between LG1 rearrangement groups by region revealed a decline in LG1 homozygous inverted and heterozygous individuals driven by the decline in sites around Labrador, suggesting spatial variation in selection intensity (fig. S3).
Fig. 3. Comparison of recent relative effective population size (Ne) in LG1 homozygous noninverted, LG1 homozygous inverted, and heterozygous individuals and measured and historically reconstructed stock abundance of Northern cod.
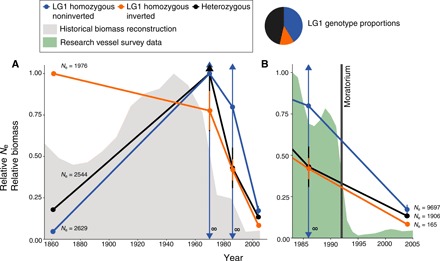
(A) Recent effective population size relative to maximum effective population size within each group of LG1 homozygous noninverted, LG1 homozygous inverted, and heterozygous Northern cod groupings calculated using LinkNe, plotted with reconstructed estimates of historical biomass measured from 1850 to 2004, relative to the maximum value. Confidence intervals with arrows indicate a very large Ne. (B) Recent proportional effective population size relative to maximum effective population size of LG1 homozygous noninverted, homozygous LG inverted, and heterozygous Northern cod calculated using LinkNe compared to measurement of stock abundance from 1983 to 2004 relative to the maximum value.
The capacity of genomic data to detect the recent collapse of Northern cod was also supported by coalescent bottleneck tests that identify patterns of excess heterozygosity predicted following recent bottlenecks (31). These signatures of heterozygosity excess were observed across LG1 inverted homozygous, LG1 noninverted homozygous, and heterozygous groups, inferred from significant sign tests (P < 10−5) and standardized difference tests (P < 10−5), as well as shifted modes in allele frequency distributions in both homozygous groups.
Our results provide advances in understanding biocomplexity at a genomic level in marine species. We demonstrate that intraspecific diversity in migratory behavior is associated with a chromosomal rearrangement consisting of two adjacent inversions spanning LG1 and illustrate that this rearrangement enables extensive differentiation among individuals within Northern cod. Suppressed recombination between opposite orientations of the LG1 rearrangement can promote coordinated inheritance of migration-associated loci, allowing the two adjacent inversions to function as a single supergene protected from gene flow (2, 9, 10, 18). We found genomic evidence of extensive decline among cod populations coinciding with the expansion of industrial fishing; individuals homozygous for the migration-associated supergene showed a different demographic history than heterozygous or noninverted individuals, resulting in markedly reduced effective size for this group following overharvest of Northern cod. The collapse of the Atlantic cod has resulted in massive restructuring of coastal shelf ecosystems (32). Continued overharvesting may lead to loss of the migration-associated LG1 rearrangement, potentially resulting in widespread altered ecological function and composition in the Northwest Atlantic through changing cod distribution and site persistence. Removal of this genomic diversity through overfishing may also reduce the buffering effect provided by phenotypic diversity within these populations, increasing the risk of future collapse (5). Management of Northern cod stocks may thus require consideration of the genomic architectural variation within these populations to ensure maintenance of biocomplexity across the Northwest Atlantic.
MATERIALS AND METHODS
Sampling and genotyping
We combined Atlantic cod (G. morhua) genotype data from three studies conducted by Sinclair-Waters et al. (7, 15) and Berg et al. (10) (table S1) and included unanalyzed samples from two sites in coastal Newfoundland: Triton Harbour and Smith Sound. The combined dataset comprised 511 individuals in 24 sample groups collected between 2001 and 2015 from across 18 sites in the Northwest Atlantic. Sample collection and preparation methods were described by Sinclair-Waters et al. (7, 15) and Berg et al. (10); methods used for samples from the two new sampling locations matched those from Sinclair-Waters et al. (15). All sampled individuals were genotyped using a 12 K SNP array developed for Atlantic cod at the Centre for Integrative Genetics (CIGENE), Norwegian University of Life Sciences in Ås, Norway. Following correction for strand flips, we retained a subset of 6669 informative SNPs with complete mapping information across all datasets for further analysis.
Genetic structure detection
To characterize genomic sources of individual variation within Northern cod, we used the pcadapt (33) R package to conduct PCA on the 330 individuals from sampling sites around Newfoundland and Labrador. Using 6030 SNPs with minor allele frequency greater than 0.025, we carried out PCA retaining the first PC axis to identify the largest source of individual genomic variation. We plotted P values from transformed Mahalanobis distances, measuring the significance of correlation with each SNP and this PC axis across each linkage group using the R package ggman (34).
Fine-scale characterization of genetic structure
We then analyzed whole-genome data to confirm the utility of the 12 K SNP chip in detecting signatures of differentiation consistent with the presence of the LG1 rearrangement identified by Kirubakaran et al. (14). We obtained whole-genome data from 31 individuals from the Gulf of Maine and Georges Bank analyzed by Barney et al. (19). We extracted LG1 SNP genotypes from whole-genome data using vcftools (35), conducted LD filtering in 50 SNP windows for an r2 value exceeding 0.5, and then down-sampled to 20,000 SNPs using PLINK 1.90 (36). We then repeated PCA in pcadapt and calculated mean r2 among SNPs in 100 SNP windows generated in PLINK.
Range-wide phylogeny and genetic structure analysis
To identify intraspecific variation in LG1 rearrangement frequency relative to the genome-wide average across the Northwest Atlantic, we then calculated individual neighbor-joining trees using all 511 samples. We calculated separate trees for the 237 SNPs with map coordinates within the LG1 rearrangement and for 2500 randomly selected SNPs outside of the known chromosomal rearrangements found in Atlantic cod on linkage groups LG1, LG2, LG7, and LG12 (10). Trees were calculated using POPULATIONS v1.2.33 (37) on the basis of Cavalli-Sforza and Edwards’ (38) chord distances with 1000 bootstrap replicates on loci. Neighbor-joining trees were visualized using FigTree v1.74 (39). Subsampling of genome-wide and inversion loci was conducted using the genepopedit R package (40).
Next, we conducted Bayesian clustering analysis of all 511 sampled individuals with three replicate Markov chain Monte Carlo runs on LG1 rearrangement genotypes in STRUCTURE v2.3.4 (41). We implemented these runs using parallelstructure (42) in R, assuming K = 2 clusters, and conducted 100,000 burn-ins and 500,000 iterations. We visualized individual genetic ancestry proportions using CLUMPAK (43). Conversion of genotypes to STRUCTURE format was carried out using PGDSpider (44).
Genome-wide association analysis of migratory phenotype
We assigned migratory behavioral phenotypes to all individuals in each sampling site from previously identified behavioral classes categorized in Atlantic cod by Robichaud and Rose (20) (table S1). When multiple locations within the range of a sampling site exhibited variable phenotypes, we assigned the most frequent phenotype identified across these locations. Because of the potential variation in sampling and behavioral measurement methods between studies, we binned three migratory behavioral classes together (accurate homers, inaccurate homers, and dispersers) as migratory and categorized sedentary individuals as nonmigratory. We then conducted genome-wide association using two polygenic methods, RF (45) and partial RDA (46), and a single-locus LFMM (47).
To find polygenic associations with migratory phenotype, we first used RF (45) classification, a nonparametric, decision tree–based algorithm. We produced a matrix with individual migratory phenotypes as the response variable and SNP genotypes at each locus as predictor variables. Random forest was run using the R package randomForest (48) after imputing missing genotypes using the rfImpute function. We ran 5000 trees as predictor rank stabilization was ensured with this quantity of trees. We set the mtry parameter to default following testing of optimal mtry parameter values using the tuneRF function. Stratified sampling was applied to ensure unbiased representation of migratory phenotype class within each tree. Out-of-bag mean decrease in accuracy was calculated across all trees for each SNP as an estimate of predictor importance. We retained all SNPs with a mean decrease in accuracy greater than 0.0015; values below this limit exhibited a sharp drop in mean decrease in accuracy, indicating relatively limited utility of these SNPs in classifying migratory and nonmigratory individuals.
We then carried out a constrained ordination using partial RDA. This method maximizes the proportion of variance explained in linear combinations of response variables by linear combinations of predictor variables (49). To detect polygenic associations, we used a matrix of diploid genotypes for each individual at each locus as dependent variables and migratory phenotype as the predictor variable and carried out RDA using the vegan R package (50). Missing genotype calls were imputed for each individual using LinkImpute (51). To account for population structure due to geography, we conditioned the relationship between genotype and migratory phenotype by latitude and longitude. Significance of genotype-phenotype association was calculated with 999 permutations (P < 0.001). We categorized SNPs with absolute values of canonical redundancy axis scores, exceeding the 99th percentile on the first axis (RDA1) as significantly associated with migratory phenotype. LD was calculated between migration-associated SNPs identified across all three methods in PLINK using an r2 cutoff of 0.2.
Detection of SNP association in the LFMM was conducted by using a matrix factorization algorithm to calculate loadings of K latent factors, similar to a reduced set of PC, that each described a different source of population structure (47). We estimated the value of K with sparse nonnegative matrix factorization (52) using the sNMF function on all 6669 SNPs in the LEA R package (53). We then used the lfmm function in LEA to identify SNP association with migratory phenotype using K = 4 latent factors. We converted P values for all loci to q values to control for false discovery rate (54) and retained loci with q < 0.05.
Detection of selective sweeps
To identify genomic regions exhibiting signatures of selection distinct to LG1 homozygous inverted and noninverted individuals, we calculated CLR (21) and iHS (22) across LG1 separately within groups of individuals with different inversion genotypes. We calculated CLR using the SweeD (55) software package and iHS using selscan (56). Individuals were separated by rearrangement genotype to reduce the possibility of confounding presence of inversions with signatures of selection; although residual LD within rearrangements will still occur through reduced recombination within heterozygotes, both CLR and iHS have been shown to be robust to recombination rate variation (23, 24). Because inversion genotypes were not known a priori, and SNP genotypes do not allow for direct contiguous sequence comparison to identify rearrangements from shifts in orientation, ordination-based identification of inversions has been used for both human SNP data and to identify the four known rearrangements in SNPs from Atlantic cod (57, 58). Using this approach, we separated individuals from Newfoundland and Labrador sampling sites into LG1 homozygous noninverted and LG1 homozygous inverted groups based on individual PC scores. We calculated CLR for windows of 20 SNPs within inverted (n = 41) and noninverted (n = 136) individuals, assuming a folded site frequency spectrum, and selected regions exhibiting CLR exceeding the 95th percentile as potential targets of selection. To calculate iHS, we first extracted SNP genotypes from LG1 only using vcftools and then phased vcf files using Beagle 5.0 (59). We then calculated iHS in selscan for each group with the following parameters: --gap-scale 100000 --max-gap 1000000 --max-extend 0 --keep-low-freq -trunc-ok. Next, we normalized these values using the norm software distributed with selscan. We selected regions exhibiting an absolute value of iHS exceeding the 95th percentile as potentially under selection.
Contemporary and historical effective population size estimation
To detect the recent genetic history of Northern cod, we used the linkage-based method implemented in LinkNe (29) to estimate effective population size (Ne). We calculated Ne across a 150-year period in the 330 Northern cod samples from Newfoundland and Labrador. We separated LG1 homozygous inverted, heterozygous (n = 153), and LG1 homozygous noninverted individuals based on PC score. Before Ne estimation, we filtered genome-wide SNP datasets for these individuals to remove SNPs within the LG1 and LG12 rearrangements identified within Northern cod, resulting in a final dataset of 6270 SNPs. These regions were removed as selection and reduced recombination within rearrangements may skew LD inference of historical Ne. For each group, we calculated Ne separately in LinkNe in bins of 0.05 Morgans for all SNPs with minor allele frequency values greater than 0.05. We binned Ne estimates into generations calculated from recombination rate, assuming a mean generation time of 6 years (13). We assigned a contemporary sampling date of 2010, corresponding to an intermediate estimate of sampling dates from all Northern cod samples. To compare changes in effective population size between groups that exhibited large differences in Ne, we calculated relative Ne by scaling Ne values within each group by the maximum observed Ne for that group. We then plotted relative Ne estimates as proportions against estimates of cod abundance measured from 1983 to 2004 and historical reconstructions of stock biomass from 1850 to 2004 (11).
We also estimated Ne using a bin size of 0.01 to identify both finer-scale and long-term demographic variation. We found similar estimates of contemporary Ne, and again uncovered similar patterns of 20th-century expansion and decline to those identified using a bin size of 0.05 (fig. S4), but found that absolute values of historical Ne at intermediate time points varied from those found using the larger bin size (table S3). Simulations by Hollenbeck et al. (29) indicate that the detection of demographic changes over longer time periods will be limited by the number of genomic markers available, and previous analyses have suggested that long-term estimates may be affected by mutation over extended time frames (60), which may bias LD estimates of effective size. Our estimates with smaller bin sizes are thus expected to exhibit lower precision than those obtained from a larger sample of markers and likely account for the discrepancy in absolute values of Ne observed between intermediate time frames with different bin size parameters.
To account for the possibility of an observed decline in LG1 homozygous inverted individuals driven by small sample size (n = 41), we generated 10 subsamples of 41 LG1 homozygous noninverted individuals and conducted replicate effective size estimation. We observed similar starting effective sizes across replicates (mean, Ne = 2577.449, SD = 170.1235) and identified expansions coinciding with stock recovery in all 10 samples. We did not observe patterns of decline matching the pattern observed in the sample of homozygous inverted individuals, indicating that this decline is likely not an artifact of small sample size (fig. S5). We identified a recent, post-1980 decline across 3 of the 10 subsamples, which could indicate spatial heterogeneity in recent decline patterns across LG1 homozygous noninverted Northern cod samples (fig. S5). To test this hypothesis, we split LG1 homozygous noninverted, LG1 homozygous inverted, and heterozygous groups into population groups corresponding to coastal Labrador samples, offshore samples, and samples from coastal Newfoundland. We then conducted LinkNe analysis on these samples, revealing a recent decline of LG1 homozygous noninverted and heterozygous individuals within the coastal Labrador grouping (fig. S3). Together, these results indicate that small sample sizes have likely not produced false signals of decline and demonstrate potential spatial variation in cod stock decline across Newfoundland and Labrador.
Bottleneck detection
To provide secondary genetic confirmation of observed stock collapses observed in abundance data and using effective size reconstruction, we also tested for the presence of recent genetic bottlenecks in LG1 homozygous noninverted, heterozygous, and LG1 homozygous inverted groups. We analyzed 6270 genome-wide SNPs excluding LG1 and LG12 rearrangements in each group using the BOTTLENECK (31) software to detect bottlenecks assuming an infinite alleles model. We inferred bottlenecks from presence of a mode shift in allele frequency toward reduction in the number of low frequency alleles or significant sign and standardized differences test results.
Supplementary Material
Acknowledgments
We thank C. Hollenbeck for assistance with LinkNe software and Z. Szpiech for help with selscan. We also thank L. Weir, D. Heath, B. Star, and R. Waples for helpful comments on the manuscript and the CIGENE for genotyping of Atlantic cod used in this study. Funding: This study was supported by the Genomics Research and Development Initiative (GRDI) of the Department of Fisheries and Oceans Canada (DFO) and the Natural Sciences Engineering and Research Council Canada (NSERC). Author contributions: T.K., S.J.L., I.R.B., and P.B. designed the study. T.K., S.J.L., and E.V.A.S. performed statistical analyses. P.B., I.R.B., M.S.-W., M.P.K., and S.L. provided molecular data for the study. P.R. provided cod abundance survey data. T.K. drafted the manuscript, and all authors contributed to discussion, writing, and editing of the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Genomic data archiving information is available in source publications (7, 10, 15, 19), and individual genotypes, inversion haplotype assignment, and assigned migratory phenotype will be available on Dryad (doi:10.5061/dryad.p6m4340). Migratory category and stock reconstruction data were obtained from tables in source publications (11, 20). Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav2461/DC1
Fig. S1. Fine-scale analysis of individual divergence and LD within LG1 using whole-genome data obtained from Barney et al. (19).
Fig. S2. Significance of association per SNP with migration phenotype in a LFMM, categorizing SNPs with a false discovery rate q value of <0.05 as significant.
Fig. S3. Comparison of historical and recent (2004) effective size of LG1 homozygous noninverted, LG1 homozygous inverted, and heterozygous individuals in Labrador, Newfoundland, and offshore sites and proportion of each genotype group at each site.
Fig. S4. Recent effective population size relative to maximum effective population size within each group of LG1 homozygous noninverted, LG1 homozygous inverted, and heterozygous Northern cod groupings calculated using LinkNe assuming a bin size of 0.01 M, plotted with reconstructed estimates of historical biomass measured from 1500 to 2004 (11) relative to the maximum value.
Fig. S5. Comparison of historical and recent (2004) effective size in subsamples of 41 LG1 homozygous noninverted individuals.
Table S1. Site code, geographic location (latitude, longitude, name), dataset source, number of genotyped individuals, migratory behavior, and sites used to infer migratory behavior from Robichaud and Rose (20) for 511 Atlantic cod (G. morhua) samples.
Table S2. Single Nucleotide Polymorphism Database (dbSNP) information for all 24 migration-associated SNPs identified across all genome-wide association methods, and gene names and functions of closest known gene for each SNP as described by Berg et al. (10).
Table S3. Effective size estimates calculated by LG1 rearrangement genotype using LinkNe.
REFERENCES AND NOTES
- 1.Mimura M., Yahara T., Faith D. P., Vázquez-Domínguez E., Colautti R. I., Araki H., Javadi F., Núñez-Farfán J., Mori A. S., Zhou S., Hollingsworth P. M., Neaves L. E., Fukano Y., Smith G. F., Sato Y.-I., Tachida H., Hendry A. P., Understanding and monitoring the consequences of human impacts on intraspecific variation. Evol. Appl. 10, 121–139 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Des Roches S., Post D. M., Turley N. E., Bailey J. K., Hendry A. P., Kinnison M. T., Schweitzer J. A., Palkovacs E. P., The ecological importance of intraspecific variation. Nat. Ecol. Evol. 2, 57–64 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Farkas T. E., Mononen T., Comeault A. A., Hanski I., Nosil P., Evolution of camouflage drives rapid ecological change in an insect community. Curr. Biol. 23, 1835–1843 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Hilborn R., Quinn T. P., Schindler D. E., Rogers D. E., Biocomplexity and fisheries sustainability. Proc. Natl. Acad. Sci. U.S.A. 100, 6564–6568 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schindler D. E., Hilborn R., Chasco B., Boatright C. P., Quinn T. P., Rogers L. A., Webster M. S., Population diversity and the portfolio effect in an exploited species. Nature 465, 609–612 (2010). [DOI] [PubMed] [Google Scholar]
- 6.Joron M., Frezal L., Jones R. T., Chamberlain N. L., Lee S. F., Haag C. R., Whibley A., Becuwe M., Baxter S. W., Ferguson L., Wilkinson P. A., Salazar C., Davidson C., Clark R., Quail M. A., Beasley H., Glithero R., Lloyd C., Sims S., Jones M. C., Rogers J., Jiggins C. D., ffrench-Constant R. H., Chromosomal rearrangements maintain a polymorphic supergene controlling butterfly mimicry. Nature 477, 203–206 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sinclair-Waters M., Bradbury I. R., Morris C. J., Lien S., Kent M. P., Bentzen P., Ancient chromosomal rearrangement associated with local adaptation of a postglacially colonized population of Atlantic cod in the Northwest Atlantic. Mol. Ecol. 27, 339–351 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Allendorf F. W., Hohenlohe P., Luikart G., Genomics and the future of conservation genetics. Nat. Rev. Genet. 11, 697–709 (2010). [DOI] [PubMed] [Google Scholar]
- 9.Bradbury I. R., Hubert S., Higgins B., Bowman S., Borza T., Paterson I. G., Snelgrove P. V. R., Morris C. J., Gregory R. S., Hardie D., Hutchings J. A., Ruzzante D. E., Taggart C. T., Bentzen P., Genomic islands of divergence and their consequences for the resolution of spatial structure in an exploited marine fish. Evol. Appl. 6, 450–461 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Berg P. R., Star B., Pampoulie C., Bradbury I. R., Bentzen P., Hutchings J. A., Jentoft S., Jakobsen K. S., Trans-oceanic genomic divergence of Atlantic cod ecotypes is associated with large inversions. Heredity 119, 418–428 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rose G. A., Reconciling overfishing and climate change with stock dynamics of Atlantic cod (Gadus morhua) over 500 years. Can. J. Fish. Aquat. Sci. 61, 1553–1557 (2004). [Google Scholar]
- 12.Hutchings J. A., Life history consequences of overexploitation to population recovery in Northwest Atlantic cod (Gadus morhua). Can. J. Fish. Aquat. Sci. 62, 824–832 (2005). [Google Scholar]
- 13.Olsen E. M., Heino M., Lilly G. R., Morgan M. J., Brattey J., Ernande B., Dieckmann U., Maturation trends indicative of rapid evolution preceded the collapse of Northern cod. Nature 428, 932–935 (2004). [DOI] [PubMed] [Google Scholar]
- 14.Kirubakaran T. G., Grove H., Kent M. P., Sandve S. R., Baranski M., Nome T., De Rosa M. C., Righino B., Johansen T., Otterå H., Sonesson A., Lien S., Andersen Ø., Two adjacent inversions maintain genomic differentiation between migratory and stationary ecotypes of Atlantic cod. Mol. Ecol. 25, 2130–2143 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Sinclair-Waters M., Bentzen P., Morris C. J., Ruzzante D. E., Kent M. P., Lien S., Bradbury I. R., Genomic tools for management and conservation of Atlantic cod in a coastal marine protected area. Can. J. Fish. Aquat. Sci. 75, 1915–1925 (2018). [Google Scholar]
- 16.Hutchings J. A., Spatial and temporal variation in the density of northern cod and a review of the hypotheses for the stock collapse. Can. J. Fish. Aquat. Sci. 53, 943–962 (1996). [Google Scholar]
- 17.Morgan M. J., Koen-Alonso M., Rideout R. M., Buren A. D., Maddock Parsons D., Growth and condition in relation to the lack of recovery of northern cod. ICES J. Mar. Sci. 75, 631–641 (2018). [Google Scholar]
- 18.Hoffmann A. A., Rieseberg L. H., Revisiting the impact of inversions in evolution: From population genetic markers to drivers of adaptive shifts and speciation? Annu. Rev. Ecol. Evol. Syst. 39, 21–42 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barney B. T., Munkholm C., Walt D. R., Palumbi S. R., Highly localized divergence within supergenes in Atlantic cod (Gadus morhua) within the Gulf of Maine. BMC Genomics 18, 271 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robichaud D., Rose G. A., Migratory behaviour and range in Atlantic cod: Inference from a century of tagging. Fish Fish. 5, 185–214 (2004). [Google Scholar]
- 21.Nielsen R., Williamson S., Kim Y., Hubisz M. J., Clark A. G., Bustamante C., Genomic scans for selective sweeps using SNP data. Genome Res. 15, 1566–1575 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Voight B. F., Kudaravalli S., Wen X., Pritchard J. K., A map of recent positive selection in the human genome. PLOS Biol. 4, e72 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williamson S. H., Hubisz M. J., Clark A. G., Payseur B. A., Bustamante C. D., Nielsen R., Localizing recent adaptive evolution in the human genome. PLOS Genet. 3, e90 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.al Basatena N.-K. S., Hoggart C. J., Coin L. J., O’Reilly P. F., The effect of genomic inversions on estimation of population genetic parameters from SNP data. Genetics 193, 243–253 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.COSEWIC, Assessment and status report on the Atlantic cod Gadus morhua in Canada (COSEWIC, 2010).
- 26.Ruzzante D. E., Taggart C. T., Doyle R. W., Cook D., Stability in the historical pattern of genetic structure of Newfoundland cod (Gadus morhua) despite the catastrophic decline in population size from 1964 to 1994. Conserv. Genet. 2, 257–269 (2001). [Google Scholar]
- 27.Jakobsdóttir K. B., Pardoe H., Magnússon Á., Björnsson H., Pampoulie C., Ruzzante D. E., Marteinsdóttir G., Historical changes in genotypic frequencies at the Pantophysin locus in Atlantic cod (Gadus morhua) in Icelandic waters: Evidence of fisheries-induced selection? Evol. Appl. 4, 562–573 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Therkildsen N. O., Hemmer-Hansen J., Als T. D., Swain D. P., Morgan M. J., Trippel E. A., Palumbi S. R., Meldrup D., Nielsen E. E., Microevolution in time and space: SNP analysis of historical DNA reveals dynamic signatures of selection in Atlantic cod. Mol. Ecol. 22, 2424–2440 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Hollenbeck C. M., Portnoy D. S., Gold J. R., A method for detecting recent changes in contemporary effective population size from linkage disequilibrium at linked and unlinked loci. Heredity 117, 207–216 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Waples R. K., Larson W. A., Waples R. S., Estimating contemporary effective population size in non-model species using linkage equilibrium across thousands of loci. Heredity 117, 233–240 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Piry S., Luikart G., Cornuet J.-M., BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 90, 502–503 (1999). [Google Scholar]
- 32.Frank K. T., Petrie B., Choi J. S., Leggett W. C., Trophic cascades in a formerly cod-dominated ecosystem. Science 308, 1621–1623 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Luu K., Bazin E., Blum M. G. M., pcadapt: An R package to perform genome scans for selection based on principal component analysis. Mol. Ecol. Resour. 17, 67–77 (2017). [DOI] [PubMed] [Google Scholar]
- 34.V. M. Rajagopal, ggman: R package for Manhattan plots, GitHub repository (2017); https://github.com/drveera/ggman.
- 35.Danecek P., Auton A., Abecasis G., Albers C. A., Banks E., DepRisto M. A., Handsaker R. E., Lunter G., Marth G. T., Sherry S. T., McVean G., Durbin R.; 1000 Genomes Project Analysis Group , The variant call format and VCFtools. Bioinformatics 27, 2156–2158 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang C. C., Chow C. C., Tellier L. C. A. M., Vattikuti S., Purcell S. M., Lee J. J., Second-generation PLINK: Rising to the challenge of larger and richer datasets. GigaScience 4, 7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O. Langella, POPULATIONS 1.2.28. Population genetic software (individuals or populations distances, phylogenetic trees) (2012); https://launchpad.net/~olivier-langella/+archive/ubuntu/ppa.
- 38.Cavalli-Sforza L. L., Edwards A. W. F., Phylogenetic analysis. Models and estimation procedures. Am. J. Hum. Genet. 19, 233–257 (1967). [PMC free article] [PubMed] [Google Scholar]
- 39.A. Rambaut, FigTree v1.4 (2012); http://tree.bio.ed.ac.uk/software/figtree/.
- 40.Stanley R. R., Jeffery N. W., Wringe B. F., DiBacco C., Bradbury I. R., genepopedit: A simple and flexible tool for manipulating multilocus molecular data in R. Mol. Ecol. Resour. 17, 12–18 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Pritchard J. K., Stephens M., Donnelly P., Inference of population structure using multilocus genotype data. Genetics 155, 945–959 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Besnier F., Glover K. A., ParallelStructure: A R package to distribute parallel runs of the population genetics program STRUCTURE on multi-core computers. PLOS ONE 8, e70651 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kopelman N. M., Mayzel J., Jakobsson M., Rosenberg N. A., Mayrose I., Clumpak: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Resour. 15, 1179–1191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lischer H. E. L., Excoffier L., PGDSpider: An automated data conversion tool for connecting population genetics and genomics programs. Bioinformatics 28, 298–299 (2012). [DOI] [PubMed] [Google Scholar]
- 45.Breiman L., Random forests. Mach. Learn. 45, 5–32 (2001). [Google Scholar]
- 46.Rao C. R., The use and interpretation of principal component analysis in applied research. Sankhyā Ser. A 26, 329–358 (1964). [Google Scholar]
- 47.Frichot E., Schoville S. D., Bouchard G., François O., Testing for associations between loci and environmental gradients using latent factor mixed models. Mol. Biol. Evol. 30, 1687–1699 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liaw A., Wiener M., Classification and regression by randomForest. R News 2, 18–22 (2002). [Google Scholar]
- 49.Lasky J. R., Des Marais D. L., McKay J. K., Richards J. H., Juenger T. E., Keitt T. H., Characterizing genomic variation of Arabidopsis thaliana: The roles of geography and climate. Mol. Ecol. 21, 5512–5529 (2012). [DOI] [PubMed] [Google Scholar]
- 50.J. Oksanen, F. Guillaume Blanchet, M. Friendly, R. Kindt, P. Legendre, D. McGlinn, P. R. Minchin, R. B. O’Hara, G. L. Simpson, P. Solymos, M. Henry H. Stevens, E. Szoecs, H. Wagner, vegan: Community Ecology Package. R package version 1.17-6 (2011); http://cran.r-project.org/package=vegan.
- 51.Money D., Gardner K., Migicovsky Z., Schwaninger H., Zhong G. Y., Myles S., LinkImpute: Fast and accurate genotype imputation for nonmodel organisms. G3 (Bethesda) 5, 2383–2390 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frichot E., Mathieu F., Trouillon T., Bouchard G., François O., Fast and efficient estimation of individual ancestry coefficients. Genetics 196, 973–983 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Frichot E., François O., LEA: An R package for landscape and ecological association studies. Methods Ecol. Evol. 6, 925–929 (2015). [Google Scholar]
- 54.Benjamini Y., Hochberg Y., Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. Methodol. 57, 289–300 (1995). [Google Scholar]
- 55.Pavlidis P., Živkovic D., Stamatakis A., Alachiotis N., SweeD: Likelihood-based detection of selective sweeps in thousands of genomes. Mol. Biol. Evol. 30, 2224–2234 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Szpiech Z. A., Hernandez R. D., selscan: An efficient multithreaded program to perform EHH-based scans for positive selection. Mol. Biol. Evol. 31, 2824–2827 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ma J., Amos C. I., Investigation of inversion polymorphisms in the human genome using principal components analysis. PLOS ONE 7, e40224 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sodeland M., Jorde P. E., Lien S., Jentoft S., Berg P. R., Grove H., Kent M. P., Arnyasi M., Olsen E. M., Knutsen H., “Islands of Divergence” in the Atlantic cod genome represent polymorphic chromosomal rearrangements. Genome Biol. Evol. 8, 1012–1022 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Browning B. L., Zhou Y., Browning S. R., A one-penny imputed genome from next generation reference panels. Am. J. Hum. Genet. 103, 338–348 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corbin L. J., Liu A. Y. H., Bishop S. C., Woolliams J. A., Estimation of historical effective population size using linkage disequilibria with marker data. J. Anim. Breed. Genet. 129, 257–270 (2012). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav2461/DC1
Fig. S1. Fine-scale analysis of individual divergence and LD within LG1 using whole-genome data obtained from Barney et al. (19).
Fig. S2. Significance of association per SNP with migration phenotype in a LFMM, categorizing SNPs with a false discovery rate q value of <0.05 as significant.
Fig. S3. Comparison of historical and recent (2004) effective size of LG1 homozygous noninverted, LG1 homozygous inverted, and heterozygous individuals in Labrador, Newfoundland, and offshore sites and proportion of each genotype group at each site.
Fig. S4. Recent effective population size relative to maximum effective population size within each group of LG1 homozygous noninverted, LG1 homozygous inverted, and heterozygous Northern cod groupings calculated using LinkNe assuming a bin size of 0.01 M, plotted with reconstructed estimates of historical biomass measured from 1500 to 2004 (11) relative to the maximum value.
Fig. S5. Comparison of historical and recent (2004) effective size in subsamples of 41 LG1 homozygous noninverted individuals.
Table S1. Site code, geographic location (latitude, longitude, name), dataset source, number of genotyped individuals, migratory behavior, and sites used to infer migratory behavior from Robichaud and Rose (20) for 511 Atlantic cod (G. morhua) samples.
Table S2. Single Nucleotide Polymorphism Database (dbSNP) information for all 24 migration-associated SNPs identified across all genome-wide association methods, and gene names and functions of closest known gene for each SNP as described by Berg et al. (10).
Table S3. Effective size estimates calculated by LG1 rearrangement genotype using LinkNe.