

Computer-assisted strategy for drug delivery leads to discovery of polymeric micelle formulations for poorly soluble drugs.
Abstract
Many drug candidates fail therapeutic development because of poor aqueous solubility. We have conceived a computer-aided strategy to enable polymeric micelle-based delivery of poorly soluble drugs. We built models predicting both drug loading efficiency (LE) and loading capacity (LC) using novel descriptors of drug-polymer complexes. These models were employed for virtual screening of drug libraries, and eight drugs predicted to have either high LE and high LC or low LE and low LC were selected. Three putative positives, as well as three putative negative hits, were confirmed experimentally (implying 75% prediction accuracy). Fortuitously, simvastatin, a putative negative hit, was found to have the desired micelle solubility. Podophyllotoxin and simvastatin (LE of 95% and 87% and LC of 43% and 41%, respectively) were among the top five polymeric micelle-soluble compounds ever studied experimentally. The success of the strategy described herein suggests its broad utility for designing drug delivery systems.
INTRODUCTION
One of the major obstacles for the development of highly potent pharmaceuticals is their poor aqueous solubility, which is characteristic of approximately 40% of drug candidates (1). This undesired property could substantially delay or even halt the progression of drug candidates to the clinic. Various drug delivery systems based on liposomes (2), nanoparticles (3), nanogels (4), and polymeric micelles (5) have been studied intensely to improve the solubilization of drugs and drug candidates (6), but relatively few of them have been advanced to clinical products. Various characteristics of these systems have been considered such as physiological barriers, physicochemical properties of drugs, and carrier-forming materials. However, despite certain progress in developing practically useful delivery systems, this experimental approach has remained time-consuming and expensive. The need to use rational, computer-aided approaches to designing delivery systems for drug molecules has been previously articulated in the literature (7). These approaches can enable early decisions to streamline the development process and decrease the attrition of drug candidates by matching them with their preferred delivery systems. However, while computational methods have found broad application in the field of drug discovery, they have not yet become equally popular in the area of drug delivery. Most computational studies have relied on molecular docking and molecular dynamics to offer insights concerning molecular interactions between drugs and carriers (8–10). For instance, molecular dynamics approaches have been applied to better understand the micelle structure of polymers (11) and to simulate drug loading into a delivery system (12), while mathematical modeling has been applied to investigate the hydrogel drug release (13). Shi et al. (14) have applied molecular docking to identify small molecules as optimal building blocks for designing an optimal telodendrimer for doxorubicin. The authors synthesized a series of nanocarriers and experimentally validated their findings. One of the nanocarriers has shown improved delivery properties, lower toxicity, and superior anticancer effects. More recently, another docking-based method to predict the drug affinity for PLA [poly(lactic acid)]–PEG [poly(ethylene glycol)] nanoparticles and their effective drug loading was reported (15). While targeting mechanistic aspects of drug loading into delivery systems or drug release from these systems, these approaches are computationally expensive, which makes it difficult to expect their routine application in pharmaceutics; besides, these approaches do not target directly the prediction of drug loading efficiency (LE) and/or loading capacity (LC).
There have also been some studies using statistical approaches as applied to modeling and design of drug delivery systems. These approaches, known as quantitative structure-property relationships (QSPR) modeling, found especially prolific use in both medicinal chemistry and chemical toxicology (16, 17) but much less so in drug delivery, perhaps mostly due to the scarcity of experimental data. A recent study reported the development of a series of QSPR models to assess the loading of doxorubicin in polymeric micelles using the genetic function approximation algorithm (18), but since these models were developed for one drug only, they are not generalizable across multiple drugs. In another recent study, the authors predicted fouling release activity for polymer coating materials (19). Transgene expression efficacy of polymers obtained from aminoglycoside antibiotics has been modeled using an online web tool named “Support vector regression-based Online Learning Equipment” (SOLE) (20).
Previously, we have successfully developed (21) and applied (22) QSPR models to predict loading of amphiphilic drugs into liposomes. However, liposomes by design are not the best system for incorporation of various poorly water-soluble molecules since loading of these molecules is constrained by the structure of the lipid bilayers. Recently, we have developed a novel polymeric micelle system formed by amphiphilic block copolymers of hydrophilic poly(2-methyl-2-oxazoline) (PMeOx) and hydrophobic poly(2-butyl-2-oxazoline) (PBuOx). This system exhibited an exceptionally high solubilization for some hydrophobic drugs such as taxanes (23, 24). However, poly(2-oxazoline) (POx) micelles could not solubilize every poorly water-soluble drug equally well. Mechanisms of encapsulation of poorly water-soluble drugs into the polymeric micelle systems have been previously studied (25, 26), but they continue to be poorly understood, and so far, there have been no approaches that would assure success of loading experiments for any selected drugs or drug candidates.
Our previous studies have led us to assert that POx micelles with very high solubilization of some drugs and very poor solubilization of others represent both practically important and descriptive example to evaluate a computer-aided approach to rational design of a polymeric micelle-based delivery systems for poorly soluble drugs. Here, as a proof of concept, we have (i) rationally selected a set of about 21 poorly soluble and chemically diverse drugs from the Selleck Chemicals library (www.selleckchem.com/) and tested them for LE and LC to supplement previously collected data on 20 compounds; (ii) compiled, curated, and integrated all LE and LC data for all drugs tested experimentally in one of our laboratories; (iii) developed novel chemical descriptors for polymers and drug-polymer complexes; (iv) generated and interpreted QSPR models for drug loading into polymeric micelle-based delivery systems; (v) identified, by virtual screening, drugs with poorly aqueous solubility predicted to have either high or low LE and LC; and (vi) experimentally measured LE and LC values of selected virtual screening hits and successfully validated model predictions. To the best of our knowledge, this is the first study on rational design of drug delivery systems that combines, in a single workflow, rationally designed experimental data collection to enable model development, computational modeling of drug loading into polymeric micelles, and effective experimental validation of predicted formulation properties for the studied drug delivery systems. The success of this investigation suggests that computational approaches could substantially streamline and accelerate the development of novel and effective drug delivery systems.
RESULTS
Rational design of a diverse set of poorly soluble drugs
In the absence of rational approaches to the experimental design, the discovery of suitable drug-POx systems is left to serendipity, implying high cost and time-consuming effort. Thus, we endeavored to develop computational QSPR models capable of accurate prediction of drug molecules with high LE and LC values in POx micelles that could be formulated using this drug delivery system and thereby achieve much greater therapeutic efficacy (see Fig. 1 and the “Study design” section). Before this study, only 20 compounds were tested for solubilization in POx micelles. Among these, we have serendipitously discovered several drugs with good or excellent solubilization in POx micelles, whereas at the same time many compounds were found to have poor micelle solubilization properties. These data were not sufficient for building predictive QSPR models. Furthermore, chemical diversity of these compounds was limited as compared to that of a drug library represented by 768 chemicals from the Selleck collection of U.S. Food and Drug Administration (FDA)–approved drugs (Fig. 2). Therefore, using a diversity sampling approach, we rationally selected a set of chemically diverse drug molecules that were poorly soluble or insoluble. The resulting expanded set of 61 molecules was chemically diverse and structurally representative of the chemical space of FDA-approved drugs (Fig. 2). From these 61 compounds, 21 drugs were selected on the basis of their clinical indications and respective biological pathways, as well as price and availability, followed by their testing using our standard experimental protocols to ensure data consistency. Combining the results of testing obtained on these 21 compounds as well as on 20 compounds tested previously, we thus obtained a unique training set of 41 compounds comprising 408 experimental data points. The complete micelle solubilization data for single and binary drug combination are given in data file S1. These data were used for molecular modeling studies (see the next section).
Fig. 1. Study design.
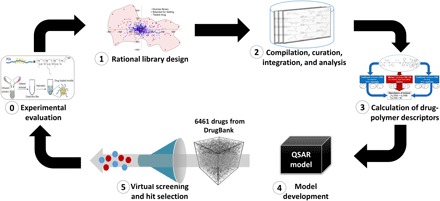
Fig. 2. Coverage of chemical space by previously tested drugs and compounds rationally selected to increase structural diversity.
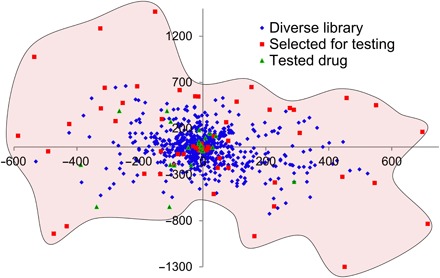
Barycentric coordinates are calculated using two-dimensional SiRMS (molecular fragments) descriptors differentiated by atom type.
Cheminformatics analysis
We have compiled a dataset of 41 compounds investigated in several concentrations and under different experimental conditions. Originally, our collection included 408 data points for these 41 compounds, reflecting different drug concentrations, structural diversity of POx polymers, and experimental conditions (see Materials and Methods). In our previous studies, we have used standard chemical descriptors of molecules and/or certain experimental conditions to establish a correlation with drug loading or bioactivity, respectively. Modeling of drug loading into polymeric micelles proved to be a more challenging exercise, as drug chemical descriptors and experimental conditions were found insufficient to enable the development of statistically significant models.
To address this challenge in this study, we have developed novel descriptors of drug-polymer systems reflective of the chemical structures of both small molecules and polymers. These new descriptors were developed on the basis of SiRMS (simplex representation of molecular structure) descriptors (27) originally devised for small organic molecules and later adapted for mixtures of organic molecules (28, 29). These new descriptors were obtained by considering drug-POx systems as stoichiometric mixtures of polymers (represented by unique monomeric blocks used for their synthesis) and drug molecules (see Materials and Methods for more details).
We have also observed (see the “Data curation” section) that drug concentration and other experimental conditions had a strong influence on both LE and LC. For instance, although drug concentration does not influence the encapsulation effectiveness, most of the compounds that could be solubilized had both high LE and high LC values for low drug concentration. When the polymeric micelle is saturated with the drug and if the drug concentration is higher than the saturation point, then the polymer may collapse. Therefore, to achieve the best LE and LC, compounds were tested using a variety of concentrations and experimental conditions. All the concentrations and experimental conditions were used as additional descriptors of drug-polymer complexes for both model building and virtual screening to improve model accuracy.
To illustrate the influence of polymer structure, experimental conditions, molecular weight (MW), and lipophilicity (LogP) on the compound solubilization in POx micelles, we have identified clusters of drugs tested at 8 mg/ml, i.e., at the highest concentration used for testing the largest number of drugs. For this analysis, original SiRMS descriptors were normalized and low variance descriptors (threshold = 0.1) were removed. Hierarchical clustering was performed using SciPy package (www.scipy.org/) in Python 3.6 (www.python.org/) on the basis of Euclidean distance and the Ward method (30). A heat map of proximity matrix and dendrogram are reported in fig. S1. The summary of clusters showing the LE and LC (mean, SD, and maximum value) for the drugs tested at 8 mg is shown in table S1.
As can be seen in table S1, similar compounds belonging to the same cluster could have considerably different LE values. For instance, the cisplatin prodrug derivatives (cluster 1) had optimal chain length of six carbons [cisplatin prodrug (C6)], with maximum LE = 85.1%. The analogs with 4 and 10 carbons had similar LE, while the one with eight carbons had the lowest LE. VE-822 and NVP-BEZ235 have similar LogP and MW, which, in turn, were different from those of the remaining compounds (LY364947 and imiquimod) in cluster 2. There, only VE-822 showed high LE max (83.34%), but imiquimod, LY364947, and NVP-BEZ235 were not soluble in POx micelles. Compounds in cluster 3 contain similar chemical features, but only AZD5363 shows high LE (63.8%, respectively). In cluster 4, LY294002 and EFV also have similar LogP and MW, but only EFV presents high LE max (86.23). In cluster 7, ABT-263 and ABT-737 have similar chemical structures and similar MW and LogP, but the LE max values for both compounds are drastically different with a maximal LE of 100% for ABT-263 and a maximal LE of 7.3% for ABT-737.
These results show that the knowledge of chemical structure alone is not sufficient to evaluate whether a compound has a good chance to be highly soluble in polymeric micelles. Moreover, even the same drug combined with different polymers or even with the same polymer but under different experimental conditions may exhibit a very different LE. Variable importance estimated from all developed models indicated that some experimental conditions (hydration solvent, hydration temperature, and total solvent volume before evaporation) had very high scores (fig. S2). For instance, combination of docetaxel (DTX) (10 mg/ml) and polymer P6 has been tested twice under the same experimental conditions, varying only the hydration solvent. When DTX was dissolved in deionized water, an LE of 81.8% and an LC of 44.9% were observed; using a mix of deionized water, saline, and phosphate-buffered saline led to the increase of both LE and LC to 90 and 47.4%, respectively.
This analysis illustrates the need to consider the experimental conditions that define the outcome of the loading process as important descriptors of the system. To reflect on this point further, we shall highlight several factors that need to be considered to enable predictive models with much higher accuracy than historical success rate of purely experimental investigations: (i) close interaction between experimental and computational groups; (ii) rational design of the training set; (iii) special descriptors of drug-polymer complexes reflecting the interactions between drugs and micelles; (iv) the use of experimental conditions available for our datasets as descriptors, such as solvents, in which both polymer and drug samples were prepared, their volumes before evaporation, hydration solvent, and hydration temperature; and (v) experimental validation of drug delivery properties for selected both negative and positive hits. Our analysis further highlights the importance of recording and using all parameters/characteristics related to the experiment. For instance, we have traced the size and morphology data for 15 drug-polymer complexes (see data file S1). Although these data are insufficient to be used for model building, our preliminary observations show that most drug-polymer complexes look like worms or spheres, while the specific polymer used in this study alone only forms worm-like structures. The particle size also changes upon the transition from worms to spheres induced by the drug, and the transition point depends on the selected drug. In addition, at a certain point, which is different for each drug, we have observed saturation and sediment formation. The particle size and morphology along with the micelle stability and drug release characteristics are important parameters for the pharmacological performance of these drug delivery systems (31). We anticipate that as we generate and collect new data on size and morphology and other parameters, we will be able to explicitly incorporate these data into our models.
Our data analysis showed that the optimal combination of the experimental conditions varies from case to case. Overall, this indicates the following: (i) the importance of experimental conditions for drug solubilization; (ii) the necessity of choosing the optimal solvent, temperature, etc., for each specific drug-polymer combination; (iii) the need to use experimental conditions as descriptors during modeling and virtual screening; and (iv) the requirement to select not only the computational hits for experimental confirmation but also the optimal experimental conditions (solvent, temperature, etc.) to improve the success rate.
In addition, the type and length of block chains of polymers are shown to be important, as we shall discuss below. Table S2 presents summary data for three drugs tested at 8 mg/ml with the highest experimental variability. As one can see, LE values for DTX vary from 1.56 to 80.06%. Three variables that change their values include polymer batch, mass of the polymer, and hydration temperature. It is possible to see that, when tested in different polymers, DTX is more soluble in polymer P8 (LE = 57.59%) than in polymer P2 (LE = 1.56%) when the mass of the latter polymer is 10 mg. However, when the mass of polymer P2 increased to 20 mg, an LE of 80.06% was achieved. For LDN-57444, the variation in the polymer and drug solvent, the total solvent volume before evaporation, and hydration temperature led to a difference of 26% in the LE. Last, the LE of paclitaxel (PTX) varied from 2.44 to 100%. In this case, the difference is mostly due to different polymers. Both P2 (LE = 2.44%) and P8 (35.72%) presented low LE, while P1 (LE = 86.1%), P4 (100%), and P6 (LE = 91.18%) presented high LE.
Most of the POx polymers used in our studies (P1, P3, P4, P5, and P6) are triblock copolymers: poly(2-methyl-2-oxazoline)-block-poly(2-butyl-2-oxazoline)-block-poly(2-methyl-2-oxazoline) [P(MeOx-b-BuOx-b-MeOx)], differing in the chain length of each block. These differences, however, all within 10 to 15% variability typical for batch-to-batch variations, were not expected to result in any substantial difference in solubilization. Polymer P2 is a diblock copolymer, P(MeOx-b-BuOx); it had a decreased ability to solubilize PTX and DTX compared to the respective triblock, which illustrated the effect of the copolymer architecture (32). The triblock polymer P8 contains a few aromatic 2-benzyl-2-oxazoline (BzOx) units copolymerized with aliphatic BuOx [P(MeOx-b-co-BuOx/BzOx-b-MeOx)]. Another polymer (P7) is a triblock containing 2-nonyl-2-oxazoline (NOx) units instead of BuOx units in the hydrophobic block [P(MeOx-b-NOx-b-MeOx)]. Both modifications obtained by adding aromatic groups or long-chain alkyl groups to the core of the POx micelle appear to have adverse effects on the solubilization of PTX (33). These observations reinforce the importance of building QSPR models incorporating all available information about both chemical structures of drugs and polymers as well as the experimental conditions to predict putative positive hits with high confidence.
The results of the cluster analysis confirmed that LogP and MW alone are not sufficient to predict drug loading. Thus, we developed a series of robust and externally predictive [correct classification rate (CCR), 0.76 to 0.85] binary QSPR models for forecasting LC and LE. Corresponding statistical characteristics estimated by fivefold external cross-validation are summarized in table S3. All models showed both high sensitivity (>70%) and specificity (>76%), as well as high positive predictive (PPV; >75%) and negative predictive (NPV; >76%) values.
Virtual screening of DrugBank and experimental evaluation
We have used our QSPR models for virtual screening of the DrugBank database to identify drugs predicted to have both high LE and high LC for POx micelles. Aqueous solubility of drugs was used for initial filtering. Only compounds classified as poorly soluble (<10 mg/ml) including those defined as practically insoluble (<0.1 mg/ml) (34) were selected. All remaining compounds were paired with the polymer of interest, and the LE and LC values for the drug loading into polymeric micelles were predicted by respective models. Rational design of the training set allowed all DrugBank compounds chosen for virtual screening to be inside models’ applicability domains. Selected hits were dissimilar from the training set, but they were still found inside the applicability domain of the model, which increased our confidence in predictions.
Then, four compounds (podophyllotoxin, rutin, teniposide, and diosmin) predicted to have high solubilization in POx micelles and four compounds (olanzapine, simvastatin, spironolactone, and tamibarotene) predicted to be insoluble in POx were selected for experimental validation. Low aqueous solubility of these compounds was confirmed experimentally before the experimental evaluation of the LE and LC in POx.
Experimental results for these eight drug-POx complexes are shown in Table 1. Overall, we have reached 75% experimental hit rate. Thus, three of four drugs predicted as positive hits displayed moderate to excellent solubilization in POx micelles. Podophyllotoxin, rutin, and teniposide could be solubilized under certain experimental conditions (i.e., feed ratio of polymer, 10 mg/ml) at concentrations as high as 8 mg/ml. Podophyllotoxin presented exceptional ability for incorporation into POx micelles, as it could be solubilized under the experimental conditions at concentrations as high as 8 mg/ml, with LE = 95.2% and LC = 43.2%. Teniposide showed LE = 85% and LC = 14.5 at 8 mg/ml, while rutin presented LE = 45.1% and LC = 26.5%. Diosmin was a false positive, i.e., insoluble in POx micelles. Conversely, one of the predicted negative hits, olanzapine, showed very low or negligible LC and LE at all studied drug feed concentrations. Specifically, the concentration of olanzapine did not exceed 1 mg/ml. The very low LC and LE of this drug implies that at least about 90% or even 99% of the drug is lost upon formulation. Spironolactone and tamibarotene showed high solubilization, but only at low concentrations. These drugs were solubilized at 2 mg/ml with an LE of 89.7 and 82.9%, respectively, and an LC of 14.2 and 15.2%, respectively, but both featured very low solubilization when tested at 8 mg/ml (LE = 20.9% and LC = 14.3% for spironolactone and LE = 9.9% and LC = 7.4% for tamibarotene). As an instance of fortuitous misprediction, simvastatin was found to be a false negative, i.e., it was soluble in POx micelles at concentrations as high as 7 mg/ml, with an LE of 87.2% and an LC of 41.1%. Most likely, simvastatin was mispredicted because its nearest neighbor, wortmannin (Tc = 0.72), has poor solubility in POx micelles. Although wortmannin presents moderate solubility in POx micelles when tested mixed with PTX, this drug alone has both low LE (2.3 to 5.8%) and low LC (0.9 to 2.2%). Irrespective of the reasons, misprediction of simvastatin represents a fortuitous prediction error, as this drug appears to greatly benefit from POx solubilization.
Table 1. List of positive and negative hits with experimental values.
NA, not available.
| Name | Water solubility | Concentration (mg) | Predicted by QSPR | Experimental | ||||
| LE 80 | LC 10 | LC 20 | LC 30 | LE (%) | LC (%) | |||
| Positive hits | ||||||||
| Podophyllotoxin | Very slightly soluble | 15 | NA | NA | NA | NA | 23.8 | 26.3 |
| 10 | 1 | 1 | 1 | 1 | 58.7 | 37.0 | ||
| 8 | 1 | 1 | 1 | 0 | 95.2 | 43.2 | ||
| 4 | 1 | 1 | 0 | 0 | 95.6 | 27.7 | ||
| 2 | 1 | 1 | 0 | 0 | 100.0 | 16.7 | ||
| Rutin | Slightly soluble | 15 | NA | NA | NA | NA | 3.9 | 5.6 |
| 10 | 1 | 1 | 1 | 1 | 6.5 | 6.1 | ||
| 8 | 1 | 1 | 1 | 0 | 45.1 | 26.5 | ||
| 4 | 1 | 1 | 1 | 0 | 60.3 | 19.5 | ||
| 2 | 1 | 1 | 0 | 0 | 74.5 | 13.0 | ||
| Teniposide | Insoluble | 15 | NA | NA | NA | NA | 1.5 | 2.2 |
| 10 | 1 | 1 | 1 | 1 | 6.1 | 5.7 | ||
| 8 | 1 | 1 | 1 | 1 | 85.0 | 14.5 | ||
| 4 | 1 | 1 | 1 | 0 | 76.1 | 23.3 | ||
| 2 | 1 | 1 | 0 | 0 | 85.0 | 14.5 | ||
| Diosmin | Slightly soluble | 15 | NA | NA | NA | NA | Insoluble | Insoluble |
| 10 | 1 | 1 | 1 | 1 | Insoluble | Insoluble | ||
| 8 | 1 | 1 | 1 | 0 | Insoluble | Insoluble | ||
| 4 | 1 | 1 | 1 | 0 | Insoluble | Insoluble | ||
| 2 | 1 | 1 | 0 | 0 | Insoluble | Insoluble | ||
| Negative hits | ||||||||
| Olanzapine | Insoluble | 15 | NA | NA | NA | NA | 9.7 | 12.7 |
| 10 | 0 | 0 | 0 | 0 | 6.1 | 5.8 | ||
| 8 | 0 | 0 | 0 | 0 | 4.1 | 3.2 | ||
| 4 | 0 | 0 | 0 | 0 | 7.0 | 2.7 | ||
| 2 | 0 | 0 | 0 | 0 | 42.3 | 7.8 | ||
| Simvastatin | Insoluble | 15 | NA | NA | NA | NA | 5.0 | 7.0 |
| 10 | 0 | 0 | 0 | 0 | 19.9 | 16.6 | ||
| 8 | 0 | 0 | 0 | 0 | 87.2 | 41.1 | ||
| 4 | 0 | 0 | 0 | 0 | 74.6 | 23.0 | ||
| 2 | 0 | 0 | 0 | 0 | 87.2 | 14.8 | ||
| Spironolactone | Insoluble | 15 | NA | NA | NA | NA | 3.4 | 4.9 |
| 10 | 0 | 0 | 0 | 0 | 31.8 | 24.1 | ||
| 8 | 0 | 0 | 0 | 0 | 20.9 | 14.3 | ||
| 4 | 0 | 0 | 0 | 0 | 53.8 | 17.7 | ||
| 2 | 0 | 0 | 0 | 0 | 82.9 | 14.2 | ||
| Tamibarotene | Insoluble | 15 | NA | NA | NA | NA | 0.9 | 1.3 |
| 10 | 0 | 0 | 0 | 0 | 2.0 | 1.9 | ||
| 8 | 0 | 0 | 0 | 0 | 9.9 | 7.4 | ||
| 4 | 0 | 0 | 0 | 0 | 87.3 | 25.9 | ||
| 2 | 0 | 0 | 0 | 0 | 89.7 | 15.2 | ||
Overall, four compounds showed good solubility, with both LE and LC values among the top 15 compounds ever tested by our group (Table 2). Podophyllotoxin and simvastatin demonstrated exceptional ability for incorporation into POx micelles. Podophyllotoxin and its analogs have shown several important biological activities (e.g., cytotoxic, antiviral, and antifungal) (35); therefore, the discovery of previously unidentified formulations described here may have a substantial impact on the development of this drug candidate. The case of simvastatin (negative hit) with high LE and LC is an example of a fortuitous error of prediction, since this drug appears to be a great candidate for POx solubilization. Simvastatin depends on solubilization enhancement techniques to achieve optimal bioavailability, and the improved solubilization with POx could potentially improve its bioavailability and pharmacological response (36). Overall, three of four positive hits and one negative hit showed highly desirable solubilization properties.
Table 2. Top 15 compounds ranked by LE and LC for 8-mg drug versus 10-mg polymer.
| Compound name | LE % (mean) | LC % (mean) |
| ABT-263 | 100 | 44.4 |
| Podophyllotoxin | 95.2 | 43.2 |
| Etoposide | 91.83 ± 2.92 | 42.33 ± 0.75 |
| Simvastatin | 87.2 | 41.1 |
| Efavirenz | 86.23 | 40.82 |
| Cisplatin prodrug (C6) | 84.8 | 40.4 |
| VE-822 | 80.17 ± 4.48 | 26.65 ± 18.88 |
| PTX | 63.09 ± 42.16 | 30.38 ± 18.54 |
| AZD5363 | 62.27 ± 1.93 | 33.27 ± 0.68 |
| Cisplatin prodrug (C4) | 58.5 | 31.9 |
| Teniposide | 57.2 | 31.4 |
| Cisplatin prodrug (C10) | 53.65 | 23.85 |
| AZD8055 | 50.8 | 28.9 |
| DTX | 46.40 ± 40.43 | 18.99 ± 15.81 |
| Rutin | 45.1 | 26.5 |
As one can see from Table 2, variation of both LE and LC values is small for almost all drugs. At the same time, both PTX and DTX had much higher SD than the other compounds. In the course of preclinical development, these two compounds have been studied very extensively in a variety of experimental conditions. This observation reinforces the high impact of experimental conditions on the studied properties.
DISCUSSION
We have developed and successfully used a computer-aided strategy for the rational design of novel drug-polymeric micelle combinations. Our approach used special, novel descriptors of drug-polymer complexes for building predictive models of drug solubility in polymeric micelles, virtual screening of drug library, and experimental validation of selected hits. Another unique aspect of this investigation was that, in addition to previously collected data, we have generated new experimental data for compounds selected rationally from the library of approved drugs. This was done solely to enable model development for a sufficiently large and chemically diverse dataset. In total, 41 drugs tested in different concentrations both individually and in binary combinations for loading into four different polymeric micelles (408 data points) were used for modeling. This allowed us to develop the set of binary QSPR models for predicting both LE and LC of drugs in polymeric micelles.
The high predictive power of the developed models (external balanced accuracy of 76 to 85%) was confirmed by the “mixtures out” approach (28, 29) especially designed for estimating true predictivity of the QSPR model obtained for compound mixtures (see Materials and Methods for additional details). The developed models were used for virtual screening of the DrugBank database, and four drugs with high (positive hits) along with four drugs with low (negative hits) predicted LE and LC were prioritized for testing. Predicted LE and LC values for three positive and three negative computational hits were confirmed experimentally. Luckily, the remaining negative hit, simvastatin, with an LE of 87% and an LC of 41%, had desired delivery properties. Moreover, simvastatin and podophyllotoxin (LE = 95% and LC = 43%) were among the top five compounds ever studied in POx loading experiments. This is especially important because simvastatin’s solubilization rate is too low to achieve optimal bioavailability (36), and podophyllotoxin has several desired biological properties (e.g., cytotoxic, antiviral, and antifungal) (35); therefore, the discovery of new formulations described here may have a significant impact on the further development of both drugs.
Another significant advantage of the proposed computer-aided strategy for rational design of formulations for poorly soluble drugs is the significant increase in success rate. Thus, our modeling set of 41 compounds tested in advance of model development included 20 compounds with significant solubility in POx micelles (that were used to develop models reported here), implying an experimental hit rate of ca. 48%. In contrast, the use of models developed with this modeling set to design new formulations increased the hit rate from 48 to 75%, i.e., nearly twofold. The success of this study illustrates the power of computer-aided design of novel drug delivery systems and calls for a broader application of computational modeling approaches in drug delivery.
MATERIALS AND METHODS
Study design
The overall workflow for computer-aided design of novel polymeric micelle-based delivery systems for poorly soluble drugs is shown in Fig. 1. Before this study, 20 compounds were tested for solubilization in POx micelles (step 0), which was not enough for building predictive QSPR models. Therefore, we rationally selected an additional set of 21 poorly soluble and chemically diverse drugs from the Selleck Chemicals library and tested them for LE and LC to supplement previously collected data (step 1). The full dataset for all drugs tested experimentally was compiled, curated, integrated, and analyzed (step 2), and chemical descriptors for polymers and drug-polymer complexes that were developed specifically for this study were calculated (step 3). Then, QSPR models for drug loading into polymeric micelle-based delivery systems were generated and validated (step 4). Last, we applied these models for virtual screening of the available drug library to identify compounds with poor aqueous solubility predicted to have either high or low LE and LC. We selected four putatively positive and four putatively negative hits for the experimental validation. In summary, this workflow combines rational design of the experimental data collection to enable model development, computational modeling of drug loading into polymeric micelles, and experimental validation of predicted formulation properties for selected drug delivery systems.
Polymeric micelle preparation
POx micelles loaded with single drug or multiple drugs were prepared via the thin-film hydration method (23). Predetermined amounts of polymer and drugs were solubilized in an organic solvent [e.g., acetone, acetonitrile (ACN), and ethanol] and mixed together. The organic solvent was then removed under a stream of nitrogen gas or air (40°C) to produce a thin film of intrinsically mixed drug-polymer blend. To completely remove the residual solvents and obtain a dry film, the films were deposited in the vacuum chamber (approximately 0.2 mbar) overnight. Subsequently, the formed thin films were rehydrated with the desired amounts of aqueous saline or bidistilled water and then solubilized either at room temperature or upon heating at 50° to 60°C for 5 to 20 min to produce drug-loaded polymeric micelle solutions. The rehydration time was dependent on either the drug concentration or the composition of the drugs or the multidrug mixtures. The polymeric micelles loaded with the single drug were prepared accordingly with a final polymer concentration of 10 g/liter and each drug feed concentration of 2, 4, 6, 8, 10, and sometimes 15 g/liter. The polymeric micelles coloaded with multiple drugs were prepared using the same final polymer concentration (10 g/liter) and predetermined concentrations of each drug components of multiple drug mixtures. The polymers used in this work are presented in table S4.
In every case, the formulations were stable for at least 24 hours when the analysis of the drug incorporation was done. Prepared micelle samples were allowed to cool to room temperature and centrifuged at 10,000 rpm for 3 min (Sorvall Legend Micro 21R Microcentrifuge, Thermo Fisher Scientific) to remove precipitates. The transparent supernatant solutions of micelle samples were used for the quantification of the amounts of drugs solubilized in the polymeric micelle. The amounts of drugs encapsulated in polymeric micelles were analyzed with a high-performance liquid chromatography (HPLC) system (Agilent Technologies 1200 series). The micelle samples were diluted with mobile phase (specified below) and injected (10 μl) into the HPLC column [Agilent Eclipse Plus C18, 3.5 μm column (4.6 mm × 150 mm)]. Predetermined mixtures of ACN/water (v/v) were used as the mobile phase. For PTX, AZD8055, olaparib, imiquimod, NVP-BEZ235, ABT-263, ABT-737, sabutoclax, LY2109761, AZD5363, LY364947, and the combination of each of these drugs with PTX, a mixture of ACN/water (50%/50%, v/v; 0.01% trifluoroacetic acid) was used as the mobile phase. For VE-822, vismodegib, and their combination, a mixture of ACN/water (35%/65%, v/v; 0.01% trifluoroacetic acid) was used as the mobile phase. For PTX, wortmannin, LY294002, LY294002 HCl, etoposide (ETO), cisplatin prodrug (C6) (37), and the combination of wortmannin/PTX, LY294002/PTX, LY294002 HCl/PTX, and ETO/cisplatin prodrug (C6), a mixture of ACN/water (50%/50%, v/v) was used as the mobile phase. For PTX, brefeldin, cisplatin prodrug (C6), and the combination of brefeldin/PTX and cisplatin prodrug (C6)/PTX, a mixture of ACN/water (40%/60%, v/v) was used as the mobile phase. For PTX, KU55933, LDN-57444, and the combination of KU55933/PTX and LDN-57444/PTX, a mixture of ACN/water (70%/30%, v/v) was used as the mobile phase. For PTX, ETO, VE-822, and the combination of PTX/ETO/VE-822, a stepwise gradient was used. First, the analyte was eluted for 13 min with ACN/water (30%/70%, v/v; 0.01% trifluoroacetic acid) followed by a second 2-min elution change from ACN/water (30%/70%, v/v; 0.01% trifluoroacetic acid) to ACN/water (60%/40%, v/v; 0.01% trifluoroacetic acid). Then, the analyte was eluted for 15 min. These measurements produced each drug concentration for each polymeric micelle composition (mg/ml). The flow rate was 1 ml/min, and the column temperature was 40°C. Detection wavelengths were determined by the drugs solubilized. The full description of platinum complexes with sufficient hydrophobicity for encapsulation in POx micelles is described in the Supplementary Materials (“Description of platinum complexes” section), as well as the compilation of all experimental data on solubilization of drugs in POx micelles (“Compiling all experimental data” section).
LE (Eq. 1) and LC (Eq. 2) were calculated as follows:
| (1) |
| (2) |
Datasets
Creation of drug-polymer micellar solubilization dataset
Before this study, we had collected LE and LC data on 20 drugs chosen from the DrugBank (www.drugbank.ca/) that belonged to different structural classes. All these compounds had considerable issues with aqueous solubility. Many of these compounds were not approved by the FDA or failed as treatments for solid tumors because of their toxicity. We hypothesized that solubilization of these compounds using our POx micelle system would greatly improve their anticancer efficacy. However, although we have demonstrated a very high solubilization capacity of some of the drugs using POx micelles, we have also seen compounds where this technology was less helpful (23, 24).
Rational design of a chemically diverse library of poorly soluble drugs
The chemical space formed by 20 previously tested compounds combined with the Selleck library of FDA-approved drugs was analyzed by plotting the barycentric coordinates in the space of SiRMS descriptors of all the 788 drugs. Barycentric coordinates correspond to the location of points of a simplex (a triangle, tetrahedron, etc.) in the space defined by the vertices (38). In this case, a simplex is defined by all the SiRMS descriptors of a particular chemical substance. Barycentric coordinates were determined using the Methods of Data Analysis module of the HiT QSAR software (39). Then, we selected the insoluble or poorly soluble drugs as preliminary candidates for solubilization in POx polymeric micelle delivery systems. The selected compounds were subject to further chemical diversity sampling following a procedure similar to that described by Kuz’min et al. (40). Ultimately, a collection of 61 molecules covering maximal chemical space for investigation of their LE was obtained. Twenty-one compounds were selected from this collection on the basis of diversity of both clinical applications and mechanisms of action as well as availability and cost, and these compounds were tested for LC and LE. Thus, the total experimental dataset for model building included 20 compounds tested previously and 21 new compounds selected from the Selleck library as described above.
Selleck database
This dataset containing 853 FDA-approved drugs was retrieved from www.selleckchem.com/. After curation, 768 compounds remained, and a diverse subset of 61 molecules was selected from this dataset as described above (see also step 1 in Fig. 1).
DrugBank database
This dataset containing 7133 drug entries, including FDA-approved small-molecule drugs, nutraceuticals, as well as illicit, withdrawn, and experimental drugs, was retrieved from the DrugBank website (www.drugbank.ca/). After curation, 6461 drugs were kept for virtual screening.
Data curation
We compiled all the data on drug loading into polymeric micelles generated over the years in our experimental laboratory. Originally, the dataset consisted of 408 records for 41 compounds tested in different concentrations and combinations for loading into micelles made of seven different polymers. Most of the compounds were tested in different concentrations and under different laboratory conditions. As part of the data curation procedure, each record was manually inspected. Chemical structures were retrieved from either ChemSpider (www.chemspider.com/) or SciFinder (https://scifinder.cas.org) databases using the Chemical Abstracts Service (CAS) registry numbers and chemical names. The dataset was thoroughly curated according to the workflows developed by our group (41–43). Briefly, structural normalization of specific chemotypes, such as aromatic and nitro groups, was performed using ChemAxon Standardizer (v. 16.10.24.0, ChemAxon, Budapest, Hungary; www.chemaxon.com). Organometallic compounds and mixtures were kept. After structure standardization, the structural duplicates were identified using HiT QSAR (39). During this process, we identified 33 records that appeared more than once (up to 12 times), totaling 108 duplicates. The records describing the mixtures containing three drugs (nine records) and eight cases of real duplicates, where the experiment was performed more than once for the same drug-polymer complex, were removed from the modeling process. The concordance of property values for duplicated records was very high (average deviation was equal to 7.6%); thus, only one record associated with the averaged property value was kept for modeling. The high concordance between values for true duplicative measurements indicated high experimental reproducibility. The following experimental conditions were retained and used as descriptors for model building: polymer and drug solvent, total solvent volume before evaporation, hydration solvent, and hydration temperature. The final curated dataset of 391 records is available in data file S1.
Molecular descriptors
SiRMS descriptors
Two-dimensional (2D) SiRMS descriptors (27) (number of tetratomic fragments with fixed composition and topological structure) were generated by the HiT QSAR software (39). At the 2D level, the connectivity of atoms in a simplex, atom type, and bond nature (single, double, triple, or aromatic) was considered. SiRMS descriptors account not only for the atom type but also for other atomic characteristics that may influence biological activity of molecules, e.g., partial charge, lipophilicity, refraction, and atom ability for being a donor/acceptor in hydrogen-bond formation (H-bond). For atom characteristics with continuous values (charge, lipophilicity, and refraction), the division of the entire value range into definite discrete groups was carried out. The atoms were divided into four groups corresponding to their (i) partial charge A ≤ −0.05 < B ≤ 0 < C ≤ 0.05 < D, (ii) lipophilicity A ≤ −0.5 < B ≤ 0 < C ≤ 0.5 < D, and (iii) refraction A ≤ 1.5 < B ≤ 3 < C ≤ 8 < D. For the H-bond characteristic, the atoms were divided into three groups: A (acceptor of hydrogen in the H-bond), D (donor of hydrogen in the H-bond), and I (indifferent atom). The usage of sundry variants of differentiation of simplex vertexes (atoms) represents the principal feature of the SiRMS approach (44). Detailed description of HiT QSAR and SiRMS can be found elsewhere (27, 39).
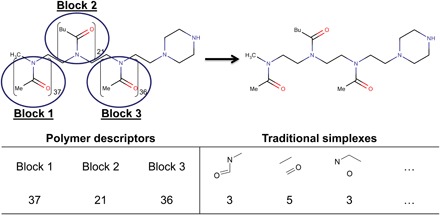
Polymer descriptors
Each block of the polymer was described by the number of its repetitions in the polymer. In addition, traditional SiRMS descriptors were calculated for simplified polymer representation as a pseudo small molecule, with all repetitive monomers introduced only once. Overall scheme of descriptor calculation for polymers developed for this study for the first time is shown in Fig. 3.
Fig. 3. General scheme of descriptor calculation for polymers.
Descriptors for drug-polymer complexes
We modified the SiRMS approach developed earlier to calculate descriptors for organic compound mixtures (28, 29) to make it suitable for the QSPR analysis of drug-polymer complexes as follows. Each complex was represented as a binary mixture consisting of the drug molecule and a simplified representation of a polymer as a pseudo small molecule as described in the previous section. Then, the simplex descriptors were calculated as usual. Bounded simplexes describe only single components of the mixture (compound A or B), when unbounded simplexes can describe both the constituent parts and the mixture as a whole. It is necessary to indicate whether the parts of unbounded simplexes belong to the same molecule or to different ones. In the latter case, these unbounded simplexes will not reflect the structure of a single molecule but will characterize a pair of different molecules. Simplexes of this kind are specific for a given drug-polymer complex (Fig. 4). Special mark was used during descriptor generation to distinguish these “mixture” simplexes from ordinary ones. The mixture composition was taken into account, i.e., descriptors of constituent parts (compounds A and B) were weighted according to their molar fraction and mixture descriptors were multiplied by the doubled molar fraction of the minor component. If in the same task both drug-polymer complexes (mixtures) and pure compounds had been considered, pure compounds were considered as a mixture with composition A1B0. In this case, only descriptors of the pure compound A would be generated with the weight equal to 1. Thus, the structure of every mixture was characterized by both descriptors of the mixture and descriptors of its individual constituents.
Fig. 4. Descriptor calculation of drug-polymer complexes.
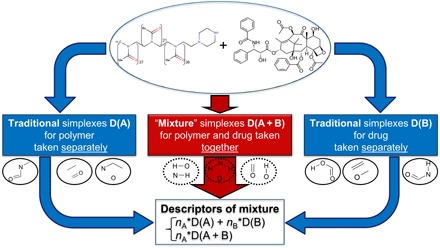
nA and nB are molar fractions of components A and B (nA < nB).
A simpler approach was used for complexes consisting of a polymer and a mixture of drugs. Here, the polymer was represented in the same way, but descriptors for all the members of such mixture of drug-polymer complex were calculated separately, weighted according to their concentration, and then summarized in one string corresponding to a given complex. This approach was used for datasets containing both drug-polymer and mixture of drug-polymer complexes. It allowed the use of the maximal amount of available experimental data for model building. In the end, constant, near-constant, and cross-correlated variables (r ≥ 0.9) were removed to reduce the dimensionality of the chemical space without loss of important information.
Experimental conditions
Certain experimental conditions were used as features, in addition to molecular descriptors, to describe the system under the investigation. Specifically, we considered solvents used to prepare both polymer and drug samples, total solvent volume before evaporation, hydration solvent, and hydration temperature.
Cluster analysis
Chemical clusters were generated by the sequential agglomerative hierarchical nonoverlapping method implemented in the ISIDA/Cluster software (45). Briefly, the software generates a dendrogram of the parent-child relationships between clusters and a heat map of the proximity matrix colored according to the pairwise chemical similarity between compounds. This approach is well known; it has been extensively used by our (46–48) and other groups (45, 49). Of course, clusters are data specific, e.g., if the new data would be introduced to the dataset, clusters might change. However, repeating cluster analysis for the same dataset will result in the same clusters. In this study, we used clustering only to analyze whether LogP and MW are relevant for drug loading of similar compounds. We did not use cluster analysis to predict loading parameters, which was done by QSPR models.
QSPR modeling
Binary QSPR models were developed and rigorously validated according to the best practices of QSPR modeling (50). Models were developed with random forest (RF) algorithm (51). One thousand trees were built for each forest, and the outputs of all trees were aggregated to obtain one final prediction. In each tree, about one-third of the set of N compounds were sampled by bootstrap as out-of-bag (OOB) set and the remaining compounds were used as a training set. The best split by the CART algorithm (52) among the m randomly selected descriptors from the entire pool in each node was chosen, and each tree was then grown to the largest possible extent; there was no pruning. The predicted classification values are defined by the majority voting for one of the classes. Thus, each tree predicts values for only those compounds that are not included in the training set of that tree (for OOB set only). The final model is chosen by the lowest error for prediction of the OOB set.
Models were built using R (www.r-project.org/) and implemented in a KNIME (www.knime.com/) workflow (available at https://figshare.com/s/69c56ca431c4963b0ecf). We followed the mixtures out procedure for the validation of QSAR models of mixtures described by Muratov et al. (28). In this method, all data points corresponding to mixtures composed of the same constituents, but in different ratios, are simultaneously removed and placed in the same external fold. Thus, every mixture is present in either the training or external set, but never in both sets. This approach allows one to minimize the influence of known information on the prediction and obtain reliable results for predicting novel drug-polymer complexes created by a known polymer and a new drug. Thus, we combined the mixtures out strategy with a fivefold external cross-validation procedure (16, 53).
Briefly, the full set of compounds with known experimental activity was divided into five subsets of equal size. Each subset (20% of the compounds) was selected once as a test set, while the other subsets (80% of the compounds) were merged into a training set to develop a model. This procedure was repeated with the other subsets, allowing each of the five subsets to be used once as a test set. In addition, 30 rounds of Y-randomization test (40) were performed for each dataset to ensure that the accuracy of models was not obtained due to chance correlations.
The QSPR models were built for two endpoints: LE and LC. For LE, the model was generated using a threshold of 80%. Thresholds of 10, 20, 30, and 40% were separately applied to build four models for LC. This system of binary models would allow us to predict LC within certain ranges (0 to 10%, 10 to 20%, 20 to 30%, 30 to 40%, and 40 to 50%), which is more informative than standard binary prediction. Compounds predicted above the threshold by all the individual models were selected as positive hits, while those predicted below the threshold were selected as negative hits. The applicability domain of the models was calculated as Dcutoff = <D> + Zs, where Z is a similarity threshold parameter defined by a user (0.5 in this study) and <D> and s are the average and SD, respectively, of all Euclidian distances in the multidimensional descriptor space between each compound and its nearest neighbors for all compounds in the training set (54).
Statistical analysis
The following statistical metrics were used to assess different aspects of performance of classification models (Eqs. 3 to 7):
CCR
| (3) |
Sensitivity (Se)
| (4) |
Specificity (Sp)
| (5) |
PPV
| (6) |
NPV
| (7) |
Supplementary Material
Acknowledgments
Funding: This study was supported, in part, by Carolina Center of Cancer Nanotechnology Excellence (U54CA198999) of the National Cancer Institute Alliance for Nanotechnology in Cancer, the NIH (grants 1U01CA207160 and GM5105946), the NC TraCS Institute (award 4DR11404), and The Carolina Partnership, a strategic partnership between the UNC Eshelman School of Pharmacy and The University Cancer Research Fund through the Lineberger Comprehensive Cancer Center. V.M.A., E.M., and C.H.A. thank CNPq (grant 400760/2014-2). V.M.A. also thanks CAPES for PhD scholarship. Author contributions: E.M., A.K., and A.T. conceived the study. V.M.A., E.M., E.V., C.H.A., and A.T. contributed to the computational part. D.H., M.S.-P., N.V., C.L., and A.K. contributed to the experimental part. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. Competing interests: A.K. is an inventor on a U.S. patent related to this work filed by DelAQUA Pharmaceuticals Inc. (no. 9,402,908, filed on 2 August 2016). A.K. and M.S.-P. declare a financial relationship to DelAQUA Pharmaceuticals Inc. that has license rights to U.S. patent no. 9,402,908. The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav9784/DC1
Description of platinum complexes
Compiling all experimental data
Fig. S1. Results of cluster analysis of 25 compounds tested alone at 8 mg.
Fig. S2. Variable importance for the five models developed.
Table S1. List of 25 compounds tested alone at 8 mg with their respective clusters, LE and LC (minimum, maximum, mean, and SD values), LogP, MW, and number of performed experiments.
Table S2. List of three drugs with highest LE variability tested at 8 mg/ml.
Table S3. Statistical characteristics of LC and LE QSPR models based on fivefold external cross-validation.
Table S4. List of eight polymers used in this study with specification of block sizes and end group.
Data file S1. Curated solubilization data for a single drug or a two-drug combination loaded into polymeric micelles. This file contains all the data on compound solubilization generated in this study subject to curation protocols as described in Materials and Methods.
REFERENCES AND NOTES
- 1.Lipinski C. A., Poor aqueous solubility—An industry wide problem in drug discovery. Am. Pharm. Rev. 5, 82–85 (2002). [Google Scholar]
- 2.Yang T., Cui F.-D., Choi M.-K., Cho J.-W., Chung S.-J., Shim C.-K., Kim D.-D., Enhanced solubility and stability of PEGylated liposomal paclitaxel: In vitro and in vivo evaluation. Int. J. Pharm. 338, 317–326 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Slowing I. I., Vivero-Escoto J. L., Wu C.-W., Lin V. S.-Y., Mesoporous silica nanoparticles as controlled release drug delivery and gene transfection carriers. Adv. Drug Deliv. Rev. 60, 1278–1288 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Kabanov A. V., Vinogradov S. V., Nanogels as pharmaceutical carriers: Finite networks of infinite capabilities. Angew. Chem. Int. Ed. 48, 5418–5429 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kabanov A. V., Batrakova E. V., Alakhov V. Y., Pluronic® block copolymers as novel polymer therapeutics for drug and gene delivery. J. Control. Release 82, 189–212 (2002). [DOI] [PubMed] [Google Scholar]
- 6.T. Heimbach, D. Fleisher, A. Kaddoumi, in Prodrugs, V. J. Stella, R. T. Borchardt, M. J. Hageman, R. Oliyai, H. Maag, J. W. Tilley, Eds. (Springer New York, 2007), pp. 157–215. [Google Scholar]
- 7.A. Nag, B. Dey, Computer-Aided Drug Design and Delivery Systems (McGraw-Hill, 2011). [Google Scholar]
- 8.Huynh L., Neale C., Pomès R., Allen C., Computational approaches to the rational design of nanoemulsions, polymeric micelles, and dendrimers for drug delivery. Nanomedicine 8, 20–36 (2012). [DOI] [PubMed] [Google Scholar]
- 9.Frederix P. W. J. M., Patmanidis I., Marrink S. J., Molecular simulations of self-assembling bio-inspired supramolecular systems and their connection to experiments. Chem. Soc. Rev. 47, 3470–3489 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thota N., Jiang J., Computational amphiphilic materials for drug delivery. Front. Mater. 2, 64 (2015). [Google Scholar]
- 11.Kuramochi H., Andoh Y., Yoshii N., Okazaki S., All-Atom molecular dynamics study of a spherical micelle composed of N-acetylated poly(ethylene glycol)−Poly(γ-benzyl l-glutamate) block copolymers: A potential carrier of drug delivery systems for cancer. J. Phys. Chem. B 113, 15181–15188 (2009). [DOI] [PubMed] [Google Scholar]
- 12.Badalkhani-Khamseh F., Ebrahim-Habibi A., Hadipour N. L., Atomistic computer simulations on multi-loaded PAMAM dendrimers: A comparison of amine- and hydroxyl-terminated dendrimers. J. Comput. Aided Mol. Des. 31, 1097–1111 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Caccavo D., Barba A. A., d’Amore M., De Piano R., Lamberti G., Rossi A., Colombo P., Modeling the modified drug release from curved shape drug delivery systems – Dome Matrix®. Eur. J. Pharm. Biopharm. 121, 24–31 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Shi C., Guo D., Xiao K., Wang X., Wang L., Luo J., A drug-specific nanocarrier design for efficient anticancer therapy. Nat. Commun. 6, 7449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meunier M., Goupil A., Lienard P., Predicting drug loading in PLA-PEG nanoparticles. Int. J. Pharm. 526, 157–166 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Cherkasov A., Muratov E. N., Fourches D., Varnek A., Baskin I. I., Cronin M., Dearden J., Gramatica P., Martin Y. C., Todeschini R., Consonni V., Kuz’min V. E., Cramer R., Benigni R., Yang C., Rathman J., Terfloth L., Gasteiger J., Richard A., Tropsha A., QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 57, 4977–5010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dearden J. C., The history and development of Quantitative Structure-Activity Relationships (QSARs). Int. J. Quant. Struct. Prop. Relat. 1, 1–44 (2016). [Google Scholar]
- 18.Wu W., Zhang C., Lin W., Chen Q., Guo X., Qian Y., Zhang L., Quantitative Structure-Property Relationship (QSPR) modeling of drug-loaded polymeric micelles via genetic function approximation. PLOS ONE 10, e0119575 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rasulev B., Jabeen F., Stafslien S., Chisholm B. J., Bahr J., Ossowski M., Boudjouk P., Polymer coating materials and their fouling release activity: A cheminformatics approach to predict properties. ACS Appl. Mater. Interfaces 9, 1781–1792 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Potta T., Zhen Z., Grandhi T. S. P., Christensen M. D., Ramos J., Breneman C. M., Rege K., Discovery of antibiotics-derived polymers for gene delivery using combinatorial synthesis and cheminformatics modeling. Biomaterials 35, 1977–1988 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cern A., Golbraikh A., Sedykh A., Tropsha A., Barenholz Y., Goldblum A., Quantitative structure - property relationship modeling of remote liposome loading of drugs. J. Control. Release 160, 147–157 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cern A., Marcus D., Tropsha A., Barenholz Y., Goldblum A., New drug candidates for liposomal delivery identified by computer modeling of liposomes’ remote loading and leakage. J. Control. Release 252, 18–27 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Luxenhofer R., Schulz A., Roques C., Li S., Bronich T. K., Batrakova E. V., Jordan R., Kabanov A. V., Doubly amphiphilic poly(2-oxazoline)s as high-capacity delivery systems for hydrophobic drugs. Biomaterials 31, 4972–4979 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.He Z., Schulz A., Wan X., Seitz J., Bludau H., Alakhova D. Y., Darr D. B., Perou C. M., Jordan R., Ojima I., Kabanov A. V., Luxenhofer R., Poly(2-oxazoline) based micelles with high capacity for 3rd generation taxoids: Preparation, in vitro and in vivo evaluation. J. Control. Release 208, 67–75 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yamamoto T., Yokoyama M., Opanasopit P., Hayama A., Kawano K., Maitani Y., What are determining factors for stable drug incorporation into polymeric micelle carriers? Consideration on physical and chemical characters of the micelle inner core. J. Control. Release 123, 11–18 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Lu J., Zheng M., Wang Y., Shen Q., Luo X., Jiang H., Chen K., Fragment-based prediction of skin sensitization using recursive partitioning. J. Comput. Aided Mol. Des. 25, 885–893 (2011). [DOI] [PubMed] [Google Scholar]
- 27.Muratov E. N., Artemenko A. G., Varlamova E. V., Polischuk P. G., Lozitsky V. P., Fedchuk A. S., Lozitska R. L., Gridina T. L., Koroleva L. S., Sil’nikov V. N., Galabov A. S., Makarov V. A., Riabova O. B., Wutzler P., Schmidtke M., Kuz’min V. E., Per aspera ad astra: Application of Simplex QSAR approach in antiviral research. Future Med. Chem. 2, 1205–1226 (2010). [DOI] [PubMed] [Google Scholar]
- 28.Muratov E. N., Varlamova E. V., Artemenko A. G., Polishchuk P. G., Kuz’min V. E., Existing and developing approaches for QSAR analysis of mixtures. Mol. Inform. 31, 202–221 (2012). [DOI] [PubMed] [Google Scholar]
- 29.Oprisiu I., Varlamova E., Muratov E., Artemenko A., Marcou G., Polishchuk P., Kuz’min V., Varnek A., QSPR approach to predict nonadditive properties of mixtures. Application to bubble point temperatures of binary mixtures of liquids. Mol. Inform. 31, 491–502 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Ward J. H., Jr., Hierarchical grouping to optimize an objective function. J. Am. Stat. Assoc. 58, 236–244 (1963). [Google Scholar]
- 31.Wan X., Min Y., Bludau H., Keith A., Sheiko S. S., Jordan R., Wang A. Z., Sokolsky-Papkov M., Kabanov A. V., Drug Combination synergy in worm-like polymeric micelles improves treatment outcome for small cell and non-small cell lung cancer. ACS Nano 12, 2426–2439 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Seo Y., Schulz A., Han Y., He Z., Bludau H., Wan X., Tong J., Bronich T. K., Sokolsky M., Luxenhofer R., Jordan R., Kabanov A. V., Poly(2-oxazoline) block copolymer based formulations of taxanes: Effect of copolymer and drug structure, concentration, and environmental factors. Polym. Adv. Technol. 26, 837–850 (2015). [Google Scholar]
- 33.Schulz A., Jaksch S., Schubel R., Wegener E., Di Z., Han Y., Meister A., Kressler J., Kabanov A. V., Luxenhofer R., Papadakis C. M., Jordan R., Drug-induced morphology switch in drug delivery systems based on poly(2-oxazoline)s. ACS Nano 8, 2686–2696 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stegemann S., Leveiller F., Franchi D., de Jong H., Lindén H., When poor solubility becomes an issue: From early stage to proof of concept. Eur. J. Pharm. Sci. 31, 249–261 (2007). [DOI] [PubMed] [Google Scholar]
- 35.Yu X., Che Z., Xu H., Recent advances in the chemistry and biology of podophyllotoxins. Chemistry 23, 4467–4526 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Murtaza G., Solubility enhancement of simvastatin: A review. Acta Pol. Pharm. 69, 581–590 (2012). [PubMed] [Google Scholar]
- 37.Dhar S., Kolishetti N., Lippard S. J., Farokhzad O. C., Targeted delivery of a cisplatin prodrug for safer and more effective prostate cancer therapy in vivo. Proc. Natl. Acad. Sci. U.S.A. 108, 1850–1855 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vityuk N., Voskresenskaja E., Kuz’min V., The synergism of methods barycentric coordinates and trend-vector for solution—Structure-property tasks. Pattern Recognit. Image Anal. 3, 521–528 (1999). [Google Scholar]
- 39.Kuz’min V. E., Artemenko A. G., Muratov E. N., Hierarchical QSAR technology based on the Simplex representation of molecular structure. J. Comput. Aided Mol. Des. 22, 403–421 (2008). [DOI] [PubMed] [Google Scholar]
- 40.Kuz’min V. E., Muratov E. N., Artemenko A. G., Varlamova E. V., Gorb L., Wang J., Leszczynski J., Consensus QSAR modeling of phosphor-containing chiral AChE inhibitors. QSAR Comb. Sci. 28, 664–677 (2009). [Google Scholar]
- 41.Fourches D., Muratov E., Tropsha A., Trust, but verify: On the importance of chemical structure curation in cheminformatics and QSAR modeling research. J. Chem. Inf. Model. 50, 1189–1204 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fourches D., Muratov E., Tropsha A., Curation of chemogenomics data. Nat. Chem. Biol. 11, 535 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Fourches D., Muratov E., Tropsha A., Trust, but verify II: A practical guide to chemogenomics data curation. J. Chem. Inf. Model. 56, 1243–1252 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kuz’min V. E., Artemenko A. G., Muratov E. N., Volineckaya I. L., Makarov V. A., Riabova O. B., Wutzler P., Schmidtke M., Quantitative structure–activity relationship studies of [(biphenyloxy)propyl]isoxazole derivatives. Inhibitors of human rhinovirus 2 replication. J. Med. Chem. 50, 4205–4213 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Varnek A., Fourches D., Horvath D., Klimchuk O., Gaudin C., Vayer P., Solov’ev V., Hoonakker F., Tetko I., Marcou G., ISIDA—Platform for virtual screening based on fragment and pharmacophoric descriptors. Curr. Comput. Aided Drug Des. 4, 191–198 (2008). [Google Scholar]
- 46.Alves V. M., Muratov E. N., Zakharov A., Muratov N. N., Andrade C. H., Tropsha A., Chemical toxicity prediction for major classes of industrial chemicals: Is it possible to develop universal models covering cosmetics, drugs, and pesticides? Food Chem. Toxicol. 112, 526–534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Alves V. M., Capuzzi S. J., Muratov E. N., Braga R. C., Thornton T. E., Fourches D., Strickland J., Kleinstreuer N., Andrade C. H., Tropsha A., QSAR models of human data can enrich or replace LLNA testing for human skin sensitization. Green Chem. 18, 6501–6515 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Alves V. M., Muratov E., Fourches D., Strickland J., Kleinstreuer N., Andrade C. H., Tropsha A., Predicting chemically-induced skin reactions. Part I: QSAR models of skin sensitization and their application to identify potentially hazardous compounds. Toxicol. Appl. Pharmacol. 284, 262–272 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.G. M. Downs, J. M. Barnard, in Reviews in Computational Chemistry, K. B. Lipkowitz, D. B. Boyd, Eds. (John Wiley & Sons Inc., 2003), vol. 18, pp. 1–40. [Google Scholar]
- 50.Tropsha A., Best practices for QSAR model development, validation, and exploitation. Mol. Inform. 29, 476–488 (2010). [DOI] [PubMed] [Google Scholar]
- 51.Breiman L., Random forests. Mach. Learn. 45, 5–32 (2001). [Google Scholar]
- 52.L. Breiman, J. H. Friedman, R. A. Olshen, C. J. Stone, Classification and Regression Trees, vol. 19 of Statistics/Probability Series (Chapman and Hall/CRC, 1984).
- 53.Zakharov A. V., Varlamova E. V., Lagunin A. A., Dmitriev A. V., Muratov E. N., Fourches D., Kuz’min V. E., Poroikov V. V., Tropsha A., Nicklaus M. C., QSAR modeling and prediction of drug–drug interactions. Mol. Pharm. 13, 545–556 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Golbraikh A., Shen M., Xiao Z., Xiao Y.-D., Lee K.-H., Tropsha A., Rational selection of training and test sets for the development of validated QSAR models. J. Comput. Aided Mol. Des. 17, 241–253 (2003). [DOI] [PubMed] [Google Scholar]
- 55.Schulz A., Han Y., He Z., Bronich T. K., Kabanov A. V., Luxenhofer R., Jordan R., Poly (2-oxazoline)s: An all-around drug delivery system? Polym. Prepr. 53, 354 (2012). [Google Scholar]
- 56.Han Y., He Z., Schulz A., Bronich T. K., Jordan R., Luxenhofer R., Kabanov A. V., Synergistic combinations of multiple chemotherapeutic agents in high capacity poly(2-oxazoline) micelles. Mol. Pharm. 9, 2302–2313 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav9784/DC1
Description of platinum complexes
Compiling all experimental data
Fig. S1. Results of cluster analysis of 25 compounds tested alone at 8 mg.
Fig. S2. Variable importance for the five models developed.
Table S1. List of 25 compounds tested alone at 8 mg with their respective clusters, LE and LC (minimum, maximum, mean, and SD values), LogP, MW, and number of performed experiments.
Table S2. List of three drugs with highest LE variability tested at 8 mg/ml.
Table S3. Statistical characteristics of LC and LE QSPR models based on fivefold external cross-validation.
Table S4. List of eight polymers used in this study with specification of block sizes and end group.
Data file S1. Curated solubilization data for a single drug or a two-drug combination loaded into polymeric micelles. This file contains all the data on compound solubilization generated in this study subject to curation protocols as described in Materials and Methods.