

Abstract
Alcohol oxidation reactions are widely used for the preparation of aldehydes and ketones. The electrolysis of alcohols to carbonyl compounds have been underutilized owing to low efficiency. Herein, we report an electrochemical oxidation of various alcohols in a continuous-flow reactor without external oxidants, base or mediators. The robust electrochemical oxidation is performed for a variety of alcohols with good functional group tolerance, high efficiency and atom economy, whereas mechanistic studies support the benzylic radical intermediate formation and hydrogen evolution. The electrochemical oxidation proves viable on diols with excellent levels of selectivity for the benzylic position.
Subject terms: Flow chemistry, Electrocatalysis, Sustainability, Synthetic chemistry methodology
Alcohol oxidation to carbonyl compounds is a very useful functional group transformation in organic synthesis. Here, the authors perform the direct electrochemical oxidation of various alcohols to the corresponding ketones in a continuous-flow reactor without external oxidants, base or mediators.
Introduction
Aldehydes or ketones are not only essential functionalities of various biologically active compounds, but also important reagents in modern organic synthesis1,2. In fact, the high frequently used of alcohol oxidations to carbonyl compounds leading to a ranking of 3rd of strongly prefer better reagents for pharmaceutical manufacturers3. So, developing oxidation of alcohols under environmentally benign and economic conditions is highly demanded4–7 for pharmaceutical and chemical industries. Stoichiometric oxidants such as chromium, manganese, and ruthenium salts8–15 are relatively not environmental friendly (Fig. 1a). Molecular oxygen, air, or hydrogen peroxide in combination with appropriate transition metal catalysts (Cu, Ru, Pd, Au, Fe, V, or Ir)16–22 represent superior alternatives according to the principles of green and sustainable chemistry. However, the oxidation remains challenging for a broad group of alcohols. In contrast, oxidant-free alcohol oxidation with hydrogen evolution is apparently an ideal process to reach the higher atom economy (Fig. 1b). This concept has been achieved with transition metal catalysis23–30, which normally involves expensive transition metals, sophisticated ligands, and generally requires high reaction temperatures. On the other hand, alcohol oxidation with hydrogen release via photocatalysis31,32 or electrocatalysis33–40 have the merit of running reactions under mild conditions.
Fig. 1.
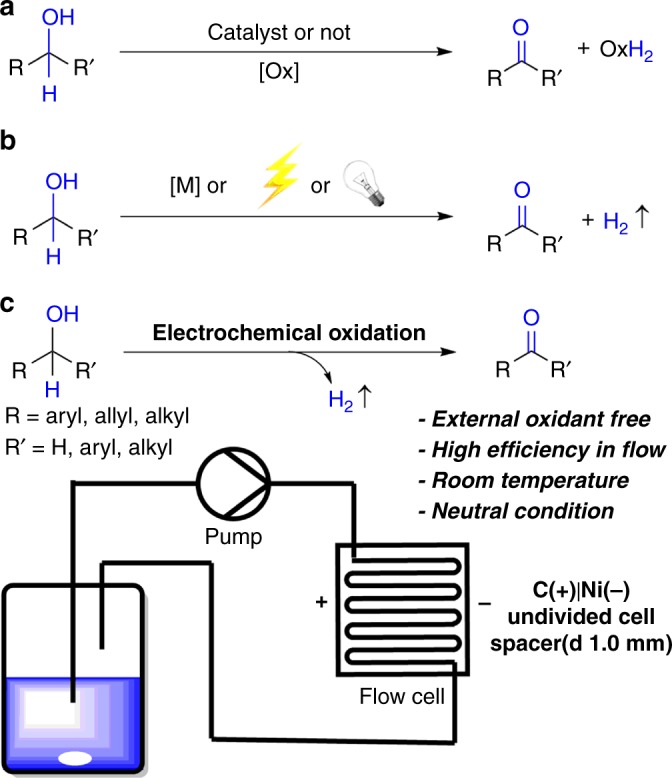
Alcohol oxidation conditions. a Alcohols oxidation with oxidant. b Alcohols oxidation with dehydrogenation. c Alcohol oxidation under continuous-flow set-up
Direct electrolysis of alcohols to carbonyl compounds is an idealized goal, which produces only hydrogen without product contamination. The direct electro-oxidation of alcohol can be tracked back to the pioneering work of Lund and Mayeda et al.41–43 using undivided electrolysis cells. However, prior attempts gave only the desired aldehydes or ketones in low selectivity and efficiency. We envisioned that electrolysis in a continuous-flow set-up would be a sustainable method to replace stoichiometric oxidants and improve both energy efficiency and productivity. Large surface-to-volume ratio, enhanced mass transfer, operation at quasi-isothermal condition, and lower resistances are the advantages of using flow electrochemistry set-up, which often make it superior than batch mode44–46. In addition, the electrode surface can be regenerated efficiently to avoid low conductivity for the transformations with gas evolution. Furthermore, flow cells are easy to scale up and employed in industry47–49. Recently, electrochemical flow cells have been successfully used in a variety of organic transformations50–64. Herein, we report the direct electrolysis of alcohols to afford the aldehydes or ketones with high efficiency without any mediators or catalysts under neutral conditions by using a flow reactor (Fig. 1c and Supplementary Fig. 1).
Results
Investigation of reaction conditions
Our studies commenced with benzyl alcohol (1aa), which was subjected to a batch condition in an undivided cell (Table 1, entry 1), yielding 37% of benzaldehyde (2aa) at 100 mA constant current electrolysis for 1 h (Supplementary Fig. 2). In order to improve the solubility of the starting material, CH3CN and water (1:1) were used as solvents in the continuous-flow set-up, and benzaldehyde (2aa) was isolated in 93% yield under the same current (10 mA, 10 h, Table 1, entry 2). Encouraged by these results, we investigated the magnitude of current from 10 mA to 1000 mA (entries 2–8). When 50 mA was applied for the oxidation, 92% of benzaldehyde was isolated in 2 h (Table 1, entry 3). Among a set of studies, 800 mA enabled effective oxidation up to 99% yield in 10 min (Table 1, entry 7). However, when the current was increased to 1000 mA, 2aa was provided in lower yield (81%, entry 8). The flow rate was another parameter, which was evaluated at the current of 800 mA. Accordingly, 0.10 mL s−1 was the optimum flow rate (Table 1, entries 9–11). Then the reaction conditions of 800 mA, 10 min and 0.10 mL s−1 were employed for the further researches.
Table 1.
Optimization of electrochemical oxidation of benzyl alcohol 1aa
|
| |||||
|---|---|---|---|---|---|
| Entry | Current | Reaction time | Current density | Flow rate | Yield (%)a |
| 1b | 100 mA | 1 h | 44.44 mA cm−2 | Undivided cell | 37 |
| 2 | 10 mA | 10 h | 0.64 mA cm−2 | 0.10 mL s−1 | 93 |
| 3 | 50 mA | 2 h | 3.19 mA cm−2 | 0.10 mL s−1 | 92 |
| 4 | 100 mA | 1 h | 6.38 mA cm−2 | 0.10 mL s−1 | 89 |
| 5 | 500 mA | 12 min | 31.89 mA cm−2 | 0.10 mL s−1 | 90 |
| 6 | 800 mA | 8 min | 51.02 mA cm−2 | 0.10 mL s−1 | 91 |
| 7 | 1000 mA | 6 min | 63.78 mA cm−2 | 0.10 mL s−1 | 81 |
| 8 | 800 mA | 10 min | 51.02 mA cm−2 | 0.10 mL s−1 | 99 |
| 9 | 800 mA | 10 min | 51.02 mA cm−2 | 0.05 mL s−1 | 67 |
| 10 | 800 mA | 10 min | 51.02 mA cm−2 | 0.15 mL s−1 | 95 |
| 11 | 800 mA | 10 min | 51.02 mA cm−2 | 0.20 mL s−1 | 84 |
Reaction conditions: carbon paper (93 × 93 × 0.2 mm) anode (contact area 1.6 cm2), Ni plate (93 × 93 × 0.3 mm) cathode (contact area 1.6 cm2), 1aa (2.0 mmol), nBu4NBF4 (0.20 mmol), CH3CN/H2O (1:1, 30 mL), N2, room temperature, flow cell (2.49 F mol−1)
aIsolated yield
bCarbon cloth (15 × 15 × 0.2 mm) anode, Ni plate (15 × 15 × 0.5 ) cathode, 1aa (2.0 mmol), nBu4NBF4 (0.20 mmol), CH3CN/H2O (1:1, 30 mL), N2, room temperature, undivided cell (1.86 F mol−1)
At the flow rate of 0.10 mL s−1, the solution passed through the flow cell multiple times by using peristaltic pump. We think that it would be possible to achieve full conversion by employing large current and small flow rate. By using this strategy, the 2 mmol benzyl alcohol in 30 mL mixed solvent could be quantitatively transformed to benzyaldehyde at 800 mA and 0.02 mL s−1 in 25 min (Fig. 2a and Supplementary Fig. 3). Furthermore, the reaction scale can be extended to 100.0 mmol at the same current and the flow rate still in quantitative yield (Fig. 2b and Supplementary Fig. 4). These results illustrated the potential applicability of this method.
Fig. 2.
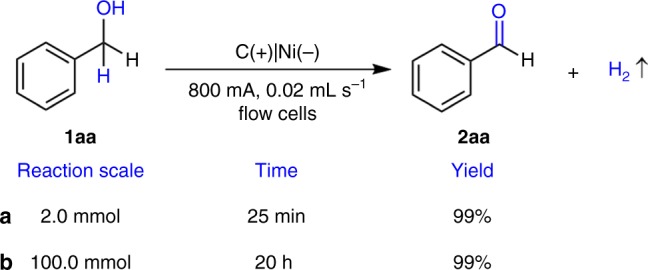
Slow flow rate of electrochemical oxidation of benzyl alcohol 1aa. a The amount of 1aa is 2.0 mmol. b The amount of 1aa is 100.0 mmol
Substrate scope
With the optimized reaction conditions in hand, we next explored various benzylic and allylic alcohols 1ab-1ap (2.0 mmol) under galvanostatic conditions (Fig. 3). Electron-rich benzylic alcohols with ortho-, meta-, and para-substitution could be converted to the corresponding aldehydes 2ab-2ah in nearly quantitative yields (96–99%) without over oxidation. Halogenated benzylic alcohols could be oxidized to afford the corresponding aldehydes 2ai-2ak in excellent yield (98–99%), which could be further functionalized. Owing to weak C–I bond, 10 mA cell current had to be adapted and 4-iodo benzaldehyde (2al) was obtained in moderate yield (60%). Moreover, electron-deficient benzylic alcohols gave less satisfactory results. For example, compound 2am was obtained in only 35% yield. On the other hand, 1-naphthyl methanol (1an) was converted to the corresponding aldehyde 2an in 88% yield. The flow conditions could also be applied to thiophene derivative 1ao to afford aldehyde 2ao in 76% yield without significant side product formation. Additionally, we explored the possibility of electrolysis of allylic alcohol 1ap, thus obtaining the corresponding unsaturated aldehyde 2ap in 64% yield.
Fig. 3.

Substrate scopes of electrochemical oxidation of primary alcohols. Reaction conditions: carbon paper (93 × 93 × 0.2 mm) anode (contact area 1.6 cm2), Ni plate (93 × 93 × 0.3 mm) cathode (contact area 1.6 cm2), constant current = 800 mA, flow rate = 0.10 mL s−1, 10 min, 1a (2.0 mmol), nBu4NBF4 (0.20 mmol), CH3CN/H2O (1:1, 30 mL), N2, room temperature, flow cell (2.49 F mol−1), isolated yield. a10 mA, 10 h. b10 mA, 20 h
The reaction conditions were subsequently employed to oxidize a range of secondary alcohols (Fig. 4). Excellent results were observed for oxidation of alcohols 1ba-1bf to the corresponding ketones 2ba-2bf in 83–97% yield. There was no significant difference in the reactivity of primary and secondary alcohols in the continuous-flow reactor. The oxidation of heterocyclic alcohol 1bg proceeded smoothly under 10 mA cell current condition to afford ketone 2bg in 70% yield. Oxidation of allylic alcohols 1bh−1bi afforded the desired ketones in 50–75% yield. However, aliphatic alcohol was not oxidized smoothly under electrochemical oxidation condition and gave 2bj in 25% yield.
Fig. 4.

Substrate scopes of electrochemical oxidation of secondary alcohols. Reaction conditions: carbon paper (93 × 93 × 0.2 mm) anode (contact area 1.6 cm2), Ni plate (93 × 93 × 0.3 mm) cathode (contact area 1.6 cm2), constant current = 800 mA, flow rate = 0.10 mL s−1, 10 min, 1b (2.0 mmol), nBu4NBF4 (0.20 mmol), CH3CN/H2O (1:1, 30 mL), N2, room temperature, flow cell (2.49 F mol−1), isolated yield. a10 mA, 20 h. b10 mA, 10 h. c1be: benzoin used as starting material
This observation led us to explore the selective oxidation of diols 3 and 5 as shown in Fig. 5. For both substrates, benzylic hydroxyl groups were oxidized selectively in the presence of aliphatic primary or secondary hydroxyl groups to afford hydroxyl ketones 4 and 6 in 78% and 85% yields, respectively. This oxidation could be complementary to Swern oxidation, which is selective for primary or less steric hindered alcohols 65–67.
Fig. 5.

Selective oxidation of benzylic alcohols in the presence of aliphatic alcohols. a Selective oxidation of 1-phenylbutane-1,3-diol. b Selective oxidation of 1-phenylbutane-1,4-diol
The continuous-flow electrolysis was further extended to pharmaceutical relevant substrates (Fig. 6). Rosuvastatin precursor 1aq68 could be oxidized to the corresponding aldehyde in 76% yield within only 10 min, which presented the potential application prospect of this protocol. Fluorenol (1bk)69 was oxidized to the corresponding ketones in good yields (86%).
Fig. 6.

Electrochemical oxidation of biologically relevant substrates. a Electrochemical oxidation of Rosuvastatin precursor. b Electrochemical oxidation of 9H-fluoren-9-ol
The possibility of using water as solvent was also estimated. However, benzaldehyde was obtained in only 82% yield due to the poor solubility of alcohols in water. Thus, the surfactant was employed and we noted that the ionic surfactant could also be the supporting electrolyte. By using this strategy, benzaldehyde could be prepared in quantitative yield in water (Fig. 7). Six primary alcohols in Fig. 3 and four secondary alcohols in Fig. 4 have been chosen to re-evaluate the yields in the presence of surfactant in pure water. In general, aqueous conditions provided the desired products in comparable yields with our standard conditions, which showed that the combination of water and surfactant would be a good choice for this protocol.
Fig. 7.

Substrate scopes of electrochemical oxidation of alcohols in water. Reaction conditions: carbon paper (93 × 93 × 0.2 mm) anode (contact area 1.6 cm2), Ni plate (93 × 93 × 0.3 mm) cathode (contact area 1.6 cm2), constant current = 800 mA, flow rate = 0.10 mL s−1, 10 min, 1 (2.0 mmol), N,N,N-trimethylhexadecan-1-ammonium sulfate (0.10 mmol), H2O (15 mL), N2, room temperature, flow cell (2.49 F mol−1), isolated yield. aLiClO4 instead of N,N,N-trimethylhexadecan-1-ammonium sulfate. b10 mA, 20 h. c10 mA, 10 h
Since the oxidation of alcohol in the anode means the loss of electron, the existence of radical intermediate is highly probable. To gain insight into the reaction mechanism, electron paramagnetic resonance (EPR) experiments were performed by adding the radical spin trapping agent DMPO (5, 5-dimethyl-1-pyrroline N-oxide). No radical signal was detected in the absence of 1aa (Fig. 8a, blank line). When DMPO was added to the reaction under constant current conditions, a radical signal (g = 2.0069, AN = 14.82, AH = 21.42) was identified (Fig. 8a, red line). According to the fitting result, this radical signal came from benzyl radical captured by DMPO.
Fig. 8.

Mechanistic studies experiments. a EPR results of benzyl alcohol oxidation. b D2O and H218O labeling experiments. c Kinetic profiles under different concentrations of 1aa. d Kinetic profiles under different current
No deuterium or 18O was incorporated into benzaldehyde when H2O was substituted with D2O or H218O in the reaction system (Fig. 8b and Supplementary Figs. 5 and 6). This result indicated that water did not react with the intermediate under the reaction conditions. This was in accordance with no over oxidation to benzoic acid was observed even when benzaldehyde was used as starting material for this electrolysis (Supplementary Fig. 8). In addition, no over oxidation of the aldehyde products may also benefit from the fact that no extra base was added in our reaction system.
The influence of the concentration, current, and flow rate to the electrochemical oxidation of benzyl alcohol have been evaluated as shown in Fig. 8. Reaction rate increased with the increasing of the concentration of benzyl alcohol (Fig. 8c). It was the same that large current meant high reaction rate (Fig. 8d). The reaction rate kept unchanged when the flow rate was larger than 0.10 mL s−1 (Supplementary Fig. 7). These results suggested that the electrochemical oxidation was likely to be the rate-limiting step during electrolysis.
On the basis of our mechanistic studies and literature reports41–43, a possible mechanism for the oxidation of alcohols to carbonyl functionality is depicted in Fig. 9. The oxidation of benzyl alcohol was initiated by anodic oxidation to afford intermediate B. The consequent deprotonation of the radical cation B resulted in the formation of benzylic radical C, which has been detected by EPR. The following fast single-electron oxidation and deprotonation of D produce the desired benzaldehyde (2aa). Lower efficiency of oxidation with electron deficient benzylic alcohols could be explained by considering that electron withdrawing groups destabilized intermediates. In the meantime, water underwent cathodic reduction to generate hydroxide accompanied by releasing hydrogen70. The in situ formed hydroxide acted as base to trap the protons. For the whole reaction, no external oxidant was needed, which is in accordance with the idea of green chemistry.
Fig. 9.

Proposed mechanism of benzyl alcohol electrochemical oxidation. A tentative reaction mechanism involves anodic oxidation of benzyl alcohol to afford intermediate B, deprotonation, single electron oxidation and deprotonation to form the desired benzaldehyde (2aa). Water underwent cathodic reduction to generate hydroxide accompanied by releasing hydrogen
In summary, a direct electrochemical oxidation of alcohols to the corresponding carbonyl compounds has been accomplished efficiently by the continuous-flow reactor just using carbon anode. Reactions were performed without external oxidant, mediator or additive and no over oxidation was observed, which make this method an ideal transformation from alcohols to aldehydes or ketones. Even water can be used as solvent in the presence of surfactant. The reaction conditions have been applied for selective oxidation and biologically relevant substrates. The reaction can be adjusted conveniently from milligram to gram scale based on demand by using the flow set-up. Further research to broaden the substrate scope of alcohol oxidation will be reported in due course.
Methods
General procedures for the electrolysis in acetonitrile and water
In an oven-dried schlenck tube (100 mL) equipped with a stir bar, alcohol 1a (2.0 mmol), nBu4NBF4 (65.9 mg, 0.2 mmol) and CH3CN/H2O (1:1, 30 mL) were added. The flow cell was equipped with carbon paper (9.3 cm × 9.3 cm × 0.2 mm) as the anode (contact area 1.6 cm2) and nickel plate (9.3 cm × 9.3 cm × 0.3 mm) as the cathode (contact area 1.6 cm2). In order to preclude the possibility that air was involved in the oxidation of alcohol, we flushed the whole system with nitrogen before the direct electrolysis. The reaction mixture was pumped into the electrochemical reactor at the flow rate of 0.10 mL s−1 (Supplementary Fig. 1). Method A: A constant current of 800 mA was employed during the electrolysis under room temperature for 10 min. (Method B: A constant current of 10 mA was employed during the electrolysis under room temperature for 10 h. Method C: A constant current of 10 mA was employed during the electrolysis under room temperature for 20 h.) When the reaction was finished, the reaction mixture was washed with water and extracted with dichloromethane (10 mL x 3). The organic layers were combined, dried over Na2SO4, and concentrated. The pure product was obtained by flash column chromatography on silica gel using petroleum ether and ethyl acetate as the eluent.
General procedures for the electrolysis in water with surfactant
Method D: In an oven-dried schlenck tube (100 mL) equipped with a stir bar, alcohol 1a (2.0 mmol), N,N,N-trimethylhexadecan-1-ammonium sulfate (69.6 mg, 0.10 mmol) and H2O (15 mL) were added. The flow cell was equipped with carbon paper (9.3 cm × 9.3 cm × 0.2 mm) as the anode (contact area 1.6 cm2) and nickel plate (9.3 cm × 9.3 cm × 0.3 mm) as the cathode (contact area 1.6 cm2). In order to preclude the possibility that air was involved in the oxidation of alcohol, we flushed the whole system with nitrogen before the direct electrolysis. The reaction mixture was pumped into the electrochemical reactor in a flow rate of 0.10 mL s−1 (Supplementary Fig. 1). A constant current of 800 mA was employed during the electrolysis under room temperature for 10 min. When the reaction was finished, the reaction mixture was washed with water and extracted with dichloromethane (10 mL x 3). The organic layers were combined, dried over Na2SO4, and concentrated. The pure product was obtained by flash column chromatography on silica gel using petroleum ether and ethyl acetate as the eluent.
Supplementary information
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21390402, 21520102003, 21272180) and the Natural Science Foundation of Hubei Province (2017CFA010, 2016CFB571). The Program of Introducing Talents of Discipline to Universities of China (111 Program) is also appreciated. The numerical calculations in this paper have been done on the supercomputing system in the Supercomputing Center of Wuhan University. We thank Prof. Xuefeng Li for providing surfactant as gift.
Author contributions
D.W., P.D. and S.-C.W. performed and analyzed experiments. A.L., Y.-H.C., H.Z. and D.W. conceived the project and designed the experiments. A.L., Y.-H.C., H.Z. and D.W. wrote the manuscript. All the authors discussed the results and commented on the manuscript.
Data availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files.
Competing interests
The authors declare no competing interests.
Footnotes
Peer review information: Nature Communications thanks Carlos Ponce de Leon and other anonymous reviewer(s) for their contribution to the peer review of this work.
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Yi-Hung Chen, Email: yihungchen@whu.edu.cn.
Heng Zhang, Email: hengzhang@whu.edu.cn.
Aiwen Lei, Email: aiwenlei@whu.edu.cn.
Supplementary information
Supplementary Information accompanies this paper at 10.1038/s41467-019-10928-0.
References
- 1.Tojo G, Fernandez M. Basic Reactions in Organic Synthesis: Oxidation of Alcohols to Aldehydes and Ketones. New York: Springer; 2010. [Google Scholar]
- 2.March, J. in March’s Advanced Organic Chemistry: Reactions, Mechanisms and Structures, 7th edn, (ed. Smith, M. B.) 1442–1452 (Wiley-VCH, Weinheim, 2013).
- 3.Constable DJC, et al. Key green chemistry research areas-a perspective from pharmaceutical manufacturers. Green Chem. 2007;9:411–420. doi: 10.1039/B703488C. [DOI] [Google Scholar]
- 4.Sheldon RA, Arends I, Ten Brink GJ, Dijksman A. Green, catalytic oxidations of alcohols. Acc. Chem. Res. 2002;35:774–781. doi: 10.1021/ar010075n. [DOI] [PubMed] [Google Scholar]
- 5.Faisca Phillips AM, Pombeiro AJL, Kopylovich MN. Recent advances in cascade reactions initiated by alcohol oxidation. ChemCatChem. 2017;9:217–246. doi: 10.1002/cctc.201601176. [DOI] [Google Scholar]
- 6.Siddiki SMAH, Toyao T, Shimizu K-i. Acceptorless dehydrogenative coupling reactions with alcohols over heterogeneous catalysts. Green Chem. 2018;20:2933–2952. doi: 10.1039/C8GC00451J. [DOI] [Google Scholar]
- 7.Anastas, P. & Warner, J. Green Chemistry: Theory and Practice (Oxford University Press, New York, 1998).
- 8.Takemoto, T., Yasuda, K. & Ley, S. V. Solid-supported reagents for the oxidation of aldehydes to carboxylic acids. Synlett10, 1555–1556 (2001).
- 9.Lou JD, Xu ZN. Selective oxidation of primary alcohols with chromium trioxide under solvent free conditions. Tetrahedron Lett. 2002;43:6095–6097. doi: 10.1016/S0040-4039(02)01333-3. [DOI] [Google Scholar]
- 10.Kumar A, Jain N, Chauhan SMS. Oxidation of benzylic alcohols to carbonyl compounds with potassium permanganate in ionic liquids. Synth. Commun. 2004;34:2835–2842. doi: 10.1081/SCC-200026242. [DOI] [Google Scholar]
- 11.Gonzalez-Nunez ME, Mello R, Olmos A, Acerete R, Asensio G. Oxidation of alcohols to carbonyl compounds with CrO3 center dot SiO2 in supercritical carbon dioxide. J. Org. Chem. 2006;71:1039–1042. doi: 10.1021/jo052137j. [DOI] [PubMed] [Google Scholar]
- 12.Hampton PD, Whealon MD, Roberts LM, Yaeger AA, Boydson R. Continuous organic synthesis in a spinning tube-in-tube reactor: TEMPO-catalyzed oxidation of alcohols by hypochlorite. Org. Process Res. Dev. 2008;12:946–949. doi: 10.1021/op800051t. [DOI] [Google Scholar]
- 13.De Crisci AG, Chung K, Oliver AG, Solis-Ibarra D, Waymouth RM. Chemoselective oxidation of polyols with chiral palladium catalysts. Organometallics. 2013;32:2257–2266. doi: 10.1021/om4001549. [DOI] [Google Scholar]
- 14.Manzini S, Urbina-Blanco CA, Nolan SP. Chemoselective oxidation of secondary alcohols using a ruthenium phenylindenyl complex. Organometallics. 2013;32:660–664. doi: 10.1021/om301156v. [DOI] [Google Scholar]
- 15.Jing Y, Daniliuc CG, Studer A. Direct conversion of alcohols to α-chloro aldehydes and α-chloro ketones. Org. Lett. 2014;16:4932–4935. doi: 10.1021/ol5024568. [DOI] [PubMed] [Google Scholar]
- 16.Zheng, N. & Stucky, G. D. Promoting gold nanocatalysts in solvent-free selective aerobic oxidation of alcohols. Chem. Commun. 3862–3864 (2007). [DOI] [PubMed]
- 17.Jiang B, Feng Y, Ison EA. Mechanistic investigations of the iridium(III)-catalyzed aerobic oxidation of primary and secondary alcohols. J. Am. Chem. Soc. 2008;130:14462–14464. doi: 10.1021/ja8049595. [DOI] [PubMed] [Google Scholar]
- 18.Parmeggiani C, Cardona F. Transition metal based catalysts in the aerobic oxidation of alcohols. Green Chem. 2012;14:547–564. doi: 10.1039/c2gc16344f. [DOI] [Google Scholar]
- 19.Shi Z, Zhang C, Tang C, Jiao N. Recent advances in transition-metal catalyzed reactions using molecular oxygen as the oxidant. Chem. Soc. Rev. 2012;41:3381–3430. doi: 10.1039/c2cs15224j. [DOI] [PubMed] [Google Scholar]
- 20.Ryland BL, Stahl SS. Practical aerobic oxidations of alcohols and amines with homogeneous copper/TEMPO and related catalyst systems. Angew. Chem. Int. Ed. 2014;53:8824–8838. doi: 10.1002/anie.201403110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gunasekaran N. Aerobic oxidation catalysis with air or molecular oxygen and ionic liquids. Adv. Synth. Catal. 2015;357:1990–2010. doi: 10.1002/adsc.201400989. [DOI] [Google Scholar]
- 22.Crotti C, Farnetti E. Selective oxidation of glycerol catalyzed by iron complexes. J. Mol. Catal. A: Chem. 2015;396:353–359. doi: 10.1016/j.molcata.2014.10.021. [DOI] [Google Scholar]
- 23.Zhang J, Gandelman M, Shimon LJW, Rozenberg H, Milstein D. Electron-rich, bulky ruthenium PNP-type complexes. Acceptorless catalytic alcohol dehydrogenation. Organometallics. 2004;23:4026–4033. doi: 10.1021/om049716j. [DOI] [Google Scholar]
- 24.Nielsen M, et al. Efficient hydrogen production from alcohols under mild reaction conditions. Angew. Chem. Int. Ed. 2011;50:9593–9597. doi: 10.1002/anie.201104722. [DOI] [PubMed] [Google Scholar]
- 25.Kawahara R, Fujita K-i, Yamaguchi R. Dehydrogenative oxidation of alcohols in aqueous media using water-soluble and reusable Cp*Ir catalysts bearing a functional bipyridine ligand. J. Am. Chem. Soc. 2012;134:3643–3646. doi: 10.1021/ja210857z. [DOI] [PubMed] [Google Scholar]
- 26.Kawahara R, Fujita K-i, Yamaguchi R. Cooperative catalysis by iridium complexes with a bipyridonate ligand: versatile dehydrogenative oxidation of alcohols and reversible dehydrogenation-hydrogenation between 2-propanol and acetone. Angew. Chem. Int. Ed. 2012;51:12790–12794. doi: 10.1002/anie.201206987. [DOI] [PubMed] [Google Scholar]
- 27.Nielsen M, et al. Low-temperature aqueous-phase methanol dehydrogenation to hydrogen and carbon dioxide. Nature. 2013;495:85. doi: 10.1038/nature11891. [DOI] [PubMed] [Google Scholar]
- 28.Gunanathan C, Milstein D. Applications of acceptorless dehydrogenation and related transformations in chemical synthesis. Science. 2013;341:1229712. doi: 10.1126/science.1229712. [DOI] [PubMed] [Google Scholar]
- 29.Song H, Kang B, Hong SH. Fe-catalyzed acceptorless dehydrogenation of secondary benzylic alcohols. ACS Catal. 2014;4:2889–2895. doi: 10.1021/cs5007316. [DOI] [Google Scholar]
- 30.Fujita K-i, Kawahara R, Aikawa T, Yamaguchi R. Hydrogen production from a methanol-water solution catalyzed by an anionic iridium complex bearing a functional bipyridonate ligand under weakly basic conditions. Angew. Chem. Int. Ed. 2015;54:9057–9060. doi: 10.1002/anie.201502194. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Zhang P, Tian B, Zhang J. Core-shell structural CdS@SnO2 nanorods with excellent visible-light photocatalytic activity for the selective oxidation of benzyl alcohol to benzaldehyde. ACS Appl. Mater. Interfaces. 2015;7:13849–13858. doi: 10.1021/acsami.5b04128. [DOI] [PubMed] [Google Scholar]
- 32.Zhao L-M, et al. Photocatalysis with quantum dots and visible light: selective and efficient oxidation of alcohols to carbonyl compounds through a radical relay process in water. Angew. Chem. Int. Ed. 2017;56:3020–3024. doi: 10.1002/anie.201700243. [DOI] [PubMed] [Google Scholar]
- 33.Bianchini C, Shen PK. Palladium-based electrocatalysts for alcohol oxidation in half cells and in direct alcohol fuel cells. Chem. Rev. 2009;109:4183–4206. doi: 10.1021/cr9000995. [DOI] [PubMed] [Google Scholar]
- 34.Hickey DP, McCammant MS, Giroud F, Sigman MS, Minteer SD. Hybrid enzymatic and organic electrocatalytic cascade for the complete oxidation of glycerol. J. Am. Chem. Soc. 2014;136:15917–15920. doi: 10.1021/ja5098379. [DOI] [PubMed] [Google Scholar]
- 35.Trincado M, Banerjee D, Gruetzmacher H. Molecular catalysts for hydrogen production from alcohols. Energy Environ. Sci. 2014;7:2464–2503. doi: 10.1039/C4EE00389F. [DOI] [Google Scholar]
- 36.Cha HG, Choi K-S. Combined biomass valorization and hydrogen production in a photoelectrochemical cell. Nat. Chem. 2015;7:328–333. doi: 10.1038/nchem.2194. [DOI] [PubMed] [Google Scholar]
- 37.Rafiee M, Miles KC, Stahl SS. Electrocatalytic alcohol oxidation with TEMPO and bicyclic nitroxyl derivatives: driving force trumps steric effects. J. Am. Chem. Soc. 2015;137:14751–14757. doi: 10.1021/jacs.5b09672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Badalyan A, Stahl SS. Cooperative electrocatalytic alcohol oxidation with electron-proton-transfer mediators. Nature. 2016;535:406–410. doi: 10.1038/nature18008. [DOI] [PubMed] [Google Scholar]
- 39.Choi Y, Sinev I, Mistry H, Zegkinoglou I, Roldan Cuenya B. Probing the dynamic structure and chemical state of Au nanocatalysts during the electrochemical oxidation of 2-propanol. ACS Catal. 2016;6:3396–3403. doi: 10.1021/acscatal.6b00057. [DOI] [Google Scholar]
- 40.Nutting JE, Rafiee M, Stahl SS. Tetramethylpiperidine N-Oxyl (TEMPO), phthalimide N-Oxyl (PINO), and related N-Oxyl species: electrochemical properties and their use in electrocatalytic reactions. Chem. Rev. 2018;118:4834–4885. doi: 10.1021/acs.chemrev.7b00763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lund H. Electroorganic preparations II. Oxidation carbinols. Acta Chem. Scand. 1957;11:491–498. doi: 10.3891/acta.chem.scand.11-0491. [DOI] [Google Scholar]
- 42.Mayeda EA, Miller LL, Wolf JF. Electrooxidation of benzylic ethers, esters, alcohols, and phenyl epoxides. J. Am. Chem. Soc. 1972;94:6812–6816. doi: 10.1021/ja00774a039. [DOI] [Google Scholar]
- 43.Mayeda EA. Anodic cleavages of secondary and tertiary alkylphenylcarbinols. J. Am. Chem. Soc. 1975;97:4012–4015. doi: 10.1021/ja00847a024. [DOI] [Google Scholar]
- 44.Plutschack MB, Pieber B, Gilmore K, Seeberger PH. The Hitchhiker’s guide to flow chemistry(II) Chem. Rev. 2017;117:11796–11893. doi: 10.1021/acs.chemrev.7b00183. [DOI] [PubMed] [Google Scholar]
- 45.Atobe M, Tateno H, Matsumura Y. Applications of flow microreactors in electrosynthetic processes. Chem. Rev. 2018;118:4541–4572. doi: 10.1021/acs.chemrev.7b00353. [DOI] [PubMed] [Google Scholar]
- 46.Pletcher D, Green RA, Brown RCD. Flow electrolysis cells for the synthetic organic chemistry laboratory. Chem. Rev. 2018;118:4573–4591. doi: 10.1021/acs.chemrev.7b00360. [DOI] [PubMed] [Google Scholar]
- 47.Bebelis S, et al. Highlights during the development of electrochemical engineering. Chem. Eng. Res. Des. 2013;91:1998–2020. doi: 10.1016/j.cherd.2013.08.029. [DOI] [Google Scholar]
- 48.Gütz C, Stenglein A, Waldvogel SR. Highly modular flow cell for electroorganic synthesis. Org. Process Res. Dev. 2017;21:771–778. doi: 10.1021/acs.oprd.7b00123. [DOI] [Google Scholar]
- 49.Laudadio G, de Smet W, Struik L, Cao Y, Noël T. Design and application of a modular and scalable electrochemical flow microreactor. J. Flow. Chem. 2018;8:157–165. doi: 10.1007/s41981-018-0024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fol-gueiras-Amador AA, Wirth T. Perspectives in flow electrochemistry. J. Flow. Chem. 2017;7:94–95. doi: 10.1556/1846.2017.00020. [DOI] [Google Scholar]
- 51.Folgueiras-Amador AA, Philipps K, Guilbaud S, Poelakker J, Wirth T. An easy-to-machine electrochemical flow microreactor: efficient synthesis of isoindolinone and flow functionalization. Angew. Chem. Int. Ed. 2017;56:15446–15450. doi: 10.1002/anie.201709717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Arai K, Wirth T. Rapid electrochemical deprotection of the isonicotinyloxycarbonyl group from carbonates and thiocarbonates in a microfluidic reactor. Org. Process Res. Dev. 2014;18:1377–1381. doi: 10.1021/op500155f. [DOI] [Google Scholar]
- 53.Horcajada, R., Okajima, M., Suga, S. & Yoshida, J. Microflow electroorganic synthesis without supporting electrolyte. Chem. Commun. 1303–1305 (2005). [DOI] [PubMed]
- 54.Green RA, Pletcher D, Leach SG, Brown RCD. N-Heterocyclic carbene-mediated microfluidic oxidative electrosynthesis of amides from aldehydes. Org. Lett. 2016;18:1198–1201. doi: 10.1021/acs.orglett.6b00339. [DOI] [PubMed] [Google Scholar]
- 55.Laudadio G, et al. An environmentally benign and selective electrochemical oxidation of sulfides and thiols in a continuous-flow microreactor. Green Chem. 2017;19:4061–4066. doi: 10.1039/C7GC01973D. [DOI] [Google Scholar]
- 56.Arai K, Watts K, Wirth T. Difluoro- and trifluoromethylation of electron-deficient alkenes in an electrochemical microreactor. ChemistryOpen. 2014;3:23–28. doi: 10.1002/open.201300039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kabeshov MA, Musio B, Murray PRD, Browne DL, Ley SV. Expedient preparation of nazlinine and a small library of indole alkaloids using flow electrochemistry as an enabling technology. Org. Lett. 2014;16:4618–4621. doi: 10.1021/ol502201d. [DOI] [PubMed] [Google Scholar]
- 58.Kashiwagi T, Elsler B, Waldvogel SR, Fuchigami T, Atobe M. Reaction condition screening by using electrochemical microreactor: application to anodic phenol-arene C,C cross-coupling reaction in high acceptor number media. J. Electrochem. Soc. 2013;160:G3058–G3061. doi: 10.1149/2.011307jes. [DOI] [Google Scholar]
- 59.Tateno H, Matsumura Y, Nakabayashi K, Senboku H, Atobe M. Development of a novel electrochemical carboxylation system using a microreactor. RSC Adv. 2015;5:98721–98723. doi: 10.1039/C5RA19289G. [DOI] [Google Scholar]
- 60.HillCousins JT, et al. TEMPO-mediated electrooxidation of primary and secondary alcohols in a microfluidic electrolytic cell. ChemSusChem. 2012;5:326–331. doi: 10.1002/cssc.201100601. [DOI] [PubMed] [Google Scholar]
- 61.Xu F, Qian X-Y, Li Y-J, Xu H-C. Synthesis of 4H−1,3-benzoxazines via metal- and oxidizing reagent-free aromatic C–H oxygenation. Org. Lett. 2017;19:6332. doi: 10.1021/acs.orglett.7b03152. [DOI] [PubMed] [Google Scholar]
- 62.Folgueiras-Amador AA, Qian X-Y, Xu H-C, Wirth T. Catalyst- and supporting-electrolyte-free electrosynthesis of benzothiazoles and thiazolopyridines in continuous flow. Chem. Eur. J. 2018;24:487–491. doi: 10.1002/chem.201705016. [DOI] [PubMed] [Google Scholar]
- 63.Huang C, Qian X-Y, Xu H-C. Continuous-flow electrosynthesis of benzofused S-heterocycles by dehydrogenative C–S cross-coupling. Angew. Chem. Int. Ed. 2019;58:6650–6653. doi: 10.1002/anie.201901610. [DOI] [PubMed] [Google Scholar]
- 64.Hosokawa Y-Y, Hakamata H, Murakami T, Kusu F. Electrosynthesis of cholesta-4,6-dien-3-one from cholesterol on a laboratory synthetic scale. Tetrahedron Lett. 2010;51:129–132. doi: 10.1016/j.tetlet.2009.10.106. [DOI] [Google Scholar]
- 65.Huang SL, Omura K, Swern D. Oxidation of sterically hindered alcohols to carbonyls with dimethyl sulfoxide-trifluoracetic anhydride. J. Org. Chem. 1976;41:3329–3331. doi: 10.1021/jo00882a030. [DOI] [Google Scholar]
- 66.Huang SL, Omura K, Swern D. Further studies on the oxidation of alcohols to carbonyl compounds by dimethyl sulfoxide/trifluoroacetic anhydride. Synthesis. 1978;4:297–299. doi: 10.1055/s-1978-24729. [DOI] [Google Scholar]
- 67.Mancuso AJ, Huang SL, Swern D. Oxidation of long-chain and related alcohols to carbonyls by dimethyl sulfoxide “activated” by oxalyl chloride. J. Org. Chem. 1978;43:2480–2482. doi: 10.1021/jo00406a041. [DOI] [Google Scholar]
- 68.Das A, Stahl SS. Noncovalent immobilization of molecular electrocatalysts for chemical synthesis: efficient electrochemical alcohol oxidation with a pyrene-TEMPO conjugate. Angew. Chem. Int. Ed. 2017;56:8892–8897. doi: 10.1002/anie.201704921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li H, et al. Characterization of a new class of androgen receptor antagonists with potential therapeutic application in advanced prostate cancer. Mol. Cancer Ther. 2013;12:2425–2435. doi: 10.1158/1535-7163.MCT-13-0267. [DOI] [PubMed] [Google Scholar]
- 70.Wang P, Tang S, Lei A. Electrochemical intramolecular dehydrogenative C-S bond formation for the synthesis of benzothiazoles. Green Chem. 2017;19:2092–2095. doi: 10.1039/C7GC00468K. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files.