

Abstract
Although ruxolitinib improves symptoms and splenomegaly in patients with advanced myelofibrosis, whether this agent is truly disease-modifying remains unclear. Histone deacetylase inhibitors (HDACi) down-regulate JAK2 via interference with chaperone function. Pracinostat, a pan-HDACi, has modest single-agent activity in myelofibrosis. We conducted a single-institution, phase 2, investigator-initiated trial of ruxolitinib plus pracinostat (begun after 12 weeks of ruxolitinib) in 25 patients with myelofibrosis, of whom 20 received both agents. Sixteen (80%) patients had objective responses (all “clinical improvement”). The rate of spleen response (by palpation) was 74%, and that of symptom response 80%. Most responses occurred prior to pracinostat initiation. Three patients experienced improvement in bone marrow fibrosis, and one a near-complete molecular response after two years on study treatment. All patients discontinued pracinostat and are currently off-study. Pracinostat interruptions and dose reductions were frequent, often due to worsening anemia. These findings do not support continued development of pracinostat in myelofibrosis.
Keywords: myelofibrosis, JAK2 inhibitors, HDAC inhibitors, rational combinations, targeted therapies
Introduction
The Janus kinase 1/2 (JAK1/2) inhibitor, ruxolitinib, first licensed in 2011 for the treatment of myeloproliferative neoplasm (MPN)-associated myelofibrosis (MF), remains the only agent to have received regulatory approval for this disease.[1] The most pronounced benefits of ruxolitinib in MF are reduction in splenomegaly and amelioration of symptoms, and the drug broadly suppresses cytokines, the levels of many of which are increased in patients with MF.[2] Ruxolitinib was approved based on the results of the pivotal COMFORT trials conducted in patients with intermediate-2 or high risk MF, and long-term follow-up shows a survival advantage for the ruxolitinib-treated patients in these trials, despite crossover.[3] However, despite the survival benefit seen with ruxolitinib, controversy persists over whether or not this drug is truly disease-modifying, in large part because of the its rather modest effects on bone marrow fibrosis and driver mutation allele burden.[4] Indeed, it has been proposed that the improvement in survival with ruxolitinib could potentially be attributed to indirect effects, such as improved weight, appetite, energy level and overall well-being.[5] The emergence of clinical resistance to ruxolitinib is also of concern: in the COMFORT trials, the median duration of spleen response was about 3 years,[6, 7] and spleen responses to ruxolitinib have been shown to correlate with survival.[8, 9] One major mechanism of therapeutic resistance to JAK2 inhibition is the phenomenon of JAK2 inhibitor “persistence”, where JAK2 is activated in trans via heterodimerization with other members of the JAK family despite the presence of a JAK2 inhibitor, such as ruxolitinib.[10] For all these reasons, there has been considerable interest in developing rational, ruxolitinib-based combinations for patients with MF, in hopes of altering the underlying disease biology, extending ruxolitinib’s survival benefit, and circumventing resistance.
Histone deacetylase inhibitors (HDACi) exert pleiotropic effects selectively in transformed cells, that include promoting a more open chromatin configuration that favors gene transcription, reactive oxygen species (ROS) generation and induction of DNA damage, inhibition of DNA repair, induction of the endogenous cyclin-dependent kinase (CDK) inhibitor p21, and disruption of chaperone function via acetylation of heat shock protein 90 (HSP90), among many other actions (reviewed in ref.[11]). Through the last mechanism, HDACi have been shown to down-regulate several oncoproteins of critical importance in leukemogenesis, including JAK2.[12] Several non-canonical actions of JAK2V617F that broadly impact gene expression through epigenetic mechanisms, such as phosphorylation of histone H3[13] and the arginine methyltransferase PRMT5,[14] provide further support for studying histone modifying drugs in MPN. Inhibitors of HSP90 have also been shown to degrade JAK2 and overcome JAK2 inhibitor persistence,[15] but this class of agents has been difficult to develop for clinical use to date. In contrast, several HDACi are approved for the treatment of T-lymphoid and plasma cell malignancies. A number of HDACi have been tested in clinical trials in MPN and displayed promising single-agent activity (reviewed in ref.[16]). Preclinical studies have demonstrated clear evidence of synergism between JAK2 inhibitors and HDACi against MPN cell lines and primary cells, both in vitro and in vivo.[17–19]
Pracinostat (MEI Pharma, San Diego, CA; Helsinn Group, Lugano, Switzerland;) is an orally available inhibitor of class I histone deacetylases currently in phase 3 of clinical development for acute myeloid leukemia in combination with azacitidine,[20] for which indication it enjoys “breakthrough” designation from the Food and Drug Administration (FDA). We previously conducted a phase 2 study of pracinostat, administered at a dose of 60 mg every other day for three out of every four weeks, in 22 patients with MF.[21] Eight (36%) patients had clinical benefit, with six (27%) experiencing spleen shrinkage. Two (9%) patients had International Working Group (2006)-defined[22] clinical improvement (CI) in anemia. Based upon these single-agent data and the preclinical rationale and synergism discussed above, we conducted a phase 2 clinical trial of the combination of ruxolitinib and pracinostat in patients with MF (clinicaltrials.gov identifier NCT02267278).
Methods
This was a phase 2, investigator-initiated, single-institution study in adult patients with primary MF (PMF), post-polycythemia vera MF (post-PV MF) or post-essential thrombocythemia MF (post-ET MF). Key eligibility criteria included the presence of palpable splenomegaly (≥5 cm below the left costal margin), an absolute neutrophil count (ANC) ≥1 × 109/L and a platelet count ≥50 × 109/L. Patients could be treatment-naïve or previously treated; if the former, patients had to have intermediate-1, intermediate-2 or high risk disease according to the International Prognostic Scoring System (IPSS).[23] A corrected QT interval ≤470 milliseconds was required. Prior JAK inhibitor therapy was not permitted, except for ruxolitinib for <3 months’ duration and ongoing. Ruxolitinib was administered twice daily, continuously, in 28-day cycles and the starting dose was based on the platelet count, per the US prescribing information; the starting dose of pracinostat was 60 mg every other day for three out of every four weeks. Ruxolitinib was administered alone in the first 3 cycles, i.e., for the first 12 weeks, with pracinostat added on cycle 4, day 1. This was in order to avoid any added toxicity from pracinostat during the period of maximum symptomatic benefit from ruxolitinib. The primary endpoint of the study was the overall response rate (ORR), i.e., the proportion of patients achieving complete response (CR), partial response (PR) or CI according to the 2013 criteria of the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT).[24] Response assessments occurred after cycle 3 and cycle 6, and every six cycles, thereafter. Bone marrow biopsies were required after 6 and 12 cycles of therapy, and after that per the discretion of the treating physician. The MPN Symptom Assessment Form Total Symptom Score (MPN-SAF TSS) questionnaire[25] was administered every cycle in cycles 1 through 7, and every 3 cycles after that. Organomegaly was assessed by palpation. The study planned to accrue 25 patients. The method of Thall, Simon and Estey[26] was used for futility and toxicity moitoring. The study was approved by the MD Anderson Cancer Center (MDACC) Institutional Review Board (IRB), and all patients provided written informed consent. The study was conducted in accordance with the Declaration of Helsinki and the principles of Good Clinical Practice. The full study protocol is available in the Supplementary Appendix. Pracinostat was provided by MEI Pharma, while ruxolitinib was obtained through commercial supply. The study was monitored by the MDACC Investigational New Drugs (IND) office and registered at clinicaltrials.gov (NCT02267278).
Results
Patients
Of the 25 patients enrolled on the study, five never began pracinostat and are not considered further. These five patients came off the study prior to pracinostat initiation for the following reasons: one proceeded to allogeneic hematopoietic cell transplantation (allo-HCT), one experienced disease transformation to acute myeloid leukemia (AML), two had new cancer diagnoses after study enrollment, and one was taking a prohibited medication that could not be discontinued. Baseline characteristics of the 20 patients who received both study drugs are summarized in Table 1. The diagnosis was PMF in three quarters of the patients; no patient had post-ET MF. A driver mutation was identified in all but one patient. Just over half the patients were previously untreated. Eighty five percent had intermediate-2 or high risk disease by the IPSS.
Table 1.
Baseline Characteristics of the 20 Patients who Received Pracinostat.
| Variable | n (%), [range] |
|---|---|
| Median age, in years | 66 [56–78] |
| Male sex | 13 (65) |
| Myelofibrosis subtype | |
| Primary myelofibrosis | 15 (75) |
| Post-polycythemia vera myelofibrosis | 5 (25) |
| Post-essential thrombocythemia myelofibrosis | 0 |
| IPSS risk status | |
| High | 11 (55) |
| Intermediate-2 | 6 (30) |
| Intermediate-1 | 3 (15) |
| Previously treated¶ | 9 (45) |
| Bone marrow fibrosis grade | |
| MF-1 | 3 (15) |
| MF-2 | 8 (40) |
| MF-3 | 8 (40) |
| Not available | 1 (5) |
| Median WBC count, K/μL | 10.2 [3.5–54.3] |
| Median platelet count, K/μL | 253 [107–698] |
| Median hemoglobin, g/dL | 10.9 [7.4–16.2] |
| Median palpable spleen length, cm | 13 [5–20] |
| Driver mutation status | |
| JAK2 V617F | 17 (85) |
| MPL* | 1 (5) |
| CALR‡ | 1 (5) |
| Triple negative | 1 (5) |
Abbreviations: IPSS; International prognostic scoring system; WBC; White Blood Cell.
6 patients had received 1 prior therapy; 1 patient each had received 2, 3 and 5 prior therapies.
17/20 patients were tested for MPL gene mutation.
14/20 were tested for CALR gene mutation.
Pracinostat dose interruption, modification and discontinuation
Pracinostat was held in 16 patients, and never restarted in six. Of the ten patients in whom pracinostat was resumed, the dose was lowered to 45 mg in nine; one patient, in whom the drug was held for back surgery, resumed pracinostat at 60 mg. Reasons for interruption/dose reduction or pracinostat in these nine patients included anemia/increased red cell transfusion requirement in three, fatigue in two, diarrhea in one, thrombocytopenia in one, anemia and thrombocytopenia in one, and anemia and nausea in one. One patient began pracinostat at a dose of 45 mg based on investigator decision because of advanced age, anemia and erythrocyte transfusion requirements. All patients eventually discontinued pracinostat, and are currently off study. Reasons for discontinuation of pracinostat were cytopenias in eight, disease progression in three, non-hematologic toxicity, unrelated medical complications, financial constraints and physician/patient perception of lack of benefit in two patients each, and allo-HCT in one (Table 2). The median time on pracinostat was 5.3 (0.4–28.4) months. The median number of cycles on study was 11.5 (5–34). The median dose of ruxolitinib was 20 mg twice daily. Ruxolitinib dose reductions to offset pracinostat-induced anemia did not occur.
Table 2.
Reasons for Pracinostat Discontinuation.
| Patient disposition | n (%) |
|---|---|
| Cytopenias | |
| Anemia/increasing transfusion requirements | 6 (30) |
| Thrombocytopenia | 1 (5) |
| Anemia and thrombocytopenia | 1 (5) |
| Disease progression | |
| Progressive myelofibrosis | 2 (10) |
| Transformation to acute myeloid leukemia | 1 (5) |
| Allogenic stem cell transplant | 1 (5) |
| Non-hematological toxicity | |
| Acute kidney injury | 1 (5) |
| Allergic reaction | 1 (5) |
| Physician/patient perception of lack of benefit | 2 (10) |
| Financial | 2 (10) |
| Unrelated medical reasons | |
| Myocardial infarction | 1 (5) |
| Recurrence of skin cancer requiring therapy | 1 (5) |
Efficacy
Sixteen patients (80%) had objective responses, all CI. No patient experienced a PR or CR. Ten patients experienced CI in terms of spleen and symptoms, three spleen only, two symptoms only, and one had patient had CI-spleen as well as a partial molecular response. One patient had a baseline palpable spleen length of <5 cm and was, therefore, not evaluable for spleen response; 14 of 19 (74%) evaluable patients had CI-spleen at any time on the study. Similarly, five patients were not evaluable for symptoms response (TSS not calculated at baseline in two and TSS <12 in three); thus, the rate of CI-symptoms was 80% (12 of 15 evaluable patients). There were no anemia or cytogenetic responses. The median time to response was 1.6 (0.9–15.9) months. Fourteen patients had their earliest response prior to starting pracinostat, i.e. on ruxolitinib alone. The median duration of (earliest) response was 7.5 (3.6–25.7) months. A total of five patients had IWG-MRT responses after initiation of pracinostat, a median of 2.8 (0.9–13.1) months after starting. In two of these patients, their earliest response occurred after commencing therapy with pracinostat; in two others, spleen responses occurred after introduction of pracinostat, while symptom responses had occurred earlier, and one patient had a spleen response to ruxolitinib alone, while their symptom response occurred after therapy with pracinostat was begun. Two patients with spleen responses to ruxolitinib alone experienced resolution of palpable splenomegaly after the addition of pracinostat.
Bone marrow fibrosis grade improved in three patients, all after pracinostat initiation: from MF-3 to MF-1 in two patients (at 24.9 months and 14 months) and from MF-3 to MF-2 in one at 6.2 months, according to the European classification.[27] The patient whose bone marrow fibrosis grade went from MF-3 to MF-1 in 14 months had achieved a grade of MF-2 at 5.5 months. Bone marrow fibrosis grade worsened in three patients, and remained unchanged in eleven. Three patients were not evaluable for change in bone marrow fibrosis grade due to missing information at baseline or follow-up. One patient had a 99.6% reduction (from 65% to 0.27%) in the variant allele frequency of JAK2V617F at 24 months; interestingly, this patient also had an improvement in bone marrow fibrosis grade from MF-3 to MF-1 over approximately the same period. Serial assessments (twice) of JAK2V617F allelic burden were performed in only 3 patients, however, and no meaningful changes were noted in the other two. A >50% decrease in bone marrow blasts was noted in two patients, at six and eleven months, respectively.
Survival
At the data cutoff time point (May 22nd, 2018), six patients had died. The median overall survival (OS) was 33.8 months (Figure 1). The median follow-up on surviving patients was 21.4 (12.5–39.1) months. Progressive MF was the cause of death in three patients, while two patients died of pneumonia, one with acute hypoxemic respiratory failure. Mycobacterium avium intracellulare was cultured from the bronchoalveolar lavage fluid from the other patient. The sixth patient was transitioned to hospice because of progressive MF and expired soon thereafter.
Figure 1 –
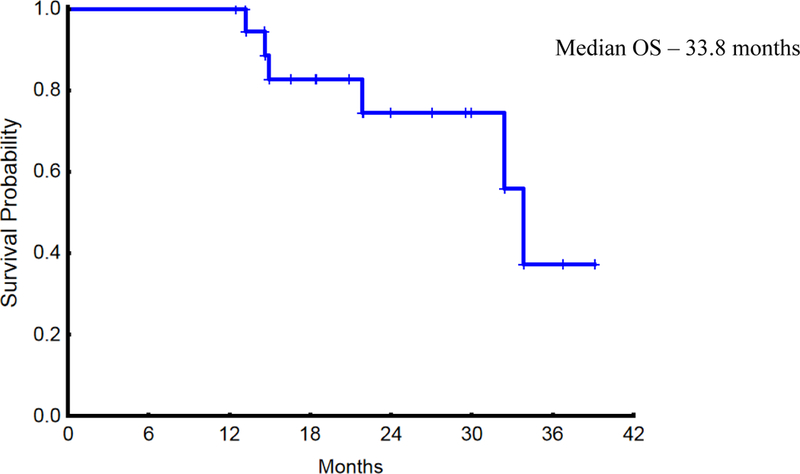
Overall Survival of the 20 Study Patients who Received Pracinostat.
Safety
Adverse events (AEs) at least possibly attributable to the study drugs are listed in Table 3. Most AEs were grade 1 or 2; however, anemia was very common and was grade 3 in nine patients. Grade 4 thrombocytopenia occurred in one patient. As alluded to above, six patients discontinued pracinostat because of worsening anemia/increasing red cell transfusion requirements, one because of thrombocytopenia, and one due to both anemia and thrombocytopenia. Shingles and weight gain, recognized complications of ruxolitinib treatment, were observed in three and four patients, respectively. Two patients discontinued pracinostat because of non-hematologic toxicity: acute kidney injury (reversible upon pracinostat discontinuation) in one and an allergic reaction to pracinostat in another.
Table 3.
Adverse Events at least Possibly Related to the Study Drugs (n = 20).
| Adverse Events* | Grade | |||
|---|---|---|---|---|
| 1 | 2 | 3 | 4 | |
| n (%) | ||||
| Hematological toxicity | ||||
| Anemia | 2 (10) | 6 (30) | 9 (45) | 0 |
| Thrombocytopenia | 5 (25) | 2 (10) | 2 (10) | 1 (5) |
| Neutropenia | 0 | 1 (5) | 0 | 0 |
| Non hematological toxicity | ||||
| Weight gain | 3 (15) | 1 (5) | 0 | 0 |
| Shingles | 0 | 3 (15) | 0 | 0 |
| Diarrhea | 1 (5) | 0 | 1 (5) | 0 |
| Fatigue | 0 | 2 (10) | 0 | 0 |
| Nausea | 1 (5) | 0 | 0 | 0 |
| Vomiting | 1 (5) | 0 | 0 | 0 |
| Flatulence | 1 (5) | 0 | 0 | 0 |
| Mouth sores | 1 (5) | 0 | 0 | 0 |
| Pruritus | 1 (5) | 0 | 0 | 0 |
| AST elevation | 1 (5) | 0 | 0 | 0 |
Adverse events were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.0)
Discussion
The development of HDACi for the treatment of MPN has been difficult. While these agents are clearly active, chronic, predominantly low-grade toxicities make them difficult to administer over long periods in these relatively indolent malignancies.[16] Prolonged treatment appears necessary for disease-modifying effects to emerge, at least in MF.[28] Nevertheless, at least one HDACi, givinostat, remains in clinical development,[29] and a phase 3 registration trial is planned in patients with PV. In MF, the path of clinical development for HDACi has been checkered, with limited single-agent activity observed, both in our own experience with pracinostat[21] and that of others with a “pan”-HDACi, panobinostat.[30, 31]
In a phase 2 trial reported by DeAngelo and colleagues,[30] panobinostat (40 mg three times a week) was associated with a high discontinuation rate, and only one patient achieved an objective (IWG-MRT) response. Although correlative studies revealed inhibition of JAK/signal transducer and activator of transcription (STAT) signaling, decreased intracellular cytokine levels and JAK2V617F allelic burden, tolerance was poor with high rates of thrombocytopenia and diarrhea. Mascarenhas et al reported an ORR of 36% to panobinostat in a separate, single-institution phase 2 trial employing a dose of 25 mg three times a week.[31] One patient obtained a complete molecular response, although the mean reduction in JAK2V617F allele burden was only 6.8%. Treatment discontinuation was frequent because of physician/patient perception of therapy ineffectiveness, but six patients remained on treatment for a median of 18 months.
In our study, pracinostat was poorly tolerated, with anemia/worsening red cell transfusion requirements being a major reason for discontinuation of pracinostat. While it is tempting to speculate that this may, at least in part, be due to suppression of erythropoietin production through pracinostat-mediated down-regulation of hypoxia-inducible factor 1-alpha (HIF1-α),[32, 33] only one patient had their erythropoietin level measured on the study, prior to initiation of pracinostat, and this patient did not have a baseline erythropoietin level measured. Another patient had an erythropoietin level checked 5.5 months after coming off the study (because of anemia attributed to pracinostat), and it was higher than the baseline value for that patient. Despite the ORR (80%) and rates of spleen response (74%) and symptom improvement (80%) being high, most patients had their initial response before the introduction of pracinostat, and although additional responses, as well as deepening of initial responses, occurred in some patients after beginning therapy with pracinostat, it was difficult to quantify the added benefit of pracinostat as response rates to ruxolitinib do improve over time.[6, 7] Inclusion of a ruxolitinib-only comparator arm may help clarify the added benefit, if any, of a second agent in the upfront setting, but such designs are often impractical in small, early phase proof-of-concept trials of rational, ruxolitinib-based combination regimens. Indeed, many current trials, e.g., ref.[34], are exploring an “add-on” strategy, where a novel agent is added in patients with an insufficient response to ruxolitinib. All patients in our study eventually discontinued pracinostat, along the lines of the above experience with panobinostat. A phase 1/2 study in Europe evaluating the combination of ruxolitinib and panobinostat identified the recommended phase 2 dose for the combination and reported promising rates of ≥35% spleen volume reduction (SVR) by imaging at 24 and 48 weeks, as well as improved bone marrow fibrosis and ≥20% decrease in JAK2V617F allele burden at 48 weeks in some patients but that trial was halted early.[35] The attainment of near-CMR in one of our patients, who remained on study for over two years, accompanied by a robust decrease in bone marrow fibrosis, is intriguing and raises the possibility of a disease-modifying effect of prolonged HDACi treatment, but such responses are also occasionally seen with ruxolitinib treatment alone.[36, 37] However, given the small number of patients in our study, that three patients had objective improvements in degree of bone marrow fibrosis at relatively early time points is somewhat striking and suggests a biological effect of pracinostat in this regard. In COMFORT-2, only 15.8% of patients randomized to ruxolitinib had improvement in bone marrow fibrosis after five years of follow-up.[7] The lack of spleen volume assessment by imaging was an obvious limitation of our investigator-initiated study. Additionally, very few of our patients had serial assessment of driver mutation allele burden.
In general, the development of rational, ruxolitinib-based combinations for clinical use in MF has been challenging. Clinical trials of ruxolitinib in combination with inhibitors of phosphatidylinositol-3-kinase[38] and the hedgehog pathway (smoothened antagonists)[39] were stopped for lack of a significant advantage over ruxolitinib alone, at least at early time points. Other ruxolitinib combinations based on laboratory evidence of synergism, such as those with HSP90 inhibitors[15], BH3-mimetics[40] and bromodomain extra-terminal (BET) inhibitors[41] have just entered the clinic, while other concepts, e.g., combinations with inhibitors of mutant isocitrate dehydrogenase[42] and poly (ADP-ribose) polymerase (PARP) inhibitors[43] await translation. The present study does not support continued development of pracinostat for MF. Whether any of the other laboratory-based combinations being explored will succeed in the clinic remains to be seen. In contrast, encouraging early results have been reported with novel agents that counteract ruxolitinib-induced anemia, potentially enabling patients to stay on ruxolitinib for longer periods of time and optimize dosing of the only agent thus far to prolong survival in MF.[44] Bone marrow fibrosis reduction and improvement in cytopenias have also been reported with PRM-151 (recombinant pentraxin-2),[45] and data from a larger trial of this agent are eagerly awaited.
In summary, in this small, investigator-initiated trial, the addition of pracinostat to ruxolitinib resulted in only modestly increased efficacy that was difficult to attribute to pracinostat versus longer exposure to ruxolitinib. Additionally, pracinostat appeared to worsen anemia in a number of patients, and discontinuation for this and other reasons was frequent. In contrast to AML, for which pracinostat has received “breakthrough” designation from the FDA, our results do not support continued development of this agent for MF.
Supplementary Material
Acknowledgements:
This work was supported in part by MEI Pharma, who provided pracinostat and financial support for the study, as well as the MD Anderson Cancer Center Support Grant P30 CA016672 from the National Cancer Institute (National Institutes of Health).
References
- [1].Deisseroth A, Kaminskas E, Grillo J, et al. U.S. food and drug administration approval: Ruxolitinib for the treatment of patients with intermediate and high-risk myelofibrosis. Clin Cancer Res 2012;18:3212–7. [DOI] [PubMed] [Google Scholar]
- [2].Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med 2010;363:1117–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Verstovsek S, Gotlib J, Mesa RA, et al. Long-term survival in patients treated with ruxolitinib for myelofibrosis: COMFORT-I and -II pooled analyses. J Hematol Oncol 2017;10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Cervantes F, Pereira A. Does ruxolitinib prolong the survival of patients with myelofibrosis?. Blood 2017;129:832–7. [DOI] [PubMed] [Google Scholar]
- [5].Mascarenhas J, Hoffman R. A comprehensive review and analysis of the effect of ruxolitinib therapy on the survival of patients with myelofibrosis. Blood 2013;121:4832–7. [DOI] [PubMed] [Google Scholar]
- [6].Verstovsek S, Mesa RA, Gotlib J, et al. Long-term treatment with ruxolitinib for patients with myelofibrosis: 5-year update from the randomized, double-blind, placebo-controlled, phase 3 COMFORT-I trial. J Hematol Oncol 2017;10: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Harrison CN, Vannucchi AM, Kiladjian JJ, et al. Long-term findings from COMFORT-II, a phase 3 study of ruxolitinib vs best available therapy for myelofibrosis. Leukemia 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Verstovsek S, Kantarjian HM, Estrov Z, et al. Long-term outcomes of 107 patients with myelofibrosis receiving JAK1/JAK2 inhibitor ruxolitinib: Survival advantage in comparison to matched historical controls. Blood 2012;120:1202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Vannucchi AM, Kantarjian HM, Kiladjian JJ, et al. A pooled analysis of overall survival in COMFORT-I and COMFORT-II, 2 randomized phase 3 trials of ruxolitinib for the treatment of myelofibrosis. Haematologica 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Koppikar P, Bhagwat N, Kilpivaara O, et al. Heterodimeric JAK-STAT activation as a mechanism of persistence to JAK2 inhibitor therapy. Nature 2012;489:155–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Bose P, Dai Y, Grant S. Histone deacetylase inhibitor (HDACI) mechanisms of action: Emerging insights. Pharmacol Ther 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Bali P, Pranpat M, Bradner J, et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J Biol Chem 2005;280:26729–34. [DOI] [PubMed] [Google Scholar]
- [13].Dawson MA, Bannister AJ, Gottgens B, et al. JAK2 phosphorylates histone H3Y41 and excludes HP1alpha from chromatin. Nature 2009;461:819–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Liu F, Zhao X, Perna F, et al. JAK2V617F-mediated phosphorylation of PRMT5 downregulates its methyltransferase activity and promotes myeloproliferation. Cancer Cell 2011;19:283–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bhagwat N, Koppikar P, Keller M, et al. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood 2014;123:2075–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bose P, Verstovsek S. Investigational histone deacetylase inhibitors (HDACi) in myeloproliferative neoplasms. Expert Opin Investig Drugs 2016;25:1393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Wang Y, Fiskus W, Chong DG, et al. Cotreatment with panobinostat and JAK2 inhibitor TG101209 attenuates JAK2V617F levels and signaling and exerts synergistic cytotoxic effects against human myeloproliferative neoplastic cells. Blood 2009;114:5024–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Civallero M, Cosenza M, Pozzi S, Sacchi S. Ruxolitinib combined with vorinostat suppresses tumor growth and alters metabolic phenotype in hematological diseases. Oncotarget 2017;8:103797–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Novotny-Diermayr V, Hart S, Goh KC, et al. The oral HDAC inhibitor pracinostat (SB939) is efficacious and synergistic with the JAK2 inhibitor pacritinib (SB1518) in preclinical models of AML. Blood Cancer J 2012;2:e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Garcia-Manero G, Atallah E, Khaled SK, et al. A phase 2 study of pracinostat and azacitidine in elderly patients with acute myeloid leukemia (AML) not eligible for induction chemotherapy: Response and long-term survival benefit. Blood 2016;128:100-. [Google Scholar]
- [21].Quintas-Cardama A, Kantarjian H, Estrov Z, Borthakur G, Cortes J, Verstovsek S. Therapy with the histone deacetylase inhibitor pracinostat for patients with myelofibrosis. Leuk Res 2012;36:1124–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Tefferi A, Barosi G, Mesa RA, et al. International working group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for myelofibrosis research and treatment (IWG-MRT). Blood 2006;108:1497–503. [DOI] [PubMed] [Google Scholar]
- [23].Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the international working group for myelofibrosis research and treatment. Blood 2009;113:2895–901. [DOI] [PubMed] [Google Scholar]
- [24].Tefferi A, Cervantes F, Mesa R, et al. Revised response criteria for myelofibrosis: International working group-myeloproliferative neoplasms research and treatment (IWG-MRT) and european LeukemiaNet (ELN) consensus report. Blood 2013;122:1395–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Emanuel RM, Dueck AC, Geyer HL, et al. Myeloproliferative neoplasm (MPN) symptom assessment form total symptom score: Prospective international assessment of an abbreviated symptom burden scoring system among patients with MPNs. J Clin Oncol 2012;30:4098–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Thall PF, Simon RM, Estey EH. Bayesian sequential monitoring designs for single-arm clinical trials with multiple outcomes. Stat Med 1995;14:357–79. [DOI] [PubMed] [Google Scholar]
- [27].Thiele J, Kvasnicka HM, Facchetti F, Franco V, van der Walt J, Orazi A. European consensus on grading bone marrow fibrosis and assessment of cellularity. Haematologica 2005;90:1128–32. [PubMed] [Google Scholar]
- [28].Mascarenhas J, Lu M, Li T, et al. A phase I study of panobinostat (LBH589) in patients with primary myelofibrosis (PMF) and post-polycythaemia vera/essential thrombocythaemia myelofibrosis (post-PV/ET MF). Br J Haematol 2013;161:68–75. [DOI] [PubMed] [Google Scholar]
- [29].Finazzi G, Martino B, Pezzuto A, et al. A two-part study of givinostat in patients with polycythemia vera: Maximum tolerated dose definition and preliminary efficacy results. Blood 2016;128:4261-. [Google Scholar]
- [30].DeAngelo DJ, Mesa RA, Fiskus W, et al. Phase II trial of panobinostat, an oral pan-deacetylase inhibitor in patients with primary myelofibrosis, post-essential thrombocythaemia, and post-polycythaemia vera myelofibrosis. Br J Haematol 2013;162:326–35. [DOI] [PubMed] [Google Scholar]
- [31].Mascarenhas J, Sandy L, Lu M, et al. A phase II study of panobinostat in patients with primary myelofibrosis (PMF) and post-polycythemia vera/essential thrombocythemia myelofibrosis (post-PV/ET MF). Leuk Res 2016;53:13–9. [DOI] [PubMed] [Google Scholar]
- [32].Kong X, Lin Z, Liang D, Fath D, Sang N, Caro J. Histone deacetylase inhibitors induce VHL and ubiquitin-independent proteasomal degradation of hypoxia-inducible factor 1alpha. Mol Cell Biol 2006;26:2019–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Qian DZ, Kachhap SK, Collis SJ, et al. Class II histone deacetylases are associated with VHL-independent regulation of hypoxia-inducible factor 1 alpha. Cancer Res 2006;66:8814–21. [DOI] [PubMed] [Google Scholar]
- [34].Moyo TK, Palmer J, Huang Y, et al. Resurrecting response to ruxolitinib: A phase I study of ruxolitinib and umbralisib (TGR-1202) in ruxolitinib-experienced myelofibrosis. Haematologica 2018:S133-. [Google Scholar]
- [35].Harrison CN, Kiladjian JJ, Heidel FH, et al. Efficacy, safety, and confirmation of the recommended phase 2 starting dose of the combination of ruxolitinib (RUX) and panobinostat (PAN) in patients (pts) with myelofibrosis (MF). Blood 2015;126:4060-. [Google Scholar]
- [36].Masarova L, Wang W, Newberry KJ, Kantarjian H, Verstovsek S. Complete remission in a patient with JAK2- and IDH2-positive myelofibrosis. Blood 2016;128:877–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Deininger M, Radich J, Burn TC, Huber R, Paranagama D, Verstovsek S. The effect of long-term ruxolitinib treatment on JAK2p.V617F allele burden in patients with myelofibrosis. Blood 2015;126:1551–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Durrant S, Nagler A, Vannucchi AM, et al. An open-label, multicenter, 2-arm, dose-finding, phase 1b study of the combination of ruxolitinib and buparlisib (BKM120) in patients with myelofibrosis: Results from HARMONY study. Blood 2015;126:827-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Gupta V, Harrison CN, Hasselbalch HC, et al. Phase 1b/2 study of the efficacy and safety of sonidegib (LDE225) in combination with ruxolitinib (INC424) in patients with myelofibrosis. Blood 2015;126:825.26473195 [Google Scholar]
- [40].Waibel M, Solomon VS, Knight DA, et al. Combined targeting of JAK2 and bcl-2/Bcl-xL to cure mutant JAK2-driven malignancies and overcome acquired resistance to JAK2 inhibitors. Cell Rep 2013;5:1047–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kleppe M, Koche R, Zou L, et al. Dual targeting of oncogenic activation and inflammatory signaling increases therapeutic efficacy in myeloproliferative neoplasms. Cancer Cell 2018;33: [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].McKenney AS, Lau AN, Somasundara AVH, et al. JAK2/IDH-mutant-driven myeloproliferative neoplasm is sensitive to combined targeted inhibition. J Clin Invest 2018;128:789–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Nieborowska-Skorska M, Maifrede S, Dasgupta Y, et al. Ruxolitinib-induced defects in DNA repair cause sensitivity to PARP inhibitors in myeloproliferative neoplasms. Blood 2017;130:2848–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Bose P, Daver N, Pemmaraju N, et al. Sotatercept (ACE-011) alone and in combination with ruxolitinib in patients (pts) with myeloproliferative neoplasm (MPN)-associated myelofibrosis (MF) and anemia. Blood 2017;130:255-. [Google Scholar]
- [45].Verstovsek S, Mesa RA, Foltz LM, et al. PRM-151 in myelofibrosis: Durable efficacy and safety at 72 weeks. Blood 2015;126:56. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.