

Abstract
Objective:
In response to tissue injury, the appropriate progression of events in angiogenesis is controlled by a careful balance between pro- and anti-angiogenic factors. We aimed to identify and characterize microRNAs that regulate angiogenesis in response to tissue injury.
Approach and Results:
We show that in response to tissue injury, miR-615–5p is rapidly induced and serves as an anti-angiogenic miRNA by targeting endothelial cell (EC) VEGF-AKT/eNOS signaling in vitro and in vivo. MiR-615–5p expression is increased in wounds of diabetic db/db mice, in plasma of human subjects with acute coronary syndromes, and in plasma and skin of human subjects with diabetes. Ectopic expression of miR-615–5p markedly inhibited EC proliferation, migration, network tube formation in matrigel, and the release of nitric oxide, whereas miR-615–5p neutralization had the opposite effects. Mechanistic studies using transcriptomic profiling, bioinformatics, 3’-UTR reporter and miRNP-IP assays, and siRNA dependency studies demonstrate that miR-615–5p inhibits the VEGF-AKT/eNOS signaling pathway in ECs by targeting IGF2 and RASSF2. Local delivery of miR-615–5p inhibitors, markedly increased angiogenesis, granulation tissue thickness, and wound closure rates in db/db mice, whereas miR-615–5p mimics impaired these effects. Systemic miR-615–5p neutralization improved skeletal muscle perfusion and angiogenesis after hindlimb ischemia in db/db mice. Finally, modulation of miR-615–5p expression dynamically regulated VEGF-induced AKT signaling and angiogenesis in human skin organoids as a model of tissue injury.
Conclusion:
These findings establish miR-615–5p as an inhibitor of VEGF-AKT/eNOS-mediated EC angiogenic responses, and that manipulating miR-615–5p expression could provide a new target for angiogenic therapy in response to tissue injury.
Keywords: angiogenesis, endothelial cells, microRNAs, tissue repair, AKT/eNOS signaling
Subject codes: MicroRNA, Angiogenesis, Cell Signaling/Signal Transduction, endothelial cell, tissue repair
Graphical Abstract
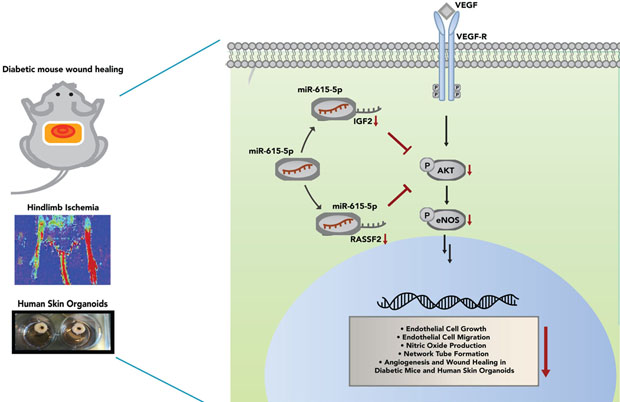
Introduction
The induction and orchestration of new blood vessels is critical for tissue repair in response to injury in ischemic cardiovascular diseases such as myocardial infarction (MI), peripheral artery disease (PAD), and diabetic wound healing.1–4 In response to proangiogenic stimuli, activated vascular endothelial cells (ECs) migrate to distant sites to proliferate and generate new primary capillaries necessary for tissue repair. Impairment of these cellular and physiologic processes can contribute to disease progression, and have been linked to poor cardiovascular function and outcomes.5
Over the years, the importance of angiogenesis has ignited numerous clinical investigations that have sought to stimulate angiogenesis to combat ischemic pathologies and tissue injury.6, 7 These studies have primarily focused on the intramuscular and intra-arterial introduction of a range of angiogenic growth factors such as vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), hepatocyte growth factor (HGF), hypoxia-inducible factor-1a, developmental endothelial locus-1 (Del-1), to promote neovascularization and tissue perfusion in subjects with PAD or chronic stable angina.8–13 Unfortunately, clinical trials using these growth factors neither succeeded in improving patient outcomes nor led to effective therapeutic angiogenesis.14–16 The failure of these trials have been attributed to a plethora of reasons, including challenges in translating preclinical models to clinical disease, selection of patient cohorts, delivery routes, and therapeutic dosing.17
However, accumulating studies have recently suggested that these failures in therapeutic angiogenesis may not be due to a deficiency of angiogenic growth factors, but rather to an impairment in downstream signaling.18–21 In support, circulating pro-angiogenic growth factors (e.g. VEGF-A) are increased in patients that present with myocardial ischemia with the highest levels observed in patients with the most severe myocardial ischemic burden.22, 23 Similarly, VEGF-A, Tie2, and Angiopoietin-2 were also expressed higher in subjects with PAD compared to without PAD. Similarly, patients with critical limb ischemia had the highest levels of these pro-angiogenic circulating factors compared to those with intermittent claudication or without PAD.24 These findings highlight that growth factor deficiency may not be a major contributor for the impaired neovascularization in response to ischemic disease states. Rather, impaired angiogenic signaling or “angiogenic resistance” may be a more dominant contributor through mechanisms analogous to the process by which impaired insulin signaling drives the development of insulin resistance in diabetic subjects.
Growth factors such as VEGF activate a broad range of canonical pathways including AKT, eNOS, p38, and MAPK to promote the proliferative, migratory, and organizational processes that are necessary for effective angiogenesis.25 Thus, reduced activation of key mediators of these pathways within the context of ischemic disease and tissue injury may significantly impede successful neovascularization. For example, despite no differences in VEGF expression, there is significantly reduced phosphorylation of AKT/eNOS in diabetic limbs compared to non-diabetic limbs.26 In addition, decreased AKT/eNOS phosphorylation in the vascular endothelium promoted vascular dysfunction and reduced angiogenesis in murine models of hindlimb ischemia and myocardial infarction.27–30 Together, theses studies suggest that defects in angiogenesis can be overcome by focusing on approaches to augment downstream effects of pro-angiogenic growth factor signaling pathways.
MicroRNAs (miRNAs) are small, single-stranded, non-coding RNAs that suppress the expression of target genes at the post-transcriptional level and are involved in a variety of pathophysiological processes.31, 32 The role of miRNAs in angiogenesis is well-established, and has been implicated as important regulators of a range of pathophysiological processes such as myocardial and limb ischemia and diabetic wound healing through their pro-angiogenic and anti-angiogenic properties.18, 19, 33–38 The capacity of a few of these miRNAs, such as miR-126, miR-200b, miR-503, and miR-26a, to impede angiogenesis has been attributed to their ability to silence important downstream effectors of growth factor signaling in ECs.19, 39–41 Thus, the identification of miRNAs and downstream growth factor targets that govern the angiogenic response may provide novel therapeutic approaches to improve tissue repair and ischemic cardiovascular pathologies.
In this study, we identified miR-615–5p as a novel regulator of pathological and physiological angiogenesis in ECs. MiR-615–5p expression is increased in response to tissue injury or a range of ischemic disease states and confers anti-angiogenic properties by specifically targeting Insulin-like Growth factor 2 (IGF2) and Ras Associating Domain Family Member 2 (RASSF2) to decrease AKT/eNOS signaling in the presence of pro-angiogenic stimuli. Consequently, miR-615–5p impairs EC functions critical for angiogenesis and wound healing. Conversely, neutralization of miR-615–5p can successfully restore angiogenesis and promote wound healing in diabetic mice and human skin organoid models by increasing activation of AKT signaling. Collectively, these findings establish a new approach to overcome impaired angiogenic signaling in response to tissue injury or ischemic cardiovascular disease.
Materials and Methods
All supporting data are available within the article and its online-only Data Supplement
Tube-like network formation on Matrigel (in vitro)
Matrigel (BD Bioscience) basement membrane matrix was added to 96-well culture plates and incubated at 37°C until gelation occurred. Network tube formation was assessed 14 hours post-plating and quantitated by counting the number of tubes formed per high power field as described.19, 42, 43
Chemotaxis assays
Migration assay was performed using ChemTX multiwell system (Neuro probe Inc, MD). The number of cells migrating to the lower chamber was counted using a hemocytometer after 5 hours.44
MiRNP immunoprecipitation (MiRNP-IP)
MiRNP-IP was performed as we previously described.45
Microarray transcriptomic profiling and bioinformatics
For DNA microarray gene chip analysis, HUVECs were transfected with 30 nM miRNA negative control or miR-615–5p mimics for 24 hours. Cells were collected into RNeasy mini kit (Qiagen) and sent for Two-Color, 4 × 44 K format (Agilent Technologies), Human Whole Genome Oligo Microarray Service (ArrayStar, Inc.).
Differentially expressed genes that were identified as being at least 1.5-fold repressed (p-value<0.05) were subjected to gene set enrichment analyses (GSEA). Gene Ontology (GOs) pathways were explored to determine whether a known biological network, process, or molecular function was suppressed by overexpression of miR-615–5p (Ingenuity Pathway Analysis (IPA, Qiagen)). The top ten GOs for molecular and cellular functions and the top ten GOs for signaling networks were ranked using p-value or score, respectively. The primers used for real time-qPCR analyses are listed in Online Supplementary Table 1.
Scratch assay for EC migration
Scratch wound assay was performed using Culture-Insert 2 Well 35mm μ-Dishes (ibidi). HUVECs transfected with miR-615–5p mimic, miR-615–5p inhibitor, non-specific miRNA controls, IGF2 siRNA, RASSF2 siRNA, or control siRNA were cultured for 60 hours in 12-well plates and plated at 21K cells per well into the μ-Dishes. Inserts were lifted at 72 hours after transfection, and cells were imaged using an Eclipse TE2000-U inverted microscope (Nikon) at 2× and 4× over time to assess for wound closure. Three technical replicates were performed per condition. Significance was determined by student’s two-tailed t-test, p<0.05.
In vivo miR-615–5p inhibition or over-expression and mouse experiments
Animal protocols were approved by the Laboratory Animal Care at Harvard Medical School and Brigham and Women’s Hospital. For mouse dermal wound studies, male, 8–10 weeks old, db/db mice (Jackson Laboratories) were used for local intradermal injections of either scrambled control LNA-anti-miR or LNA-anti-miR-615–5p (Exiqon, Inc) at 0.63 mg/kg 48h and 24h prior to surgery. On day 0, dorsal full thickness skin wounds (1cm2) were generated and covered with semi-occlusive dressing (Tegaderm). Images of the wounds were immediately acquired after surgery (day 0) and on day 9 following the removal of the Tegaderm dressing. Mice were euthanized 9 days post-surgery and the 1×1 cm2 sections of skin surrounding the wound were excised down to fascia. Angiogenesis in wounds was analyzed by mouse CD31 staining (DIA-310; Dianova, Inc) and DAPI (H21492; Invitrogen, Inc) of the paraffin embedded wound sections. Granulation tissue thickness was measured on day 9 using H&E stained sections obtained from the center of the wound. Granulation tissue thickness is defined as the distance of intact tissue from the bottom of the epidermis to the top of the subcutaneous fat layer and was quantified using Image J. Maximal thickness of granulation tissue was quantified at a fixed distance 200 micrometers from the wound edge. Fluorescent images were acquired by Olympus Fluoview FV1000 confocal microscope.
Hindlimb Ischemia
Ischemic injury was produced by unilateral femoral artery ligation in C57BL/6 mice as previously described44. Mice were injected via tail vein with either phosphorodithioate (PS2)-anti-miR-615 inhibitor or a scrambled PS2 anti-miR control (1mg/kg, AM Biotech, Inc) on the day prior and at 2 and 6 days post-hindlimb ischemia. Mice were imaged immediately after surgery over 15 days on a 785-nm near-infrared Laser Doppler Imager-2 (Moor Instruments Inc.). Skeletal muscles were harvested for CD31 staining as described.44
Circulating miR-615–5p levels in patients with acute coronary syndromes or diabetes mellitus
Patient plasma samples were collected in EDTA-containing tubes as part of a prospectively enrolled cohort of patients that underwent either cardiac catheterization or coronary artery bypass graft surgery in accordance with the Institutional Review Board-approved protocols at Brigham and Women’s Hospital or at the Regional Ethics Committee in Lillehamer Hospital, Norway, or Ethics Committee in Research at the Instituto de Cardiologia, Fundação Universitária de Cardiologia - Porto Alegre, Brazil. Written informed consent was obtained from all participants or their appropriate surrogates. Control patients were defined as without clinically significant coronary atherosclerosis (<20% stenosis in any epicardial coronary artery determined by angiography) and had no elevation of cardiac biomarkers. Patients with acute coronary syndromes (ACS) were defined as acute atherothrombotic coronary artery occlusion resulting in either an NSTEMI (with >70% occlusion of an epicardial artery) or an STEMI (complete occlusion of an epicardial coronary artery determined by angiography) with elevation of cardiac biomarkers. Diabetes was defined as diabetes diagnosis registered in medical records. Anonymized plasma samples were generated from blood collected in EDTA-containing tubes at the time of the procedure and stored at −80°C. Diabetic patient plasma and skin samples collected as part of of Lillehamer study (obtained from human subjects undergoing cardiac surgery) were generated from blood collected in EDTA-containing tubes or skin samples preserved in Allprotect tissue reagent (Qiagen). Plasma was isolated from whole blood and skin samples were homogenized for respective RT-qPCR analyses. Anonymized plasma samples were generated from blood collected in EDTA-containing tubes at the time of the procedure and skin samples were collected into Allprotect tissue reagent (Qiagen) and stored at −80°C. Plasma was isolated from whole blood at 1500g for 15 minutes at room temperature. 60% of the upper phase was collected. Samples that were red in color were excluded from the analyses and only clear light yellow plasma samples were utilized for RNA extraction. Total RNA was isolated from plasma by using total RNA purification kit from Norgen Biotek Corporation46, 47 and reverse transcription and real time qPCR was performed as described in the Section of “Real-time qPCR”.
Nitric Oxide (NO) Assay
Nitrite content in cell culture supernatant was measured to indirectly reflect the content of NO using Griess reagent.48–50 Briefly, NO release from HUVECs was detected by enzymatic conversion of nitrate to nitrite by the enzyme nitrate reductase, followed by the Griess reaction to form a colored azo dye product per manufacturers protocols (Enzo, ADI-917–020). HUVECs transfected with miR-615–5p mimic or non-specific miRNA controls were cultured for 60 hours in 12-well plates and stimulated with 50ng/ml VEGF for 1 or 3 hrs as indicated. Supernatant from ECs were collected to detect total NO release.
Generation of human skin organoids
Full-thickness circular (6-mm) human skin organoids were taken from surgical skin samples and 3-mm full-thickness wounds were created as described previously.51 Briefly, human skin organoids were then embedded in collagen I matrix and maintained in Dulbecco’s Modified Eeagle’s Media (Life Technologies) supplemented with 10mm 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 50 μg/mL ascorbic acid, 100 μm adenine, 0.5 μm hydrocortisone, 0.1 nm cholera toxin, 100 μU/mL penicillin, and 10 μg/mL streptomycin (Sigma-Aldrich, St. Louis, MO). The ex-vivo organ cultures were cultured at the air–liquid interface and maintained in the cell culture incubator at 37°C with 5% CO2. The media were changed every other day. The viability of cultured explants was validated by histologic evaluation. Human skin organoids were transfected at the indicated time points using 30nM miR-615–5p mimics or non-specific controls or 100nM miR-615–5p inhibitors or non-specific controls. Neovascularization were measured at days 3 and 7 to determine miR-615–5p effects on wound healing.
Statistical Analysis
Data are presented as mean ± SEM. All experiments are representative of 3 independent experiments unless indicated otherwise. Sample sizes for mouse and human organoid studies were chosen based upon pilot studies or similar well-characterized studies in the literature. For 2 group comparisons, data were subjected to unpaired two-sided Student’s t-test if it passed normality and equal variance tests. If data for either normality or variance tests failed, then nonparametric Mann-Whitney U test was used. For 2 or more group comparisons, if data passed normality and variance testing, one-way ANOVA with Bonferroni correction was used. If data did not pass either test, then nonparametric Kruskal Wallis test with Dunn post hoc test was used. P<0.05 was considered statistically significant.
Results
To identify microRNAs (miRNAs) that regulate angiogenesis in response to tissue injury, microRNA (miRNA) microarray profiling studies52 were undertaken using plasma from human subjects with acute coronary syndromes (ACS) with coronary angiograms showing >70% stenotic lesions compared with non-ACS human subjects with coronary angiograms with lesions <20% stenosis and increased expression of miR-615–5p was noted with ACS (data not shown). Upon further investigation using real-time PCR (RT-PCR) analysis, we found that miR-615–5p was reduced significantly by both proangiogenic stimuli VEGF or bFGF over the course of 24 hrs (Figure 1A), whereas anti-angiogenic stimuli TSP1 and TSP2 significantly increased miR-615–5p expression (Online Figure 1A). Furthermore, co-stimulation of ECs with VEGF and TSP1 resulted in the loss of VEGF-mediated suppression of miR-615–5p expression (Online Figure 1B). VEGF represses target genes via an HDAC Class IIa dependent mechanism.53, 54 Pretreatment of ECs with an inhibitor of HDAC Class IIa blocked the VEGF repression of miR-615–5p suggesting that VEGF-mediated repression of miR-615–5p was mediated in part by HDAC class IIa (Online Figure 1C). We also verified by RT-PCR that miR-615–5p expression levels were increased in a larger cohort of human subjects with ACS. As shown in Figure 1B, circulating levels of miR-615–5p increased by 2.2-fold in ACS subjects with coronary angiograms bearing >70% stenotic lesions compared to non-ACS human subjects with coronary angiograms with lesions <20% stenosis. Similarly, miR-615–5p was significantly increased in the skin and plasma of patients with diabetes mellitus (DM) by 2.2- and 2-fold, respectively (Figure 1C–D). We also examined miR-615–5p expression in response to tissue injury from dermal wounds generated by punch biopsy of diabetic db/db mice. Under basal conditions, miR-615–5p was expressed 4.6 fold higher in the skin of db/db mice compared to WT mice (Day 0). MiR-615–5p expression remained elevated in wounds of db/db mice by 2.4-fold on day 7 and 1.95-fold on day 9 post-wounding compared to WT controls (Figure 1E). Collectively, these data suggest that miR-615–5p is dynamically regulated by pro-angiogenic stimuli in ECs and its expression may correlate with acute ischemic injury or diabetic states, raising the possibility that targeting this miRNA may facilitate the induction of angiogenesis.
Figure 1.

MiR-615–5p is regulated by pro-angiogenic stimuli and inhibits endothelial cell growth. (A) Real-time qPCR analysis of miR-615–5p expression in response to VEGF and bFGF in HUVECs. * P < 0.005 compared to Ctrl. Data is representative of n=4 experiments. (B) Circulating miR-615–5p levels are increased in plasma from human subjects with acute coronary syndrome ((ACS) (n = 40; 28 male, 12 female)) compared to subjects with normal coronary angiograms ((NCA) (n = 20; 9 male, 11 female)). * P < 0.05 compared to normal coronary angiogram. (C-D) Expression of miR-615–5p is increased in skin and plasma of patients with diabetes (n = 13; 9 male, 4 female) compared to controls (n=10; 8 male, 2 female). * P < 0.05 compared to controls. (E) WT and db/db mice (n=4 – 6/group) underwent punch-biopsy wounding of the dorsal skin, and wounds were collected for qPCR analyses for miR-615–5p on the indicated days post-wounding. (F) HUVECs transfected with miR negative control (NSm), miR-615–5p mimics (miR-615–5pm), miR inhibitor negative control (NSi), or miR-615–5p inhibitor (miR-615–5pi) and stimulated with VEGF or bFGF as indicated were subjected to BrdU cell proliferation assay. * P < 0.05 compared to controls. All data represent mean ± s.e.m.
To assess the potential role of miR-615–5p in endothelial angiogenic functions, we examined the effect of miR-615–5p on EC growth by gain- and loss-of-function experiments. Overexpression of miR-615–5p ‘mimics’ (miR-615–5pm) in HUVECs inhibited cell growth by 32% under basal conditions and by 62% and 53% in response to VEGF and bFGF stimulation, respectively, whereas miR-615–5p inhibitors (miR-615–5pi, complementary antagonist) increased EC growth by 3.2-fold under basal conditions and 2-fold and 1.85-fold in response to VEGF and bFGF stimulation, respectively (Figure 1F). To further characterize the role of miR-615–5p in HUVECs, we assessed vascular network formation assays in matrigel. Overexpression of miR-615 inhibited network tube formation in matrigel in vitro (Figure 2A, top) by 40%, whereas miR-615–5p inhibition significantly increased tube formation in matrigel in vitro (Figure 2A, bottom) by 28%. In addition, miR-615–5p overexpression decreased EC migration by ~17%, compared to the nonspecific (NS) control, whereas miR-615–5p inhibition increased migration by 20% compared to the NS control under basal conditions (Figure 2B). Interestingly, overexpression of miR-615–5p in the presence of VEGF (50 ng/mL) or bFGF (50 ng/mL) stimulation further inhibited the EC migration by 68% and 60%, respectively, while inhibition of miR-615–5p increased EC migration by 18% and 24%, respectively (Figure 2B). Finally, consistent with its effects on EC growth and migration, HUVECs overexpressing miR-615–5p decreased wound closure in scratch assays by 32% under basal conditions and by 81% in the presence of VEGF, whereas inhibition of miR-615–5p increased wound closure compared to the NS control by 20% under basal conditions and by 23% in the presence of VEGF (Figure 2C). Taken together, these data indicate that miR-615–5p inhibited EC angiogenic functions in vitro.
Figure 2. MiR-615–5p inhibits pro-angiogenic functions in ECs in vitro.

HUVECs transfected with miR negative control (NSm), miR-615–5p mimics (miR-615–5pm), miR inhibitor negative control (NSi), or miR-615–5p inhibitor (miR-615–5pi) were subjected to (A) tube-like network formation in matrigel; (B) EC migration in transwell Boyden chambers; (C) scratch assay. Data is representative of n = 6 per group and 3 independent experiments. * P < 0.05 compared to NSm or NSi ** P < 0.001 compared to NSi. Scale bars, 150μm (A) and 100μm (D). All data represent mean ± s.e.m.
To identify potential target signaling pathways of miR-615–5p, we performed a microarray gene chip profiling approach from HUVECs overexpressing miR-615–5p followed by gene ontology (GO) analyses. In accordance with our in vitro findings from ECs, cellular growth and proliferation was predicted to be one of the top biological pathways to be regulated by miR-615–5p (Figure 3A). In addition, the AKT signaling pathway is predicted to be the top regulated signaling pathway by miR-615–5p in ECs (Figure 3B–C). Based on the GO analyses, we first verified that AKT phosphorylation was significantly reduced by ~2 fold in response to 15, 30, and 60 minutes of VEGF stimulation in HUVECs overexpressing miR-615–5p (Figure 4A), whereas miR-615–5p inhibition increased AKT phosphorylation by 40% under baseline conditions and by ~2 fold in response to 15, 30, and 60 mins of VEGF stimulation (Figure 4B). Because activation of eNOS in ECs is dependent upon AKT phosphorylation, we assessed for phosphorylation of eNOS at Ser 1177. Similarly, overexpression of miR-615–5p decreased eNOS phosphorylation by ~2 fold (Figure 4A), whereas miR-615–5p inhibition increased eNOS phosphorylation by 48% under basal conditions and by ~2 fold in response to VEGF stimulation (Figure 4B). This regulation was specific to AKT/eNOS signaling and not other signaling pathways including p38 and ERK1/2 (Figure 4 and Online Figure 2). Furthermore, miR-615–5p overexpression reduced nitric oxide (NO) release from HUVECs by 60% under baseline conditions and by 62% and 50% in response to 1 or 3 hrs, respectively, of VEGF treatment (Figure 4C).
Figure 3. Bioinformatics and miR-615–5p gene profiling predicts AKT as a targeted signaling pathway.

(A) Gene ontology analysis of 337 genes repressed by miR-615–5p overexpression in endothelial cells (ECs) identified from transcriptomic profiling. (B-C) AKT signaling pathway is predicted to be the top regulated signaling network regulated by miR-615–5p. GO, gene ontology.
Figure 4. MiR-615–5p regulates the expression of downstream AKT/eNOS signaling in ECs.

HUVECs transfected with (A) miR negative control (NSm) or miR-615–5p mimics (miR-615–5pm) or (B) miR inhibitor negative control (NSi) or miR-615–5p inhibitor (miR-615–5pi) were subjected to Western analysis using antibodies to p-AKT, p-eNOS, AKT, eNOS, p-p38, p38, p-ERK1/2, ERK1/2 and β-actin (n = 3 to 5 experiments). (C) NO release was measured by Griess assay. All data represent mean ± s.e.m. * P < 0.05 compared to controls.
To identify a direct target of miR-615–5p and narrow down the potential targets from the microarray gene chip profiling data, we took a rigorous, systematic approach using a combination of bioinformatics and prediction algorithms (e.g. miRWalk, Micro T4, miRNAMAP, RNAhybrid, and TargetScan) and validation by expression on the mRNA and protein levels (Figure 5A). From 337 genes repressed by 2-fold, 24 genes contained at least 1 potential binding site in the 3’-UTR and 11 genes showed significantly decreased mRNA expression in HUVECs overexpressing miR-615–5p (Figure 5A and data not shown). Of these, overexpression of miR-615–5p in HUVECs decreased the mRNA (Figure 5B) and protein expression (Figure 5C) and 3‘-UTR activity (Figure 5D) of only 2 genes – IGF2 and RASSF2. In contrast, inhibition of miR-615–5p increased IGF2 and RASSF2 expression (Figure 5B–C). Overexpression of miR-615–5p in ECs inhibited the activity of luciferase reporter constructs containing the IGF2 or RASSF2 3’-UTR by 72% and 30% respectively; in contrast, inhibition of miR-615–5p increased IGF2 and RASSF2 3’-UTR reporter activity by 2 and ~3.9 fold respectively (Figure 5D). In addition, deletion of the miR-615–5p binding sites on the IGF2 or RASSF2 3’UTRs blocked the miR-615–5p-mediated inhibition (Figure 5E and Online Figure 3). To further verify that miR-615–5p directly targets IGF2 and RASSF2 in ECs, we performed Argonaute2 (AGO2) microribonucleoprotein IP (miRNP-IP) studies to assess whether IGF2 and RASSF2 mRNA is enriched in the RNA-induced silencing complex following miR-615–5p overexpression in HUVECs. An approximately 7-fold enrichment of IGF2 and a 1.7-fold enrichment of RASSF2 mRNA were observed after AGO2 miRNP-IP in the presence of miR-615–5p, as compared with the miRNA-negative control. In contrast, AGO2 miRNP-IP did not enrich the mRNA for SMAD1, a gene that was not predicted to be a miR-615–5p target (Figure 5F). In addition, overexpression if miR-615–5p decreased the protein expression of IGF2 in mouse skeletal muscle microvascular ECs (Online Figure 4A). SiRNA silencing of RASSF2 (Figure 6A) and IGF2 (Figure 6D), ‘phenocopied’ the effects of miR-615–5p overexpression on AKT phosphorylation (Figure 6B, 6E), eNOS phosphorylation (Online Figure 4F–G), EC proliferation by BrdU assay (Online Figure 4B, 4C), and wound closure in scratch assays (Online Figure 4D, 4E). Furthermore, siRNA knockdown of both RASSF2 and IGF2 had a cooperative inhibitory effect on both AKT and eNOS phosphorylation (Online Figure 4F–H) and exacerbated endothelial proliferation in EC scratch assays compared to RASSF2 or IGF2 knockdown alone (Figure 6G). To explore whether the miR-615–5p-mediated inhibitory effects on EC proliferation were dependent on RASSF2 and IGF2, we performed siRNA knockdown studies using EC scratch assays and quantified EC wound closure. Functionally in the absence of RASSF2 (Figure 6C) or IGF2 (Figure 6F), miR-615–5p overexpression had markedly impaired ability to inhibit EC wound closure showing that the miR-615–5p-mediated effects are dependent in part on RASSF2 and IGF2. In addition, siRNA-mediated knockdown of AKT or eNOS blocked the inhibitory effect of miR-615 on EC proliferation (Online Figure 5A) and migration (Online Figure 5B). Collectively, these data indicate that IGF2 and RASSF2 are bona fide targets of miR-615–5p in ECs and raise the possibility that miR-615–5p may be a ‘molecular switch’ in which increased levels of miR-615–5p reduce IGF2 and RASSF2 expression, thereby suppressing endothelial cell growth and angiogenesis.
Figure 5. IGF2 and RASSF2 are bona fide targets of miR-615–5p in ECs.

(A) Discovery and validation of MiR-615–5p target genes. HUVECs transfected with miR negative control (NSm) and miR-615–5p mimics (miR-615–5pm) were subjected to microarray gene profiling. Potential gene targets were further narrowed down by sequential use of bioinformatics and prediction algorithms, RT-qPCR, Western blot analyses, 3’-UTR reporter studies, and microribonucleoprotein immunoprecipitation (miRNP-IP) analysis. (B-C) HUVECs transfected with NSm or miR-615–5pm were subjected RT-qPCR for IGF2 and RASSF2 expression (B) or Western blot analyses using antibodies to IGF2, RASSF2, and GAPDH (n = 3 experiments) (C). (D) Luciferase activity of IGF2 3’-untranslated region (UTR) and RASSF2 3’-UTR normalized to total protein was quantified in HUVECs transfected with NSm, miR-615–5pm, NSi, or miR-615–5pi (n = 3 experiments). (E) Luciferase activity of IGF2 or RASSF2 3’-UTRs bearing a deletion of the miR-615 binding site (miR-615 DEL) normalized to total protein was quantified in HUVECs transfected with NSm or miR-615m (n=3 experiments). (F) miRNP-IP analysis of enrichment of IGF2 and RASSF2 mRNA in HUVECs transfected with NSm or miR-615–5pm. *P < 0.01. RT-qPCR was performed to detect IGF2, RASSF2 or SMAD1. Results are representative of n = 3 replicates per group and 2 independent experiments. *P < 0.01. All data represent means ± s.e.m.
Figure 6. SiRNA-mediated knockdown of IGF2 and RASSF2 recapitulates miR-615–5p functional effects in ECs.

(A) HUVECs were transfected with siRNA to RASSF2 (A-C), IGF2 (D-F), IGF2 and RASSF2 (G) or scrambled control (ctrl) siRNA. Protein expression was determined by Western analysis under baseline conditions (A, D) or in response to VEGF treatment (B,E) using antibodies to RASSF2, IGF2, p-Akt, Akt and GAPDH (n = 2 experiments). *P < 0.01. (C,F,G) migration of ECs were quantified by scratch assay. *P < 0.01. Results are representative of n = 3 replicates per group. All data represent means ± s.e.m.
Diabetic wound healing represents a complex disease state associated with significant morbidity and mortality.18 Accumulating studies reveal that impaired angiogenesis is a hallmark in the pathogenesis of such wounds.55–58 On the basis that miR-615–5p inhibition promoted features of EC angiogenesis in vitro (Figure 1F–2), we explored the effect of inhibiting miR-615–5p on angiogenesis in a diabetic db/db model of dermal wound healing generated by punch biopsy of the skin on the dorsal suface of the mice (Figure 7A). Compared to scrambled non-specific control anti-miRs (NSi), local delivery of LNA-anti-miR-615–5p (MiR-615–5pi) not only significantly improved wound closure (Figure 7B), but also increased granulation tissue thickness (GTT) by 2.6-fold (Figure 7C) and robust induction of angiogenesis as measured by CD31 by 1.9-fold (Figure 7D). In contrast, overexpression of miR-615–5p not only significantly delayed wound closure by 68% (Figure 7E) and decreased granulation tissue thickness (GTT) (Figure 7F), but also reduced angiogenesis in wounds by 35% compared to mice that received local intradermal delivery of control LNA-anti-miRs (Figure 7G). In addition, while miR-615–5p neutralization had no effect on the expression of epithelial markers keratin 10 or 14 (Online Figure 6A, 6B) or accumulation of M1 or M2 macrophages in wounds (Online Figure 6C), there was a significantly increase in VE-cadherin and α-smooth muscle actin (α-SMA) expression (Online Figure 7A, 7B). Finally, using a mouse model of femoral artery ligation in diabetic db/db mice, miR-615–5p neutralization significantly improved blood flow recovery and skeletal muscle neovascularization after 15 days compared to controls (Online Figure 8). Thus, targeting miR-615–5p induced angiogenesis and wound healing under diabetic conditions.
Figure 7. Local delivery of LNA-anti-miR-615–5p promotes wound healing in db/db mice.

(A) After two local injections in mice of LNA-anti-miR-615–5p (MiR-615–5pi) or scrambled non-specific control LNA-anti-miRs (NSi) (n =11–12 per group), mice underwent dorsal skin wounding. (B-D) Wound analyses included: (B) wound closure areas (C) granulation tissue thickness (GTT) and (D) confocal immunofluorescence staining for CD31. (E-G) After two local injections in mice of miR-615–5p mimics (MiR-615–5pm) or scrambled non-specific control miRs (NSm) (n =10 per group), mice underwent dorsal skin wounding. (E-G) Wound analyses included: (E) wound closure areas (F) granulation tissue thickness (GTT) and (G) confocal immunofluorescence staining for CD31. Scale bars, 5mm (B,E), 500 μm (C,F) and 100 μm (D,G) All data represent means ± s.e.m. * P <0.05, ** P<0.001.
To evaluate whether neutralization of miR-615–5p may regulate angiogenesis from human tissues, we developed a modified human skin organoid assay in which a 6mm circular full-thickness punch biopsy is generated. In the middle of each biopsy a 3mm full-thickness wound is created using a 3mm punch biopsy which can be maintained at the air-liquid interface in culture for several days (Figure 8A).51 Human skin organoids can be transduced by microRNAs as we have shown previously using a NS-Cy3 control.52 To explore whether miR-615–5p also regulated angiogenesis in human skin organoids, we transduced human skin organoids with anti-miR-615–5p (MiR-615–5pi) or scrambled non-specific control antimiRs (NSi) or miR-615–5p mimics (MiR-615–5pm) or scrambled non-specific control antimiRs (NSm) (Figure 8A). Remarkably, inhibition of miR-615–5p induced angiogenesis as measured by CD31 by ~35% (Figure 8B), whereas overexpression of miR-615–5p reduced angiogenesis by ~1.8 fold (Figure 8C). Additionally, inhibition of miR-615–5p increased AKT phosphorylation by ~1.5 fold and ~1.2 fold, under basal conditions and in response to VEGF treatment, respectively (Figure 8D); in contrast, overexpression of miR-615–5p decreased AKT phosphorylation by 25% and 40%, under baseline and in response to VEGF treatment respectively (Figure 8E). Finally, we investigated the downstream effects of miR-615–5p “knockdown” or overexpression on RASSF2 or IGF2 in human skin organoids. In accordance with the in vitro findings in ECs, overexpression of miR-615–5p in human skin organoids significantly decreased RASSF2 mRNA (Online Figure 9A, left) and protein expression (Online Figure 9B, left), whereas the miR-615 inhibitor had the opposite effects (Online Figure 9A, 9B right). Similarly, overexpression of miR-615–5p in human skin organoids significantly decreased IGF2 mRNA (Online Figure 9C, left) and protein expression (Online Figure 9C, left), whereas the miR-615–5p inhibitor had the opposite effects (Online Figure 9C, 9D right). Collectively, these data indicate that increased miR-615–5p expression adversely affects angiogenesis in response to tissue injury, whereas its neutralization can potently promote angiogenesis in human tissue.
Figure 8. Inhibition of miR-615–5p promotes angiogenesis in human skin organoids.

(A) Punch biopsies of human skin were embedded into a collagen matrix, transfected with miR inhibitor negative control (NSi), miR-615–5p inhibitor (miR-615–5pi), miR negative control (NSm) or miR-615–5p mimics (miR-615–5pm), and cultured for indicated number of days. (B-C) Human skin organoids were transduced with the indicated miRNAs and cultured for 9 days followed by confocal immunofluorescence staining for CD31. (D-E) Human skin organoids (n=3–6) were transduced with the indicated miRNAs and cultured for 3 days with or without VEGF and followed by Western blot analyses for p-AKT, AKT, and β-actin at the indicated times (D-E). Scale bars, 50 μm (B-C). All data represent means ± s.e.m. * P <0.05.
DISCUSSION
Impaired neovascularization after tissue injury is a major hallmark in a range of conditions such as ischemic cardiovascular disease and diabetic wound healing, and can lead to maladaptive or delayed tissue recovery. The inability of ECs to perform their physiological functions (known as EC dysfunction) including EC proliferation, migration, lumen formation, and formation of new basement membranes is a major obstacle in therapeutic angiogenesis.5, 59–62 Over the past few decades, experimental treatment strategies have focused on addressing EC dysfunction and angiogenesis by delivering recombinant growth factors such as VEGF and a diverse set of molecules that can modestly or indirectly target angiogenic signaling pathways such as free radical scavengers63–65, receptors such as angiotensin receptor66 or angiopoetin 1 (Ang1)67, and many others68, 69 – all have shown limited efficacy with transitory effects.70 Indeed, an emerging concept in the field of therapeutic angiogenesis is the concept of “angiogenic resistance” supported by the observations that: 1) circulating angiogenic factors are actually increased (not deficient) in several studies of subjects with ischemic cardiovascular disease states22, 23; 2) human subjects with higher ischemic burden have higher circulating levels of pro-angiogenic growth factors24; and 3) experimental animal models demonstrate that despite sufficiency of pro-angiogenic growth factors (e.g. VEGF), there is markedly reduced activation of downstream signaling (e.g. AKT phosphorylation)26. These findings support the notion of impaired angiogenic signaling or “angiogenic resistance” in an analogous manner as how insulin resistance is associated with hyperinsulinemia with impaired insulin signaling/action in target tissues. Furthermore, these studies raise the possibility that defects in angiogenesis can be overcome by focusing on approaches to augment defects in downstream signaling of pro-angiogenic growth factors, rather than delivery of growth factors themselves.
MiRNAs are highly expressed in ECs. Emerging studies indicate that there is an imbalance of miRNAs in diverse disease states thereby making them attractive candidates for the development of therapeutic strategies.71 For example, we previously identified that the expression of miR-26a, an anti-angiogenic miR, was increased under acute myocardial infarction (MI) and in human subjects with ACS19 and in skin wounds of db/db mice, and that its neutralization could effectively promote angiogenesis by targeting Smad1, thereby promoting downstream effects of the bone morphogenic protein signaling pathway.18 Here, we identified a new miRNA, miR-615–5p, acting as a negative modulator of angiogenesis under pathological conditions by regulating the VEGF-AKT/eNOS signaling pathway through targeting the downstream genes RASSF2 and IGF2 (Schema, Online Figure 10). Treatment of ECs with pro-angiogenic stimuli such as VEGF or bFGF significantly reduced miR-615–5p expression while HDACIIa inhibitor MC-1568 blocked the VEGF mediated repression (Figure 1 and Online Figure 1). Local neutralization of miR-615–5p improved diabetic wound healing in mice and induced angiogenesis (Figure 7) without effecting keratinocyte growth or alteration of M1/M2 macrophage polarization (Online Figure 6). Systemic inhibition of miR-615–5p followed by femoral artery ligation increased blood flow recovery and promoted neoangiogenesis in diabetic mice (Online Figure 8). In human skin organoids, inhibition of miR-615–5p markedly increased AKT signaling and angiogenesis associated with increased VE-Cadherin and α-SMA expression for microvessel formation (Figure 8 and Online Figure 7). In contrast, miR-615–5p overexpression had the opposite effects in both diabetic wounds in mice and in human skin organoids. Our findings can potentially be utilized to develop new therapeutic approaches to address impaired neoangiogenesis observed in response to tissue injury such as diabetic wound healing or ischemic cardiovascular disease states.
The PI3/AKT signaling pathway is known to be involved in key EC functions such as cell growth, migration, vascular tone, survival and angiogenesis. Of the 2 isoforms of AKT, AKT1 phosphorylates eNOS and promotes NO release.72 Furthermore, genetic deletion of AKT1 under ischemic conditions resulted in impairment in ischemia-induced arteriogenesis and VEGF-induced post-natal angiogenesis.73–75 Endothelium-secreted NO not only plays an important role in the maintenance of vascular tone through activating smooth muscle relaxation, but is also known to drive EC angiogenic responses.76, 77 For example, vascular endothelium with decreased AKT/eNOS phosphorylation resulted in vascular dysfunction and reduced angiogenesis in mouse models of hindlimb ischemia and myocardial infarction.27–30 Our findings showing that miR-615–5p regulates EC growth and angiogenesis through the selective regulation of VEGF-induced AKT/eNOS pathway and not the p38 or pERK pathways (Figure 3, 4 and Online Figure 1) is consistent with the above paradigms. Moreover, siRNA-mediated knockdown of AKT or eNOS significantly attenuated the inhibitory effect of miR-615 on EC proliferation (Online Fig 4A) and migration (Online Figure 4B)., thereby highlighting miR-615–5p dependency on the AKT/eNOS signaling pathway.
We used an unbiased transcriptomic profiling approach in combination with bioinformatics to identify and validate miR-615–5p target genes. After filtering 337 genes repressed by at least 2-fold by miR-615–5p in transcriptomic profiling, only 2 target genes, IGF2 and RASSF2, were verified by expression, 3’-UTR reporter assays, miRNP-IP studies, and siRNA dependency (Figure 5). Interestingly, IGF2 was identified as a target of miR-615–5p in pancreatic ductal adenocarcinoma (PDAC) where overexpression of miR-615–5p decreased cell proliferation, migration, and cell invasion.78 Our findings demonstrate that miR-615–5p overexpression significantly decreased EC growth, migration, network tube formation in matrigel, and NO release (Figures 1–2) are consistent with the observed effects in this study. Moreover, our findings implicate that miR-615–5p is dependent in part on IGF2 for exerting downstream EC functional effects (Figure 6F). In addition, IGF2 deficiency is associated with reduced VEGF-induced activation of AKT in an analogous manner as miR-615–5p overexpression (Figure 6E), an effect consistent with this gene serving as a miR-615–5p target (Online Figure 6).
Our study also identified a previously unknown target of miR-615–5p, RASSF2. Interestingly, RASSF2 is known to function as a tumor suppressor inhibiting growth of lung cancer cells via its association with farnesylated K-RAS.79, 80 In addition, in 293T embryonic kidney cells, expression of RASSF2 resulted in growth inhibition enhanced by activated K-Ras.81 Our findings in ECs are distinct from the reported effects of RASSF2 in tumor cells and in 293T embryonic kidney cells. We and others have shown that these miRNA functional differences can be attributed to cell type-specific miRNA-mediated effects that are often dependent on the relative expression of the proteins targeted by the miRNA.19 In addition, differences in basal miRNA expression may be different in primary and transformed cells lines, resulting in distinct effects on downstream signaling events.82–84 Similar to IGF2, our findings demonstrate that miR-615–5p is dependent in part upon RASSF2 for exerting downstream EC functional effects (Figure 6C). RASSF2 deficiency is associated with reduced VEGF-induced activation of AKT in an analogous manner as miR-615–5p overexpression (Figure 6B), an effect consistent with this gene serving as a miR-615–5p target. In addition, combined RASSF2 and IGF2 siRNA knockdown further reduced p-AKT and p-eNOS expression in ECs (Online Figure 4G) and exacerbated endothelial proliferation in EC scratch assays compared to RASSF2 or IGF2 knockdown alone (Figure 6G), implicating potential cooperativity of these targets in mediating miR-615–5p downstream effects.
In addition to miR-615–5p, a handful of other miRNAs have been implicated in regulating the VEGF-AKT/eNOS signaling pathway. However, most studies have not identified direct miRNA target genes that may underlie these observations. For example, miR-126 is an endothelial-enriched miRNA that was found in human cardiac microvascular ECs to protect against hypoxia/reoxygenation-induced injury by activating the AKT/eNOS signaling pathway, albeit no miR-126 direct targets were identified that may account for these observations.85 Using retinal endothelial cells, miR-152 was found to repress VEGF by altering prorenin receptor expression, although effects on AKT/eNOS signaling were not assessed.86 Finally, miR-223 overexpression attenuated VEGF-induced EC proliferation and inhibited VEGF-induced phosphorylation of VEGFR2 and AKT activation by targeting β1 integrin. It will be of interest to determine whether miR-615–5p in combination with these or other miRNAs that activate VEGF-AKT/eNOS signaling can more effectively overcome “angiogenic resistance” as a basis for therapeutic intervention.
There are several limitations that we wish to acknowledge in this study. It is unclear whether miR-615–5p may serve as a biomarker of ischemic injury in other clinical contexts, or may be associated with specific baseline demographics including gender. Future studies will be required to prospectively assess the clinical utility of this miRNA as a biomarker or therapeutic. For the human skin organoid studies, it is possible that effects of miR-615–5p inhibitors may be more pronounced in specific patient subgroups or in skin derived from specific locations; however, these were anonymzed samples and future focused studies will be needed to clarify this point futher. For the human skin organoid studies, Finally, while we have demonstrated that miR-615–5p mediates EC angiogenic functional responses dependent on the 2 targets IGF2 and RASSF2, other targets may certainly be involved in response to divergent pathophysiological stimuli as we have observed for other microRNAs.45, 87, 88
In summary, we identified miR-615–5p as a novel negative regulator of angiogenesis by regulating the VEGF-AKT/eNOS pathway and direct targeting of IGF2 and RASSF2 genes. Inhibition of miR-615–5p improved wound healing and angiogenesis under diabetic conditions in addition to markedly increasing angiogenesis in human skin organoids. Because miR-615–5p is increased under conditions associated with impaired microvascular disease, therapies focusing on neutralization of miR-615–5p may be beneficial for promoting neovascularization and tissue repair.
Supplementary Material
Highlights.
MiR-615–5p expression is rapidly increased in response to diverse stimuli including in wounds of diabetic db/db mice, in plasma of human subjects with acute coronary syndromes, and in plasma and skin of human subjects with diabetes.
MiR-615–5p serves as an anti-angiogenic miRNA by targeting RASSF2 and IGF2 and subsequently endothelial cell VEGF-AKT/eNOS signaling.
Local miR-615–5p neutralization markedly increased angiogenesis, granulation tissue thickness, and wound closure rates in diabetic db/db mice, whereas local delivery of miR-615–5p mimics impaired these effects.
Systemic miR-615–5p neutralization improved skeletal muscle perfusion and angiogenesis after hindlimb ischemia in db/db mice.
Gain and loss-of-function studies of miR-615–5p in human skin organoids demonstrated dynamic regulation of VEGF-induced AKT signaling and angiogenesis.
ACKNOWLEDGMENTS
We thank Lay-Hong Ang and Aniket Gad for Confocal Microscopy technical assistance (NIH P30DK034854).
Sources of Funding
This work was supported by the National Institutes of Health (HL115141, HL117994, HL134849, and GM115605 to M.W.F.), the Arthur K. Watson Charitable Trust (to M.W.F.), the Dr. Ralph & Marian Falk Medical Research Trust, Bank of America, N.A. Trustee (to M.W.F.), an American Heart Association grant 18SFRN33900144 (to M.W.F.), an American Diabetes Association grant #1–16-JDF-046 (to BI), a Watkins Discovery Award (to BI), a Lerner Young Investigator Award (to BI), a TUBITAK Predoctoral Scholarship (to D.Oz) and a grant from South-Eastern Regional Health Authorities and from Norwegian Womeńs Public Health Association, Norway (to IH).
Non-standard Abbreviations and Acronyms
- ACS
Acute Coronary Syndrome
- DM
Diabetes Mellitus
- EC
Endothelial cell
- eNOS
endothelial nitric oxide synthase
- HUVECs
human umbilical vein ECs
- IGF2
Insulin-like Growth factor 2
- MI
myocardial infarction
- miRNA
microRNA
- NO
nitric oxide
- NS
nonspecific
- RASSF2
Ras Associating Domain Family Member 2
- RT-PCR
Real-time polymerase chain reaction
- siRNA
small interfering RNA
- 3’-UTR
3’-untranslated region
- VEGF
vascular endothelial growth factor
Footnotes
Disclosures
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Blakytny R, Jude E. The molecular biology of chronic wounds and delayed healing in diabetes. Diabet Med. 2006;23:594–608 [DOI] [PubMed] [Google Scholar]
- 2.Falanga V Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–1743 [DOI] [PubMed] [Google Scholar]
- 3.Krishna SM, Moxon JV, Golledge J. A review of the pathophysiology and potential biomarkers for peripheral artery disease. Int J Mol Sci. 2015;16:11294–11322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: From inflammation to fibrosis. Circ Res. 2016;119:91–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–887 [DOI] [PubMed] [Google Scholar]
- 6.Folkman J Seminars in medicine of the beth israel hospital, boston. Clinical applications of research on angiogenesis. N Engl J Med. 1995;333:1757–1763 [DOI] [PubMed] [Google Scholar]
- 7.Folkman J Therapeutic angiogenesis in ischemic limbs. Circulation. 1998;97:1108–1110 [DOI] [PubMed] [Google Scholar]
- 8.Belch J, Hiatt WR, Baumgartner I, Driver IV, Nikol S, Norgren L, Van Belle E, Committees T, Investigators. Effect of fibroblast growth factor nv1fgf on amputation and death: A randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet. 2011;377:1929–1937 [DOI] [PubMed] [Google Scholar]
- 9.Kusumanto YH, van Weel V, Mulder NH, Smit AJ, van den Dungen JJ, Hooymans JM, Sluiter WJ, Tio RA, Quax PH, Gans RO, Dullaart RP, Hospers GA. Treatment with intramuscular vascular endothelial growth factor gene compared with placebo for patients with diabetes mellitus and critical limb ischemia: A double-blind randomized trial. Hum Gene Ther. 2006;17:683–691 [DOI] [PubMed] [Google Scholar]
- 10.Powell RJ, Simons M, Mendelsohn FO, Daniel G, Henry TD, Koga M, Morishita R, Annex BH. Results of a double-blind, placebo-controlled study to assess the safety of intramuscular injection of hepatocyte growth factor plasmid to improve limb perfusion in patients with critical limb ischemia. Circulation. 2008;118:58–65 [DOI] [PubMed] [Google Scholar]
- 11.Rajagopalan S, Mohler ER 3rd, Lederman RJ, Mendelsohn FO, Saucedo JF, Goldman CK, Blebea J, Macko J, Kessler PD, Rasmussen HS, Annex BH. Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: A phase ii randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation. 2003;108:1933–1938 [DOI] [PubMed] [Google Scholar]
- 12.Grossman PM, Mendelsohn F, Henry TD, Hermiller JB, Litt M, Saucedo JF, Weiss RJ, Kandzari DE, Kleiman N, Anderson RD, Gottlieb D, Karlsberg R, Snell J, Rocha-Singh K. Results from a phase ii multicenter, double-blind placebo-controlled study of del-1 (vlts-589) for intermittent claudication in subjects with peripheral arterial disease. Am Heart J. 2007;153:874–880 [DOI] [PubMed] [Google Scholar]
- 13.Creager MA, Olin JW, Belch JJ, Moneta GL, Henry TD, Rajagopalan S, Annex BH, Hiatt WR. Effect of hypoxia-inducible factor-1alpha gene therapy on walking performance in patients with intermittent claudication. Circulation. 2011;124:1765–1773 [DOI] [PubMed] [Google Scholar]
- 14.Jones WS, Annex BH. Growth factors for therapeutic angiogenesis in peripheral arterial disease. Curr Opin Cardiol. 2007;22:458–463 [DOI] [PubMed] [Google Scholar]
- 15.Ouma GO, Jonas RA, Usman MH, Mohler ER 3rd. Targets and delivery methods for therapeutic angiogenesis in peripheral artery disease. Vasc Med. 2012;17:174–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cooke JP, Losordo DW. Modulating the vascular response to limb ischemia: Angiogenic and cell therapies. Circ Res. 2015;116:1561–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Iyer SR, Annex BH. Therapeutic angiogenesis for peripheral artery disease: Lessons learned in translational science. JACC Basic Transl Sci. 2017;2:503–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Icli B, Nabzdyk CS, Lujan-Hernandez J, Cahill M, Auster ME, Wara AK, Sun X, Ozdemir D, Giatsidis G, Orgill DP, Feinberg MW. Regulation of impaired angiogenesis in diabetic dermal wound healing by microrna-26a. J Mol Cell Cardiol. 2016;91:151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Icli B, Wara AK, Moslehi J, Sun X, Plovie E, Cahill M, Marchini JF, Schissler A, Padera RF, Shi J, Cheng HW, Raghuram S, Arany Z, Liao R, Croce K, MacRae C, Feinberg MW. Microrna-26a regulates pathological and physiological angiogenesis by targeting bmp/smad1 signaling. Circ Res. 2013;113:1231–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin Y, Tymen SD, Chen D, Fang ZJ, Zhao Y, Dragas D, Dai Y, Marucha PT, Zhou X. Microrna-99 family targets akt/mtor signaling pathway in dermal wound healing. PLoS One. 2013;8:e64434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Annex BH, Beller GA. Towards the development of novel therapeutics for peripheral artery disease. Trans Am Clin Climatol Assoc. 2016;127:224–234 [PMC free article] [PubMed] [Google Scholar]
- 22.Hojo Y, Ikeda U, Zhu Y, Okada M, Ueno S, Arakawa H, Fujikawa H, Katsuki T, Shimada K. Expression of vascular endothelial growth factor in patients with acute myocardial infarction. J Am Coll Cardiol. 2000;35:968–973 [DOI] [PubMed] [Google Scholar]
- 23.Nakajima K, Tabata S, Yamashita T, Kusuhara M, Arakawa K, Ohmori R, Yonemura A, Higashi K, Ayaori M, Nakamura H, Ohsuzu F. Plasma vascular endothelial growth factor level is elevated in patients with multivessel coronary artery disease. Clin Cardiol. 2004;27:281–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Findley CM, Mitchell RG, Duscha BD, Annex BH, Kontos CD. Plasma levels of soluble tie2 and vascular endothelial growth factor distinguish critical limb ischemia from intermittent claudication in patients with peripheral arterial disease. J Am Coll Cardiol. 2008;52:387–393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Abhinand CS, Raju R, Soumya SJ, Arya PS, Sudhakaran PR. Vegf-a/vegfr2 signaling network in endothelial cells relevant to angiogenesis. J Cell Commun Signal. 2016;10:347–354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hazarika S, Dokun AO, Li Y, Popel AS, Kontos CD, Annex BH. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: Differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ Res. 2007;101:948–956 [DOI] [PubMed] [Google Scholar]
- 27.Bell RM, Yellon DM. Bradykinin limits infarction when administered as an adjunct to reperfusion in mouse heart: The role of pi3k, akt and enos. J Mol Cell Cardiol. 2003;35:185–193 [DOI] [PubMed] [Google Scholar]
- 28.Gao F, Gao E, Yue TL, Ohlstein EH, Lopez BL, Christopher TA, Ma XL. Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: The roles of pi3-kinase, akt, and endothelial nitric oxide synthase phosphorylation. Circulation. 2002;105:1497–1502 [DOI] [PubMed] [Google Scholar]
- 29.Lee MY, Gamez-Mendez A, Zhang J, Zhuang Z, Vinyard DJ, Kraehling J, Velazquez H, Brudvig GW, Kyriakides TR, Simons M, Sessa WC. Endothelial cell autonomous role of akt1: Regulation of vascular tone and ischemia-induced arteriogenesis. Arterioscler Thromb Vasc Biol. 2018;38:870–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu ZY, Li RL, Zhou HS, Huang JJ, Su ZX, Qi J, Zhang L, Li Y, Shi YQ, Hao CN, Duan JL. Therapeutic ultrasound reverses peripheral ischemia in type 2 diabetic mice through pi3k-akt-enos pathway. Am J Transl Res. 2016;8:3666–3677 [PMC free article] [PubMed] [Google Scholar]
- 31.Bonauer A, Boon RA, Dimmeler S. Vascular micrornas. Curr Drug Targets. 2010;11:943–949 [DOI] [PubMed] [Google Scholar]
- 32.Sayed D, Abdellatif M. Micrornas in development and disease. Physiol Rev. 2011;91:827–887 [DOI] [PubMed] [Google Scholar]
- 33.Araldi E, Chamorro-Jorganes A, van Solingen C, Fernandez-Hernando C, Suarez Y. Therapeutic potential of modulating micrornas in atherosclerotic vascular disease. Curr Vasc Pharmacol. 2013 [PMC free article] [PubMed] [Google Scholar]
- 34.Okonkwo UA, DiPietro LA. Diabetes and wound angiogenesis. Int J Mol Sci. 2017;18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Welten SM, Goossens EA, Quax PH, Nossent AY. The multifactorial nature of micrornas in vascular remodelling. Cardiovasc Res. 2016;110:6–22 [DOI] [PubMed] [Google Scholar]
- 36.Icli B, Feinberg MW. Micrornas in dysfunctional adipose tissue: Cardiovascular implications. Cardiovasc Res. 2017;113:1024–1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang Y, Sun X, Icli B, Feinberg MW. Emerging roles for micrornas in diabetic microvascular disease: Novel targets for therapy. Endocr Rev. 2017;2017:1–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ozdemir D, Feinberg MW. Micrornas in diabetic wound healing: Pathophysiology and therapeutic opportunities. Trends Cardiovasc Med. 2019;29:131–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caporali A, Meloni M, Vollenkle C, Bonci D, Sala-Newby GB, Addis R, Spinetti G, Losa S, Masson R, Baker AH, Agami R, le Sage C, Condorelli G, Madeddu P, Martelli F, Emanueli C. Deregulation of microrna-503 contributes to diabetes mellitus-induced impairment of endothelial function and reparative angiogenesis after limb ischemia. Circulation. 2011;123:282–291 [DOI] [PubMed] [Google Scholar]
- 40.Chan YC, Roy S, Khanna S, Sen CK. Downregulation of endothelial microrna-200b supports cutaneous wound angiogenesis by desilencing gata binding protein 2 and vascular endothelial growth factor receptor 2. Arterioscler Thromb Vasc Biol. 2012;32:1372–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang S, Aurora AB, Johnson BA, Qi X, McAnally J, Hill JA, Richardson JA, Bassel-Duby R, Olson EN. The endothelial-specific microrna mir-126 governs vascular integrity and angiogenesis. Dev Cell. 2008;15:261–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Melero-Martin JM, Bischoff J. Chapter 13. An in vivo experimental model for postnatal vasculogenesis. Methods Enzymol. 2008;445:303–329 [DOI] [PubMed] [Google Scholar]
- 43.Wara AK, Croce K, Foo S, Sun X, Icli B, Tesmenitsky Y, Esen F, Rosenzweig A, Feinberg MW. Bone marrow-derived cmps and gmps represent highly functional proangiogenic cells: Implications for ischemic cardiovascular disease. Blood. 2011;118:6461–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wara AK, Foo S, Croce K, Sun X, Icli B, Tesmenitsky Y, Esen F, Lee JS, Subramaniam M, Spelsberg TC, Lev EI, Leshem-Lev D, Pande RL, Creager MA, Rosenzweig A, Feinberg MW. Tgf-beta1 signaling and kruppel-like factor 10 regulate bone marrow-derived proangiogenic cell differentiation, function, and neovascularization. Blood. 2011;118:6450–6460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sun X, Icli B, Wara AK, Belkin N, He S, Kobzik L, Hunninghake GM, Vera MP, Registry M, Blackwell TS, Baron RM, Feinberg MW. Microrna-181b regulates nf-kappab-mediated vascular inflammation. J Clin Invest. 2012;122:1973–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Elsafadi M, Manikandan M, Alajez NM, Hamam R, Dawud RA, Aldahmash A, Iqbal Z, Alfayez M, Kassem M, Mahmood A. Microrna-4739 regulates osteogenic and adipocytic differentiation of immortalized human bone marrow stromal cells via targeting lrp3. Stem Cell Res. 2017;20:94–104 [DOI] [PubMed] [Google Scholar]
- 47.Wang C, Saji M, Justiniano SE, Yusof AM, Zhang X, Yu L, Fernandez S, Wakely P Jr., La Perle K, Nakanishi H, Pohlman N, Ringel MD. Rcan1–4 is a thyroid cancer growth and metastasis suppressor. JCI Insight. 2017;2:e90651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang Y, Wang SJ, Han ZH, Li YQ, Xue JH, Gao DF, Wu XS, Wang CX. Pi3k/akt signaling pathway plays a role in enhancement of enos activity by recombinant human angiotensin converting enzyme 2 in human umbilical vein endothelial cells. Int J Clin Exp Pathol. 2014;7:8112–8117 [PMC free article] [PubMed] [Google Scholar]
- 49.Wang L, Wu B, Sun Y, Xu T, Zhang X, Zhou M, Jiang W. Translocation of protein kinase c isoforms is involved in propofol-induced endothelial nitric oxide synthase activation. Br J Anaesth. 2010;104:606–612 [DOI] [PubMed] [Google Scholar]
- 50.Versari S, Villa A, Bradamante S, Maier JA. Alterations of the actin cytoskeleton and increased nitric oxide synthesis are common features in human primary endothelial cell response to changes in gravity. Biochim Biophys Acta. 2007;1773:1645–1652 [DOI] [PubMed] [Google Scholar]
- 51.Balaji S, Moles CM, Bhattacharya SS, LeSaint M, Dhamija Y, Le LD, King A, Kidd M, Bouso MF, Shaaban A, Crombleholme TM, Bollyky P, Keswani SG. Comparison of interleukin 10 homologs on dermal wound healing using a novel human skin ex vivo organ culture model. J Surg Res. 2014;190:358–366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Icli B, Wu W, Ozdemir D, Li H, Haemmig S, Liu X, Giatsidis G, Cheng HS, Avci SN, Kurt M, Lee N, Guimaraes RB, Manica A, Marchini JF, Rynning SE, Risnes I, Hollan I, Croce K, Orgill DP, Feinberg MW. Microrna-135a-3p regulates angiogenesis and tissue repair by targeting p38 signaling in endothelial cells. FASEB J. 2019:fj201802063RR [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sacilotto N, Chouliaras KM, Nikitenko LL, Lu YW, Fritzsche M, Wallace MD, Nornes S, Garcia-Moreno F, Payne S, Bridges E, Liu K, Biggs D, Ratnayaka I, Herbert SP, Molnar Z, Harris AL, Davies B, Bond GL, Bou-Gharios G, Schwarz JJ, De Val S. Mef2 transcription factors are key regulators of sprouting angiogenesis. Genes Dev. 2016;30:2297–2309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nebbioso A, Dell’Aversana C, Bugge A, Sarno R, Valente S, Rotili D, Manzo F, Teti D, Mandrup S, Ciana P, Maggi A, Mai A, Gronemeyer H, Altucci L. Hdacs class ii-selective inhibition alters nuclear receptor-dependent differentiation. J Mol Endocrinol. 2010;45:219–228 [DOI] [PubMed] [Google Scholar]
- 55.Erba P, Ogawa R, Ackermann M, Adini A, Miele LF, Dastouri P, Helm D, Mentzer SJ, D’Amato RJ, Murphy GF, Konerding MA, Orgill DP. Angiogenesis in wounds treated by microdeformational wound therapy. Ann Surg. 2011;253:402–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Greene AK, Puder M, Roy R, Arsenault D, Kwei S, Moses MA, Orgill DP. Microdeformational wound therapy: Effects on angiogenesis and matrix metalloproteinases in chronic wounds of 3 debilitated patients. Ann Plast Surg. 2006;56:418–422 [DOI] [PubMed] [Google Scholar]
- 57.Peng C, Chen B, Kao HK, Murphy G, Orgill DP, Guo L. Lack of fgf-7 further delays cutaneous wound healing in diabetic mice. Plast Reconstr Surg. 2011;128:673e–684e [DOI] [PubMed] [Google Scholar]
- 58.Song G, Nguyen DT, Pietramaggiori G, Scherer S, Chen B, Zhan Q, Ogawa R, Yannas IV, Wagers AJ, Orgill DP, Murphy GF. Use of the parabiotic model in studies of cutaneous wound healing to define the participation of circulating cells. Wound Repair Regen. 2010;18:426–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sun L, Bai Y, Du G. Endothelial dysfunction--an obstacle of therapeutic angiogenesis. Ageing Res Rev. 2009;8:306–313 [DOI] [PubMed] [Google Scholar]
- 60.Goligorsky MS. Endothelial cell dysfunction: Can’t live with it, how to live without it. Am J Physiol Renal Physiol. 2005;288:F871–880 [DOI] [PubMed] [Google Scholar]
- 61.Avogaro A, Albiero M, Menegazzo L, de Kreutzenberg S, Fadini GP. Endothelial dysfunction in diabetes: The role of reparatory mechanisms. Diabetes Care. 2011;34 Suppl 2:S285–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goveia J, Stapor P, Carmeliet P. Principles of targeting endothelial cell metabolism to treat angiogenesis and endothelial cell dysfunction in disease. EMBO Mol Med. 2014;6:1105–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: Results of the hope study and micro-hope substudy. Heart outcomes prevention evaluation study investigators. Lancet. 2000;355:253–259 [PubMed] [Google Scholar]
- 64.Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin e after myocardial infarction: Results of the gissi-prevenzione trial. Gruppo italiano per lo studio della sopravvivenza nell’infarto miocardico. Lancet. 1999;354:447–455 [PubMed] [Google Scholar]
- 65.Heart Protection Study Collaborative G. Mrc/bhf heart protection study of antioxidant vitamin supplementation in 20,536 high-risk individuals: A randomised placebo-controlled trial. Lancet. 2002;360:23–33 [DOI] [PubMed] [Google Scholar]
- 66.Schmieder RE, Delles C, Mimran A, Fauvel JP, Ruilope LM. Impact of telmisartan versus ramipril on renal endothelial function in patients with hypertension and type 2 diabetes. Diabetes Care. 2007;30:1351–1356 [DOI] [PubMed] [Google Scholar]
- 67.Koh YJ, Kim HZ, Hwang SI, Lee JE, Oh N, Jung K, Kim M, Kim KE, Kim H, Lim NK, Jeon CJ, Lee GM, Jeon BH, Nam DH, Sung HK, Nagy A, Yoo OJ, Koh GY. Double antiangiogenic protein, daap, targeting vegf-a and angiopoietins in tumor angiogenesis, metastasis, and vascular leakage. Cancer Cell. 2010;18:171–184 [DOI] [PubMed] [Google Scholar]
- 68.Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Welti J, Loges S, Dimmeler S, Carmeliet P. Recent molecular discoveries in angiogenesis and antiangiogenic therapies in cancer. J Clin Invest. 2013;123:3190–3200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lee R, Channon KM, Antoniades C. Therapeutic strategies targeting endothelial function in humans: Clinical implications. Curr Vasc Pharmacol. 2012;10:77–93 [DOI] [PubMed] [Google Scholar]
- 71.Landskroner-Eiger S, Moneke I, Sessa WC. Mirnas as modulators of angiogenesis. Cold Spring Harb Perspect Med. 2013;3:a006643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by akt-dependent phosphorylation. Nature. 1999;399:601–605 [DOI] [PubMed] [Google Scholar]
- 73.Lee MY, Luciano AK, Ackah E, Rodriguez-Vita J, Bancroft TA, Eichmann A, Simons M, Kyriakides TR, Morales-Ruiz M, Sessa WC. Endothelial akt1 mediates angiogenesis by phosphorylating multiple angiogenic substrates. Proc Natl Acad Sci U S A. 2014;111:12865–12870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ackah E, Yu J, Zoellner S, Iwakiri Y, Skurk C, Shibata R, Ouchi N, Easton RM, Galasso G, Birnbaum MJ, Walsh K, Sessa WC. Akt1/protein kinase balpha is critical for ischemic and vegf-mediated angiogenesis. J Clin Invest. 2005;115:2119–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ju R, Zhuang ZW, Zhang J, Lanahan AA, Kyriakides T, Sessa WC, Simons M. Angiopoietin-2 secretion by endothelial cell exosomes: Regulation by the phosphatidylinositol 3-kinase (pi3k)/akt/endothelial nitric oxide synthase (enos) and syndecan-4/syntenin pathways. J Biol Chem. 2014;289:510–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820 [DOI] [PubMed] [Google Scholar]
- 77.Bir SC, Xiong Y, Kevil CG, Luo J. Emerging role of pka/enos pathway in therapeutic angiogenesis for ischaemic tissue diseases. Cardiovasc Res. 2012;95:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gao W, Gu Y, Li Z, Cai H, Peng Q, Tu M, Kondo Y, Shinjo K, Zhu Y, Zhang J, Sekido Y, Han B, Qian Z, Miao Y. Mir-615–5p is epigenetically inactivated and functions as a tumor suppressor in pancreatic ductal adenocarcinoma. Oncogene. 2015;34:1629–1640 [DOI] [PubMed] [Google Scholar]
- 79.Akino K, Toyota M, Suzuki H, Mita H, Sasaki Y, Ohe-Toyota M, Issa JP, Hinoda Y, Imai K, Tokino T. The ras effector rassf2 is a novel tumor-suppressor gene in human colorectal cancer. Gastroenterology. 2005;129:156–169 [DOI] [PubMed] [Google Scholar]
- 80.Zhang Z, Sun D, Van do N, Tang A, Hu L, Huang G. Inactivation of rassf2a by promoter methylation correlates with lymph node metastasis in nasopharyngeal carcinoma. Int J Cancer. 2007;120:32–38 [DOI] [PubMed] [Google Scholar]
- 81.Vos MD, Ellis CA, Elam C, Ulku AS, Taylor BJ, Clark GJ. Rassf2 is a novel k-ras-specific effector and potential tumor suppressor. J Biol Chem. 2003;278:28045–28051 [DOI] [PubMed] [Google Scholar]
- 82.Johnson SM, Grosshans H, Shingara J, Byrom M, Jarvis R, Cheng A, Labourier E, Reinert KL, Brown D, Slack FJ. Ras is regulated by the let-7 microrna family. Cell. 2005;120:635–647 [DOI] [PubMed] [Google Scholar]
- 83.Mollainezhad H, Eskandari N, Pourazar A, Salehi M, Andalib A. Expression of microrna-370 in human breast cancer compare with normal samples. Adv Biomed Res. 2016;5:129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iorio MV, Ferracin M, Liu CG, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Menard S, Palazzo JP, Rosenberg A, Musiani P, Volinia S, Nenci I, Calin GA, Querzoli P, Negrini M, Croce CM. Microrna gene expression deregulation in human breast cancer. Cancer Res. 2005;65:7065–7070 [DOI] [PubMed] [Google Scholar]
- 85.Yang HH, Chen Y, Gao CY, Cui ZT, Yao JM. Protective effects of microrna-126 on human cardiac microvascular endothelial cells against hypoxia/reoxygenation-induced injury and inflammatory response by activating pi3k/akt/enos signaling pathway. Cell Physiol Biochem. 2017;42:506–518 [DOI] [PubMed] [Google Scholar]
- 86.Haque R, Hur EH, Farrell AN, Iuvone PM, Howell JC. Microrna-152 represses vegf and tgfbeta1 expressions through post-transcriptional inhibition of (pro)renin receptor in human retinal endothelial cells. Mol Vis. 2015;21:224–235 [PMC free article] [PubMed] [Google Scholar]
- 87.Sun X, He S, Wara AKM, Icli B, Shvartz E, Tesmenitsky Y, Belkin N, Li D, Blackwell TS, Sukhova GK, Croce K, Feinberg MW. Systemic delivery of microrna-181b inhibits nuclear factor-kappab activation, vascular inflammation, and atherosclerosis in apolipoprotein e-deficient mice. Circ Res. 2014;114:32–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lin J, He S, Sun X, Franck G, Deng Y, Yang D, Haemmig S, Wara AK, Icli B, Li D, Feinberg MW. Microrna-181b inhibits thrombin-mediated endothelial activation and arterial thrombosis by targeting caspase recruitment domain family member 10. FASEB J. 2016;30:3216–3226 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.