

Abstract
Aims
Rivaroxaban exposure is considerably increased by drugs that are combined P‐glycoprotein (P‐gp) and strong cytochrome P450 (CYP) 3A inhibitors (e.g. ketoconazole). The aim of the present study was to investigate the effects of the potent P‐gp inhibitor ciclosporin and its combination with the moderate CYP3A inhibitor fluconazole on rivaroxaban pharmacokinetics and on CYP3A activity.
Methods
Twelve healthy volunteers received 20 mg rivaroxaban orally alone, in combination with ciclosporin (dose‐individualized oral regimen), and in combination with ciclosporin and fluconazole (400 mg day−1 orally). CYP3A4 activity was estimated using a midazolam microdose. Pharmacokinetics was analysed using noncompartmental and compartmental methods.
Results
Compared to baseline, ciclosporin increased rivaroxaban average exposure by 47% (90% confidence interval 28–68%), maximum concentration by 104% (70–146%), and decreased CYP3A4 activity by 34% (25–42%). Ciclosporin combined with fluconazole increased rivaroxaban average exposure by 86% (58–119%) and maximum concentration by 115% (83–153%), which was considerably stronger than observed in historical controls receiving rivaroxaban with fluconazole alone, and decreased CYP3A4 activity by 79% (76–82%).
Conclusion
Patients treated with rivaroxaban in combination with single modulators of multiple elimination pathways or multiple modulators of single elimination pathways (CYP3A, P‐gp) require particular care.
Keywords: ciclosporin, cytochrome P450 CYP3A, drug–drug interaction, fluconazole, midazolam, pharmacokinetics, rivaroxaban
What is already known about this subject
Rivaroxaban exposure is considerably increased during coadministration of drugs with combined P‐gp and strong CYP3A4 inhibitor properties.
What this study adds
The combination of 2 perpetrator drugs (ciclosporin and fluconazole) had stronger effects on a victim drug (rivaroxaban) than each perpetrator drug alone.
Ciclosporin increased average rivaroxaban exposure only by 47%, but maximum rivaroxaban concentrations were doubled. The addition of fluconazole further increased average rivaroxaban exposure.
Combinations of 2 perpetrator drugs should be considered carefully, both in daily practice and also in drug labels.
1. INTRODUCTION
Rivaroxaban, a direct factor Xa inhibitor, is not very susceptible to clinically relevant drug–drug interactions because of its diverse elimination pathways. These pathways include renal excretion of unchanged rivaroxaban (glomerular filtration and active secretion, mediated by P‐glycoprotein [P‐gp] and breast cancer resistance protein [BCRP]), metabolism by cytochrome P450 (CYP) 3A4/3A5 and CYP2J2 isozymes, and CYP‐independent hydrolytic cleavage.1 Nevertheless, drugs with combined inhibitory effects on drug‐metabolizing enzymes and drug transporters can lead to clinically relevant increases in rivaroxaban exposure. For example, the antifungal ketoconazole increased rivaroxaban exposure 2.58‐fold.1 In contrast, inducers, such as rifampicin and St John's Wort, can lead to clinically relevant decreases in rivaroxaban exposure.2, 3, 4 Thus, in patients treated with rivaroxaban, cotreatment with drugs that are combined P‐gp and strong CYP3A4 inhibitors and drugs that are P‐gp and strong CYP3A inducers should be avoided.2, 3, 4
The immunosuppressant ciclosporin inhibits several drug transporters (including P‐gp, BCRP, organic anion transporting polypeptide [OATP] 1B1, and OATP1B3) and probably also weakly CYP3A5, 6 suggesting that it might interact with rivaroxaban. The effects of ciclosporin on rivaroxaban pharmacokinetics have not been studied so far. The antifungal fluconazole is a moderate inhibitor of CYP3A6 and possibly BCRP,1 but is not known as a clinically relevant inhibitor of P‐gp.7, 8 Fluconazole (400 mg day–1) increased average rivaroxaban exposure 1.42‐fold,1 which is considered to be clinically not relevant.
Cotreatment with inhibitors such as ciclosporin or fluconazole is not restricted in patients treated with rivaroxaban according to current drug approval documents3, 4 and might be used, for example, in patients with organ transplants. However, in the presence of several modulating factors, the interaction may be more pronounced. The macrolide antibiotic erythromycin increased rivaroxaban exposure only 1.37‐fold, whereas the combined effects of erythromycin and moderate renal impairment doubled rivaroxaban exposure.9 The calcium channel blocker verapamil increased rivaroxaban exposure only 1.39‐fold, whereas the combined effects of verapamil and mild renal impairment increased rivaroxaban exposure 1.58‐fold.10 The combined effects of 2 drugs, 1 being a P‐gp inhibitor and 1 being a CYP3A inhibitor, on rivaroxaban pharmacokinetics are expected to be considerable, but have not been studied so far.
Fluconazole dose‐dependently affects ciclosporin pharmacokinetics.11 Fluconazole 200 mg day−1 increased ciclosporin exposure 1.88‐fold12 whereas fluconazole 100 mg day−1 had no effect.13 The effects of fluconazole 400 mg day−1 on ciclosporin pharmacokinetics are unknown, but may be more pronounced as indicated by the 3.72‐fold exposure increase of another CYP3A4 paradigm substrate (midazolam) by this dose.14
The primary aims of this trial were to evaluate the effects of ciclosporin on rivaroxaban pharmacokinetics alone and when combined with fluconazole. Secondary aims were to analyse potential modulators of the individual extent of the drug–drug interaction, such as sex, CYP3A5 genotype, perpetrator drug exposure and CYP3A4 phenotype (determined by using a midazolam microdose) and to evaluate the effects of fluconazole 400 mg day−1 on ciclosporin pharmacokinetics.
2. METHODS
2.1. Trial design
Twelve healthy volunteers should be included into the trial, including 6 with at least 1 (functional) CYP3A5*1 allele. Inclusion criteria were good general state of health, age between 18 and 64 years, body mass index between 18 and 30 kg m−2, body weight ≥50 kg, willingness to avoid citrus products (especially grapefruit) and follow specified contraceptive measures, and ability to communicate well with the investigator and to understand and comply with the trial requirements. Exclusion criteria included regular drug intake (with the exception of iodine, thyroid hormones and contraceptive drugs, which should not be changed during the trial), intake of a substance known to induce or inhibit drug metabolizing enzymes or drug transporters within a period of <10 times the respective elimination half‐life or 2 weeks, whichever was longer, any physical disorder that could interfere with the participant's safety during the clinical trial or with the trial objectives, any acute or chronic illness, or clinically relevant findings in the pretrial examination, especially any condition known or expected to modify absorption, distribution, metabolism, or excretion of the drugs under investigation, known or suspected inability to provide complete urine collections, regular smoking and unwilling/unable to refrain from smoking for the duration of the trial, excessive alcohol drinking and further safety‐related criteria addressing specific risks of the trial drugs.
Three single oral doses of 20 mg rivaroxaban (Xarelto; Bayer Pharma AG, Berlin, Germany) were administered. At baseline, rivaroxaban was administered alone, in the ciclosporin and the ciclosporin+fluconazole phase, rivaroxaban was administered during ciclosporin and fluconazole steady‐state. The washout period between rivaroxaban doses was at least 7 d (Figure S1). Participants were fasting overnight (water was allowed) and until 4 h after rivaroxaban administration, which is generally recommended for drug–drug interaction trials and was also done in previous rivaroxaban interaction trials.1 Rivaroxaban was administered together with the ciclosporin morning dose and with the fluconazole dose. Administration of ciclosporin and fluconazole was continued during blood sampling for rivaroxaban pharmacokinetics.
In the ciclosporin phase, participants started with 100 mg ciclosporin (Sandimmun optoral, Novartis Pharma GmbH, Nuremberg, Germany) twice daily, separated by approximately 12 h. A ciclosporin predose concentration was obtained on the 3rd day and the dose individualized, aiming at a predose concentration of 70 to 100 μg L−1. A further predose concentration could be obtained on the 6th day, if deemed necessary, but the dose was only to be changed in case of pronounced deviation from the target range, because otherwise a ciclosporin steady‐state could not be assumed on day 7 when ciclosporin pharmacokinetics was determined.
In the ciclosporin+fluconazole phase, participants started with 2 fluconazole (Fluconazol HEXAL 200 mg, Hexal AG, Holzkirchen, Germany) 400 mg doses on the 1st day, followed by 400 mg day−1 for 10 more days. On the 2nd day of fluconazole intake, ciclosporin was restarted at a reduced dose. A ciclosporin predose concentration was obtained on day 4 of the combination and the dose was individualized, again aiming at a predose concentration of 70–100 μg L−1.
Midazolam (Dormicum V, Roche Pharma AG, Grenzach‐Wyhlen, Germany) was administered as single oral 30 μg doses together with each rivaroxaban administration using an established microdosing procedure.15 In brief, midazolam, 0.03 mL of midazolam (5 mg/5 mL) parenteral solution was mixed with 100 mL of drinking water and administered orally.
Venous blood samples were drawn into lithium–heparin tubes before and 0.5, 1, 2, 3, 4, 6, 8, 10, 24 and 48 hours after rivaroxaban administration. For fluconazole determination, samples up to 24 hours were used. For midazolam, an additional sample 2.5 hours after midazolam administration was obtained. Plasma was separated after centrifugation and kept frozen at −20°C until analysis. For ciclosporin, venous blood samples were drawn into EDTA tubes before and 0.5, 1, 2, 3, 4, 6, 8 and 10 hours after administration. Ciclosporin whole blood concentrations were quantified on the same day or the next day. For coagulation parameters, venous blood samples were drawn into citrate tubes before and 4 hours after administration. Coagulation parameters were quantified on the same day.
Complete urine collections were obtained up to 48 hours after drug administration. Participants emptied their bladder shortly before rivaroxaban administration and collected all urine thereafter. Participants emptied their bladder after 24 hours and 48 hours in order to achieve accurate 24‐hour collections. The collected urine was stirred, the volume documented and samples frozen at −20°C until analysis.
For QTc assessment, 12‐lead electrocardiogram recordings were obtained at screening, 5 days after starting fluconazole, and at the trial end. QTc with heart rate correction using Fridericia's rule (QTcF) was used.
To ascertain adherence to the trial procedures, blood glucose was checked after overnight fasting and drug administration was supervised on the trial days. All ciclosporin and fluconazole doses and administration times were documented in a diary by the participants. A fluconazole predose concentration was obtained at the 5th day of the ciclosporin+fluconazole phase. Participants were informed on the particular importance of complete urine collections and were asked about completeness of the sampling when returning collected urine.
The trial was approved by the responsible Ethics Committee of the Medical Faculty of Heidelberg University (ethical approval number AFmo‐532/2016), authorized by the Federal Institute for Drugs and Medical Devices (BfArM, Bonn, Germany; EudraCT No: 2016–003120‐23), and prospectively registered in the German clinical trials register (DRKS00011528). It was planned and conducted according to the principles of the Declaration of Helsinki, the rules of good clinical practice, and the specific legal requirements in Germany. It was conducted at the Clinical Research Centre (KliPS) of the Department of Clinical Pharmacology and Pharmacoepidemiology, which is certified according to DIN EN ISO 9001:2015. Before inclusion, written informed consent was obtained from each participant.
2.2. Analytical and pharmacogenetic assays
Rivaroxaban, fluconazole and midazolam concentrations in plasma were analysed with ultra‐performance liquid chromatography coupled to tandem mass spectrometry (UHPLC–MS/MS) according to previously described assays2, 16, 17 or as described below. The lower limits of quantification were 1 μg L−1 for rivaroxaban, 0.025 mg L−1 for fluconazole and 93.0 fg mL−1 for midazolam. All assays were validated according to the Food and Drug Administration and European Medicines Agency guidelines for bioanalytical method validation18, 19 and fulfilled the respective criteria for accuracy/precision, linearity, recovery, matrix effects, and stability. For the quantification of rivaroxaban in urine, urine samples were diluted (1:10) with plasma and analysed according to the aforementioned plasma assay.16
The unbound fraction (fu) of rivaroxaban was determined at maximum concentration (Cmax) using rapid equilibrium dialysis (Thermo Fisher Scientific, Karlsruhe, Germany). Equal volumes of plasma and buffer were prepared and analysed according to the published plasma assay.16
Plasma samples containing fluconazole concentrations were extracted by liquid–liquid sample preparation at pH 9 and subsequently quantified with a stable isotope labelled internal standard (2H5‐fluconazole) using UHPLC–MS/MS system (Acquity UPLC‐TQD; Waters Eschborn, Germany). The chromatographic separation was performed on an Acquity UPLC BEH Phenyl column (Waters) with an organic solvent of pure acetonitrile and an aqueous solvent composed of 5 mM ammonium formate in water including acetonitrile (5% v/v). After positive electrospray ionization, mass‐to‐charge transitions of m/z 307 > 220 and m/z 312 > 223 were used for the MS/MS analysis of fluconazole and its internal standard, respectively (Z‐spray ionization, capillary voltage of 3 kV, source temperature of 150°C, desolvation temperature of 400°C, cone gas flow of 20 L h−1, desolvation gas flow of 900 L h−1, and collision gas flow of 0.15 mL min−1). Within‐ and between‐day accuracies ranged between 91.8–102% and precisions were constantly below 9.5%.
Ciclosporin whole‐blood concentrations, coagulation parameters (international normalized ratio, activated partial thromboplastin time) and safety parameters were measured in the accredited central laboratory of Heidelberg University Hospital. For ciclosporin, the commercial assay MassTox Immunosuppressants in Whole Blood—LC–MS/MS was used, which was validated according to the manufacturer instructions using the 6PLUS1 Multilevel Calibrator Set Immunosuppressants (Chromsystems Instruments & Chemicals GmbH, Gräfelfing, Germany). The lower limit of quantification was 25 μg L−1 and the assay was linear up to 2000 μg L−1. For plasma creatinine an enzymatic method was used. Creatinine clearance was estimated using Cockcroft and Gault's equation.20
The CYP3A5 genotype of the participants was known from a previous study (ethical approval number 026/2004). In brief, genomic DNA was isolated from whole blood using the NucleoSpin Blood Quick Pure Kit (Macherey‐Nagel, Düren, Germany) according to the manufacturer's instructions. Genotyping for the CYP3A5*3 allele (rs776746, A6986G in intron 3), leading to a functionally inactive truncated protein, was performed using the hybridization probe format on a LightCycler 480 (Roche Applied Sciences, Mannheim Germany) according to a previously published method.21
2.3. Pharmacokinetics
Pharmacokinetics were analysed using standard noncompartmental methods. Predose concentration (C0), Cmax, and time of maximum concentration (tmax) were obtained directly from the data. The terminal elimination rate (λz) was calculated by linear regression of log‐transformed concentrations from the terminal concentration decline. The area under the curve (AUC) was calculated by the trapezoidal rule (linear up, log down). For rivaroxaban and midazolam the AUC was extrapolated to infinity, for ciclosporin the AUC was extrapolated to 12 h, and for fluconazole the AUC measured up to 24 h was used. The apparent clearance after oral administration (CL/F) was calculated as dose divided by AUC, apparent terminal volume of distribution after oral administration (V/F) as CL/F divided by λz, half‐life (t1/2) as ln(2) divided by λz, and renal clearance (CLR) as amount recovered unchanged in 24‐h urine divided by AUC(0–24). The renal clearance by filtration (CLRf) was calculated as glomerular filtration rate (assessed via creatinine clearance) multiplied by fu and renal clearance by secretion (CLRs) as CLR minus CLRf.
Rivaroxaban pharmacokinetics were modelled using a linear 2‐compartment pharmacokinetic model with different functions for drug absorption (0‐order, 1st‐order, saturable, combined 0‐order and 1st‐order). The various models were compared by graphical methods and by the Akaike information criterion. Steady‐state rivaroxaban concentrations were predicted on the basis of the determined parameter values.
2.4. Statistics
Pharmacokinetic parameters were analysed assuming log‐normally distributed data unless indicated otherwise. Thus, pharmacokinetic parameter values and ratios are reported as geometric mean and 90% confidence intervals (90%CI). Demographic, coagulation and safety parameters were analysed assuming normally distributed data. Thus, demographic and safety parameters are reported as arithmetic mean and standard deviation. Parameters from pharmacokinetic modelling are reported as median and interquartile range.
Repeated‐measurements 1‐way analysis of variance (ANOVA) and Tukey's multiple comparisons test were used to analyse differences, using log‐transformed or linear values as appropriate. In case of tmax, Friedman's test and Dunn's multiple comparisons test were used. A 2‐sided paired t‐test was used to analyse ciclosporin parameter differences without and with fluconazole, using log‐transformed values. For analyses of ciclosporin dose and tmax, Wilcoxon's signed‐rank test was used. A 1‐sample t‐test was used to compare the relative change in rivaroxaban exposure due to ciclosporin+fluconazole with the published values for relative change in rivaroxaban exposure during fluconazole alone,1 to compare rivaroxaban AUC and Cmax ratios to 1 (i.e. no change), and to compare the relative change in ciclosporin exposure during fluconazole 400 mg day−1 with the value published for fluconazole 200 mg day−1.12 For analysis of potential modulators, a 2‐sided unpaired t‐test was used to analyse differences in rivaroxaban or ciclosporin clearance within a trial phase and relative change in rivaroxaban or ciclosporin exposure between trial phases according to sex or CYP3A5 genotype. Spearman correlation and linear regression were used to analyse associations with potential modulators on a continuous scale (e.g. perpetrator drug exposure, CYP3A4 phenotype = midazolam CL/F).
Pharmacokinetic calculations and statistical analyses were conducted using Phoenix WinNonlin 7.0 (Certara, Princeton, NJ, USA) and GraphPad Prism 5.01 (GraphPad Software, San Diego, CA, USA). A P‐value <.05 was considered statistically significant.
2.5. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY,22 and are permanently archived in the Concise Guide to PHARMACOLOGY 2017/18.23, 24
3. RESULTS
Eight males and 4 females were included in the trial (age 33 ± 11 years [range 21–52], weight 74 ± 17 kg [52–104] and body mass index 23.9 ± 3.8 kg m−2 [18.2–29.9]). Six had a CYP3A5 nonexpressor genotype (*3/*3) and 6 were heterozygous CYP3A5 expressors (*1/*3).
Rivaroxaban exposure was significantly increased by ciclosporin (Figure 1, Table 1) with a geometric mean AUC ratio of 1.47 (90%CI 1.28–1.68) and a Cmax ratio of 2.04 (1.70–2.46). Rivaroxaban exposure was further increased by ciclosporin+fluconazole with an AUC ratio of 1.86 (1.58–2.19) and a Cmax ratio of 2.15 (1.83–2.53) as compared to baseline and with an AUC ratio of 1.27 (1.14–1.41) and a Cmax ratio of 1.05 (0.93–1.20) compared to ciclosporin alone (Figure 2). The increase in rivaroxaban exposure due to ciclosporin+fluconazole was also significantly larger than the published AUC ratio of 1.42 and Cmax ratio of 1.28 for fluconazole alone (both P < .01).1 Coagulation markers 4 hours after drug administration were significantly higher when compared to baseline, but not when comparing the ciclosporin+fluconazole phase with the ciclosporin phase (Table 1). The relationship between rivaroxaban concentrations and relative change in coagulation markers showed no apparent influence of ciclosporin or fluconazole (Figure 3).
Figure 1.

Rivaroxaban plasma concentrations (mean ± standard error of the mean) after a single oral dose of rivaroxaban 20 mg (solid line) and during treatment with ciclosporin (dashed line) and ciclosporin in combination with fluconazole (dotted line) in healthy volunteers
Table 1.
Rivaroxaban single‐dose pharmacokinetics and pharmacodynamics after a 20 mg oral dose (baseline) and during treatment with ciclosporin without and with fluconazole in 12 healthy volunteers
| Parameter | Rivaroxaban alone | Rivaroxaban + ciclosporin | Rivaroxaban + ciclosporin + fluconazole | P (ANOVA) |
|---|---|---|---|---|
| AUC(0‐∞) (h μg L−1) | 2088 (1789–2437) | 3060a (2823–3316) | 3883b , c (3467–4350) | <.001 |
| Cmax (μg L−1) | 235 (203–273) | 480a (446–516) | 506b (451–567) | <.001 |
| tmax (h) | 2.0 (0.5, 6.0) | 3.0 (1.0, 4.0) | 2.5 (1.0, 4.0) | .34 |
| CL/F (L h−1) | 9.6 (8.2–11.2) | 6.5a (6.0–7.1) | 5.2b , c (4.6–5.8) | <.001 |
| V/F (L) | 128 (97–169) | 61a (52–71) | 53b (46–61) | <.001 |
| t1/2 (h) | 9.3 (7.3–11.9) | 6.5a (5.6–7.5) | 7.1 (6.5–7.9) | .02 |
| CLR (L h−1)d | 3.0 (2.6–3.5) | 2.2a (1.8–2.8) | 2.0b (1.6–2.5) | <.001 |
| CLRf (L h−1)d | 0.26 (0.21–0.32) | 0.25 (0.22–0.28) | 0.27 (0.21–0.33) | .88 |
| CLRs (L h−1)d | 2.76 (2.36–3.23) | 1.95a (1.48–2.57) | 1.71b (1.31–2.24) | <.001 |
| fu | 0.033 (0.028–0.040) | 0.033 (0.030–0.037) | 0.036 (0.031–0.043) | .57 |
| INR ratio (4 h) | 1.17 (1.13–1.21) | 1.37a (1.32–1.42) | 1.39b (1.35–1.43) | <.001 |
| aPTT ratio (4 h) | 1.31 (1.24–1.38) | 1.59a (1.52–1.67) | 1.62b(1.57‐1.68) | <.001 |
All values are reported as geometric mean and 90% confidence interval, except tmax, which is reported as median and range. aPTT, activated partial thromboplastin time; AUC, area under the curve; CL/F, apparent clearance after oral administration; CLR, renal clearance; CLRf, renal clearance by filtration; CLRs, renal clearance by secretion; Cmax, maximum concentration; fu, fraction unbound; INR, international normalized ratio; t1/2, half‐life; tmax, time of maximum concentration; V/F, apparent terminal volume of distribution after oral administration.
Statistically significant difference between ciclosporin phase and baseline phase (post‐test).
Statistically significant difference between ciclosporin+fluconazole phase and baseline phase (post‐test).
Statistically significant difference between ciclosporin+fluconazole phase and ciclosporin phase (post‐test).
n = 10, after exclusion of 2 participants because of collection errors.
Figure 2.

Change in rivaroxaban exposure in healthy volunteers during treatment with ciclosporin (CsA, squares) and ciclosporin in combination with fluconazole (CsA + FLC, triangles). Horizontal lines indicate geometric means. P values refer to the difference between the mean log‐transformed ratio and a hypothetical ratio of 1
Figure 3.

Correlation between rivaroxaban concentration 4 h after rivaroxaban intake and relative change in international normalized ratio (INR; ρ = .85, P < .01, r2 = .71, P < .01; A) and activated partial thromboplastin time (aPTT; ρ = .81, P < .01, r2 = .62, P < .01; B) at baseline (circles), during cotreatment with ciclosporin (open squares), and during cotreatment with ciclosporin and fluconazole (triangles)
In exploratory analyses of potential modulators, no significant differences in rivaroxaban clearance or relative change in rivaroxaban clearance according to sex or CYP3A5 genotype were found (Figure S2). Considering ciclosporin as the perpetrator drug, no significant correlations between ciclosporin dose, AUC, C0, Cmax and rivaroxaban clearance, or relative change in rivaroxaban clearance were found (Figure S3). Considering fluconazole as the perpetrator drug, a significant correlation between fluconazole AUC, C0, and rivaroxaban clearance during ciclosporin+fluconazole treatment was found, but not with the relative change in rivaroxaban clearance as compared to ciclosporin alone (Figure S3). Considering CYP3A4 phenotype, a significant correlation between midazolam clearance and rivaroxaban clearance was found, when combining data from all phases. A significant correlation between relative change in midazolam clearance and relative change in rivaroxaban clearance was found only when comparing ciclosporin+fluconazole to ciclosporin alone (Figure S4).
In pharmacokinetic modelling of rivaroxaban, a 2‐compartment model with a sequential 0‐order and 1st‐order absorption process performed best (parameter values in Table S1). Predictions of rivaroxaban concentrations after the first dose and during steady‐state suggest only slight accumulation. Ciclosporin and ciclosporin+fluconazole led to higher rivaroxaban exposure also at steady‐state, but predose rivaroxaban concentrations are predicted to be essentially unchanged (Figure 4, Figure S5).
Figure 4.
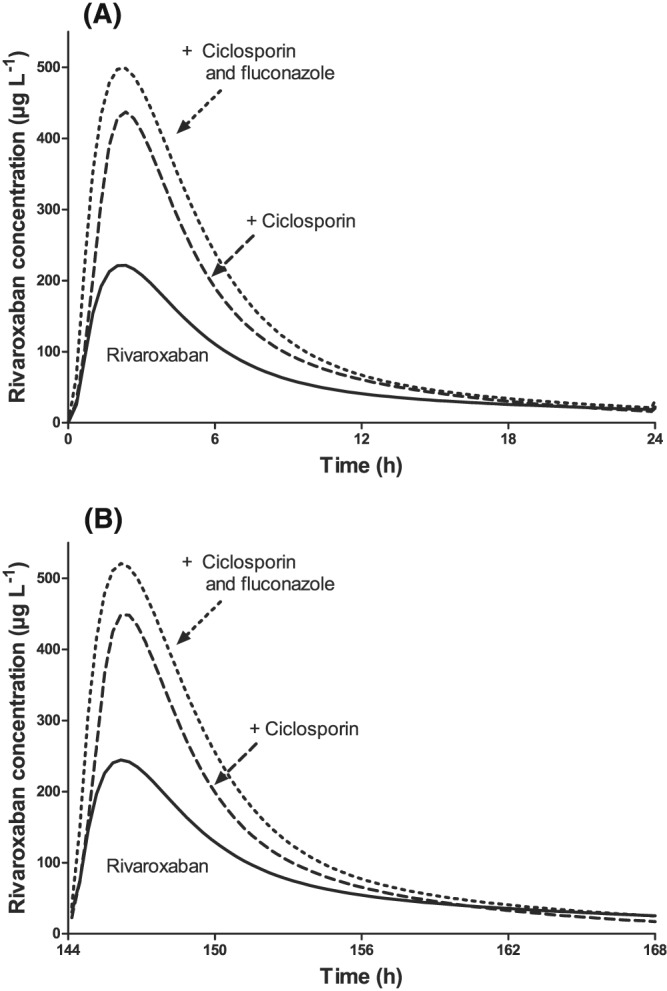
Predicted rivaroxaban plasma concentrations after the first dose (A) and during steady‐state (B) of rivaroxaban 20 mg day−1 in healthy volunteers
Ciclosporin predose concentrations were similar during treatment with ciclosporin and ciclosporin+fluconazole. Expectedly, ciclosporin dose, AUC(0–12) and Cmax were significantly lower during treatment with fluconazole (Table 2, Figure S6). The dose‐adjusted ciclosporin AUC ratio of 1.75 (1.56–1.96), in 8 participants where a steady‐state could be assumed, was not different from the published ratio of 1.88 for the relative ciclosporin AUC change caused by 200 mg day−1 fluconazole.12 No significant differences in ciclosporin clearance or relative change in ciclosporin exposure according to sex or CYP3A5 genotype were found.
Table 2.
Ciclosporin pharmacokinetics without and with fluconazole (400 mg day−1) in 12 healthy volunteers
| Parameter | Ciclosporin | Ciclosporin + fluconazole | P |
|---|---|---|---|
| All | |||
| Dose (mg) | 125 (100–200) | 55 (40–100) | <.01 |
| C0 (μg L−1) | 87 (77–98) | 76 (70–82) | .14 |
| Cmax (μg L−1) | 900 (807–1004) | 580 (513–657) | <.01 |
| AUC(0–12) (h μg L−1)a | 3,009 (2,755‐3,287) | 2,426 (2,219‐2,652) | .02 |
| Steady‐state b | |||
| Dose (mg) | 125 (100–200) | 67.5 (40–100) | .01 |
| C0 (μg L−1) | 81.7 (69.2–96.4) | 78.8 (73.6–84.3) | .69 |
| Cmax (μg L−1) | 852 (742–979) | 584 (487–700) | <.01 |
| AUC(0–12) (h μg L−1)a | 2,976 (2,681‐3,304) | 2,539 (2,254‐2,860) | .12 |
| AUC(0–12) (h μg L−1 per mg ciclosporin) | 23.2 (19.8–27.1) | 40.7 (32.2–51.3) | <.01 |
| tmax (h) | 1.5 (1.0–2.0) | 2.0 (1.0–3.0) | .25 |
| CL/F (L h−1) | 43.0 (36.9–50.3) | 24.6 (19.5–31.0) | <.01 |
| V/F (L) | 305 (257–363) | 184 (151–226) | <.01 |
| t1/2 (h) | 4.9 (4.2–5.8) | 5.2 (4.8–5.6) | .42 |
All values are reported as geometric mean and 90% confidence interval, except dose and tmax, which are reported as median and range.
AUC, area under the curve; C0, predose concentration; CL/F, apparent clearance after oral administration; Cmax, maximum concentration; t1/2, half‐life; tmax, time of maximum concentration; V/F, apparent terminal volume of distribution after oral administration.
The results for AUC0–10 were essentially the same as reported for AUC0–12.
n = 8, after exclusion of participants requiring ciclosporin dose adjustments within 2 days before a day when ciclosporin pharmacokinetics was determined.
Midazolam concentrations increased significantly during ciclosporin and during ciclosporin+fluconazole (Table S2, Figure S7). Compared to baseline, the midazolam CL/F ratio was 0.66 (90%CI 0.58–0.75) during ciclosporin and 0.21 (0.18–0.24) during ciclosporin+fluconazole, and compared to ciclosporin alone 0.32 (0.28–0.36). Fluconazole pharmacokinetics is shown in Table 3.
Table 3.
Fluconazole steady‐state pharmacokinetics (400 mg day–1) in healthy volunteers
| Parametera | Fluconazole |
|---|---|
| C0 (mg L−1) | 17.3 (15.9–18.9) |
| Cmax (mg L−1) | 26.2 (24.1–28.4) |
| AUC(0–24) (h mg L−1) | 493 (454–536) |
| tmax (h) | 3.0 (2.0–6.0) |
| CL/F (L h−1) | 0.81 (0.75–0.88) |
| V/F (L) | 46.7 (38.9–56.0) |
| t1/2 (h) | 39.9 (33.5–47.4) |
All values are reported as geometric mean and 90% confidence interval, except tmax, which is reported as median and range.
AUC, area under the curve; C0, predose concentration; CL/F, apparent clearance after oral administration; Cmax, maximum concentration; t1/2, half‐life; tmax, time of maximum concentration; V/F, apparent terminal volume of distribution after oral administration.
n = 11, after exclusion of 1 participant who skipped some fluconazole doses after the 6th fluconazole day.
No serious adverse events were observed and all other adverse events were mild and transient (Text S1).
4. DISCUSSION
Drug–drug interactions are studied most commonly using 1 perpetrator drug and 1 victim drug, but 2 perpetrator drugs can affect a victim drug stronger than each perpetrator drug alone, as shown for gemfibrozil and itraconazole.25, 26, 27, 28 In the present trial we have shown that a combination of a potent P‐gp inhibitor and a moderate CYP3A inhibitor can affect rivaroxaban stronger than each perpetrator drug alone. Ciclosporin increased rivaroxaban AUC by 47% and doubled rivaroxaban Cmax; ciclosporin combined with fluconazole increased rivaroxaban AUC by 86% and rivaroxaban Cmax by 115%, the latter values also being significantly higher than published for fluconazole alone.1
Ciclosporin decreased rivaroxaban renal clearance but the extent of this decrease was too small to fully explain the observed decrease in rivaroxaban systemic clearance and the pronounced increase in its Cmax. Therefore, the observed change in rivaroxaban exposure is probably caused by an increase in the absorbed fraction combined with a reduced active renal elimination (as indicated by the decrease in CLRs), which could be caused by inhibition of P‐gp and BCRP by ciclosporin.29 The observed increase in Cmax is the highest value reported in rivaroxaban trials so far. Even ketoconazole, which increased rivaroxaban AUC by 158%, increased rivaroxaban Cmax only by 72%.1 Rivaroxaban half‐life was shorter during cotreatment with ciclosporin, which could not be explained by changes in rivaroxaban unbound fraction (which was unchanged), but might be explained by a reduction in rivaroxaban's distribution space due to transporter inhibition.30 The latter hypothesis would be consistent with the observed increase in Cmax.
Predicted rivaroxaban steady‐state pharmacokinetics suggests that ciclosporin and ciclosporin+fluconazole will lead to higher average and maximum concentrations while C0 will probably be unchanged. The relationship between such markers of rivaroxaban exposure and bleeding events is not fully understood. In the clinical pharmacology and biopharmaceutics review of the Food and Drug Administration, exponential relationships have been reported for both, AUC and Cmax, and the risk of major bleeding.31 In a recent analysis, higher rivaroxaban peak values (defined as anti‐Xa activity 3 h after drug intake), but not trough values (defined as predose anti‐Xa activity), were associated with major and nonmajor bleeding events.32 Thus, the increase in rivaroxaban AUC and Cmax caused by ciclosporin might be clinically relevant, despite an only modest increase in rivaroxaban AUC. Therefore, in patients treated with ciclosporin (e.g. patients with organ transplants, severe rheumatoid arthritis, or severe psoriasis), potential risks and benefits of cotreatment with rivaroxaban should be carefully weighted.
Coagulation markers 4 hours after rivaroxaban administration were also significantly increased during cotreatment with ciclosporin and there was also a (not significant) trend to higher anticoagulation during ciclosporin+fluconazole compared to ciclosporin alone. However, the present trial was not powered to detect small differences. Importantly, the observed relationship between rivaroxaban concentration and change in coagulation markers did not indicate saturation and therefore suggests that further concentration increases can further intensify anticoagulation.
After a single oral ciclosporin dose of 500 mg, the average exposure of edoxaban, another direct factor Xa inhibitor, increased more (1.73‐fold) than rivaroxaban in our trial.33 This could be explained by different elimination pathways34 or by the very high ciclosporin dose, which might have more pronounced effects on intestinal transport of a victim drug because of higher local concentrations. Differences in bioavailability alone cannot explain this difference, because bioavailability of edoxaban is approximately 62% and 66% for rivaroxaban (after a 20 mg dose without food, as in our trial).
As expected, ciclosporin clearance decreased during cotreatment with fluconazole and dose adjustment was necessary to keep ciclosporin C0 constant. Drug–drug interactions with azoles are commonly dose‐dependent. However, the observed increase in dose‐adjusted ciclosporin AUC in the present trial was similar to the value reported for fluconazole 200 mg day−1,12 suggesting that a maximum inhibition was reached.
To our knowledge, fluconazole steady‐state pharmacokinetics using fluconazole 400 mg day−1 in healthy volunteers has not been studied before. Surprisingly, fluconazole CL/F was only approximately 60% of the values reported for lower doses (1.30–1.44 L h−1)35 and of the clearance in healthy volunteers (1.32 L h−1) that can be derived from the steady‐state AUC reported after intravenous fluconazole 800 mg day−1.36 The effects of fluconazole on ciclosporin pharmacokinetics are well known, but the possible effects of ciclosporin on fluconazole pharmacokinetics have not been thoroughly studied in humans. In an animal study, fluconazole AUCs were 40–50% higher when ciclosporin was coadministered,37 indicating a reduced fluconazole clearance because bioavailability is almost complete. Fluconazole, which is not a P‐gp substrate,7, 8 is predominantly eliminated by renal excretion and the low renal clearance indicates considerable re‐absorption.38 The slight decline in creatinine clearance observed in the present trial cannot explain the low fluconazole CL/F. Fluconazole is filtered in the glomerulus and re‐absorbed in the tubules. It could thus be speculated that ciclosporin might have increased net renal fluconazole re‐absorption. Assuming that fluconazole molecules might undergo a cyclic process of passive re‐absorption and efflux into urine, by as yet undefined apical drug transporters, ciclosporin could increase re‐absorption by inhibiting such apical drug transporters.
In the present trial, ciclosporin turned out to be a weak CYP3A4 inhibitor increasing midazolam AUC 1.53‐fold. This is in agreement with an earlier study assessing midazolam pharmacokinetics (reference 112 in6) and apparently contradicting a trial in which ciclosporin increased simvastatin exposure 2.6‐fold.6, 39 However, because ciclosporin is also a strong inhibitor of hepatic uptake transporters (OATP) that also transport simvastatin,40 the results certainly overestimate the contribution of CYP3A to this interaction. Our results are also supported by a report in renal allograft recipients whose midazolam clearance was 24 and 31% lower (intravenous or oral midazolam) in patients treated with ciclosporin as compared to tacrolimus.5 Thus, the myriad of known drug–drug interactions during cotreatment with ciclosporin should be interpreted as being largely a result of drug transporter inhibition by ciclosporin rather than by inhibition of CYP3A4.
Our trial has some limitations. First, ciclosporin dose, AUC, and Cmax were lower during treatment with ciclosporin and fluconazole than during ciclosporin alone. This is generally expected when predose concentrations are used for drug dose adjustments due to drug–drug interactions,41 which was also done in the present trial in order to reflect clinical routine as well as for safety reasons. Thus, the lower ciclosporin dose (considering potential effects of lower local ciclosporin concentrations on rivaroxaban absorption in the gut) or ciclosporin exposure during treatment with ciclosporin and fluconazole might have led to lower ciclosporin effects in this phase. However, there was no correlation between ciclosporin dose, AUC or Cmax and the extent of the rivaroxaban interaction. Second, rivaroxaban was administered without food, which is generally recommended for drug–drug interaction trials and was also done in previous rivaroxaban interaction studies, from which we used parameter values for comparisons.1 Nevertheless, in clinical routine, rivaroxaban 20 mg is administered with a meal in order to increase bioavailability. Thus, the effects of ciclosporin and fluconazole on rivaroxaban exposure might be less profound in the clinical setting. However, part of the Cmax increase is probably explained by a reduced distribution space due to drug transporter inhibition, which might not be affected by food. Third, rivaroxaban was administered in all phases and might have affected the pharmacokinetics of the other drugs. However, rivaroxaban has never acted as a perpetrator in drug–drug interactions and did not affect midazolam clearance.1, 2 Fourth, sequence effects cannot be excluded, because of the fixed‐sequence design. This design was chosen because of the long fluconazole half‐life, which would have required a long washout phase. Furthermore, for safety reasons, the individual ciclosporin dose (to achieve target trough concentrations without fluconazole) should be known before starting dose titration during cotreatment with fluconazole. Fifth, rivaroxaban pharmacokinetics during treatment with fluconazole (without ciclosporin) was not studied in the present trial. However, our trial design was very similar to a published trial on rivaroxaban and fluconazole,1 from which parameter values were used for statistical comparisons.
In conclusion, the combination of 2 perpetrator drugs had stronger effects on a victim drug than each perpetrator drug alone. Ciclosporin increased average rivaroxaban exposure only by 47%, but maximum rivaroxaban concentrations were doubled. The addition of fluconazole further increased average rivaroxaban exposure. Thus, rivaroxaban patients treated with single modulators of multiple elimination pathways or multiple modulators of single elimination pathways (CYP3A, P‐gp) require particular care, both in daily practice and also in drug labels.
COMPETING INTERESTS
W.E.H. and D.C. received fees from Bayer for holding lectures not related to this trial. A.B., M.‐L.L., K.I.F., J.B., and J.W. have nothing to disclose.
Part of the data has been presented at the 16th Annual NephroPharmacology Meeting, October 2017, Heidelberg, Germany as an oral presentation.
CONTRIBUTORS
A.B., M.‐L.L., K.I.F., J.B., J.W., W.E.H., and D.C. wrote the manuscript; A.B., M.‐L.L., W.E.H., and D.C. designed the research; A.B., M.‐L.L., K.I.F., J.B., J.W., W.E.H., and D.C. performed the research; and A.B. and D.C. analyzed the data.
Supporting information
Table S1
Rivaroxaban pharmacokinetic parameters using a 2‐compartment model with a sequential 0‐order and first‐order absorption process.
Table S2 Midazolam single‐dose pharmacokinetics after a 30 μg oral dose during rivaroxaban and during treatment with ciclosporin without and with fluconazole in 12 healthy volunteers.
Figure S1 Graphical illustration of the study design. Rivaroxaban (RXa) and midazolam (MDZ) were administered as single doses. Ciclosporin (CsA) and fluconazole (FLC) were administered as multiple doses in order to reach a steady‐state. The CsA dose was individualised according to the trough concentration measured at week (w) 1/day (d) 5. On w3/d2, CsA was resumed, initially using ¼ of the previous individual dose (this fraction, ¼, was adjusted in subsequent cohorts, based on actually required doses to reach CsA target concentrations in phase 3). The CsA dose was individualised again, according to the trough concentration measured on w3/d5. Visits 4, 5–7, 8 and 9–11 could be postponed by 1 or 2 days, additional days could be included between the phases, and additional CsA concentrations could be measured on w2/d1 and w4/d1, if required.
Figure S2 Rivaroxaban clearance and relative change in rivaroxaban clearance during ciclosporin (CsA) and ciclosporin+fluconazole (CsA + FLC) in relation to sex and CYP3A5 genotype.
Figure S3 Relationship between rivaroxaban clearance values and perpetrator (fluconazole, ciclosporin) pharmacokinetics.
Figure S4 Correlation between midazolam clearance, relative change in midazolam clearance, rivaroxaban clearance and relative change in rivaroxaban clearance during ciclosporin (CsA) and ciclosporin + fluconazole (CsA + FLC).
Figure S5 Predicted rivaroxaban concentrations after oral administration of 20 mg day–1 (A), during cotreatment with ciclosporin (B), and during cotreatment with ciclosporin and fluconazole (C) in 12 healthy volunteers.
Figure S6 Ciclosporin concentrations (mean ± standard error of the mean) without (dashed line) and during treatment with fluconazole (dotted line) after repeated administration in 12 healthy volunteers. Ciclosporin doses were individualized, aiming at a predose concentration of 70 to 100 μg L–1. The median ciclosporin dose was 125 mg twice per day (range 100–200) without and 55 mg twice per day (40–100) with fluconazole.
Figure S7 Midazolam concentrations (mean ± standard error of the mean) during rivaroxaban (solid line), during treatment with ciclosporin (dashed line) and ciclosporin in combination with fluconazole (dotted line) after a single oral dose of midazolam 30 μg in 12 healthy volunteers.
Text S1 Adverse events.
ACKNOWLEDGEMENTS
The authors would like to thank Marlies Stützle‐Schnetz for her valuable assistance during the trial, Julia Schäfer for monitoring the trial, and Andrea Deschlmayr and Magdalena Longo for excellent technical support.
Brings A, Lehmann M‐L, Foerster KI, et al. Perpetrator effects of ciclosporin (P‐glycoprotein inhibitor) and its combination with fluconazole (CYP3A inhibitor) on the pharmacokinetics of rivaroxaban in healthy volunteers. Br J Clin Pharmacol. 2019;85:1528–1537. 10.1111/bcp.13934
The authors confirm that the PI for this paper is David Czock and that he had direct clinical responsibility for patients.
Trial register: German clinical trials register.
Clinical trial registration number: DRKS00011528.
REFERENCES
- 1. Mueck W, Kubitza D, Becka M. Co‐administration of rivaroxaban with drugs that share its elimination pathways: pharmacokinetic effects in healthy subjects. Br J Clin Pharmacol. 2013;76(3):455‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Huppertz A, Werntz L, Meid AD, et al. Rivaroxaban and macitentan can be coadministered without dose adjustment but the combination of rivaroxaban and St John's wort should be avoided. Br J Clin Pharmacol. 2018;84(12):2903‐2913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. EMA summary of product characteristics, last updated July 2018. (http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000944/human_med_001155.jsp&murl=menus/medicines/medicines.jsp&mid=WC0b01ac058001d125. Accessed 3 September 2018.
- 4. FDA drug label of Xarelto, last updated February 2017. https://dailymed.nlm.nih.gov/dailymed/. Accessed 3 September 2018.
- 5. de Jonge H, de Loor H, Verbeke K, Vanrenterghem Y, Kuypers DR. In vivo CYP3A activity is significantly lower in cyclosporine‐treated as compared with tacrolimus‐treated renal allograft recipients. Clin Pharmacol Ther. 2011;90:414‐422. [DOI] [PubMed] [Google Scholar]
- 6. Polasek TM, Lin FP, Miners JO, Doogue MP. Perpetrators of pharmacokinetic drug‐drug interactions arising from altered cytochrome P450 activity: a criteria‐based assessment. Br J Clin Pharmacol. 2011;71(5):727‐736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lempers VJ, van den Heuvel JJ, Russel FG, et al. Inhibitory potential of antifungal drugs on atp‐binding cassette transporters P‐glycoprotein, MRP1 to MRP5, BCRP, and BSEP. Antimicrob Agents Chemother. 2016;60(6):3372‐3379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yasuda K, Lan LB, Sanglard D, Furuya K, Schuetz JD, Schuetz EG. Interaction of cytochrome P450 3A inhibitors with P‐glycoprotein. J Pharmacol Exp Ther. 2002;303(1):323‐332. [DOI] [PubMed] [Google Scholar]
- 9. Moore KT, Vaidyanathan S, Natarajan J, Ariyawansa J, Haskell L, Turner KC. An open‐label study to estimate the effect of steady‐state erythromycin on the pharmacokinetics, pharmacodynamics, and safety of a single dose of rivaroxaban in subjects with renal impairment and normal renal function. J Clin Pharmacol. 2014;54(12):1407‐1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Greenblatt DJ, Patel M, Harmatz JS, Nicholson WT, Rubino CM, Chow CR. Impaired rivaroxaban clearance in mild renal insufficiency with verapamil coadministration: potential implications for bleeding risk and dose selection. J Clin Pharmacol. 2018;58(4):533‐540. [DOI] [PubMed] [Google Scholar]
- 11. López‐Gil JA. Fluconazole‐cyclosporine interaction: a dose‐dependent effect? Ann Pharmacother. 1993;27(4):427‐430. [DOI] [PubMed] [Google Scholar]
- 12. Canafax DM, Graves NM, Hilligoss DM, Carleton BC, Gardner MJ, Matas AJ. Interaction between cyclosporine and fluconazole in renal allograft recipients. Transplantation. 1991;51(5):1014‐1018. [DOI] [PubMed] [Google Scholar]
- 13. Krüger HU, Schuler U, Zimmermann R, Ehninger G. Absence of significant interaction of fluconazole with cyclosporin. J Antimicrob Chemother. 1989;24(5):781‐786. [DOI] [PubMed] [Google Scholar]
- 14. Ahonen J, Olkkola KT, Neuvonen PJ. Effect of route of administration of fluconazole on the interaction between fluconazole and midazolam. Eur J Clin Pharmacol. 1997;51(5):415‐419. [DOI] [PubMed] [Google Scholar]
- 15. Halama B, Hohmann N, Burhenne J, Weiss J, Mikus G, Haefeli WE. A nanogram dose of the CYP3A probe substrate midazolam to evaluate drug interactions. Clin Pharmacol Ther. 2013;93(6):564‐571. [DOI] [PubMed] [Google Scholar]
- 16. Foerster KI, Huppertz A, Müller OJ, et al. Simultaneous quantification of direct oral anticoagulants currently used in anticoagulation therapy. J Pharm Biomed Anal. 2018;148:238‐244. [DOI] [PubMed] [Google Scholar]
- 17. Burhenne J, Halama B, Maurer M, et al. Quantification of femtomolar concentrations of the CYP3A substrate midazolam and its main metabolite 1′‐hydroxymidazolam in human plasma using ultra performance liquid chromatography coupled to tandem mass spectrometry. Anal Bioanal Chem. 2012;402(7):2439‐2450. [DOI] [PubMed] [Google Scholar]
- 18. FDA guidance for Industry . Bioanalytical Method Validation. 2001.
- 19. EMA guideline on bioanalytical methods validation. 2011. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf. Accessed 25 June 2018.
- 20. Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31‐41. [DOI] [PubMed] [Google Scholar]
- 21. Fredericks S, Moreton M, MacPhee IA, et al. Genotyping cytochrome P450 3A5 using the Light Cycler. Ann Clin Biochem. 2005;42(5):376‐381. [DOI] [PubMed] [Google Scholar]
- 22. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: Updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acids Res. 2018;46:D1091–D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2017/18: Enzymes. Br J Pharmacol. 2017;174(Suppl 1):S272‐S359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alexander SPH, Kelly E, Marrion NV, et al. The Concise Guide to PHARMACOLOGY 2017/18: Transporters. Br J Pharmacol. 2017;174(Suppl 1):S360‐S446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Niemi M, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics and pharmacodynamics of repaglinide: potentially hazardous interaction between gemfibrozil and repaglinide. Diabetologia. 2003;46(3):347‐351. [DOI] [PubMed] [Google Scholar]
- 26. Jaakkola T, Backman JT, Neuvonen M, Neuvonen PJ. Effects of gemfibrozil, itraconazole, and their combination on the pharmacokinetics of pioglitazone. Clin Pharmacol Ther. 2005;77(5):404‐414. [DOI] [PubMed] [Google Scholar]
- 27. Niemi M, Backman JT, Juntti‐Patinen L, Neuvonen M, Neuvonen PJ. Coadministration of gemfibrozil and itraconazole has only a minor effect on the pharmacokinetics of the CYP2C9 and CYP3A4 substrate nateglinide. Br J Clin Pharmacol. 2005;60(2):208‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Niemi M, Tornio A, Pasanen MK, Fredrikson H, Neuvonen PJ, Backman JT. Itraconazole, gemfibrozil and their combination markedly raise the plasma concentrations of loperamide. Eur J Clin Pharmacol. 2006;62(6):463‐472. [DOI] [PubMed] [Google Scholar]
- 29. Hodin S, Basset T, Jacqueroux E, et al. In vitro comparison of the role of P‐glycoprotein and breast cancer resistance protein on direct oral anticoagulants disposition. Eur J Drug Metab Pharmacokinet. 2018;43(2):183‐191. [DOI] [PubMed] [Google Scholar]
- 30. Grover A, Benet LZ. Effects of drug transporters on volume of distribution. AAPS J. 2009;11(2):250‐261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. FDA . Clinical pharmacology and biopharmaceutics review(s). 2010. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/022406Orig1s000ClinPharmR.pdf. Accessed 3 September 2018.
- 32. Sakaguchi T, Osanai H, Murase Y, et al. Monitoring of anti‐Xa activity and factors related to bleeding events: a study in Japanese patients with nonvalvular atrial fibrillation receiving rivaroxaban. J Cardiol. 2017;70:244‐249. [DOI] [PubMed] [Google Scholar]
- 33. Parasrampuria DA, Mendell J, Shi M, Matsushima N, Zahir H, Truitt K. Edoxaban drug‐drug interactions with ketoconazole, erythromycin, and cyclosporine. Br J Clin Pharmacol. 2016;82(6):1591‐1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mikkaichi T, Yoshigae Y, Masumoto H, et al. Edoxaban transport via P‐glycoprotein is a key factor for the drug's disposition. Drug Metab Dispos. 2014;42(4):520‐528. [DOI] [PubMed] [Google Scholar]
- 35. Debruyne D, Ryckelynck JP. Clinical pharmacokinetics of fluconazole. Clin Pharmacokinet. 1993;24(1):10‐27. [DOI] [PubMed] [Google Scholar]
- 36. Sobue S, Tan K, Layton G, Eve M, Sanderson JB. Pharmacokinetics of fosfluconazole and fluconazole following multiple intravenous administration of fosfluconazole in healthy male volunteers. Br J Clin Pharmacol. 2004;58(1):20‐25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Marchetti O, Entenza JM, Sanglard D, Bille J, Glauser MP, Moreillon P. Fluconazole plus cyclosporine: a fungicidal combination effective against experimental endocarditis due to Candida albicans . Antimicrob Agents Chemother. 2000;44(11):2932‐2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gross AS, McLachlan AJ, Minns I, Beal JB, Tett SE. Simultaneous administration of a cocktail of markers to measure renal drug elimination pathways: absence of a pharmacokinetic interaction between fluconazole and sinistrin, p‐aminohippuric acid and pindolol. Br J Clin Pharmacol. 2001;51(6):547‐555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Arnadottir M, Eriksson LO, Thysell H, Karkas JD. Plasma concentration profiles of simvastatin 3‐hydroxy‐3‐methyl‐glutaryl‐coenzyme A reductase inhibitory activity in kidney transplant recipients with and without ciclosporin. Nephron. 1993;65(3):410‐413. [DOI] [PubMed] [Google Scholar]
- 40. Neuvonen PJ, Niemi M, Backman JT. Drug interactions with lipid‐lowering drugs: mechanisms and clinical relevance. Clin Pharmacol Ther. 2006;80(6):565‐581. [DOI] [PubMed] [Google Scholar]
- 41. Vogel M, Voigt E, Michaelis HC, et al. Management of drug‐to‐drug interactions between cyclosporine A and the protease‐inhibitor lopinavir/ritonavir in liver‐transplanted HIV‐infected patients. Liver Transpl. 2004;10(7):939‐944. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1
Rivaroxaban pharmacokinetic parameters using a 2‐compartment model with a sequential 0‐order and first‐order absorption process.
Table S2 Midazolam single‐dose pharmacokinetics after a 30 μg oral dose during rivaroxaban and during treatment with ciclosporin without and with fluconazole in 12 healthy volunteers.
Figure S1 Graphical illustration of the study design. Rivaroxaban (RXa) and midazolam (MDZ) were administered as single doses. Ciclosporin (CsA) and fluconazole (FLC) were administered as multiple doses in order to reach a steady‐state. The CsA dose was individualised according to the trough concentration measured at week (w) 1/day (d) 5. On w3/d2, CsA was resumed, initially using ¼ of the previous individual dose (this fraction, ¼, was adjusted in subsequent cohorts, based on actually required doses to reach CsA target concentrations in phase 3). The CsA dose was individualised again, according to the trough concentration measured on w3/d5. Visits 4, 5–7, 8 and 9–11 could be postponed by 1 or 2 days, additional days could be included between the phases, and additional CsA concentrations could be measured on w2/d1 and w4/d1, if required.
Figure S2 Rivaroxaban clearance and relative change in rivaroxaban clearance during ciclosporin (CsA) and ciclosporin+fluconazole (CsA + FLC) in relation to sex and CYP3A5 genotype.
Figure S3 Relationship between rivaroxaban clearance values and perpetrator (fluconazole, ciclosporin) pharmacokinetics.
Figure S4 Correlation between midazolam clearance, relative change in midazolam clearance, rivaroxaban clearance and relative change in rivaroxaban clearance during ciclosporin (CsA) and ciclosporin + fluconazole (CsA + FLC).
Figure S5 Predicted rivaroxaban concentrations after oral administration of 20 mg day–1 (A), during cotreatment with ciclosporin (B), and during cotreatment with ciclosporin and fluconazole (C) in 12 healthy volunteers.
Figure S6 Ciclosporin concentrations (mean ± standard error of the mean) without (dashed line) and during treatment with fluconazole (dotted line) after repeated administration in 12 healthy volunteers. Ciclosporin doses were individualized, aiming at a predose concentration of 70 to 100 μg L–1. The median ciclosporin dose was 125 mg twice per day (range 100–200) without and 55 mg twice per day (40–100) with fluconazole.
Figure S7 Midazolam concentrations (mean ± standard error of the mean) during rivaroxaban (solid line), during treatment with ciclosporin (dashed line) and ciclosporin in combination with fluconazole (dotted line) after a single oral dose of midazolam 30 μg in 12 healthy volunteers.
Text S1 Adverse events.