

Abstract
Primary spinal tumors are rare lesions that require careful clinical management due to their intimate relationship with critical neurovascular structures and the significant associated risk of morbidity. While the advent of molecular and genomic profiling is beginning to impact the management of the cranial counterparts, translation for spinal tumors has lagged behind. Maximal safe surgical resection remains the mainstay of patients with primary spinal tumors, with extent of resection and histology the only consistently identified independent predictors of survival. Adjuvant therapy has had limited impact. To develop targeted neoadjuvant and adjuvant therapies, improve prognostication, and enhance patient selection in spinal oncology, a thorough understanding of the current molecular and genomic landscape of spinal tumors is required. In this review, we detail the epidemiology, current standard-of-care, and molecular features of the most commonly encountered intramedullary spinal cord tumors (IMSCT), intradural extramedullary (IDEM) tumors, and primary spinal column malignancies (PSCM). We further discuss current efforts and future opportunities for integrating molecular advances in spinal oncology with clinical management.
Keywords: Intramedullary spinal cord tumor (IMSCT), meningioma, schwannoma, chordoma, genomics
Introduction
Surgical management of primary spinal tumors can be guided by both anatomic location and histology. Anatomically, lesions within the thecal sac are either (I) within the parenchyma of the spinal cord [intramedullary spinal cord tumors (IMSCT)], or (II) not within the parenchyma but still within the thecal sac [intradural, extramedullary (IDEM) tumors]. Tumors external to the thecal sac are simply termed extradural tumors. Of the extradural tumors, primary lesions arising from the bony structures of the spine, such as the vertebral bodies, are termed primary spinal column malignancies (PSCM). Histologically, many primary spinal tumors have cranial counterparts, and management of these tumors often draws on the principles applied to primary brain tumors (1). With the advent of genetic and genomic profiling, key molecular differences between cranial and spinal tumors of shared histology have emerged. Furthermore, targeted therapies may complement the current standard-of-care by providing patient-specific neoadjuvant and adjuvant therapies.
In this review, we will detail the current landscape of molecular alterations in primary spinal tumors, focusing on the most commonly encountered IMSCTs (astrocytoma, ependymoma, and hemangioblastoma), IDEM tumors (meningioma and schwannoma), and PSCMs (chordoma). We will also discuss current efforts at genotype-directed therapy for patients with these lesions and the implications of these results for surgical decision-making.
IMSCT
In adults, IMSCTs comprise 5–10% of all spinal tumors and are the most common spinal tumor in children (2). The most commonly encountered IMSCTs are ependymomas, astrocytomas, and hemangioblastomas. Collectively, spinal ependymomas and astrocytomas account for 80–90% of IMSCTs, with ependymomas occurring roughly twice as frequently as astrocytomas (3).
Ependymoma
Ependymomas are the most common IMSCT in adults, occurring equally often in men and women (Figure 1) (4). Spinal ependymomas are associated with neurofibromatosis type 2 (NF2) (5). These neuroepithelial lesions occur most commonly in the cervical or cervicothoracic spine. The myxopapillary variant occurs almost exclusively in the conus medullaris or filum terminale and tends to present earlier in life (Figure 2). Spinal ependymomas are histopathologically classified by the World Health Organization (WHO) as grade I (subependymoma or myxopapillary), grade II (classic), or grade III (anaplastic). Recently, a molecularly defined entity, RELA fusion-positive ependymoma, has been introduced into the WHO classification of ependymoma. To date, RELA fusion-positive ependymoma have only been reported as lesions occurring supratentorially in children and young adults (6).
Figure 1.

Spinal cord ependymoma. Preoperative sagittal T1-weighted, fat-saturated pre-gadolinium (A) and post-gadolinium (B) MRI demonstrates a discrete, enhancing C2 intramedullary mass. Sagittal T2-weighted MRI (C) reveals characteristic T2 hyperintensity. Associated hemorrhage may lead to a hypointense hemosiderin rim on T2 images.
Figure 2.
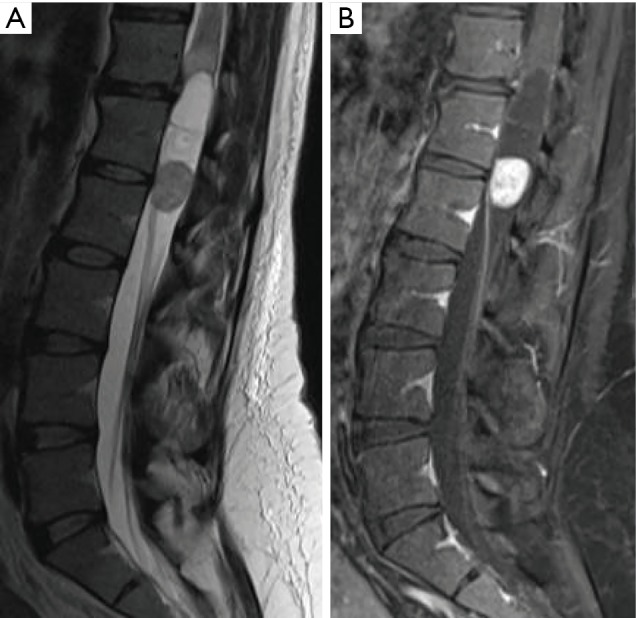
Spinal myxopapillary ependymoma. Preoperative sagittal T2-weighted (A) and T1-weighted, fat-saturated, post-gadolinium (B) MRI reveals 5.9 cm mixed cystic and solid intradural lesion with enhancing mural nodule arising from the conus medullaris at the T12-L2 levels. Associated pathologic T2 hyperintensity within the conus medullaris is observed superior to the lesion.
Standard of care
Management of spinal ependymoma is influenced by extent of spinal cord compression and symptomatic presentation. Asymptomatic patients with low concern for cord compromise may be managed with serial surveillance imaging. For patients with symptomatic ependymoma, surgical resection with the goal of achieving gross total resection (GTR) is standard-of-care. Single-institution series, population-level studies, and meta-analyses have demonstrated a survival benefit for surgical resection, particularly if GTR is achieved (7-11). GTR is achievable in the majority of cases, as a surgical plane can often be identified between the tumor and cord parenchyma (11). For myxopapillary ependymoma, en bloc resection should be attempted, as capsular violation during resection is strongly associated with local recurrence (12).
Adjuvant radiotherapy for spinal ependymoma continues to be debated and is generally reserved for high grade, difficult-to-access, and partially resected tumors (13,14). Oh et al. conducted a literature review (adjuvant radiotherapy, n=47) and found that adjuvant radiotherapy prolonged progression-free survival (PFS) among spinal ependymoma patients undergoing subtotal resection (STR), while Lee et al., used data from the Korea Spinal Oncology Research Group database (adjuvant radiotherapy, n=20), did not observe this benefit (15,16). Studies of conventional chemotherapy for spinal ependymoma are rare. A small study on recurrent spinal ependymoma (n=10) found oral etoposide is well tolerated and may benefit some patients (17), but these results have not been further validated.
Molecular characterization
Classically, spinal ependymomas are thought to arise from ependymal cells in the central canal. However, there is increasing evidence that genes regulating radial glial cell differentiation play a role in molecular pathogenesis. Compared to supratentorial and posterior fossa ependymomas, spinal ependymomas overexpressed homeobox (HOX) family genes and insulin like growth factor 1 (IGF1). Distinct malignant transformations in regional radial glial cells also can give rise to anatomically and molecularly distinct ependymomas (18). These findings underscore the notion that cranial and spinal ependymomas should be considered separately, for both clinical management and biological investigation.
Pajtler et al. extended these findings by performing DNA methylation profiling on 500 ependymal tumors (19). The authors discovered nine discrete subgroups, including three subgroups of spinal ependymoma: subependymoma (SP-SE, WHO grade I), myxopapillary ependymoma (SP-MPE, WHO grade I), and (anaplastic) ependymoma (SP-EPN, WHO grade II/III). SP-SE harbored 6q deletions, while SP-MPE and SP-EPN demonstrated chromosomal instability. Moreover, most SP-EPN tumors had loss of the 22q locus, which harbors the neurofibromin (NF2) gene. In another study, whole exome sequencing of eight spinal cord ependymomas revealed loss of heterozygosity (LOH) of chromosome 22 in all eight tumors and somatic alterations in NF2 in 4/8 tumors (20). NF2 was then sequenced in an independent validation cohort of 32 intracranial ependymoma and 11 spinal ependymoma; alterations in NF2 were found in 9 of 19 spinal ependymomas (47%) and in 0 of 40 intracranial ependymomas (20). A recent study by Witt et al. sought to validate the ependymoma DNA methylation-based subtypes and correlate molecular subtypes with histologic subtypes (21). While all tumors were assigned to a previously defined molecular subtype, there was marked reassignment of spinal ependymomas, suggesting that molecular subtyping may enable more accurate risk assessment and precise clinical trial design (21).
Microarray studies, measuring mRNA expression, have also identified molecular differences between intracranial and extracranial ependymoma (22-24). Korshunov et al. profiled 39 central nervous system ependymomas (spinal, n=10) and reported increased expression of homeobox B5 (HOXB5), phospholipase A2 group 5 (PLA2G5), inter-αtrypsin inhibitor heavy chain 2 (ITIH2), and cyclin-dependent kinase inhibitor 2A (CDKN2A) in spinal ependymoma (24) (Table 1).
Table 1. Mutations associated with intramedullary spinal cord tumors (IMSCTs).
| Tumor | Gene | Locus | Grade | Molecular finding |
|---|---|---|---|---|
| Ependymoma | HOXB5 | 17q21.3 | I–II | Increased expression in spinal versus intracranial ependymoma |
| PLA2G5 | 1p35 | I–II | ||
| ITIH2 | 10p15 | I–II | ||
| CDKN2A | – | I–II | ||
| NF2 | 22q12 | II–III | Characteristic of spinal ependymoma; absent intracranial ependymoma | |
| – | 6q | I | Associated with spinal subependymoma | |
| Astrocytoma | BRAF | 7q34 | I–II | BRAF-KIAA1549 fusion and other somatic mutations identified in low-grade spinal astrocytoma |
| CDKN2A | 9q21 | I | Deletions in 2/10 spinal pilocytic astrocytoma | |
| H3F3A | 17q25 | III–IV | Mutation that discriminates grade III–IV spinal astrocytoma from grade I–II tumors | |
| Hemangioblastomas | VHL | 3p25.3 | – | Recent WES studies find VHL mutations in sporadic, as well as familial, spinal hemangioblastomas |
WES, whole exome sequencing; VHL, von Hippel-Landau.
Genotype-directed therapy and targeted agents
Achieving GTR in spinal ependymoma offers a durable treatment option with excellent survival outcomes. However, for patients in whom GTR is not technically feasible or for patients with higher grade tumors, other therapeutic options are necessary. Taken together, the rarity of spinal ependymomas and the dearth of recurrently aberrant targets have limited efforts to develop targeted therapies. One case report describes treatment with imatinib, a PDGF-receptor-targeted agent in a patient with recurrent spinal ependymoma expressing PDGF (25). Additionally, molecularly-guided therapies include mammalian target of rapamycin (mTOR) inhibitors and bevacizumab. Merlin, the protein encoded by NF2, has been shown to interact with rapamycin, thus forming the basis of mTOR inhibitors in the management of NF2-mutated ependymoma. A phase II trial evaluating the efficacy of everolimus, an mTOR inhibitor, in children with recurrent ependymoma (NCT02155920) was scheduled to end in July 2018 though no formal results have been published. Morris et al. recently described a retrospective review of 32 NF2 patients treated with bevacizumab who had spinal ependymomas. They observed clinical improvement in seven patients, all of whom had cystic spinal ependymomas. However, no assessment of survival benefit was reported (26). A recent multi-center retrospective review attempted to compare surgical management of NF2-associated spinal ependymomas with bevacizumab (27). Despite confounding variables inherent to the study design, the authors concluded that while resection may prevent neurological deterioration, bevacizumab may be beneficial for patients with significant tumor burden that is not amenable to resection (27).
Astrocytoma
Astrocytomas are the second most commonly observed IMSCT in adults but represent the most common pediatric IMSCT (Figure 3) (28,29). The incidence of spinal astrocytomas is slightly higher among males. Spinal astrocytomas are rarely observed caudal to the thoracic spine and appear on magnetic resonance imaging (MRI) as heterogeneously enhancing lesions within the parenchyma of the spinal cord.
Figure 3.

Spinal cord astrocytoma. Preoperative sagittal (A) MRI demonstrates T2 hyperintensity within the cord at T11, measuring 1.3 cm × 0.6 cm in craniocaudal and AP dimensions. Axial (B) image reveals the lesion expanding the cord, measuring 9 mm in transverse dimension.
Standard of care
Maximal, safe surgical resection is standard-of-care for patients with symptomatic spinal astrocytomas. In contrast to ependymomas, astrocytomas tend to be diffuse and infiltrative, lacking a good plane of dissection. Thus, achieving GTR must be balanced against iatrogenic neurological damage. Current evidence suggests that higher grade spinal astrocytomas are more infiltrative, further limiting the ability to achieve GTR (30). While the benefit of extent of resection is well-established in intracranial astrocytoma, the survival benefit of more aggressive resection in spinal astrocytoma is less evident (31-35). A recent multi-institutional study suggested that GTR is associated with longer PFS and overall survival (OS) (36). The authors observed higher post-operative McCormick scores, suggesting potential iatrogenic harm; however, a subgroup analysis of the change in McCormick score by extent of resection revealed no significant difference among groups (36).
Adjuvant radiotherapy for spinal astrocytomas remains controversial and is generally reserved for patients in whom GTR cannot be achieved. Single-series studies have generally concluded non-significance for adjuvant radiotherapy, whereas a multi-institutional study by Zou et al. found that radiotherapy may improve survival outcomes when adjusting for tumor grade (36). In contrast to intracranial astrocytoma, the benefit of temozolomide for spinal astrocytoma has been limited for both low-grade spinal astrocytoma and spinal glioblastoma (37-40). Bevacizumab has also been trialed in small series of patients with recurrent spinal glioblastoma with mixed results (38,41).
Molecular characterization
Large-scale sequencing studies of supratentorial gliomas have revealed distinct genomic alterations capable of discriminating pilocytic astrocytomas, WHO grade II and III diffuse gliomas, and WHO grade IV glioblastoma, with important differences observed between adult and pediatric tumors (42-47). While these discoveries have been used to enhance WHO classification of astrocytoma (48,49), the implications for spinal astrocytoma remain unclear.
Genomic studies of spinal astrocytoma are limited due to the rarity of these lesions. Mutations in the isocitrate dehydrogenase 1 (IDH1) gene are observed in 12% of intracranial glioblastoma and 50–80% of lower grade intracranial astrocytomas (47,50,51). However, in a series of spinal cord astrocytomas (n=9), the IDH1 R132H mutation was not observed (52). Shankar et al. performed targeted sequencing of adult and pediatric spinal cord astrocytomas (n=17) and observed IDH1 or IDH2 alterations in four patients. However, none represented recurrent IDH1 or IDH2 mutations previously described in adult glioma (53). It remains unknown if IDH mutations in spinal astrocytoma confer survival benefits.
Alterations in the proto-oncogene BRAF do appear to have correlates between intracranial and spinal astrocytoma. The majority (50–70%) of intracranial pilocytic astrocytoma harbor BRAF-KIAA1549 fusion genes (54-57). Shankar et al. report this fusion in 3/10 grade I spinal astrocytomas and also detected amplifications in BRAF in 5/10 specimens (53). Another recurrent alteration in intracranial astrocytoma is the BRAF V600E missense mutation, which occurs in 10–20% of pediatric pilocytic astrocytomas and is associated with poor outcome (58,59). The importance of this mutation in spinal astrocytoma remains unclear, with Shankar et al. reporting 0/17 spinal astrocytomas harboring this alteration (53). Deletion of CDKN2A may be another recurrent alteration in spinal pilocytic astrocytoma. Horbinski et al. (59) reported LOH at the 9p21 locus (which includes CDKN2A) in nearly one-third of pilocytic astrocytomas of the midbrain, brainstem, and spinal cord (spinal cord, n=9). Shankar et al. observed deletions of CDKN2A in 2/10 pilocytic astrocytomas (53).
In higher grade spinal astrocytomas (grade III and IV), histone 3 variant H3.3 (H3F3A) K27M mutations have been reported. Shankar et al. report the presence of H3F3A K27M in 6/7 grade III and IV and absence in grade I and II (0/15) (53). This mutation was initially reported in 2012 and found to be recurrently mutated in pediatric midline gliomas (44,60). Tumor suppressor protein 53 (TP53) has also been shown to be frequently altered in spinal cord glioblastomas (80–90%) (61).
Genotype-directed therapy and targeted agents
The BRAF alterations identified in WHO grade I and II spinal astrocytomas suggests that these tumors may be amenable to BRAF-MEK inhibitors. In keeping with this concept, the recent VE-BASKET study found evidence of durable antitumor activity of selective BRAFV600 inhibition in some patients with BRAFV600-mutant gliomas, though further investigation is needed to determine which patients are most likely to benefit (62). Patients with the H3F3A K27M spinal cord astrocytoma currently have exceedingly poor outcomes. Ongoing investigations of diffuse intrinsic pontine glioma (DIPG), which also harbors this mutation, suggest that inhibition of histone deacetylase and histone demethylase may be promising therapeutic avenues (63).
Hemangioblastoma
Spinal cord hemangioblastomas are vascular lesions which constitute 8–15% of IMSCTs and occur with equal frequency in men and women (64). Hemangioblastomas can be sporadic or familial, with familial hemangioblastomas associated with von Hippel-Landau (VHL) disease. VHL-associated hemangioblastomas account for 20–40% of presenting cases (65), with 20% of these lesions presenting in the spinal cord (65,66).
Standard of care
Patients with asymptomatic spinal cord hemangioblastomas can be managed with serial MRI and clinical evaluation. Given the relatively high incidence of spinal cord hemangioblastomas in patients with VHL disease, regular MRI screening is recommended (67). Though these vascular lesions arise from the pia, and are thus considered juxtamedullary, they may possess an intramedullary component. If a spinal cord hemangioblastoma hemorrhages, space-occupying hematomas can develop and lead to neurological deficits. Therefore, surgical resection is appropriate for patients with symptomatic spinal cord hemangioblastoma (68,69). Preoperative spinal angiography may be performed for surgical planning, but embolization is rarely indicated (1). Advances in microsurgical resection have improved the ability to achieve GTR of the lesion and associated mural nodule, while limiting post-operative complications (68). Stereotactic radiosurgery (SRS) for symptomatic spinal cord hemangioblastoma has delivered promising results, though results of long-term follow up remain limited (70,71).
Molecular characterization
VHL disease is characterized by an autosomal dominant pattern of inheritance, has 90% penetrance, and is caused by germline alterations in the eponymous tumor suppressor gene, VHL (72). This gene encodes an E3 ubiquitin ligase which has been shown to degrade hypoxia-inducible factor 1a (HIF-α), a well-described effector of vascular proliferation. Alterations in VHL limit a cell’s ability to ubiquitinate and degrade HIF-α, leading to increased angiogenesis (73). The pathogenesis of VHL disease is hypothesized to occur via a “two-hit” mechanism, where patients have a germline mutation in VHL and later develop a second somatic mutation, resulting in biallelic inactivation of VHL (74,75).
Glasker et al. reported that 94% of VHL-associated hemangioblastomas (n=29; spinal, n=4) express germline mutations in VHL, with 62% of these tumors harboring LOH at the VHL locus (3p25-56) (75). Interrogating sporadic hemangioblastomas (n=13; spinal, n=2), the authors found that 23% of these tumors possessed somatic mutations in VHL. Takayanagi et al. profiled a series of sporadic (n=21; spinal, n=2) and VHL-related (n=11; spinal, n=0) CNS hemangioblastomas, identifying VHL alterations in 100% of VHL-associated tumors and 62% of sporadic lesions (74). Moreover, the authors observed alterations specific to sporadic hemangioblastomas, namely VHL promoter hypermethylation (33% of sporadic tumors) and LOH of chromosome 6 or 10 (43% of sporadic tumors) (74). Shankar et al. performed whole-exome sequencing of 32 sporadic hemangioblastomas (spinal, n=9) and detected VHL-inactivating events in the majority of cases (78%). The recent studies by Takayanagi et al. and Shankar et al., using modern molecular profiling techniques, suggest that both VHL-inactivation is a key pathogenic mechanism in both VHL-associated and sporadic hemangioblastoma.
Genotype-directed therapy and targeted agents
Microsurgical resection and SRS are likely to continue to be mainstays in the management of spinal hemangioblastomas. However, genotype-informed therapies may play a role in surgically-challenging lesions and for patients with VHL-associated disease. To this end, bevacizumab has been used effectively in case reports of patients with unresectable spinal hemangioblastoma (76,77). Thalidomide, which demonstrates anti-angiogenic effects, has also been used in the management of a patient with progressive, multifocal spinal hemangioblastomas (78). Pazopanib, a tyrosine kinase inhibitor (TKI) which blocks the VEGF and PDGF pathways, was recently shown in a single-arm phase II trial (n=31) to achieve objective responses in 42% of patients with VHL disease. There were 49 CNS hemangioblastomas, only two of which demonstrated a response (79). Case reports of VHL patients with multiple CNS hemangioblastomas treated with pazopanib have shown varying results (80,81).
Intradural extramedullary (IDEM) spinal tumors
IDEM tumors include meningiomas, schwannomas, and neurofibromas. The majority of these tumors are benign, generally becoming symptomatic due to mass effect, and are managed with surgical resection. Presentation of multiple IDEM is often characteristic of underlying a genetic syndrome.
Meningioma
Meningiomas of the spine are IDEM tumors arising from meningothelial cells of the leptomeninges of the spinal cord and are the most common primary spinal tumor in adults (2) Spinal meningiomas occur most frequently in the thoracic spine and arise more commonly in females (Figure 4) (82). WHO classification of meningiomas consists of grade I (benign), grade II (atypical), and grade III (malignant). Meningiomas can be subtyped histologically, with psammomatous, meningothelial, and transitional subtypes most commonly observed in spinal meningiomas (83,84).
Figure 4.
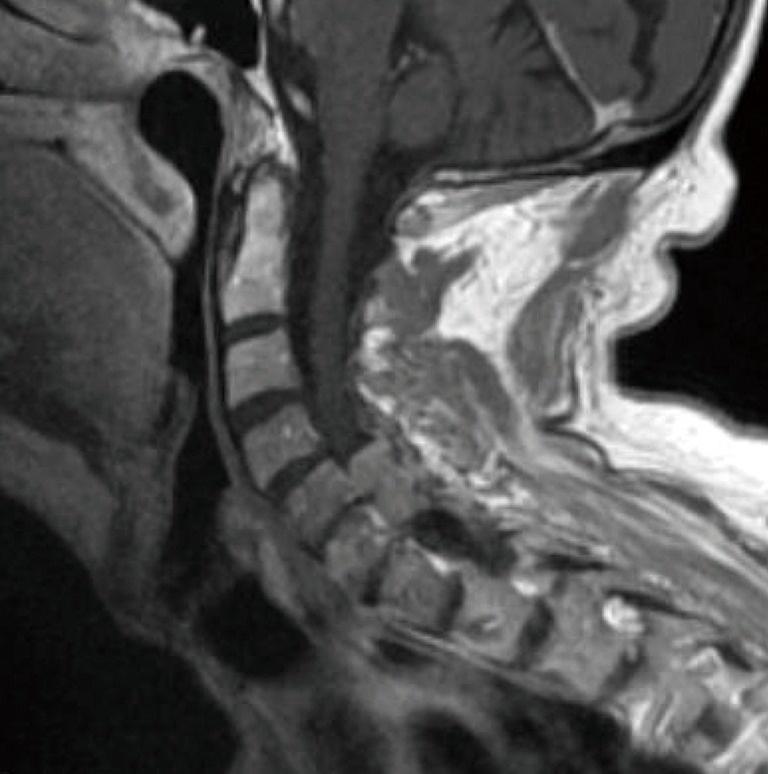
Spinal meningioma. T1-weighted MRI of the cervical spine reveals an extra-axial intraspinal soft tissue mass lesion with dural tail occupying the spinal canal at the C5–C6 level. The mass homogeneously enhances and exerts mass effect on the cord.
Current standard of care
Although spinal meningiomas are typically benign, they can result in mass effect and spinal cord compression. Therefore, surgical resection is standard-of-care and can be performed with limited rates of iatrogenic injury and low recurrence rates (1–4%) (84). The completeness of meningioma resection is evaluated by Simpson grading. Grade I is defined as complete removal of the tumor, including resection of underlying bone and associated dura. Grade II resection requires complete removal and coagulation of dural attachment. Grade III is defined as complete removal without dural resection or coagulation. Grade IV is STR and grade V is only surgical decompression. When possible, Simpson grade I resection, complete removal of the tumor and involved dura, should be the surgical goal. Nakamura et al. reported their experience with spinal meningiomas (n=62) in whom complete resection was achieved (Simpson grades I and II) and long-term follow up was available. Six patients of these patients (9.7%) recurred, all of whom had received Simpson grade II resection (85). However, extensive resection must be balanced with concern for operative morbidity. Kim et al. reported their experience with spinal meningiomas (Simpson grade I, n=21; Simpson grade II, n=20) and observed lower recurrence in the Simpson grade I group but a slightly increased rate of complications, described as neurological deterioration by the authors (86). Recently, Chin et al. reported long-term results of benign spinal tumors (meningioma, n=39) treated with SRS, observing 5-year and 10-year local failure rates of 2% and 8%, respectively (87). No SRS-related secondary malignancies were observed.
The use of adjuvant radiation for spinal meningiomas is controversial and generally limited to higher grade meningiomas or cases where complete resection cannot be achieved. Sun et al. investigated outcomes following surgical management of WHO grade II meningiomas (n=18) and found Simpson grade I–III resection without adjuvant radiation therapy could achieve symptomatic resolution and low recurrence (88).
Molecular characterization
Complete or partial loss of chromosome 22 is the most consistently reported genetic hallmark of spinal meningiomas, compared to intracranial meningiomas (89,90). Other cytogenetic abnormalities include loss of 1p, 9q, and 10q and gains of 5p and 17q (90). Notably, these cytogenetic abnormalities appear to be more frequently observed in atypical and anaplastic spinal meningiomas, compared to benign lesions. Barresi et al. used immunohistochemistry to assess hormone receptor and matrix metalloproteinase (MMP) immunoexpression in spinal meningiomas (n=58), observing that progesterone receptor immunoexpression in 86% of cases and elevated MMP-9 immunoexpression in 46% of cases (91).
Presentation with multiple meningiomas is often associated with NF2. However, Smith et al. identified cases of familial spinal meningiomas without NF2 mutations and performed whole-exome sequencing to identify germline mutations in a chromatin-remodeling gene, switch/sucrose, nonfermentable related, matrix associated, actin dependent regulator of chromatin, subfamily E, member 1 (SMARCE1) (92). Importantly, the tumors from patients with germline SMARCE1 mutations were all of clear-cell histology and had loss of SMARCE1 protein, supporting a tumor suppressor mechanism (92).
Large-scale sequencing studies of spinal meningiomas lag behind intracranial meningiomas, where potentially actionable mutations have been identified. Somatic mutations in cranial meningiomas have been identified in Smoothened (SMO), AKT1, Krüppel-like factor 4 (KLF4), TNF receptor associated factor 7 (TRAF7), and POLR2A (93-96), potentially forming the basis of targeted therapeutics. Recently, Juratli et al. identified deletions in DMD of patients with WHO grade II–III cranial meningiomas that appear to be associated with poor survival outcomes (97). However, the relevance of these findings to spinal meningiomas remains to be elucidated (Table 2).
Table 2. Mutations associated with intradural extramedullary (IDEM) tumors.
| Tumor | Gene | Locus | Grade | Molecular finding |
|---|---|---|---|---|
| Meningioma | – | 22 | I–III | Most consistently reported finding of spinal meningioma |
| MMP9 | 20q11.2 | I–III | Overexpressed in spinal meningiomas via IHC | |
| SMARCE1 | 17q21.2 | I–III | Familial spinal meningiomas in patients without NF2; all clear-cell histology | |
| Schwannoma | NF2 | 22q12 | – | Germline inactivating mutations |
| SMARCB1 | 22q11 | – | Germline alteration in ~50% of familial and ~10% of sporadic schwannomatosis cases | |
| LZTR1 | 22q11 | – | Germline LoF in majority of schwannomatosis cases lacking mutations in SMARCB1, but with loss of chromosome 22q in tumors | |
| ARID1A | 1p36.11 | – | Recurrent somatic mutations in sporadic schwannomas, identified by WES | |
| ARID1B | 6q25.3 | – | ||
| DDR1 | 6p21.33 | – | ||
| SH3PXD2A-HTRA1 | – | – | Recurrent fusion in 10% of sporadic schwannoma |
IHC, immunohistochemistry; LoF, loss-of-function; WES, whole-exome sequencing.
Genotype-directed therapy and targeted agents
While the discovery of somatic mutations in intracranial meningiomas is fostering investigation into targeted therapies (98), genotype-directed therapy of spinal meningiomas remains limited. Further studies interrogating the molecular features of spinal meningioma are likely necessary before effective neoadjuvant or adjuvant treatment strategies can be devised.
Schwannoma
Schwannomas arise from the transformation of Schwann cells, the myelin-producing cells of the peripheral nervous system and can develop in association with both cranial and spinal nerves. Neoplastic Schwann cells form two patterns: (I) compact, elongated cells with occasional nuclear palisading (Antoni A pattern) or (II) less cellular, loosely textured cells with indistinct processes (Antoni B) (99). Spinal schwannomas are most commonly sporadic and solitary (90%) but can also be familial in origin (Figure 5) (100). Multiple schwannomas increase suspicion for an underlying syndromic disorder, typically NF2 (and schwannomatosis (101). The vast majority of schwannomas are benign though malignant schwannomas have been observed and are characterized with markedly poor outcome (102).
Figure 5.

Spinal schwannoma. Preoperative axial (A) and sagittal (B) post-gadolinium MRI demonstrates a heterogeneously enhancing mass centered in the left C2–C3 neural foramen with intraspinal canal extension resulting in marked compression of cord.
Current standard of care
Maximal safe surgical resection is the standard-of-care for most cases of spinal schwannoma (103). Safaee et al. report their experience with spinal nerve sheath tumors (schwannoma, n=163), observing that GTR was achieved in 83% of patients and lower recurrence rate (4% versus 19%). Notably, GTR was less readily achieved in NF2-associated spinal schwannoma (104). SRS has also been used as first-line management of spinal schwannoma, with multiple case series demonstrating high rates of symptomatic improvement and local control with limited radiation-related adverse events (105-107). Adjuvant radiation can be considered in cases of STR, though the benefit of this therapy remains undetermined (104,108).
Molecular characterization
Patients with NF2-associated spinal schwannomas likely harbor germline inactivating mutations of NF2 and later accumulate a second alteration resulting in biallelic NF2 inactivation, providing a basis for transformation of Schwann cells into schwannomas. Familial schwannomatosis clinically presents with multiple schwannomas throughout the body, though generally sparing the vestibular nerve. This disorder is associated with constitutional alterations of SMARCB1, a tumor-suppressor gene also located also on chromosome 22 (109). SMARCB1 has been shown to be altered in roughly 50% of familial and nearly 10% of sporadic schwannomatosis cases (110). In schwannomatosis cases lacking mutations in SMARCB1, but with loss of chromosome 22q in tumors, germline loss-of-function mutations in LZTR1 were identified in 80% of cases (111).
To better characterize sporadic schwannomas, Agnihotri et al. performed DNA and RNA sequencing on 26 schwannomas (spinal, n=13) and targeted sequencing and methylation profiling of 125 schwannomas (spinal, n=61) (112). As expected loss of 22q was identified in 56% of spinal schwannomas by methylation profiling and found two methylation-based clusters, which almost uniformly segregated vestibular and spinal schwannomas. They identified recurrent somatic mutations in ARID1A, ARID1B and DDR1. RNA sequencing identified a recurrent in-frame SH3PXD2A-HTRA1 fusion in 10% (vestibular, n=7; spinal, n=5) of cases and demonstrated the fusion resulted from a balanced 19-Mb chromosomal inversion on chromosome 10q (112).
Genotype-directed therapy and targeted agents
Agnihotri et al. further observed that expression of the SH3PXD2A-HTRA1 fusion resulted in elevated phosphorylated ERK, leading to increased cellular proliferation, invasion, and in vivo tumorigenesis (112). The authors provided in vitro evidence that targeting the MEK-ERK pathway with a MEK inhibitor, trametinib, may be an effective therapeutic strategy (112). While data on NF2-associated spinal schwannomas is lacking, bevacizumab is being actively investigated in the management of NF2-associated vestibular schwannomas and is beginning to be explored in the management of patients with schwannomatosis (113-115).
PSCM
PSCM are a rare group of lesions which include chordoma, chondrosarcoma, osteosarcoma, and Ewing’s sarcoma. Chordoma and chondrosarcoma are indolent but locally invasive, while Ewing’s sarcoma and osteosarcoma are more aggressive and associated with higher rates of tumor progression and recurrence (116-118).
Chordoma
Chordomas are locally aggressive notochord-derived lesions of the axial skeleton and comprise 1–4% of all primary bone tumors (Figure 6) (119). They affect men more commonly than women and often present in the fifth and sixth decades of life (120). An analysis of the SEER database (n=400) revealed a median survival of 6.3 years with 10-year survival of 39.9% (120). Chordomas arise in the skull base (32%), mobile spine (32.8%), and sacrum (29.2%) (120).
Figure 6.

Spinal chordoma. Preoperative T2-weighted (A) and T1-weighted, fat-saturated, post-gadolinium (B) MRI reveals a large pre-vertebral mass extending from C2–C5, characterized by abnormal signal and enhancement in the C3 vertebral body.
Current standard of care
Despite chordoma’s indolent course, these lesions are locally aggressive and may present with large size at diagnosis, complicating management. Standard-of-care for spinal column chordomas is wide margin, en bloc surgical resection. Fuchs et al. demonstrated that patients with sacral chordoma who underwent radical resection had markedly longer time to local recurrence compared to those who underwent STR (2.3 years vs. 8 months, respectively) (121). These results have been confirmed in other series and in chordomas of the mobile spine (122,123). Among patients in the study by Fuchs et al. in whom a wide margin was achieved, 5-year local control rate was 100% (121). While en bloc resection has been consistently shown to have superior local control, this management approach introduces morbidity due to the critical neurovascular structures involved. En bloc resection can be achieved in roughly 50% of sacral chordomas with markedly lower rates of total resection in chordomas of the mobile spine (124). Adjuvant radiation therapy is appropriate and may have benefit when true oncologic margins have not been achieved (125). Definitive radiation therapy using high-dose photons or protons continues to be actively investigated, with recent results demonstrating 5-year local control rates of 85.4% (126). Conventional chemotherapy has shown little efficacy in the management of chordoma (124).
Molecular characterization
In 2009, Yang et al. identified unique duplications of 6q27 in four families with multiple members who had developed chordoma. The duplicated genomic region contains the transcription factor T (brachyury) gene, further providing evidence of the notochordal origin of chordoma (127). Pillay et al. conducted a genetic association study and identified a common nonsynonymous SNP, rs2305089, with a highly significant association with chordoma risk (allelic odds ratio =6.1) (128). This finding was confirmed in an independent study by Kelley et al., in both familial and sporadic chordoma. These authors identified another common variant, rs1056048, strongly associated with chordoma in families and an additional common variant, rs3816300, in sporadic cases significantly associated with risk when jointly analyzed with rs2305089 (129).
Recently, Tarpey et al. investigated the somatic mutational landscape of sporadic chordoma (n=104). They identified somatic duplications of brachyury in up to 27% of cases, PI3K signaling mutations in 16% of cases, and recurrent inactivation mutations in LYST in 10% of cases (130). An additional feature of chordomas is expression and activation of tyrosine kinase receptors and downstream signaling molecules, a finding which has been confirmed in multiple studies (131-134).
MicroRNA (miRNA) has also been explored in defining the molecular landscape of chordomas (135). miRNA is a 20–30 nucleotides, non-coding, single-stranded ribonucleic acid (RNA) molecule which can play oncogenic or tumor suppression roles by modulating gene regulation and transcription. Recently, Chen et al. profiled miRNA in chordomas, compared to fetal nucleus pulposus tissue, and found expression of hsa-miR-21-3p, hsa-miR-150-5p, hsa-miR-1290 and hsa-miR-623 to be upregulated (135). Further work is necessary to determine the mechanistic, prognostic, and therapeutic roles of miRNA in chordoma.
Genotype-directed therapy and targeted agents
The somatic mutations in PI3K signaling genes may form the basis of targeted agents for chordoma patients with these alterations. Further characterization of LYST mutations may also provide a basis for targeted chordoma therapies. Drawing on studies identifying activation of tyrosine kinase receptors in chordoma, a phase II study of imatinib in patients with advanced chordoma (n=50) found a 64% clinical benefit rate (136). Another promising targeted agent is afatinib, a TKI, as a recent in vitro study demonstrated this agent degraded both EGFR and brachyury (137). Other targeted agents that have been explored in larger studies of chordoma include lapatinib and sorafenib (138,139).
Recently, our group sought to determine the prognostic significance of the rs2305089 SNP. We genotyped this locus in 109 patients with spinal chordomas and found 102 patients (93.6%) harbored the A variant at this position. We observed that these patients had significantly improved OS compared with those lacking the variant but did not appreciate any association between SNP status and local recurrence-free survival (140). An additional molecular prognostic biomarker in spinal chordoma appears to be mutations in the hTERT promoter. Our group recently performed genotyping of 92 spinal chordomas and found that eight patients (8.7%) harbored mutations in the hTERT promoter region. Notably, all patients with hTERT mutations were alive at 10-year follow up, significantly better than patients lacking these mutations (141) (Table 3).
Table 3. Mutations associated with primary spinal column malignancies (PSCM).
| Tumor | Gene | Locus | Grade | Molecular finding |
|---|---|---|---|---|
| Chordoma | T | 6q27 | – | Multiple germline SNPs associated with development of familial and sporadic chordoma; rs2305089 shown to be prognostic for OS |
| LYST | 1q42.1-q42.2 | – | Recurrent inactivating somatic mutations in sporadic chordomas | |
| TERT | 5p15.33 | – | Mutations in TERT promoter shown to have prognostic implications |
OS, overall survival; SNP, single nucleotide polymorphism.
Conclusions
Primary spinal tumors are rare and diverse lesions that present unique clinical challenges due to their intimate relationship with the spinal cord and nerve roots. The current standard-of-care for spinal tumors involves maximal safe resection with adjuvant therapy for cases of residual tumor or recurrent disease. Dissection of molecular mechanisms and identification of genetic biomarkers has begun to influence the management of the cranial counterparts of spinal tumors by enhancing patient selection, improving prognostication, and forming the basis of targeted therapies. However, application of these discoveries to spinal tumors must proceed cautiously with rigorous validation—important differences have been identified between many cranial and spinal tumors of shared histology. Surgical resection will likely remain the mainstay of spinal tumors, but further interrogation of the molecular underpinnings of these lesions is necessary to identify improved neoadjuvant and adjuvant treatment and to better inform patient stratification and risk assessment.
Acknowledgments
None.
Footnotes
Conflicts of Interest: C Bettegowda is a consultant for Depuy-Synthes. The other authors have no conflicts of interest to declare.
References
- 1.Zadnik PL, Gokaslan ZL, Burger PC, et al. Spinal cord tumours: advances in genetics and their implications for treatment. Nat Rev Neurol 2013;9:257-66. 10.1038/nrneurol.2013.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duong LM, McCarthy BJ, McLendon RE, et al. Descriptive epidemiology of malignant and nonmalignant primary spinal cord, spinal meninges, and cauda equina tumors, United States, 2004-2007. Cancer 2012;118:4220-7. 10.1002/cncr.27390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Milano MT, Johnson MD, Sul J, et al. Primary spinal cord glioma: a Surveillance, Epidemiology, and End Results database study. J Neurooncol 2010;98:83-92. 10.1007/s11060-009-0054-7 [DOI] [PubMed] [Google Scholar]
- 4.Villano JL, Parker CK, Dolecek TA. Descriptive epidemiology of ependymal tumours in the United States. Br J Cancer 2013;108:2367-71. 10.1038/bjc.2013.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Evans DG. Neurofibromatosis type 2. Handb Clin Neurol 2015;132:87-96. 10.1016/B978-0-444-62702-5.00005-6 [DOI] [PubMed] [Google Scholar]
- 6.Parker M, Mohankumar KM, Punchihewa C, et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014;506:451-5. 10.1038/nature13109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Cai R, Wang R, et al. Outcome predictors in the management of intramedullary classic ependymoma: An integrative survival analysis. Medicine (Baltimore) 2018;97:e10870. 10.1097/MD.0000000000010870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Khalid SI, Adogwa O, Kelly R, et al. Adult Spinal Ependymomas: An Epidemiologic Study. World Neurosurg 2018;111:e53-61. 10.1016/j.wneu.2017.11.165 [DOI] [PubMed] [Google Scholar]
- 9.Harrop JS, Ganju A, Groff M, et al. Primary intramedullary tumors of the spinal cord. Spine (Phila Pa 1976) 2009;34:S69-77. 10.1097/BRS.0b013e3181b95c6f [DOI] [PubMed] [Google Scholar]
- 10.Epstein FJ, Farmer JP, Freed D. Adult intramedullary spinal cord ependymomas: the result of surgery in 38 patients. J Neurosurg 1993;79:204-9. 10.3171/jns.1993.79.2.0204 [DOI] [PubMed] [Google Scholar]
- 11.Wostrack M, Ringel F, Eicker SO, et al. Spinal ependymoma in adults: a multicenter investigation of surgical outcome and progression-free survival. J Neurosurg Spine 2018;28:654-62. 10.3171/2017.9.SPINE17494 [DOI] [PubMed] [Google Scholar]
- 12.Abdulaziz M, Mallory GW, Bydon M, et al. Outcomes following myxopapillary ependymoma resection: the importance of capsule integrity. Neurosurg Focus 2015;39:E8. 10.3171/2015.5.FOCUS15164 [DOI] [PubMed] [Google Scholar]
- 13.Volpp PB, Han K, Kagan AR, et al. Outcomes in treatment for intradural spinal cord ependymomas. Int J Radiat Oncol Biol Phys 2007;69:1199-204. 10.1016/j.ijrobp.2007.04.058 [DOI] [PubMed] [Google Scholar]
- 14.Ruda R, Reifenberger G, Frappaz D, et al. EANO guidelines for the diagnosis and treatment of ependymal tumors. Neuro Oncol 2018;20:445-56. 10.1093/neuonc/nox166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee SH, Chung CK, Kim CH, et al. Long-term outcomes of surgical resection with or without adjuvant radiation therapy for treatment of spinal ependymoma: a retrospective multicenter study by the Korea Spinal Oncology Research Group. Neuro Oncol 2013;15:921-9. 10.1093/neuonc/not038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Oh MC, Ivan ME, Sun MZ, et al. Adjuvant radiotherapy delays recurrence following subtotal resection of spinal cord ependymomas. Neuro Oncol 2013;15:208-15. 10.1093/neuonc/nos286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chamberlain MC. Etoposide for recurrent spinal cord ependymoma. Neurology 2002;58:1310-1. 10.1212/WNL.58.8.1310 [DOI] [PubMed] [Google Scholar]
- 18.Taylor MD, Poppleton H, Fuller C, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 2005;8:323-35. 10.1016/j.ccr.2005.09.001 [DOI] [PubMed] [Google Scholar]
- 19.Pajtler KW, Witt H, Sill M, et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015;27:728-43. 10.1016/j.ccell.2015.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bettegowda C, Agrawal N, Jiao Y, et al. Exomic sequencing of four rare central nervous system tumor types. Oncotarget 2013;4:572-83. 10.18632/oncotarget.964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Witt H, Gramatzki D, Hentschel B, et al. DNA methylation-based classification of ependymomas in adulthood: implications for diagnosis and treatment. Neuro Oncol 2018;20:1616-24. 10.1093/neuonc/noy118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palm T, Figarella-Branger D, Chapon F, et al. Expression profiling of ependymomas unravels localization and tumor grade-specific tumorigenesis. Cancer 2009;115:3955-68. 10.1002/cncr.24476 [DOI] [PubMed] [Google Scholar]
- 23.Lukashova-v Zangen I, Kneitz S, Monoranu CM, et al. Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol 2007;113:325-37. 10.1007/s00401-006-0190-5 [DOI] [PubMed] [Google Scholar]
- 24.Korshunov A, Neben K, Wrobel G, et al. Gene expression patterns in ependymomas correlate with tumor location, grade, and patient age. Am J Pathol 2003;163:1721-7. 10.1016/S0002-9440(10)63530-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fakhrai N, Neophytou P, Dieckmann K, et al. Recurrent spinal ependymoma showing partial remission under Imatimib. Acta Neurochir (Wien) 2004;146:1255-8. 10.1007/s00701-004-0374-5 [DOI] [PubMed] [Google Scholar]
- 26.Morris KA, Afridi SK, Evans DG, et al. The response of spinal cord ependymomas to bevacizumab in patients with neurofibromatosis Type 2. J Neurosurg Spine 2017;26:474-82. 10.3171/2016.8.SPINE16589 [DOI] [PubMed] [Google Scholar]
- 27.Kalamarides M, Essayed W, Lejeune JP, et al. Spinal ependymomas in NF2: a surgical disease? J Neurooncol 2018;136:605-11. 10.1007/s11060-017-2690-7 [DOI] [PubMed] [Google Scholar]
- 28.Azad TD, Pendharkar AV, Pan J, et al. Surgical outcomes of pediatric spinal cord astrocytomas: systematic review and meta-analysis. J Neurosurg Pediatr 2018;22:404-10. 10.3171/2018.4.PEDS17587 [DOI] [PubMed] [Google Scholar]
- 29.Luksik AS, Garzon-Muvdi T, Yang W, et al. Pediatric spinal cord astrocytomas: a retrospective study of 348 patients from the SEER database. J Neurosurg Pediatr 2017;19:711-9. 10.3171/2017.1.PEDS16528 [DOI] [PubMed] [Google Scholar]
- 30.Karikari IO, Nimjee SM, Hodges TR, et al. Impact of tumor histology on resectability and neurological outcome in primary intramedullary spinal cord tumors: a single-center experience with 102 patients. Neurosurgery 2015;76 Suppl 1:S4-13; discussion S13. [DOI] [PubMed]
- 31.Xiao R, Abdullah KG, Miller JA, et al. Molecular and clinical prognostic factors for favorable outcome following surgical resection of adult intramedullary spinal cord astrocytomas. Clin Neurol Neurosurg 2016;144:82-7. 10.1016/j.clineuro.2016.03.009 [DOI] [PubMed] [Google Scholar]
- 32.Adams H, Avendano J, Raza SM, et al. Prognostic factors and survival in primary malignant astrocytomas of the spinal cord: a population-based analysis from 1973 to 2007. Spine (Phila Pa 1976) 2012;37:E727-35. 10.1097/BRS.0b013e31824584c0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Minehan KJ, Brown PD, Scheithauer BW, et al. Prognosis and treatment of spinal cord astrocytoma. Int J Radiat Oncol Biol Phys 2009;73:727-33. 10.1016/j.ijrobp.2008.04.060 [DOI] [PubMed] [Google Scholar]
- 34.Kim MS, Chung CK, Choe G, et al. Intramedullary spinal cord astrocytoma in adults: postoperative outcome. J Neurooncol 2001;52:85-94. 10.1023/A:1010680924975 [DOI] [PubMed] [Google Scholar]
- 35.Babu R, Karikari IO, Owens TR, et al. Spinal cord astrocytomas: a modern 20-year experience at a single institution. Spine (Phila Pa 1976) 2014;39:533-40. 10.1097/BRS.0000000000000190 [DOI] [PubMed] [Google Scholar]
- 36.Zou Y, Sun J, Zhou Y, et al. Prognostic Factors and Treatment of Spinal Astrocytomas: A Multi-institutional Cohort Analysis. Spine (Phila Pa 1976) 2018;43:E565-73. 10.1097/BRS.0000000000002485 [DOI] [PubMed] [Google Scholar]
- 37.Hernandez-Duran S, Bregy A, Shah AH, et al. Primary spinal cord glioblastoma multiforme treated with temozolomide. J Clin Neurosci 2015;22:1877-82. 10.1016/j.jocn.2015.04.017 [DOI] [PubMed] [Google Scholar]
- 38.Kaley TJ, Mondesire-Crump I, Gavrilovic IT. Temozolomide or bevacizumab for spinal cord high-grade gliomas. J Neurooncol 2012;109:385-9. 10.1007/s11060-012-0905-5 [DOI] [PubMed] [Google Scholar]
- 39.Kim WH, Yoon SH, Kim CY, et al. Temozolomide for malignant primary spinal cord glioma: an experience of six cases and a literature review. J Neurooncol 2011;101:247-54. 10.1007/s11060-010-0249-y [DOI] [PubMed] [Google Scholar]
- 40.Chamberlain MC. Temozolomide for recurrent low-grade spinal cord gliomas in adults. Cancer 2008;113:1019-24. 10.1002/cncr.23677 [DOI] [PubMed] [Google Scholar]
- 41.Chamberlain MC, Johnston SK. Recurrent spinal cord glioblastoma: salvage therapy with bevacizumab. J Neurooncol 2011;102:427-32. 10.1007/s11060-010-0330-6 [DOI] [PubMed] [Google Scholar]
- 42.Jones DT, Hutter B, Jager N, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet 2013;45:927-32. 10.1038/ng.2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang J, Wu G, Miller CP, et al. Whole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet 2013;45:602-12. 10.1038/ng.2611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schwartzentruber J, Korshunov A, Liu XY, et al. Driver mutations in histone H3.3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012;482:226-31. 10.1038/nature10833 [DOI] [PubMed] [Google Scholar]
- 45.Cancer Genome Atlas Research Network , Brat DJ, Verhaak RG, et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med 2015;372:2481-98. 10.1056/NEJMoa1402121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008;455:1061-8. 10.1038/nature07385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ceccarelli M, Barthel FP, Malta TM, et al. Molecular Profiling Reveals Biologically Discrete Subsets and Pathways of Progression in Diffuse Glioma. Cell 2016;164:550-63. 10.1016/j.cell.2015.12.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eckel-Passow JE, Lachance DH, Molinaro AM, et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med 2015;372:2499-508. 10.1056/NEJMoa1407279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louis DN, Perry A, Reifenberger G, et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol 2016;131:803-20. 10.1007/s00401-016-1545-1 [DOI] [PubMed] [Google Scholar]
- 50.Tateishi K, Wakimoto H, Cahill DP. IDH1 Mutation and World Health Organization 2016 Diagnostic Criteria for Adult Diffuse Gliomas: Advances in Surgical Strategy. Neurosurgery 2017;64:134-8. 10.1093/neuros/nyx247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Parsons DW, Jones S, Zhang X, et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008;321:1807-12. 10.1126/science.1164382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ellezam B, Theeler BJ, Walbert T, et al. Low rate of R132H IDH1 mutation in infratentorial and spinal cord grade II and III diffuse gliomas. Acta Neuropathol 2012;124:449-51. 10.1007/s00401-012-1011-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shankar GM, Lelic N, Gill CM, et al. BRAF alteration status and the histone H3F3A gene K27M mutation segregate spinal cord astrocytoma histology. Acta Neuropathol 2016;131:147-50. 10.1007/s00401-015-1492-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lin A, Rodriguez FJ, Karajannis MA, et al. BRAF alterations in primary glial and glioneuronal neoplasms of the central nervous system with identification of 2 novel KIAA1549:BRAF fusion variants. J Neuropathol Exp Neurol 2012;71:66-72. 10.1097/NEN.0b013e31823f2cb0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu J, Deshmukh H, Gutmann RJ, et al. Alterations of BRAF and HIPK2 loci predominate in sporadic pilocytic astrocytoma. Neurology 2009;73:1526-31. 10.1212/WNL.0b013e3181c0664a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jones DT, Kocialkowski S, Liu L, et al. Tandem duplication producing a novel oncogenic BRAF fusion gene defines the majority of pilocytic astrocytomas. Cancer Res 2008;68:8673-7. 10.1158/0008-5472.CAN-08-2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pfister S, Janzarik WG, Remke M, et al. BRAF gene duplication constitutes a mechanism of MAPK pathway activation in low-grade astrocytomas. J Clin Invest 2008;118:1739-49. 10.1172/JCI33656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lassaletta A, Zapotocky M, Mistry M, et al. Therapeutic and Prognostic Implications of BRAF V600E in Pediatric Low-Grade Gliomas. J Clin Oncol 2017;35:2934-41. 10.1200/JCO.2016.71.8726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Horbinski C, Nikiforova MN, Hagenkord JM, et al. Interplay among BRAF, p16, p53, and MIB1 in pediatric low-grade gliomas. Neuro Oncol 2012;14:777-89. 10.1093/neuonc/nos077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu G, Broniscer A, McEachron TA, et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat Genet 2012;44:251-3. 10.1038/ng.1102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nagaishi M, Nobusawa S, Yokoo H, et al. Genetic mutations in high grade gliomas of the adult spinal cord. Brain Tumor Pathol 2016;33:267-9. 10.1007/s10014-016-0263-7 [DOI] [PubMed] [Google Scholar]
- 62.Kaley T, Touat M, Subbiah V, et al. BRAF Inhibition in BRAFV600-Mutant Gliomas: Results From the VE-BASKET Study. J Clin Oncol 2018. doi: . 10.1200/JCO.2018.78.9990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grasso CS, Tang Y, Truffaux N, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med 2015;21:555-9. 10.1038/nm.3855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Westwick HJ, Giguere JF, Shamji MF. Incidence and Prognosis of Spinal Hemangioblastoma: A Surveillance Epidemiology and End Results Study. Neuroepidemiology 2016;46:14-23. 10.1159/000441147 [DOI] [PubMed] [Google Scholar]
- 65.Glasker S. Central nervous system manifestations in VHL: genetics, pathology and clinical phenotypic features. Fam Cancer 2005;4:37-42. 10.1007/s10689-004-5347-6 [DOI] [PubMed] [Google Scholar]
- 66.Ostrom QT, Gittleman H, Farah P, et al. CBTRUS statistical report: Primary brain and central nervous system tumors diagnosed in the United States in 2006-2010. Neuro Oncol 2013;15 Suppl 2:ii1-56. 10.1093/neuonc/not151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lonser RR, Glenn GM, Walther M, et al. von Hippel-Lindau disease. Lancet 2003;361:2059-67. 10.1016/S0140-6736(03)13643-4 [DOI] [PubMed] [Google Scholar]
- 68.Mandigo CE, Ogden AT, Angevine PD, et al. Operative management of spinal hemangioblastoma. Neurosurgery 2009;65:1166-77. 10.1227/01.NEU.0000359306.74674.C4 [DOI] [PubMed] [Google Scholar]
- 69.Na JH, Kim HS, Eoh W, et al. Spinal cord hemangioblastoma: diagnosis and clinical outcome after surgical treatment. J Korean Neurosurg Soc 2007;42:436-40. 10.3340/jkns.2007.42.6.436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bridges KJ, Jaboin JJ, Kubicky CD, et al. Stereotactic radiosurgery versus surgical resection for spinal hemangioblastoma: A systematic review. Clin Neurol Neurosurg 2017;154:59-66. 10.1016/j.clineuro.2017.01.012 [DOI] [PubMed] [Google Scholar]
- 71.Pan J, Jabarkheel R, Huang Y, et al. Stereotactic radiosurgery for central nervous system hemangioblastoma: systematic review and meta-analysis. J Neurooncol 2018;137:11-22. 10.1007/s11060-017-2697-0 [DOI] [PubMed] [Google Scholar]
- 72.Latif F, Tory K, Gnarra J, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993;260:1317-20. 10.1126/science.8493574 [DOI] [PubMed] [Google Scholar]
- 73.Maxwell PH, Wiesener MS, Chang GW, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999;399:271-5. 10.1038/20459 [DOI] [PubMed] [Google Scholar]
- 74.Takayanagi S, Mukasa A, Tanaka S, et al. Differences in genetic and epigenetic alterations between von Hippel-Lindau disease-related and sporadic hemangioblastomas of the central nervous system. Neuro Oncol 2017;19:1228-36. 10.1093/neuonc/nox034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Glasker S, Bender BU, Apel TW, et al. Reconsideration of biallelic inactivation of the VHL tumour suppressor gene in hemangioblastomas of the central nervous system. J Neurol Neurosurg Psychiatry 2001;70:644-8. 10.1136/jnnp.70.5.644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Omar AI. Bevacizumab for the treatment of surgically unresectable cervical cord hemangioblastoma: a case report. J Med Case Rep 2012;6:238. 10.1186/1752-1947-6-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Riklin C, Seystahl K, Hofer S, et al. Antiangiogenic treatment for multiple CNS hemangioblastomas. Onkologie 2012;35:443-5. 10.1159/000341075 [DOI] [PubMed] [Google Scholar]
- 78.Sardi I, Sanzo M, Giordano F, et al. Monotherapy with thalidomide for treatment of spinal cord hemangioblastomas in a patient with von Hippel-Lindau disease. Pediatr Blood Cancer 2009;53:464-7. 10.1002/pbc.22065 [DOI] [PubMed] [Google Scholar]
- 79.Jonasch E, McCutcheon IE, Gombos DS, et al. Pazopanib in patients with von Hippel-Lindau disease: a single-arm, single-centre, phase 2 trial. Lancet Oncol 2018;19:1351-9. 10.1016/S1470-2045(18)30487-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Migliorini D, Haller S, Merkler D, et al. Recurrent multiple CNS hemangioblastomas with VHL disease treated with pazopanib: a case report and literature review. CNS Oncol 2015;4:387-92. 10.2217/cns.15.22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Taylor DG, Ilyas A, Mehta GU, et al. Variable response of CNS hemangioblastomas to Pazopanib in a single patient with von Hippel-Lindau disease: Case report. J Clin Neurosci 2018;50:154-6. 10.1016/j.jocn.2018.01.040 [DOI] [PubMed] [Google Scholar]
- 82.Maiuri F, De Caro ML, de Divitiis O, et al. Spinal meningiomas: age-related features. Clin Neurol Neurosurg 2011;113:34-8. 10.1016/j.clineuro.2010.08.017 [DOI] [PubMed] [Google Scholar]
- 83.Sandalcioglu IE, Hunold A, Muller O, et al. Spinal meningiomas: critical review of 131 surgically treated patients. Eur Spine J 2008;17:1035-41. 10.1007/s00586-008-0685-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Setzer M, Vatter H, Marquardt G, et al. Management of spinal meningiomas: surgical results and a review of the literature. Neurosurg Focus 2007;23:E14. 10.3171/FOC-07/10/E14 [DOI] [PubMed] [Google Scholar]
- 85.Nakamura M, Tsuji O, Fujiyoshi K, et al. Long-term surgical outcomes of spinal meningiomas. Spine (Phila Pa 1976) 2012;37:E617-23. 10.1097/BRS.0b013e31824167f1 [DOI] [PubMed] [Google Scholar]
- 86.Kim CH, Chung CK, Lee SH, et al. Long-term recurrence rates after the removal of spinal meningiomas in relation to Simpson grades. Eur Spine J 2016;25:4025-32. 10.1007/s00586-015-4306-2 [DOI] [PubMed] [Google Scholar]
- 87.Chin AL, Fujimoto D, Kumar KA, et al. Long-Term Update of Stereotactic Radiosurgery for Benign Spinal Tumors. Neurosurgery 2018. [Epub ahead of print]. 10.1093/neuros/nyy442 [DOI] [PubMed] [Google Scholar]
- 88.Sun SQ, Cai C, Ravindra VM, et al. Simpson Grade I-III Resection of Spinal Atypical (World Health Organization Grade II) Meningiomas is Associated With Symptom Resolution and Low Recurrence. Neurosurgery 2015;76:739-46. 10.1227/NEU.0000000000000720 [DOI] [PubMed] [Google Scholar]
- 89.Sayagues JM, Tabernero MD, Maillo A, et al. Microarray-based analysis of spinal versus intracranial meningiomas: different clinical, biological, and genetic characteristics associated with distinct patterns of gene expression. J Neuropathol Exp Neurol 2006;65:445-54. 10.1097/01.jnen.0000229234.13372.d8 [DOI] [PubMed] [Google Scholar]
- 90.Arslantas A, Artan S, Oner U, et al. Detection of chromosomal imbalances in spinal meningiomas by comparative genomic hybridization. Neurol Med Chir (Tokyo) 2003;43:12-8; discussion 19. 10.2176/nmc.43.12 [DOI] [PubMed] [Google Scholar]
- 91.Barresi V, Alafaci C, Caffo M, et al. Clinicopathological characteristics, hormone receptor status and matrix metallo-proteinase-9 (MMP-9) immunohistochemical expression in spinal meningiomas. Pathol Res Pract 2012;208:350-5. 10.1016/j.prp.2012.02.013 [DOI] [PubMed] [Google Scholar]
- 92.Smith MJ, O'Sullivan J, Bhaskar SS, et al. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat Genet 2013;45:295-8. 10.1038/ng.2552 [DOI] [PubMed] [Google Scholar]
- 93.Harmanci AS, Youngblood MW, Clark VE, et al. Integrated genomic analyses of de novo pathways underlying atypical meningiomas. Nat Commun 2017;8:14433. 10.1038/ncomms14433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Clark VE, Harmanci AS, Bai H, et al. Recurrent somatic mutations in POLR2A define a distinct subset of meningiomas. Nat Genet 2016;48:1253-9. 10.1038/ng.3651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Clark VE, Erson-Omay EZ, Serin A, et al. Genomic analysis of non-NF2 meningiomas reveals mutations in TRAF7, KLF4, AKT1, and SMO. Science 2013;339:1077-80. 10.1126/science.1233009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brastianos PK, Horowitz PM, Santagata S, et al. Genomic sequencing of meningiomas identifies oncogenic SMO and AKT1 mutations. Nat Genet 2013;45:285-9. 10.1038/ng.2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Juratli TA, McCabe D, Nayyar N, et al. DMD genomic deletions characterize a subset of progressive/higher-grade meningiomas with poor outcome. Acta Neuropathol 2018;136:779-92. 10.1007/s00401-018-1899-7 [DOI] [PubMed] [Google Scholar]
- 98.Gupta S, Bi WL, Dunn IF. Medical management of meningioma in the era of precision medicine. Neurosurg Focus 2018;44:E3. 10.3171/2018.1.FOCUS17754 [DOI] [PubMed] [Google Scholar]
- 99.Louis DN, Ohgaki H, Wiestler OD, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 2007;114:97-109. 10.1007/s00401-007-0243-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Antinheimo J, Sankila R, Carpen O, et al. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology 2000;54:71-6. 10.1212/WNL.54.1.71 [DOI] [PubMed] [Google Scholar]
- 101.Christiaans I, Kenter SB, Brink HC, et al. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet 2011;48:93-7. 10.1136/jmg.2010.082420 [DOI] [PubMed] [Google Scholar]
- 102.Guha D, Davidson B, Nadi M, et al. Management of peripheral nerve sheath tumors: 17 years of experience at Toronto Western Hospital. J Neurosurg 2018;128:1226-34. 10.3171/2017.1.JNS162292 [DOI] [PubMed] [Google Scholar]
- 103.Fehlings MG, Nater A, Zamorano JJ, et al. Risk Factors for Recurrence of Surgically Treated Conventional Spinal Schwannomas: Analysis of 169 Patients From a Multicenter International Database. Spine (Phila Pa 1976) 2016;41:390-8. 10.1097/BRS.0000000000001232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Safaee M, Parsa AT, Barbaro NM, et al. Association of tumor location, extent of resection, and neurofibromatosis status with clinical outcomes for 221 spinal nerve sheath tumors. Neurosurg Focus 2015;39:E5. 10.3171/2015.5.FOCUS15183 [DOI] [PubMed] [Google Scholar]
- 105.Shin DW, Sohn MJ, Kim HS, et al. Clinical analysis of spinal stereotactic radiosurgery in the treatment of neurogenic tumors. J Neurosurg Spine 2015;23:429-37. 10.3171/2015.1.SPINE14910 [DOI] [PubMed] [Google Scholar]
- 106.Marchetti M, De Martin E, Milanesi I, et al. Intradural extramedullary benign spinal lesions radiosurgery. Medium- to long-term results from a single institution experience. Acta Neurochir (Wien) 2013;155:1215-22. 10.1007/s00701-013-1756-3 [DOI] [PubMed] [Google Scholar]
- 107.Sachdev S, Dodd RL, Chang SD, et al. Stereotactic radiosurgery yields long-term control for benign intradural, extramedullary spinal tumors. Neurosurgery 2011;69:533-9; discussion 539. 10.1227/NEU.0b013e318218db23 [DOI] [PubMed] [Google Scholar]
- 108.Cavalcanti DD, Martirosyan NL, Verma K, et al. Surgical management and outcome of schwannomas in the craniocervical region. J Neurosurg 2011;114:1257-67. 10.3171/2010.5.JNS0966 [DOI] [PubMed] [Google Scholar]
- 109.Hulsebos TJ, Plomp AS, Wolterman RA, et al. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet 2007;80:805-10. 10.1086/513207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Smith MJ, Wallace AJ, Bowers NL, et al. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics 2012;13:141-5. 10.1007/s10048-012-0319-8 [DOI] [PubMed] [Google Scholar]
- 111.Piotrowski A, Xie J, Liu YF, et al. Germline loss-of-function mutations in LZTR1 predispose to an inherited disorder of multiple schwannomas. Nat Genet 2014;46:182-7. 10.1038/ng.2855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Agnihotri S, Jalali S, Wilson MR, et al. The genomic landscape of schwannoma. Nat Genet 2016;48:1339-48. 10.1038/ng.3688 [DOI] [PubMed] [Google Scholar]
- 113.Blakeley JO, Ye X, Duda DG, et al. Efficacy and Biomarker Study of Bevacizumab for Hearing Loss Resulting From Neurofibromatosis Type 2-Associated Vestibular Schwannomas. J Clin Oncol 2016;34:1669-75. 10.1200/JCO.2015.64.3817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Plotkin SR, Stemmer-Rachamimov AO, Barker FG, 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med 2009;361:358-67. 10.1056/NEJMoa0902579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Blakeley J, Schreck KC, Evans DG, et al. Clinical response to bevacizumab in schwannomatosis. Neurology 2014;83:1986-7. 10.1212/WNL.0000000000000997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Schoenfeld AJ, Hornicek FJ, Pedlow FX, et al. Chondrosarcoma of the mobile spine: a review of 21 cases treated at a single center. Spine (Phila Pa 1976) 2012;37:119-26. 10.1097/BRS.0b013e31823d2143 [DOI] [PubMed] [Google Scholar]
- 117.Schoenfeld AJ, Hornicek FJ, Pedlow FX, et al. Osteosarcoma of the spine: experience in 26 patients treated at the Massachusetts General Hospital. Spine J 2010;10:708-14. 10.1016/j.spinee.2010.05.017 [DOI] [PubMed] [Google Scholar]
- 118.Boriani S, Bandiera S, Biagini R, et al. Chordoma of the mobile spine: fifty years of experience. Spine (Phila Pa 1976) 2006;31:493-503. 10.1097/01.brs.0000200038.30869.27 [DOI] [PubMed] [Google Scholar]
- 119.Healey JH, Lane JM. Chordoma: a critical review of diagnosis and treatment. Orthop Clin North Am 1989;20:417-26. [PubMed] [Google Scholar]
- 120.McMaster ML, Goldstein AM, Bromley CM, et al. Chordoma: incidence and survival patterns in the United States, 1973-1995. Cancer Causes Control 2001;12:1-11. 10.1023/A:1008947301735 [DOI] [PubMed] [Google Scholar]
- 121.Fuchs B, Dickey ID, Yaszemski MJ, et al. Operative management of sacral chordoma. J Bone Joint Surg Am 2005;87:2211-6. [DOI] [PubMed] [Google Scholar]
- 122.Stacchiotti S, Casali PG, Lo Vullo S, et al. Chordoma of the mobile spine and sacrum: a retrospective analysis of a series of patients surgically treated at two referral centers. Ann Surg Oncol 2010;17:211-9. 10.1245/s10434-009-0740-x [DOI] [PubMed] [Google Scholar]
- 123.Hsieh PC, Xu R, Sciubba DM, et al. Long-term clinical outcomes following en bloc resections for sacral chordomas and chondrosarcomas: a series of twenty consecutive patients. Spine (Phila Pa 1976) 2009;34:2233-9. 10.1097/BRS.0b013e3181b61b90 [DOI] [PubMed] [Google Scholar]
- 124.Walcott BP, Nahed BV, Mohyeldin A, et al. Chordoma: current concepts, management, and future directions. Lancet Oncol 2012;13:e69-76. 10.1016/S1470-2045(11)70337-0 [DOI] [PubMed] [Google Scholar]
- 125.Potluri S, Jefferies SJ, Jena R, et al. Residual postoperative tumour volume predicts outcome after high-dose radiotherapy for chordoma and chondrosarcoma of the skull base and spine. Clin Oncol (R Coll Radiol) 2011;23:199-208. 10.1016/j.clon.2010.09.011 [DOI] [PubMed] [Google Scholar]
- 126.Kabolizadeh P, Chen YL, Liebsch N, et al. Updated Outcome and Analysis of Tumor Response in Mobile Spine and Sacral Chordoma Treated With Definitive High-Dose Photon/Proton Radiation Therapy. Int J Radiat Oncol Biol Phys 2017;97:254-62. 10.1016/j.ijrobp.2016.10.006 [DOI] [PubMed] [Google Scholar]
- 127.Yang XR, Ng D, Alcorta DA, et al. T (brachyury) gene duplication confers major susceptibility to familial chordoma. Nat Genet 2009;41:1176-8. 10.1038/ng.454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Pillay N, Plagnol V, Tarpey PS, et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet 2012;44:1185-7. 10.1038/ng.2419 [DOI] [PubMed] [Google Scholar]
- 129.Kelley MJ, Shi J, Ballew B, et al. Characterization of T gene sequence variants and germline duplications in familial and sporadic chordoma. Hum Genet 2014;133:1289-97. 10.1007/s00439-014-1463-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Tarpey PS, Behjati S, Young MD, et al. The driver landscape of sporadic chordoma. Nat Commun 2017;8:890. 10.1038/s41467-017-01026-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.de Castro CV, Guimaraes G, Aguiar S, Jr, et al. Tyrosine kinase receptor expression in chordomas: phosphorylated AKT correlates inversely with outcome. Hum Pathol 2013;44:1747-55. 10.1016/j.humpath.2012.11.024 [DOI] [PubMed] [Google Scholar]
- 132.Shalaby A, Presneau N, Ye H, et al. The role of epidermal growth factor receptor in chordoma pathogenesis: a potential therapeutic target. J Pathol 2011;223:336-46. 10.1002/path.2818 [DOI] [PubMed] [Google Scholar]
- 133.Tamborini E, Virdis E, Negri T, et al. Analysis of receptor tyrosine kinases (RTKs) and downstream pathways in chordomas. Neuro Oncol 2010;12:776-89. 10.1093/neuonc/noq003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tamborini E, Miselli F, Negri T, et al. Molecular and biochemical analyses of platelet-derived growth factor receptor (PDGFR) B, PDGFRA, and KIT receptors in chordomas. Clin Cancer Res 2006;12:6920-8. 10.1158/1078-0432.CCR-06-1584 [DOI] [PubMed] [Google Scholar]
- 135.Chen K, Chen H, Zhang K, et al. MicroRNA profiling and bioinformatics analyses reveal the potential roles of microRNAs in chordoma. Oncol Lett 2017;14:5533-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Stacchiotti S, Longhi A, Ferraresi V, et al. Phase II study of imatinib in advanced chordoma. J Clin Oncol 2012;30:914-20. 10.1200/JCO.2011.35.3656 [DOI] [PubMed] [Google Scholar]
- 137.Magnaghi P, Salom B, Cozzi L, et al. Afatinib Is a New Therapeutic Approach in Chordoma with a Unique Ability to Target EGFR and Brachyury. Mol Cancer Ther 2018;17:603-13. 10.1158/1535-7163.MCT-17-0324 [DOI] [PubMed] [Google Scholar]
- 138.Bompas E, Le Cesne A, Tresch-Bruneel E, et al. Sorafenib in patients with locally advanced and metastatic chordomas: a phase II trial of the French Sarcoma Group (GSF/GETO). Ann Oncol 2015;26:2168-73. 10.1093/annonc/mdv300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Stacchiotti S, Tamborini E, Lo Vullo S, et al. Phase II study on lapatinib in advanced EGFR-positive chordoma. Ann Oncol 2013;24:1931-6. 10.1093/annonc/mdt117 [DOI] [PubMed] [Google Scholar]
- 140.Bettegowda C, Yip S, Lo SL, et al. Spinal column chordoma: prognostic significance of clinical variables and T (brachyury) gene SNP rs2305089 for local recurrence and overall survival. Neuro Oncol 2017;19:405-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bettegowda C, Yip S, Jiang B, et al. Prognostic significance of hTERT (human telomerase reverse transcriptase) promoter region mutations C228T and C250T for overall survival in spinal chordomas. Neuro Oncol 2019. [Epub ahead of print]. 10.1093/neuonc/noz066 [DOI] [PMC free article] [PubMed] [Google Scholar]