

Abstract
Beta-propeller protein-associated neurodegeneration (BPAN) is a subtype of neurodegeneration with brain iron accumulation (NBIA) that presents with childhood developmental delay (especially speech delay), occasionally associated with epileptic encephalopathy, autism, or Rett-like syndrome. The majority of children described to date have been severely affected, with little to no expressive speech function, severe developmental delay, and cognitive impairment. Herein, five additional patients with BPAN identified in the same center in Canada are described, four with the typical severe phenotype and one with a milder phenotype. Our findings provide further evidence that a spectrum of severity exists for this rare and newly described condition. Challenges in identifying iron accumulation on brain MRI are also addressed. Additionally, the importance of including the WDR45 gene on epilepsy and Rett-like syndrome genetic panels is highlighted.
Keywords: Iron accumulation, Neurodegeneration, Brain, BPAN, WDR45, Rett
1. Introduction
Neurodegeneration with brain iron accumulation (NBIA) disorders are a group of heterogeneous neurological diseases with a unifying feature of brain iron accumulation in the basal ganglia [1]. Mutations in ten causative genes represent the known NBIA subtypes [2]. WDR45 is one of these genes, and was initially linked to beta-propeller protein-associated neurodegeneration (BPAN) in 2012 [3]. Of the known cases of NBIA, BPAN is estimated to comprise about 1–2% [2]. BPAN is the only X-linked form of NBIA, with a female predilection and often causes more severe disease or fatality in males [4]. Iron accumulation is eventually detected on brain MRI and is found in the substantia nigra and globus pallidus in patients with this condition [3]. Mutations in WDR45 have been mostly novel and de novo in the affected individuals to date [4].
The BPAN phenotype was originally defined in a sentinel study [4] and has since been expanded upon in several case studies and smaller series. The typical presentation is childhood-onset global developmental delay with prominent expressive speech delay, followed by progressive neurodegeneration in adolescence and adulthood with dystonia, Parkinsonism, and dementia [4]. Additionally, children may display a Rett-like phenotype, autistic features, intellectual delay or epileptic encephalopathy, and in many cases their early brain MRI studies may not show iron accumulation in the basal ganglia [4,5], making early diagnosis difficult due to a heterogeneous and non-specific clinical picture. In addition, there have been limited reports of patients displaying a milder phenotype in childhood [6,7], which can make the diagnosis even more elusive. In this report, we further the understanding of the BPAN phenotype by showing a spectrum of severity manifesting in five patients seen at the Alberta Children's Hospital (Calgary, Alberta, Canada), one of whom had a rarely described milder form of presentation.
2. Case descriptions
2.1. Case #1
A 16-year-old girl presented with a history of severe global developmental delay, with possible language regression at a young age. While she has been walking independently since 2 years of age, she is nonverbal and is dependent on her parents for all activities of daily living (ADL). She was diagnosed with Autism Spectrum Disorder (ASD) at two and a half years old and has a history of abnormal stereotypical movements and behaviors including hand rubbing, body rocking, lip smacking, spontaneous laughter, and teeth grinding, reminiscent of atypical Rett syndrome. The patient developed medication-refractory epilepsy at 15 months old, requiring multiple trials of antiepileptic drugs as well as regular intravenous immunoglobulin G (IVIG). There were features of precocious puberty at 3 years old and hypothyroidism was diagnosed at 12 years old. Family history is non-contributory. There had been extensive metabolic and genetic testing including routine and specialized biochemical testing, CGH microarray, fragile X testing, Angelman syndrome testing, and MECP2 testing, all of which were non-diagnostic. Brain MRI at ages 4 and 12 showed thinning of the corpus callosum but were otherwise normal.
At age 12 she began to have a decline in her ambulation, with an abnormal gait and a “hunched” posture of her trunk and knees, along with mild spasticity with minor flexion contractures at her lower extremities. There had been a decline in her cognition over the same time-frame. Her current exam reveals average growth parameters and head circumference, and she is not dysmorphic. A repeat brain MRI was completed at age 14, showing bilateral T2 hypointense signals in the globus pallidus and substantia nigra, consistent with iron deposition, along with the previous finding of thinning of the corpus callosum (Fig. 1). A NBIA gene panel was subsequently requested; a novel nonsense variant; c.761_762insAG (p.Cys254Ter), in WDR45 was found, classified as pathogenic.
Fig. 1.
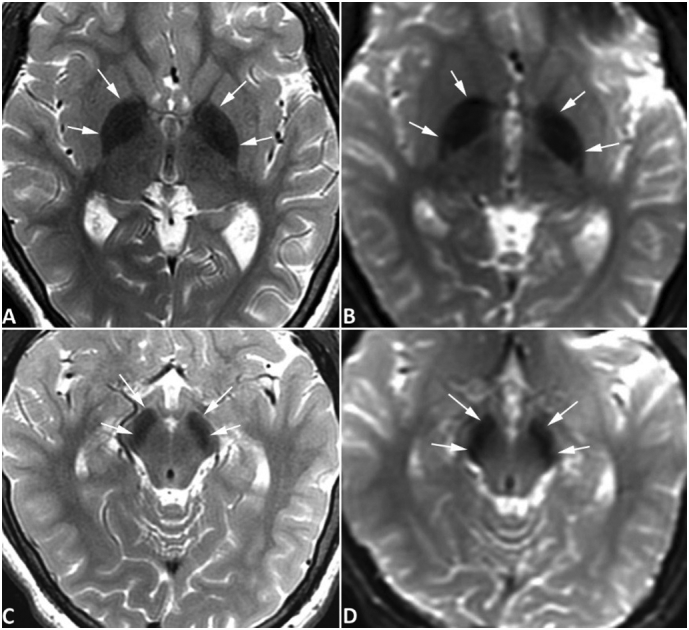
Brain MRI of case 1. MR images of case #1 were obtained at the age of 14 years with a 3-Tesla MRI scanner. Symmetrical hypointense signals are seen in the globus pallidus bilaterally (arrows, A and B) and substantia nigra (arrows, C and D) on both spin-echo T2-weighted images (A, C) and diffusion-weighted images acquired with an echo-planar sequence (B, D), all consistent with iron deposition. The degree of iron accumulation in substantia nigra is similar to that seen in the globus pallidus. No abnormal signals are evident in the red nuclei, and no abnormal signals were appreciated on T1-weighted images (not shown) surrounding or within the globus pallidus or substantia nigra. No gradient-echo T2*-weighed images or susceptibility-weighted images were obtained. The hypointense signal is more pronounced than that of age-matched normal subjects obtained with the same scanner using similar MR sequences (not shown), which confirms the iron deposition is pathological rather than physiological. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.2. Case #2
A 10-year-old girl (previously reported [8], outcome up to age 3) presented with a history of severe global developmental delay without regression. The patient was never able to walk and ambulates with assistance in a wheelchair, vocalizes but has no speech, and is dependent on her parents for all ADL. She was diagnosed with ASD at two and a half years old, has some stereotypies including repetitive rocking and repetitively flicking things with her fingers, and has spontaneous laughter. Medication refractory epilepsy began at 3 years old. Additionally, she has had excessive weight gain, mild asymmetrical leg growth, and precocious puberty beginning at 4 years of age. The patient has difficulty with sleep and frequent night-time awakenings, and in the past has had seizure-like episodes upon awakening and in sleep transition. Family history is non-contributory. On exam there are no overt dysmorphic features. She is normocephalic and height and weight are just above the 97th percentile. There is a leg-length discrepancy. Extensive metabolic and genetic testing had been completed including routine and specialized biochemical testing, muscle biopsy, CGH microarray, fragile X testing, Angelman syndrome testing, and MECP2 testing, all of which were non-diagnostic. Brain MRI at age 5 showed isolated cerebral atrophy.
At approximately 9 years of age she was showing aggressive behavior and a mild spasticity at the lower extremities was noted (patient was previously hypotonic). WDR45 gene sequencing was performed and the patient was found to be heterozygous for a variant of unknown significance; c.614G > A (p.Gly205Asp), previously unreported in the literature and predicted to be disease causing, probably damaging and deleterious by MutationTaster, PolyPhen-2 and SIFT respectively (Parental testing was not pursued). Following this result, a repeat brain MRI then showed T2 hypointensities in the globus pallidus and substantia nigra bilaterally (Fig. 2).
Fig. 2.

Brain MRI of case #2. MR images of case #2 (upper two rows) and an age-matched normal control (lower two rows) were obtained at the age of 10 years with a 3-Tesla MRI scanner. In case #2, symmetrical hypointense signals are seen in the globus pallidus bilaterally (A-C) and substantia nigra (D-F) on spin-echo T2-weighted images (SE T2W) (A, D), gradient-echo T2*-weighted images (GRE T2*W) (B, E), and diffusion-weighted images (DWI) acquired with an echo-planar sequence (C, F), all consistent with iron deposition. The hypointensities are most obvious on DWI. The degree of iron accumulation in the substantia nigra is similar to that seen in the globus pallidus. No abnormal signals are evident in the red nuclei. No abnormal signals were appreciated on T1-weighted images surrounding or within the globus pallidus or substantia nigra (not shown). The hypointense signal is more pronounced compared to images of an age-matched normal subject obtained from the same scanner using similar MR sequences (A' and D', SE T2W; B′ and E', GRE T2*W; C′ and F′, DWI), which confirms that the iron deposition is pathological rather than physiological. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.3. Case #3
A 5-year-old girl from Vietnam presented with global developmental delay at 3 years of age. She sat before 9 months but only walked independently at 3 years 2 months; fine motor skills are also delayed with frequent hand biting. Her expressive and receptive language skills are limited, and she can say only 3 words in Vietnamese. The patient attends a modified school program, making limited progress with no regression. Sleep has not been a concern. Myoclonic and rare generalized tonic-clonic seizures occurred between 4 and 5 years and are well controlled with medication. Her weight is at the 10th percentile for age, height and head circumference are just below the 3rd percentile; she is not dysmorphic. On examination, she has a wide-based, ataxic gait and stereotyped non-purposeful hand movements. CGH microarray was normal. For further investigation of global developmental delay, a brain MRI was completed and revealed bilateral hypointense signals in the globus pallidus and substantia nigra, suggestive of iron deposition (Fig. 3). NBIA gene panel testing revealed that the patient is heterozygous for the variant c.830 + 1G > A in the WDR45 gene, lying in the canonical donor splice site and all five in silico splice site prediction programs predict that this variant will completely abolish the donor splice site and result in aberrant splicing. The mother does not carry the mutation and paternal testing has not been pursued.
Fig. 3.

Brain MRI of case #3. MR images of case #3 were obtained at the age of 4 years using a 3-Tesla MRI scanner. Symmetrical hypointense signals are seen in the globus pallidus bilaterally (A-C) and substantia nigra (D-F) on spin-echo T2-weighted images (SE T2W) (A, D), gradient-echo T2*-weighted images (GRE T2*W) (B, E), and diffusion-weighted images (DWI) acquired with an echo-planar sequence (C, F), all consistent with iron deposition. In this patient, the degree of iron accumulation in the globus pallidus is greater than that in the substantia nigra. No abnormal signals are evident in the red nuclei. No abnormal signals are appreciated on T1-weighted images (not shown) surrounding or within the globus pallidus or substantia nigra. The signal abnormality on T2W is mild and similar to physiological iron accumulation in Fig. 2 but is more pronounced than age-matched normal controls (not shown). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.4. Case #4
A 6-year-old girl presented with mild motor delay and moderate speech delay, who was reported to have had normal development up to 2 and a half years old. The patient has difficulties in both receptive and expressive language but can currently say upwards of 500 words and can speak in sentences. While she has been confirmed by neuropsychiatric testing to have mild intellectual delay (full scale intelligence quotient reported to be 55) and is overall functioning at the level of a 3- to 4-year-old, she is currently in a mainstream kindergarten class. Seizures began at 12 months of age and were initially difficult to control. Various seizure types are now present, including generalized tonic-clonic, absence, and atonic seizures. Attention deficit hyperactivity disorder was diagnosed at age 5. She has difficulty sleeping and is awake many times throughout the night. On exam, the patient has normal growth parameters and head circumference and there are no dysmorphic features. Microarray testing was normal. Sequencing of the WDR45 gene revealed that the patient is heterozygous for a de novo variant of unknown significance; c.299T>C (p.Phe100Ser), not reported previously to our knowledge and predicted to be possibly damaging, deleterious, disease causing by PolyPhen-2, SIFT and MutationTaster respectively. While MRI brain scans were interpreted as normal at ages 3 and 5, a subsequent brain MRI at age 6 showed hypointensities in the substantia nigra and globus pallidus, consistent with iron deposition in these regions (Fig. 4).
Fig. 4.

Brain MRI of case #4. MR images of case #4 (upper two rows) and an age-matched normal control (lower two rows) were obtained at the age of 6 years using a 3-Tesla MRI scanner. Symmetrical hypointense signals are seen in the globus pallidus bilaterally (A-C) and substantia nigra (D-F) on spin-echo T2-weighted images (SE T2W) (A, D), gradient-echo T2*-weighted images (GRE T2*W) (B, E), and diffusion-weighted images (DWI) acquired with an echo-planar sequence (C, F), all consistent with iron deposition. The hypointensities are most obvious on DWI. In this patient, the degree of iron accumulation in the substantia nigra is greater than that in the globus pallidus, which is distinct from the pattern seen in Case #3 (Fig. 3). The degree of signal change in globus pallidus is mild and similar to physiological iron accumulation in older children as illustrated in Fig. 2. But when compared to images from an age-matched normal control obtained from the same scanner (A' and D', SE T2W; B′ and E', GRE T2*W; C′ and F′, DWI), the abnormality is readily appreciated. No abnormal signal is evident in the red nuclei. Further, no abnormal signal is appreciated on T1-weighted images (not shown) surrounding or within the globus pallidus or substantia nigra. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
2.5. Case #5
A 6-year-old girl presented with severe expressive language delay and moderate motor delay. The patient is non-verbal but responds to her name and can follow simple commands. She sat at 12 months, crawled at 24 months, and can walk with support. There is no hand lateralization and no fine pincer grasp. Some social interactions are present including a social smile, ability to express frustration, and sustained eye contact. Although mild, she has some dysmorphic features including mild hypertelorism, broad and spread teeth and small hands and feet for age; however, she is normocephalic. Growth parameters are at the 3rd percentile for height, the 50th percentile for weight and between the 25th–50th percentile for head circumference. An EEG showed rare bilateral independent as well as synchronous interictal discharges but no clinical or electroclinical seizures were reported. Karyotype and CGH microarray were both normal. MECP2 and Angelman syndrome testing were negative. Due to mild increased muscle tone and increased deep tendon reflexes, a brain MRI was performed at 6 years old, which was first reported as normal. Whole exome sequencing was performed shortly thereafter, showing a de novo splice site donor variant in WDR45, c.830 + 1G > C, classified as pathogenic and has been previously reported. Given this finding, her brain MRI was re-evaluated, and T2 hypointensity and susceptibility artifact was noted in the globi pallidi and substantia nigra, suggestive of iron deposition. In addition, the cerebral sulci are prominent, suggesting mild atrophy and the superior vermian folia were also prominent indicates mild atrophy as well (Fig. 5).
Fig. 5.
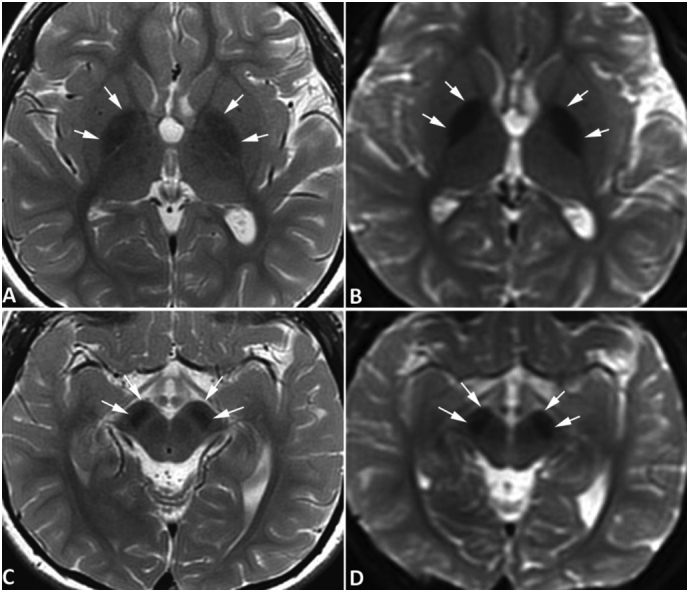
Brain MRI of case #5. MR images of case #5 were obtained at the age of 6 years using a 3-Tesla MRI scanner. Symmetrical hypointense signals are seen in the globus pallidus bilaterally (arrows, A and B) and substantia nigra (arrows, C and D) on spin-echo T2-weighted images (A, C) and diffusion-weighted images acquired with an echo-planar sequence (C, D), all consistent with iron deposition. The hypointensities are most obvious on DWI. The signal abnormality is readily appreciated when compared to images of an age-matched control subject obtained with the same scanner (Fig. 4). No abnormal signals are evident in the red nuclei. No abnormal signal is appreciated on T1-weighted images (not shown) surrounding or within the globus pallidus or substantia nigra. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3. Discussion
There have been approximately 40 published BPAN case reports since the causative gene was first identified in 2012. The majority of cases to date describe individuals who are severely delayed with prominent expressive speech impairment, epileptic encephalopathy, autism, and/or a Rett-like syndrome. Four out of five patients presented here fit this typical description. However, the phenotype of BPAN is expanding and it is important to recognize that a spectrum of severity with milder cases is emerging. Here we present one patient (case #4), with a de novo WDR45 mutation and a phenotype consisting of milder developmental delays, preserved speech, mild-to-moderate intellectual disability, and well-controlled epilepsy. There are two other case reports which clearly describe patients with milder BPAN phenotypes (see Table 1): first, a 17-year-old female who attends a basic stream in high school, is verbal but has issues with sentence structure, grammar and articulation, and with no motor delays or seizures [6].; the second, a 36-year-old female with an IQ of 45, who graduated from high school with assistance and has intelligible speech, and had epilepsy that remitted at menarche [7]. As BPAN is an X-linked condition, the existence of milder female cases is conceivable by way of possible X-inactivation. However, the existence of these mildly affected individuals could also potentially be reflective of mutations that have less of a detrimental effect on protein function.
Table 1.
Comparison of known patients with mild BPAN to case #4. ADL (activities of daily living), FSIQ (full-scale intelligence quotient), SN (substantia nigra), GP (globus pallidus)
| Case #4 | Reference #6 | Reference #7 | |
|---|---|---|---|
| Age at time of publication (yrs) | 6 | 17 | 36 |
| Motor delay | Mild (normal before seizure onset) | None | Appears to be none, but not specified |
| Expressive speech delay | 500+ words, speaks in sentences with some delays | Sentence structure, grammar, articulation difficulties | Dysarthric but intelligible speech |
| School support | Regular kindergarten class | Basic stream, age appropriate | Supports in place, details unknown |
| ADL support | Difficulties with self care | Independent | Unknown |
| FSIQ | 55 | N/A | 45 |
| Seizures (age of onset) | Generalized tonic-clonic, absence, atonic (1 year of age) | None | Febrile seizures (2 years of age) then absence seizures later (age of onset unknown), resolved with menarche |
| Sleep issues | Awake throughout the night | Restless sleeper | Unknown |
| WDR45 mutation | c.299T>C (p.Phe100Ser) | c.251A>G (p.Asp84Gly) | c.64DelT (p.Cys22Alafs*16) |
| MRI brain: T2 sequence | Hypointensities in the SN and GP | Hypointensities in the SN and GP | Hypointensities in the SN and GP |
Evidence is accumulating that BPAN should be considered in the differential for Rett-like syndrome. The possibility of Rett syndrome was considered in three of the severely-affected patients (cases #1, #2, and #5). Likewise, Rett-like features were observed in 7 out of 23 BPAN patients in the original paper by Hayflick et al. [4] and in at least 4 other BPAN patients in subsequent case reports [5,[9], [10], [11]]. These patients exhibited symptoms such as developmental regression, midline handwringing and loss of purposeful hand skills, and loss of language, some of which are features shared with the patients reported herein.
MRI is an invaluable tool in the detection of brain iron accumulation [12]. In clinical settings, a diagnosis of abnormal brain iron accumulation is made by subjective observation of hypointense signals in one or more MRI sequences. Multiple technical factors should be considered when interpreting an MRI. First, it is well known now that the commonly used clinical MR sequences have different sensitivities for detecting brain iron accumulation. While susceptibility-weighted imaging (SWI) and gradient-echo T2*-weighted imaging (T2*WI) have the highest sensitivity, they are often not routinely acquired in clinical MR studies unless there is a clinical suspicion for brain iron accumulation or intracranial hemorrhage. While diffusion-weighted imaging (DWI) acquired with an echo planar sequence is designed for detection of cytotoxic edema, its value in detecting iron accumulation has been recently recognized [4,13]. In many centres, this sequence is included in routine brain MRI protocols due to its unique value and relatively short scan time requirement. It is well known that spin-echo T2-weighted imaging (T2WI) is less sensitive than SWI, gradient-echo T2*WI in detecting brain iron accumulation. When performing diffusion studies, one needs to apply field gradients in addition to the radiofrequency used for conventional MR imaging, and MR signal were generated and collected with the single-shot echo-planar imaging technique [24]. Therefore, the DWI sequence has an inherent component of T2* weighting. In theory, and in the author's experience, the DWI sequence is more sensitive than spin-echo T2WI in detecting brain iron accumulation, but a systematic study on the difference has not been performed or published to our knowledge. Further, it is also well known that the sensitivity of MRI in brain iron accumulation also depends on the magnetic strength of the MR scanner [12]. Finally, the brain structures where abnormal iron accumulation occurs (including the globus pallidus, substantia nigra, red nuclei, and dentate nuclei) have varying normal appearances at different ages because of age-dependent physiological iron accumulation [12,14]. Recognizing these differences is paramount in order to avoid misinterpretation of a mild abnormal iron accumulation in a younger child as normal, or a normal iron accumulation in an older child or adult as mild abnormal iron accumulation.
The absence of iron accumulation in the globus pallidus and substantia nigra in early childhood brain MRI with its appearance later, either with standard or specialized MRI techniques, as was the case in patients #1 and #2, has been previously observed [[9], [10], [11],[15], [16], [17]]. Iron accumulation in the basal ganglia has been reported as early as 3 years of age using susceptibility-weighted imaging [18] and it has been observed as early as 8 years using standard T2 weighted imaging [19]. Additionally, some patients have had varying degrees of cerebral or cerebellar atrophy on brain MRI [4], as was observed in patients #2 and #5. Thinning of the corpus callosum has also been observed [4,5,11,18] and was present in patient #1. Additionally, in many reported cases with BPAN, hyperintense signals on T1-weighed images were observed surrounding regions of iron deposition [3,4,6,[19], [20], [21]]. However, abnormal T1 signals were not observed in any of the five cases described herein. The reason for this discrepancy is unknown.
BPAN is thought to be caused by an error in autophagy, the process of cellular recycling [23]. It is unknown at this time how a disruption in this process leads to iron accumulation in the brain and to the clinical phenotype of BPAN. There are no targeted therapies to date for this condition and symptomatic treatment with agents such as L-Dopa to manage dystonia are the mainstay at this time. However, we and our BPAN families remain hopeful that new targeted therapies may become available in the future as we learn more about this rare disease.
4. Conclusions
These five cases provide further evidence that there is a spectrum of clinical severity in BPAN patients and contribute to the expansion of the BPAN phenotype. BPAN should be considered in patients with mild-to-moderate childhood-onset developmental delay, especially if epilepsy or a Rett-like phenotype is present. It is important to recognize that early brain MRI may be normal in BPAN patients with iron accumulation not presenting until later in life and, as such, may not be helpful in diagnosing young patients. Additionally, If BPAN is suspected clinically, it is essential to include certain sequences that are sensitive to iron accumulation when performing the MRI. Children at different ages have remarkably different degrees of physiological brain iron accumulation, so it is crucial to compare the MR signal in the globus pallidus, substantia nigra, and red nucleus to that of age-matched controls in order to minimize false positive interpretation of the MRI.
References
- 1.Hayflick S.J. Unraveling the Hallervorden-Spatz syndrome: Pantothenate kinase-associated neurodegeneration is the name. Curr. Opin. Pediatr. 2003;15(6):572–577. doi: 10.1097/00008480-200312000-00005. [DOI] [PubMed] [Google Scholar]
- 2.Gregory A., Hayflick S. Neurodegeneration with Brain Iron Accumulation Disorders Overview. 1993. http://www.ncbi.nlm.nih.gov/pubmed/23447832 [PubMed]
- 3.Haack T.B., Hogarth P., Kruer M.C. Exome sequencing reveals de novo WDR45 mutations causing a phenotypically distinct, X-linked dominant form of NBIA. Am. J. Hum. Genet. 2012;91(6):1144–1149. doi: 10.1016/j.ajhg.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hayflick S.J., Kruer M.C., Gregory A. Beta-propeller protein-associated neurodegeneration: a new X-linked dominant disorder with brain iron accumulation. Brain. 2013;136(6):1708–1717. doi: 10.1093/brain/awt095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoffjan S., Ibisler A., Tschentscher A., Dekomien G., Bidinost C., Rosa A.L. WDR45 mutations in Rett (−like) syndrome and developmental delay: case report and an appraisal of the literature. Mol. Cell. Probes. 2016;30(1):44–49. doi: 10.1016/j.mcp.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 6.Long M., Abdeen N., Geraghty M.T., Hogarth P., Hayflick S., Venkateswaran S. Novel WDR45 mutation and pathognomonic BPAN imaging in a young female with mild cognitive delay. Pediatrics. 2015;136(3):e714–e717. doi: 10.1542/peds.2015-0750. [DOI] [PubMed] [Google Scholar]
- 7.Fonderico M., Laudisi M., Andreasi N.G. Patient affected by beta-propeller protein-associated neurodegeneration: a therapeutic attempt with iron vhelation therapy. Front. Neurol. 2017;8(AUG) doi: 10.3389/fneur.2017.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carvill G.L., Liu A., Mandelstam S., Schneider A., Lacroix A., Zemel M., McMahon J.M., Bello-Espinosa L., Mackay M., Wallace G., Waak M., Zhang J., Yang X., Malone S., Zhang Y.H., Mefford H.C., Scheffer I.E. Severe infantile onset developmental and epileptic encephalopathy caused by mutations in autophagy gene WDR45. Epilepsia. 2018 Jan;59(1):e5–e13. doi: 10.1111/epi.13957. 10.1111/epi.13957 Epub 2017 Nov 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ohba C., Nabatame S., Iijima Y. De novo WDR45 mutation in a patient showing clinically Rett syndrome with childhood iron deposition in brain. J. Hum. Genet. 2014;59(5):292–295. doi: 10.1038/jhg.2014.18. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto N., Ikeda T., Hasegawa T. Early manifestations of BPAN in a pediatric patient. Am. J. Med. Genet. Part A. 2014;164(12):3095–3099. doi: 10.1002/ajmg.a.36779. [DOI] [PubMed] [Google Scholar]
- 11.Khalifa M., Naffaa L. Exome sequencing reveals a novel WDR45 frameshift mutation and inherited POLR3A heterozygous variants in a female with a complex phenotype and mixed brain MRI findings. Eur. J. Med. Genet. 2015;58(8):381–386. doi: 10.1016/j.ejmg.2015.05.009. [DOI] [PubMed] [Google Scholar]
- 12.Dusek P., Dezortova M., Wuerfel J. Imaging of iron. Int. Rev. Neurobiol. 2013;110:195–239. doi: 10.1016/B978-0-12-410502-7.00010-7. [DOI] [PubMed] [Google Scholar]
- 13.Kruer M.C., Boddaert N., S a Schneider. Neuroimaging features of Neurodegeneration with brain Iron accumulation. AJNR Am. J. Neuroradiol. 2012 doi: 10.3174/ajnr.A2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Barkovich A., Mukherjee P. Normal development of the neonatal and infant brain, skull, and spine. In: Barkovich A., Raybaud C., editors. Pediatric Neuroimaging. 5th ed. 2012. pp. 20–80. [Google Scholar]
- 15.Verhoeven W.M.A., Egger J.I.M., Koolen D.A. Beta-propeller protein-associated neurodegeneration (BPAN), a rare form of NBIA: novel mutations and neuropsychiatric phenotype in three adult patients. Parkinsonism Relat. Disord. 2014;20(3):332–336. doi: 10.1016/j.parkreldis.2013.11.019. [DOI] [PubMed] [Google Scholar]
- 16.Rathore G.S., Schaaf C.P., Stocco A.J. Novel mutation of the WDR45 gene causing beta-propeller protein-associated neurodegeneration. Mov. Disord. 2014;29(4):574–575. doi: 10.1002/mds.25868. [DOI] [PubMed] [Google Scholar]
- 17.Abidi A., Mignon-Ravix C., Cacciagli P., Girard N., Milh M., Villard L. Early-onset epileptic encephalopathy as the initial clinical presentation of WDR45 deletion in a male patient. Eur. J. Hum. Genet. 2016;24(4):615–618. doi: 10.1038/ejhg.2015.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takano K., Shiba N., Wakui K. Elevation of neuron specific enolase and brain iron deposition on susceptibility-weighted imaging as diagnostic clues for beta-propeller protein-associated neurodegeneration in early childhood: additional case report and review of the literature. Am. J. Med. Genet. Part A. 2016;170(2):322–328. doi: 10.1002/ajmg.a.37432. [DOI] [PubMed] [Google Scholar]
- 19.Araújo R., Garabal A., Baptista M. Novel WDR45 mutation causing beta-propeller protein associated neurodegeneration (BPAN) in two monozygotic twins. J. Neurol. 2017;264(5):1020–1022. doi: 10.1007/s00415-017-8475-2. [DOI] [PubMed] [Google Scholar]
- 20.Ichinose Y., Miwa M., Onohara A. Characteristic MRI findings in beta-propeller protein-associated neurodegeneration (BPAN) Neurol. Clin. Pract. 2014 doi: 10.1212/01.CPJ.0000437694.17888.9b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryu S.W., Kim J.S., Lee S.H. Beta-propeller-protein-associated neurodegeneration: a case of mutation in WDR45. J. Clin. Neurol. 2015 doi: 10.3988/jcn.2015.11.3.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Meyer E., Kurian M.A., Hayflick S.J. Neurodegeneration with brain Iron accumulation: genetic diversity and pathophysiological mechanisms. Annu. Rev. Genomics Hum. Genet. 2015;16(1):257–279. doi: 10.1146/annurev-genom-090314-025011. [DOI] [PubMed] [Google Scholar]
- 24.Moritani T., Ekholm S., Westesson P.-L. 2nd ed. Springer-Verlag; Berlin Heidelberg: 2009. Basics of diffusion measurements by MRI. Diffusion-weighted MR imaging of the brain; pp. 1–5. [Google Scholar]