

Abstract
Aims
The aim of the present study was to characterize the pharmacokinetics and exposure–subjective response relationship of a novel oral solution of lysergic acid diethylamide (LSD) that was developed for clinical use in research and patients.
Method
LSD (100 μg) was administered in 27 healthy subjects using a placebo‐controlled, double‐blind, cross‐over design. Plasma levels of LSD, nor‐LSD, and 2‐oxo‐3‐hydroxy‐LSD (O‐H‐LSD) and subjective drug effects were assessed up to 11.5 hours.
Results
First‐order elimination kinetics were observed for LSD. Geometric mean maximum concentration (C max) values (range) of 1.7 (1.0–2.9) ng/mL were reached at a t max (range) of 1.7 (1.0–3.4) hours after drug administration. The plasma half‐life (t 1/2) was 3.6 (2.4–7.3) hours. The AUC∞ was 13 (7.1–28) ng·h/mL. No differences in these pharmacokinetic parameters were found between male and female subjects. Plasma O‐H‐LSD but not nor‐LSD (< 0.01 ng/mL) concentrations could be quantified in all subjects. Geometric mean O‐H‐LSD C max values (range) of 0.11 (0.07–0.19) ng/mL were reached at a t max (range) of 5 (3.2–8) hours. The t 1/2 and AUC∞ values of O‐H‐LSD were 5.2 (2.6–21) hours and 1.7 (0.85–4.3) ng·h/mL, respectively. The subjective effects of LSD lasted (mean ± SD) for 8.5 ± 2.0 hours (range: 5.3–12.8 h), and peak effects were reached 2.5 ± 0.6 hours (range 1.6–4.3 h) after drug administration. EC50 values were 1.0 ± 0.5 ng/mL and 1.9 ± 1.0 ng/mL for “good” and “bad” subjective drug effects, respectively.
Conclusion
The present study characterized the pharmacokinetics of LSD and its main metabolite O‐H‐LSD. The subjective effects of LSD were closely associated with changes in plasma concentrations over time.
Keywords: concentration–effect relationship, LSD, metabolism, O‐H‐LSD, pharmacodynamics, pharmacokinetics
What is already known about this subject
There is an increasing number of clinical studies using LSD in humans.
There is very limited data on the human pharmacokinetics of LSD.
There are no controlled pharmacokinetic studies with validly defined doses of LSD.
What this study adds
Subjective responses and pharmacokinetics of LSD and its metabolites are described after controlled administration of a known oral dose of LSD.
The data serves as a reference to relate plasma concentrations of LSD with its effects in clinical studies and intoxications.
1. INTRODUCTION
Lysergic acid diethylamide (LSD) is a prototypical hallucinogen that has been widely used for recreational and personal purposes.1 Additionally, LSD is increasingly used in experimental research2, 3, 4, 5, 6 and for the treatment of psychiatric patients.7, 8 However, blood plasma concentrations were not measured in most LSD studies.3, 6 Thus, unknown are the concentrations of LSD at the time points at which pharmacodynamic (PD) outcomes were collected. Only limited data are available on the pharmacokinetics (PK) of LSD. A study in five male subjects reported a mean plasma elimination half‐life of LSD of 175 minutes after intravenous administration (2 μg/kg9). Another study used non‐systematic blood sampling after the administration of 160 μg LSD in 13 subjects up to 2.5–5 hours; however, because of the sparse and short sampling, PK parameters could not be derived.10 We recently reported the first comprehensive PK data for orally administered LSD. In two studies, the PK of LSD were determined after the administration of 100 and 200 μg LSD in 24 and 16 healthy subjects, respectively.11, 12, 13 However, the formulation that was used in these studies did not have long‐term stability. Therefore, we produced a novel oral LSD solution with documented long‐term stability (single dose units) and higher content uniformity than is currently being used in experimental studies in healthy subjects (ClinicalTrials.gov no. NCT03604744, NCT03321136, and planned studies), clinical trials in patients (ClinicalTrials.gov no. NCT03153579), and in the context of individually authorized patient treatments (compassionate use in Switzerland). The primary aim of the present study was to describe the PK of this LSD formulation in healthy subjects. A second goal was to describe subjective drug effects of LSD and to link these effects to changes in plasma concentrations over time within‐subjects to derive EC50 values using PK/PD modelling. We also analysed concentrations of the LSD metabolites 2‐oxo‐3‐hydroxy LSD (O‐H‐LSD) and N‐desmethyl‐LSD (nor‐LSD) in plasma. O‐H‐LSD and nor‐LSD are the main metabolites of LSD that are detected in urine.12, 14, 15, 16, 17 However, only one previous study quantified these metabolites in human plasma.13 Finally, we compared LSD exposure in plasma between the novel solution and previous capsule formulation. The present study is different from our previous PK studies mainly because it used a novel oral formulation with documented content stability.
2. METHODS
2.1. Study design
We performed a double‐blind, placebo‐controlled, cross‐over study with four experimental 12‐hour test sessions (100 μg LSD, 125 mg methylenedioxymethamphetamine [MDMA], 40 mg D‐amphetamine and placebo) in a balanced order. The washout periods between sessions were at least 7 days. Only the novel LSD and placebo data are presented here because the PK and PD of the MDMA and D‐amphetamine formulations that were used in the study have previously been published18, 19 and because the sampling time was too short to provide full concentration–time curves for these substances with long half‐lives.
The study was conducted in accordance with the Declaration of Helsinki and approved by the local ethics committee. The use of LSD in humans was authorized by the Swiss Federal Office for Public Health, Bern, Switzerland. All of the subjects provided written consent before participating in either of the studies, and they were paid for their participation. Participants were informed about acute and potentially lasting effects of LSD.20, 21 The study was registered at ClinicalTrials.gov (NCT03019822).
2.2. Participants
Twenty‐nine healthy participants were recruited from the University of Basel campus via online advertisement. One subject stopped participation after screening and did not complete any of the test sessions, and one subject did not complete the LSD session. The final study sample included 27 subjects (13 males and 14 females) who completed the study. The participants were (mean ± SD) 28 ± 4 years old (range: 25–45 years) with a mean body weight of 71 ± 12 kg (range: 55–97 kg; 80 ± 10 kg in men and 62 ± 6 kg in women). Only healthy subjects who were between 25 and 50 years old were included in the study. The exclusion criteria were the following: pregnancy (urine pregnancy test at screening and before each test session), personal or family (first‐degree relative) history of major psychiatric disorders (assessed by the Semi‐structured Clinical Interview for Diagnostic and Statistical Manual of Mental Disorders, 4th edition, Axis I disorders by the study physician and an additional interview by a trained psychiatrist), the use of medications that may interfere with the study drug, chronic or acute physical illness (based on abnormal physical exam, electrocardiogram or haematological and chemical blood analyses or hypertension >140/90 mmHg), tobacco smoking (>10 cigarettes/day), a lifetime prevalence of illicit drug use >10 times (except for tetrahydrocannabinol [THC]), illicit drug use within the last 2 months, and illicit drug use during the study. We performed urine drug tests at screening and once randomly before one of the test sessions, and no illicit substances were detected during the study. The subjects were asked to abstain from excessive alcohol consumption between test sessions and particularly limit their alcohol use to one standard drink on the day before the test sessions. Additionally, the participants were not allowed to drink xanthine‐containing liquids after midnight before the study day. The participants did not regularly use medications that could potentially interact with the study drug. Five subjects had previously used a hallucinogen, including LSD (three participants), one to four times during their lives, and the other 22 participants were hallucinogen‐naïve. A previous study found no difference in the response to LSD between hallucinogen‐naïve and moderately experienced subjects (<10 times).5 THC use prevalence is high in Switzerland (48% in young Swiss men22) and previous experience in the absence of dependence was not an exclusion criterion. There is no data on possible interactions of THC and LSD. Subjects were not allowed to use THC during the study. Finally, the study included seven participants who smoked on average 4 ± 3 (range: 1–8) cigarettes/day and 20 non‐smokers. Tobacco smoking induces CYP1A2 function, which is involved in the metabolism of LSD in vitro.23 However, there are no human data.
2.3. Study procedures
The study included a screening visit, a psychiatric interview, four 12‐hour experimental sessions and an end‐of‐study visit. The experimental sessions were conducted in quiet standard hospital patient rooms. The participants rested in hospital beds except when going to the bathroom. Only one participant and one investigator were present during the experimental sessions per testing room. The participants could interact with the investigator, rest quietly and/or listen to music via headphones, but no other entertainment was provided. LSD or placebo was administered at 10:00 a.m. A small breakfast (two plain croissants), a small lunch (one sandwich), and a dinner (full meal) were served at 8:30 a.m., 1:30 p.m. and 6:00 p.m., respectively. The participants were never alone during the 12‐hour session and went home at 10:00 p.m.
2.4. Study drug
LSD (D‐lysergic acid diethylamide base, high‐performance liquid chromatography purity >99%, Lipomed AG, Arlesheim, Switzerland) was administered in a single oral dose. Each dose of LSD was formulated as a solution to be administered orally in 1 mL of 96% ethanol according to GMP (batch BZ17‐1) and stored free in argon‐prefilled vials in the dark at 4°C. The exact analytically confirmed LSD content (mean ± SD) of the formulation was 96.2 ± 0.3 μg (n = 6) after production. Stability of the formulation for longer than the study period was documented in an identically produced previous batch (batch BZ16) by D. Trachsel, Reseachem, Burgdorf, Switzerland. The isomerization of active LSD to inactive iso‐LSD occurred to a small degree when the solution was stored at 4°C and resulted in iso‐LSD contents (% of initial LSD content) of 0.1%, 0.1%, 1.3%, 3.2% and 3.6% after 4, 6, 12, 18 and 24 months, respectively. No other decomposition products were present. Vials that were stored at room temperature had higher iso‐LSD contents of 0%, 3.1%, 3.4%, 6.7% and 9.5% after 2, 4, 6, 12 and 24 months, respectively. The LSD base dose that was used in the present study would correspond to a dose of 118 μg of LSD tartrate (or 125 μg tartrate including crystal water), which is the form of LSD that is more likely to be used when acquired illegally (i.e., in blotter form) for recreational use.
2.5. Measures
2.5.1. Blood sampling
Blood was collected into lithium heparin tubes before and 1, 1.5, 2, 3, 3.5, 4.5, 5.5, 6.5, 7.5, 9.5 and 11.5 hours after LSD administration. The blood samples were immediately centrifuged, and the plasma was subsequently stored at −20°C. For long‐term storage (1–18 months), the samples were kept at −80°C until analysis. Long‐term stability has been shown for LSD when kept under refrigerated or frozen conditions.14, 24
2.5.2. Analysis of LSD and metabolite concentrations
LSD, O‐H‐LSD, nor‐LSD and iso‐LSD levels were analysed in human plasma by ultra‐high‐performance liquid chromatography tandem mass spectrometry (UHPLC–MS/MS). The UHPLC apparatus was from Shimadzu (Kyoto, Japan) and consisted of four pumps (LC‐30AD), a solvent degasser (DGU‐20A5R), an autosampler (SIL‐30AC), a column oven (CTO‐20AC) and a system controller (CBM‐20A). An API 5500 QTrap (AB Sciex, Concord, Canada) tandem mass spectrometer equipped with an electrospray interface was used as the detector (see Table S1 for settings).
O‐H‐LSD, O‐H‐LSD‐d10 and nor‐LSD were purchased from TRC (Ontario, Canada). Stock solutions of 1 mg/mL were prepared in dimethylsulfoxide (DMSO). LSD, LSD‐d3 and iso‐LSD solutions (1 mg/mL in ethanol) were purchased from Lipomed (Basel‐Land, Switzerland). Calibration lines of LSD, O‐H‐LSD, nor‐LSD and iso‐LSD were prepared in drug‐free plasma that contained less than 1% DMSO or ethanol, ranging from 25 (lower limit of quantification [LLOQ]) to 10 000 pg/mL. Quality control samples were prepared at 25, 100, 1000 and 10 000 pg/mL. An accuracy of 85–115% (LLOQ: 80–120%) and precision of less than 15% (LLOQ: 20%) was accepted in this study (Table S2). An aliquot of 50 μL of plasma was extracted with 150 μL acetonitrile that contained 0.1 ng/mL LSD‐d3 and 0.25 ng/mL O‐H‐LSD‐d10 of the internal standards (ISs). After rigorous mixing and 30 minutes of centrifugation at 3200g at 10°C, 10 μL of the sample's supernatant was injected into the UHPLC–MS/MS system. The sample was loaded on the analytical column (Kinetex Evo C18, 1.7 μm, 50 × 2.1 mm, Phenomenex, Torrance, CA, USA) using 10% mobile phase B (acetonitrile plus 0.1% formic acid) and 90% mobile phase A (20 mM ammonium bicarbonate adjusted to pH 9 using 25% ammonium hydroxide). The flow rate was increased from 0.1 to 0.6 mL/min within the first 0.5 minutes. In parallel, mobile phase A was delivered using a third pump to dilute the injected sample via a t‐union, which was installed before the analytical column. The flow rate of this pump was thus linearly decreased from 0.5 to 0 mL/min in the first 0.5 minutes of each run. The mobile phase B concentration was increased from 10% to 95% between 0.5 and 2.75 minutes to elute the three analytes. The analytical run was terminated by flushing the column for 0.75 minutes with 95% mobile phase B and reconditioning it for another 0.5 minutes with 10% mobile phase B. This gradient program resulted in a baseline separation of O‐H‐LSD (1.49 min), nor‐LSD (1.71 min) and LSD (1.79 min). Mobile phase B was more slowly increased from 10% to 35% between 0.5 and 4.0 minutes to separate LSD from iso‐LSD. Afterwards, the mobile phase B concentration was raised to 95% within 0.5 minutes and kept at this level for an additional minute to flush the analytical column. At the end of the run, the column was reconditioned at 10% mobile phase B for 0.5 minutes. This prolonged gradient program resulted in a baseline separation of LSD (3.8 min) and iso‐LSD (4.1 min). Concentrations of iso‐LSD were only determined in samples that were collected at 2 hours (i.e., close to the C max of LSD).
All of the analytes were detected by multiple reaction monitoring (MRM) in the positive mode. Analyte‐specific settings are given in Table S2. The mass transitions were summed for all of the analytes to increase sensitivity of the method. Gas parameters were set at medium, 20, 30 and 60 for the collision gas, curtain gas, ion source gas 1 and ion source gas 2, respectively. The interface temperature was 500°C, and the ion spray voltage was 5500 V. The system was operated with Analyst 1.6.2 software (AB Sciex, Concord, Canada). Pharmacokinetic data were quantified using MultiQuant 3.0.1 software (AB Sciex, Concord, Canada).
2.5.3. Subjective mood
Visual Analog Scales (VASs) were repeatedly used to assess subjective effects over time.4, 5, 11 The VASs included separate measures for “any drug effect,” “good drug effect,” “bad drug effect”, and “the boundaries between myself and my surroundings seemed to blur” (ego dissolution21, 25) and were presented as 100 mm horizontal lines (0–100%) marked from “not at all” on the left to “extremely” on the right. The VASs were administered before and 1, 1.5, 2, 3, 3.5, 4.5, 5.5, 6.5, 7.5, 9.5 and 11.5 hours after LSD administration.
2.6. Pharmacokinetic analyses and pharmacokinetic‐pharmacodynamic modelling
All of the analyses were performed using Phoenix WinNonlin 6.4 (Certara, Princeton, NJ, USA). Pharmacokinetic parameters were estimated using compartmental modelling. A one‐compartment model was used with first‐order input, first‐order elimination, and no lag time. Initial estimates for Vd/F and λ were derived from non‐compartmental analyses. The model fit was not relevantly improved by a two‐compartment model based on visual inspection of the plots. The one‐compartment model also resulted in smaller Akaike information criterion values in all subjects compared with a two‐compartment model. A similar one‐compartment model but with a lag time was used to determine the parameters for the metabolite O‐H‐LSD because a better fit could be obtained compared with the no‐lag‐time model. The PK model was first fitted and evaluated. The predicted concentrations were then used as an input to the PD model by treating the PK parameters as fixed and using the classic PK/PD link model module in WinNonlin. The model used a first‐order equilibrium rate constant (k eo) that related the observed PD effects of LSD to the estimated LSD concentrations at the effect site and accounted for the lag between the plasma and effect site concentration curves.11, 26 A sigmoid maximum effect (E max) model (EC50, E max, γ) was selected for all PD effects: E = (E max × C p h)/(C p h + EC 50 h), in which E is the observed effect, C p is the plasma LSD concentration, E max is the maximal effect and h is the Hill slope using WinNonlin. EC50 and E max estimates were taken from the PK/PD plots.11 Lower and upper limits for E max were set to 0% and 100%, respectively, for all of the VAS scores. The sigmoidal E max model best described the relationship between estimated effect‐site concentrations and LSD effects compared with a simple E max model (plot inspection [Figure S1] and Akaike information criteria). To compare the present PK data for the LSD solution with previous data for LSD capsules,11 the previously published data were reanalysed similarly to the present data using both compartmental and non‐compartmental analyses.
2.7. Statistical analyses
Comparisons of PK parameters between the solution and capsule were made using t‐tests (Statistica 12 software; StatSoft, Tulsa, OK, USA). The onset, t max, offset and effect duration were assessed for the model‐predicted “any drug effect” VAS effect–time plots after LSD administration using a threshold of 10% of the maximum individual response using Phoenix WinNonlin 6.4. Associations between peak concentrations and peak effects across subjects were assessed using Pearson correlations.
2.8. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY.27
3. RESULTS
3.1. Pharmacokinetics
Concentrations of LSD and O‐H‐LSD could be quantified in all of the subjects and at all time points. In contrast, nor‐LSD was detected in plasma, but concentrations could not be quantified (<25 pg/mL). Concentrations of iso‐LSD were 5% ± 2% of those of LSD, indicating no relevant isomerization/inactivation of LSD. The plasma concentration–time curves for LSD and O‐H‐LSD are shown in Figure 1A. The pharmacokinetic parameters are shown in Table 1. Individual PK/PD model‐predicted LSD concentration–time curves are shown in Figure 2A. The individual observed LSD concentrations are shown in Figure S2, together with their individual model‐predicted curves. Parameters based on non‐compartmental analysis are summarized in Table S3. No sex differences in the PK parameters were observed. There were no differences in the PK of LSD between tobacco smokers (<10 cigarettes/day) and non‐smokers.
Figure 1.
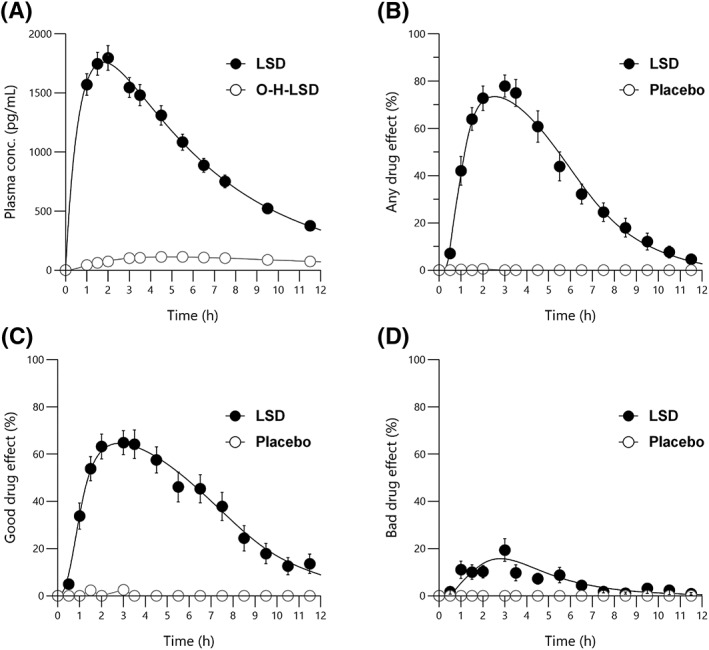
Pharmacokinetics and pharmacodynamics of LSD. (A) Plasma LSD and 2‐oxo‐3‐hydroxy LSD (O‐H‐LSD) concentration–time curves. Nor‐LSD levels were below the level of quantification. (B–D) LSD effect–time curves for visual analog scale ratings (0–100%) of (B) “any drug effect,” (C) “good drug effect,” and (D) “bad drug effect.” “Any drug effect” and “good drug effect” were robustly and markedly increased in all subjects and paralleled the changes in LSD concentrations, whereas the mean “bad drug effect” increased only slightly after LSD administration due to moderate anxiety in some but not all subjects. The data are expressed as the mean ± SEM in 27 subjects after administration of 100 μg LSD or placebo at t = 0 h. the lines represent the mean of the individual predictions based on the pharmacokinetic/pharmacodynamic model
Table 1.
Pharmacokinetic parameters for LSD and O‐H‐LSD based on compartmental modelling
| K 01 (1/h) | λ z (1/h) | V z /F (L) | C max (ng/mL) | t max (h) | t 1/2 (h) | AUC ∞ (ng·h/mL) | CL/F (L/h) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| LSD | All | 27 | Geometric mean (95% CI) | 1.2 (1.1–1.7) | 0.19 (0.18–0.22) | 39 (36–47) | 1.7 (1.6–2.0) | 1.7 (1.6–2.0) | 3.6 (3.3–4.3) | 13 (12–16) | 7.5 (6.9–9.0) |
| Range | 0.31–3.2 | 0.10–0.29 | 23–81 | 1.0–2.9 | 1.0–3.4 | 2.4–7.3 | 7.1–28 | 3.6–14 | |||
| Male | 13 | Geometric mean (95% CI) | 1.2 (1.0–1.7) | 0.18 (0.15–0.23) | 42 (34–54) | 1.7 (1.4–2.1) | 1.8 (1.5–2.2) | 3.9 (3.2–5.0) | 13 (11–18) | 7.4 (6.1–9.9) | |
| Range | 0.31–2.5 | 0.10–0.28 | 26–81 | 1.0–2.9 | 1.1–3.4 | 2.4–7.3 | 7.1–28 | 3.6–14 | |||
| Female | 14 | Geometric mean (95% CI) | 1.3 (1.0–1.9) | 0.20 (0.18–0.24) | 37 (32–46) | 1.8 (1.6–2.1) | 1.7 (1.4–2.1) | 3.4 (3.0–4.1) | 13 (12–16) | 7.6 (6.6–9.1) | |
| Range | 0.48–3.2 | 0.12–0.29 | 23–61 | 1.2–2.4 | 1.0–2.9 | 2.4–5.7 | 7.2–22 | 4.6–14 | |||
| O‐H‐LSD | All | 27a | Geometric mean (95% CI) | 0.34 (0.31–0.45) | 0.13 (0.12–0.18) | 490 (442–646) | 0.11 (0.10–0.13) | 5.0 (4.7–5.7) | 5.2 (4.2–7.8) | 1.7 (1.5–2.1) | 51 (48–69) |
| Range | 0.14–0.75 | 0.03–0.27 | 197–1294 | 0.07–0.19 | 3.2–8.0 | 2.6–21 | 0.85–4.3 | 12–118 |
AUC∞, area under the plasma concentration–time curve from time zero to infinity; CL/F, apparent total clearance; C max, estimated maximum plasma concentration; k01, first‐order absorption koefficient; t 1/2, estimated plasma elimination half‐life; t max, estimated time to reach C max; Vz/F, volume of distribution; λz, first‐order elimination coefficient.
Four subjects were not included in the the calculation of λz, t 1/2 and AUC∞ because the sampling period was too short to correctly determine λz.
Figure 2.
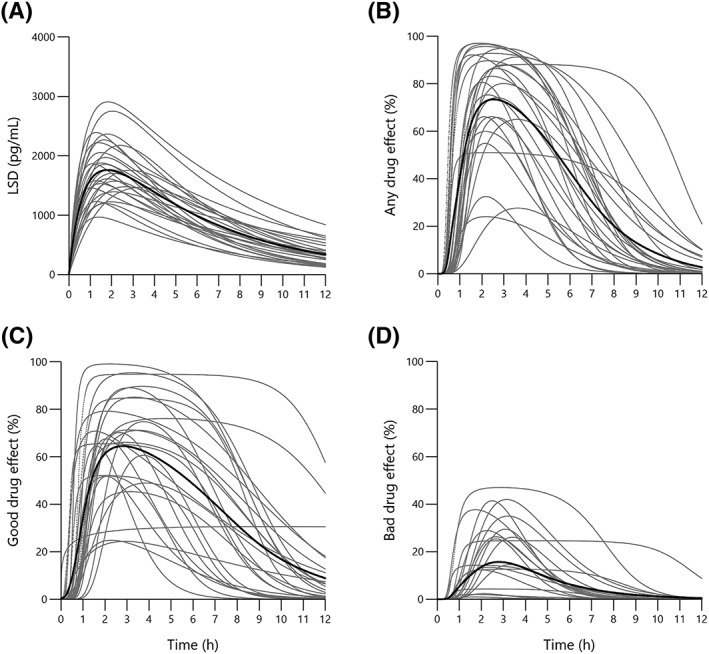
Individual pharmacokinetics and pharmacodynamics of LSD. (A) Individual plasma LSD concentration–time curves. (B–D) individual LSD effect–time curves for visual analog scale ratings (0–100%) of (B) “any drug effect,” (C) “good drug effect,” and (D) “bad drug effect.” curves represent the individual pharmacokinetic/pharmacodynamic model predictions in 27 subjects with the mean marked in bold. LSD (100 μg solution) was administered at t = 0 h
3.2. Pharmacodynamics and pharmacokinetic‐pharmacodynamic modelling
LSD produced robust increases in “any drug effect” (Figure 1B) and “good drug effect” (Figure 1C). Transient “bad drug effect” was reported in some subjects, resulting in a moderate increase in mean group ratings (Figure 1D). LSD also induced “ego dissolution” (Figure S3). The variability in intensity in subjective drug effects is illustrated in the “any drug effect,” “good drug effect” and “bad drug effect” curves in Figure 2B–D, respectively. The individual ratings for each subject and time point are shown in Figure S4–S6, respectively, together with the modelled curves. Times of onset and offset of the subjective response, assessed by the “any drug effect” VAS, were (mean ± SD) 0.7 ± 0.2 h (range: 0.3–1.0 h) and 9.1 ± 2.0 h (range: 6.0–13.2 h), respectively. The mean effect duration was 8.5 ± 2.0 h (range: 5.3–12.8 h). The time to peak drug effect was 2.5 ± 0.6 h (range: 1.6–4.3 h).
The predicted concentrations of LSD that produced half‐maximal effects (EC50 values) and E max values were 1.1 ± 0.4 ng/mL and 91% ± 17% for “any drug effect,” 1.0 ± 0.5 ng/mL and 83% ± 20% for “good drug effect,” 1.9 ± 1.0 ng/mL and 40% ± 41% for “bad drug effect,” and 1.4 ± 0.5 ng/mL and 80% ± 34% for “ego dissolution,” respectively (see Table S4 for additional parameter estimates).
The C max of LSD did not correlate with the E max values of the subjective response on any of the VAS when analysed across subjects. Thus, in contrast to the close relationship over time within‐subjects, the plasma concentrations of LSD were not associated with the subjective effects of LSD when analysed across subjects after the use of the same dose of LSD in all participants (relatively similar C max and E max values in all subjects).
3.3. Pharmacokinetic and pharmacokinetic‐pharmacodynamic comparison with LSD capsules
The model‐predicted geometric mean (CV%, 95% confidence interval [CI]) C max of the LSD solution was 1.7 (27%, 1.6–2.0) ng/mL and non‐significantly (t 1,49 = 1.6, p = 0.12) higher compared with 1.3 (60%, 1.2–1.9) ng/mL for the LSD capsule that was tested previously in different subjects.11 The geometric mean (CV%, 95% CI) LSD AUC∞ values were 13 (34%, 12–16) ng·h/mL and 8.1 (66%, 7.5–11) ng·h/mL for the LSD solution and LSD capsule, respectively, representing a significant difference in overall exposure between the two formulations (t 1,49 = 3.67, p < 0.001). Additionally, lower inter‐individual variability was observed compared with the capsule, indicated by the CV% values and plot of the individual model‐predicted LSD concentrations after administration of the two formulations (Figure S7). Additional analyses were performed to explore differences in absorption between the two formulations. Model‐estimated t max values (geometric mean [CV%, 95% CI]) were comparable for the solution and the capsule (1.7 [29%, 1.6–2.0] h and 1.4 [82%, 1.3–2.1] h, respectively; t 1,49 = 0.44, p = 0.66), although greater variability was observed for the capsule. The first‐order absorption coefficients were also not significantly different between the solution and the capsule (1.2 [59%, 1.1–1.7] and 1.4 [164%, 1.2–4.1], respectively; t 1,49 = 1.86, p = 0.07). The onset time that was needed to reach a minimum plasma LSD concentration of 0.3 ng/mL (i.e., the lowest concentration reached in all subjects) was 0.10 (48%, 0.09–0.14) h and 0.12 (164%, 0.11–0.28) h after administration of the solution and capsule, respectively, and significantly shorter (t 1,49 = 2.08, p = 0.04) and also less variable after administration of the solution compared with the capsule. The t 1/2 of the LSD solution was 3.6 (33%, 3.3–4.3) h after administration compared with 2.6 (29%, 2.4–3.0) h after capsule administration (t 1,49 = 3.70, p < 0.001). Non‐compartmental analysis confirmed the longer t 1/2 of LSD in the present study (3.7 [3.4–4.1] h) compared with our previous study that used capsules (2.6 [2.4–3.0] h). PK parameters that were derived from the non‐compartmental analysis were comparable to those that were based on the model and are shown for the LSD solution and capsule formulations in Tables S3 and S5, respectively.
The subjective‐effects PD parameter estimates for the solution and the capsule did not differ significantly, with the exception that the EC50 value for “any drug effect” was lower for the capsule compared with the solution (t 1,49 = 3.30, p < 0.01; Table S4). Times of onset and offset of the subjective response, mean effect duration, time to peak drug effect and the area under the effect–time curve did not differ significantly between the two formulations (Table S6). However, the time to effect onset presented greater variance for the capsule compared with the solution (Table S6, Figure S8), which is consistent with the PK of the two formulations (Figure S7).
4. DISCUSSION
The present study mainly characterized the PK and subjective effects of a novel LSD solution that is intended for clinical research and use. The present data on the plasma concentration–time curves of LSD are important because many experimental and therapeutic studies are currently being conducted with this formulation (ClinicalTrials.gov no. NCT03604744, NCT03321136, NCT03019822, NCT03153579, and planned). Many recent LSD studies3, 6, 7 have been published without information on the presence of LSD in the human body, and the actual exposure to LSD and exposure–time curves are unknown. The present study also allows an indirect reassessment of the doses that were reported in our previous studies.
The PK and PD parameters that were derived from the present study are generally similar to those in our previous studies that used 100 and 200 μg in capsule form,11, 12 with the exception of higher C max and AUC values. Maximum concentrations of LSD were reached an average of 1.7 hours after administration, and first‐order elimination kinetics of LSD were confirmed.9, 11 The subjective drug effects began an average of 0.7 hours after administration, lasted 8.5 hours, and declined in parallel with plasma LSD concentrations.
We expected more rapid absorption and a faster onset of action with the oral solution compared with the capsules. We found that the absorption coefficients and t max values were not significantly different between the two formulations. Thus, the rate‐limiting steps to increase LSD concentrations may not be absorption but rather other processes, such as dilution and distribution in the circulation. However, we took only a few blood samples during the expected absorption time period, and we may have missed potential differences between the two formulations. Additionally, a threshold plasma LSD level of 0.3 ng/mL was reached significantly faster after administration of the solution compared with the capsule. This could indicate faster absorption or could be explained by the overall higher LSD concentrations that were reached with the solution. Early studies indicated that the effects of LSD tartrate peak approximately 30 minutes after intravenous administration.9, 28, 29, 30 A recent study that evaluated the intravenous administration of LSD base reported that subjective drug effects began within 5–15 minutes and peaked relatively late at 45–90 minutes after infusion of the drug.3, 31 Thus, the time to the maximal response appears to be rather long after both intravenous and oral administration of LSD base. Possible reasons for the long t max may include slow rates of dilution and distribution within the circulation, slow blood–brain barrier passage, slow diffusion to target sites in the brain, and lags in the response mechanism itself.
The terminal t 1/2 value of LSD should not depend on the type of formulation that is used. The average t 1/2 of LSD was 3.6 hours in the present study, which is within the range (2.6–3.6 h) that was reported in our previous studies that used capsules.11 An older small study that evaluated the intravenous administration of LSD (2 μg/kg) calculated a t 1/2 of 2.9 hours.9
The average AUC∞ value, reflecting total LSD exposure, was 13.3 and 8.1 ng·h/mL in the present study and our previous study,11 respectively, and thus 1.6‐times greater in the present study compared with the previous study that used the same indicated dose of 100 μg of LSD base that was formulated as a capsule.11 Although the oral drinking solution may have had higher oral bioavailability than the capsule formulation of LSD, the true LSD content of the previously used LSD capsules may have been lower than reported. First, valid longer‐term stability data beyond the full study duration were unavailable for the capsules that were used in several previous studies by us and others.4, 5, 6, 11, 20, 21, 32, 33, 34, 35, 36 Second, after administration of the 200 μg dose in the form of two 100 μg capsules, iso‐LSD was detected in plasma,13 indicating that this inactive decomposition product of LSD was possibly already present in the capsules at the time of their use (although possible formation in the plasma samples cannot be completely excluded). The plasma AUC24 values of LSD and iso‐LSD of 21 and 9.2 ng·h/mL13 indicate that an average of 30% of the LSD may have isomerized to inactive iso‐LSD in the capsules. Thus, the actual administered doses of LSD may have been 70 and 140 μg LSD base rather than the indicated 100 and 200 μg, respectively. The AUC∞ values in the previous studies that used 100 and 200 μg doses were 61% and 76%, respectively, of the values that were expected based on the present confirmed 96 μg LSD dose and assuming similar bioavailability. Finally, analytical tests of four unused old LSD capsules that were performed years after study completion suggested a marked reduction of LSD content (remaining amount of LSD = 22 ± 7 μg), indicating a lack of longer‐term stability of LSD in this form and that the actual LSD doses that were used were likely already lower than indicated during the studies. Notably, a decrease in content by 15% or even 25% in single capsules would still be compatible with content uniformity, which was documented during production of the capsules.
In summary, based on the results of different quality‐control measures, analytical findings (including the present PK data), and the clinical effects of the different formulations,11 we surmise that the previous studies actually used approximately 60–70 (not 100) μg and 140–150 (not 200) μg of LSD base, corresponding to approximately 80 and 175 μg of LSD tartrate. Additionally, the solution and capsules did not produce significantly different subjective drug effects (i.e., intensity and duration) despite the differences in plasma concentrations. Thus, the actual drug effects in the present study were comparable to previous reports. Although we cannot exclude possible differences in bioavailability of the presently used oral formulation and the previously used capsules, many previously reported clinical and neuroimaging results were likely produced with LSD doses that were lower than reported. Certainly, exposure to LSD in the body, expressed by the AUC, was lower in the previous studies compared with the present study that used the same reported dose. Another consideration is that doses of LSD that were reported in previous studies may not have been very precise or may not have reflected the actual exposure of LSD in the body. This is notably also the case for recent studies that used intravenous dosing with 75 μg hydrophobic LSD base in saline because objective measures of exposure to LSD (i.e., plasma concentrations) were lacking, and the bioavailability of the solution is unknown.3, 25, 31, 37, 38 The clinical response to 75 μg of intravenous LSD was not significantly different from the oral 100 μg dose that was used in our previous studies,2, 21, 37 indirectly indicating similar exposure that is comparable to an oral dose of 60–70 μg LSD base.
The present study and discussion illustrate that PK investigations are imperative to confirm the presence and extent of the presence of LSD in the body, particularly when pharmaceutically or pharmacologically poorly characterized formulations of LSD are used in experimental research settings. The present study also described the acute subjective effects of LSD. LSD produced high subjective “good drug effect” in almost all of the subjects. “Bad drug effects” were typically smaller and not present in every subject. Mean EC50 values were 1.0 and 1.9 ng/mL for “good drug effects” and “bad drug effects,” respectively, indicating that anxiety is associated with higher LSD concentrations. The subjective LSD response was similar in intensity, onset and duration in the present study that used the novel LSD solution compared with a previously used capsule formulation,11 although a significantly higher plasma LSD concentration was reached in the present study. The only notable difference was lower variance in the time to effect onset for the solution compared with the capsule, which is consistent with the less variable time to reach a minimum plasma concentration.
The present study confirmed the previous finding11 of a close relationship between LSD concentrations and LSD effects over time within each subject. In contrast, it has previously been shown in detail that concentrations of LSD do not correlate with the response when analysed across subjects in a group of subjects who each received the same dose of LSD.11 Thus, the plasma concentrations of LSD do not predict the effects of LSD during the time it produces robust and similar effects in all subjects (i.e., little between‐subject variability).11 In contrast, there is a close relationship over time within‐subjects, as shown in the PK/PD analysis of the present study.
The present study has limitations. The comparison with the LSD capsules used data from different studies that included different subjects. Small study differences could have contributed to differences in the PK of the two LSD formulations. For example, we used a refined LC–MS/MS assay in the present study, which has higher sensitivity compared with the previous assay.11, 39 Plasma samples were collected at 1.5 hours in the present study but not in the previous study,11 whereas the previous study had a longer sampling time and included quantified LSD concentrations up to 24 hours in some subjects. Few samples were taken before C max, thus precluding good characterization of the absorption and early distribution phase. However, the half‐life of LSD is relatively short and the PK linear, allowing the determination of the PK parameters with high validity within a sampling time of 12 hours. The present study also has notable strengths. The study was relatively large and included both male and female subjects, thus allowing valid comparisons of PK between sexes. A well‐characterized formulation of LSD was also used. Quality assurance data were provided, which are typically unavailable or not reported in other studies.
In summary, we present PK data for a novel oral LSD formulation that are useful for interpreting the findings of clinical studies and LSD intoxications.
COMPETING INTERESTS
There are no competing interests to declare.
CONTRIBUTORS
F.H. and M.E.L. designed the research. F.H., U.D., P.V., F.M. and S.B. performed the research. F.H., U.D., P.V. and M.E.L. analysed the data. F.H. and M.E.L. wrote the manuscript with input from all of the other authors.
Supporting information
Figure S1. Diagnostic plots for a representative subject. Upper left panel: Observed and predicted concentrations of LSD vs. time. Upper middle panel: Observed vs. predicted concentrations of LSD. Upper right panel: Residual vs. predicted concentrations of LSD. Lower left panel: Observed and predicted effects of LSD vs. time. Lower middle panel: Observed vs. predicted effects of LSD. Lower right panel: Residual vs. predicted effects of LSD.
Figure S2. LSD plasma concentration–time curves. LSD was orally administered as a solution in ethanol at a dose of 100 μg at t = 0. The data represent individual observed LSD plasma concentrations as measured at the different time points (●) and the LSD concentrations predicted by the one‐compartment pharmacokinetic model (black lines).
Figure S3. Ego dissolution induced by LSD. (A) “Ego dissolution” was induced to variable extents. The data are expressed as the mean ± SEM in 27 subjects after administration of 100 μg LSD or placebo at t = 0 h. The line represents the mean of the individual predictions based on the pharmacokinetic/pharmacodynamic model. (B) Individual LSD effect‐time curves for Visual Analog Scale ratings (0–100%) of “ego dissolution.” Curves represent the individual pharmacokinetic/pharmacodynamic model predictions in 27 subjects with the mean marked in bold.
Figure S4. Subjective responses to LSD. LSD was orally administered as a solution in ethanol at a dose of 100 μg at t = 0 h. The data represent individual observed LSD responses on the “any drug effect” Visual Analog Scale (rated 0–100%) at the different time points (●) and the pharmacokinetic/pharmacodynamic model‐predicted effect (black lines).
Figure S5. Subjective responses to LSD. LSD was orally administered as a solution in ethanol at a dose of 100 μg at t = 0 h. The data represent individual observed LSD responses on the “good drug effect” Visual Analog Scale (rated 0–100%) at the different time points (●) and the pharmacokinetic/pharmacodynamic model‐predicted effect (black lines).
Figure S6. Subjective responses to LSD. LSD was orally administered as a solution in ethanol at a dose of 100 μg at t = 0 h. The data represent individual observed LSD responses on the “bad drug effect” Visual Analog Scale (rated 0–100%) at the different time points (●) and the pharmacokinetic‐pharmacodynamic model‐predicted effect (black lines).
Figure S7. Individual LSD plasma concentration–time curves in (A) 27 healthy subjects after 100 μg of LSD formulated as oral solution in ethanol (present study) and in (B) 24 healthy subjects after administration of 100 μg LSD formulated in gelatine capsules (Dolder et al., 2017). Higher average plasma concentrations were reached after the oral solution compared with the capsules. The geometric mean (range) C max of the LSD solution was 1.7 (1.0–2.9) ng/mL compared to 1.3 (0.3–3.7) ng/mL for the LSD capsule tested previously in different subjects (Dolder et al., 2017). The geometric mean (range) AUC∞ values were 13 (7.1–28) and 8.1 (1–19) ng·h/mL for the solution and capsule, respectively. There was less variation in the LSD concentrations between the subjects after the solution compared to the capsules. Each line indicates the LSD concentration in one subject predicted using an identical one‐compartment pharmacokinetic model for each subject and formulation. The bold line indicates the mean of all curves. LSD was administered at t = 0 h.
Figure S8. Individual LSD subjective effect–time curves (VAS any drug effect) in (A) 27 healthy subjects after 100 μg of LSD formulated as oral solution in ethanol (present study) and in (B) 24 healthy subjects after administration of 100 μg LSD formulated in gelatine capsules (Dolder et al., 2017). Times of onset and offset, effect duration and peak drug effects were comparable after the oral solution compared with the capsules. Time to onset was more variable after the capsule administration compared with the solution. Each line indicates the LSD effect in one subject predicted using an identical pharmacokinetic‐pharmacodynamic model for each subject and formulation. The bold line indicates the mean of all curves. LSD was administered at t = 0 h.
Table S1. Analyte specific settings used for the analysis of O‐H‐LSD, nor‐LSD, and LSD
Table S2. Intra‐assay accuracy and precision data of O‐H‐LSD, nor‐LSD, and LSD.
Table S3. Pharmacokinetic parameters for LSD and O‐H‐LSD based on non‐compartmental analysis
Table S4. Pharmacodynamic parameter estimates (PK‐PD link model)
Table S5. Pharmacokinetic parameters for LSD 100 μg capsules based on non‐compartmental analysis
Table S6. Characteristics of the subjective response (any drug effect) in response to the solution or capsule
ACKNOWLEDGEMENTS
The authors thank Patrick Dolder, Samuel Harder, Raoul Dürig and Laura Ley for conducting the study, Beatrice Vetter for conducting laboratory work, and Michael Arends for proofreading the manuscript. This work was supported by the Swiss National Science Foundation (grant no. 320030_170249 to ML).
Holze F, Duthaler U, Vizeli P, Müller F, Borgwardt S, Liechti ME. Pharmacokinetics and subjective effects of a novel oral LSD formulation in healthy subjects. Br J Clin Pharmacol. 2019;85:1474–1483. 10.1111/bcp.13918
The authors confirm that the PI for this paper is Matthias E. Liechti and that he had direct clinical responsibility for the participants.
REFERENCES
- 1. Krebs TS, Johansen PO. Over 30 million psychedelic users in the United States. F1000 Res. 2013;2:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Liechti ME. Modern clinical research on LSD. Neuropsychopharmacology. 2017;42(11):2114‐2127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Carhart‐Harris RL, Muthukumaraswamy S, Roseman L, et al. Neural correlates of the LSD experience revealed by multimodal neuroimaging. Proc Natl Acad Sci U S A. 2016;113(17):4853‐4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dolder PC, Schmid Y, Mueller F, Borgwardt S, Liechti ME. LSD acutely impairs fear recognition and enhances emotional empathy and sociality. Neuropsychopharmacology. 2016;41(11):2638‐2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schmid Y, Enzler F, Gasser P, et al. Acute effects of lysergic acid diethylamide in healthy subjects. Biol Psychiatry. 2015;78(8):544‐553. [DOI] [PubMed] [Google Scholar]
- 6. Preller KH, Herdener M, Pokorny T, et al. The fabric of meaning and subjective effects in LSD‐induced states depend on serotonin 2A receptor activation. Curr Biol. 2017;27(3):451‐457. [DOI] [PubMed] [Google Scholar]
- 7. Gasser P, Kirchner K, Passie T. LSD‐assisted psychotherapy for anxiety associated with a life‐threatening disease: a qualitative study of acute and sustained subjective effects. J Psychopharmacol. 2015;29(1):57‐68. [DOI] [PubMed] [Google Scholar]
- 8. Gasser P, Holstein D, Michel Y, et al. Safety and efficacy of lysergic acid diethylamide‐assisted psychotherapy for anxiety associated with life‐threatening diseases. J Nerv Ment Dis. 2014;202(7):513‐520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Aghajanian GK, Bing OH. Persistence of lysergic acid diethylamide in the plasma of human subjects. Clin Pharmacol Ther. 1964;5(5):611‐614. [DOI] [PubMed] [Google Scholar]
- 10. Upshall DG, Wailling DG. The determination of LSD in human plasma following oral administration. Clin Chim Acta. 1972;36(1):67‐73. [DOI] [PubMed] [Google Scholar]
- 11. Dolder PC, Schmid Y, Steuer AE, et al. Pharmacokinetics and pharmacodynamics of lysergic acid diethylamide in healthy subjects. Clin Pharmacokinetics. 2017;56(10):1219‐1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Dolder PC, Schmid Y, Haschke M, Rentsch KM, Liechti ME. Pharmacokinetics and concentration–effect relationship of oral LSD in humans. Int J Neuropsychopharmacol. 2015;19. pii: pyv072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Steuer AE, Poetzsch M, Stock L, Eisenbeiss L, Schmid Y, Liechti ME, Kraemer T. Development and validation of an ultra‐fast and sensitive microflow liquid chromatography–tandem mass spectrometry (MFLC–MS/MS) method for quantification of LSD and its metabolites in plasma and application to a controlled LSD administration study in humans. Drug Test Anal 2017;9(5):788–797. [DOI] [PubMed] [Google Scholar]
- 14. Klette KL, Horn CK, Stout PR, Anderson CJ. LC–MS analysis of human urine specimens for 2‐oxo‐3‐hydroxy LSD: method validation for potential interferants and stability study of 2‐oxo‐3‐hydroxy LSD under various storage conditions. J Anal Toxicol. 2002;26(4):193‐200. [DOI] [PubMed] [Google Scholar]
- 15. Poch GK, Klette KL, Anderson C. The quantitation of 2‐oxo‐3‐hydroxy lysergic acid diethylamide (O‐H‐LSD) in human urine specimens, a metabolite of LSD: comparative analysis using liquid chromatography‐selected ion monitoring mass spectrometry and liquid chromatography‐ion trap mass spectrometry. J Anal Toxicol. 2000;24:170‐179. [DOI] [PubMed] [Google Scholar]
- 16. Dolder PC, Liechti ME, Rentsch KM. Development and validation of a rapid turboflow LC–MS/MS method for the quantification of LSD and 2‐oxo‐3‐hydroxy LSD in serum and urine samples of emergency toxicological cases. Anal Bioanal Chem. 2015;407(6):1577‐1584. [DOI] [PubMed] [Google Scholar]
- 17. Canezin J, Cailleux A, Turcant A, Le Bouil A, Harry P, Allain P. Determination of LSD and its metabolites in human biological fluids by high‐performance liquid chromatography with electrospray tandem mass spectrometry. J Chromatogr B Biomed Sci Appl. 2001;765(1):15‐27. [DOI] [PubMed] [Google Scholar]
- 18. Dolder PC, Strajhar P, Vizeli P, Hammann F, Odermatt A, Liechti ME. Pharmacokinetics and pharmacodynamics of lisdexamfetamine compared with D‐amphetamine in healthy subjects. Front Pharmacol. 2017;8:617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dolder PC, Mueller F, Schmid Y, Borgwardt SJ, Liechti ME. Direct comparison of the acute subjective, emotional, autonomic, and endocrine effects of MDMA, methylphenidate, and modafinil in healthy subjects. Psychopharmacology (Berl). 2018;235(2):467‐479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schmid Y, Liechti ME. Long‐lasting subjective effects of LSD in normal subjects. Psychopharmacology (Berl). 2018;235(2):535‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liechti ME, Dolder PC, Schmid Y. Alterations in consciousness and mystical‐type experiences after acute LSD in humans. Psychopharmacology (Berl). 2017;234(9–10):1499‐1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Baggio S, Studer J, Mohler‐Kuo M, Daeppen JB, Gmel G. Profiles of drug users in Switzerland and effects of early‐onset intensive use of alcohol, tobacco and cannabis on other illicit drug use. Swiss Med Wkly 2013;143:w13805. [DOI] [PubMed] [Google Scholar]
- 23. Wagmann L, Richter LHJ, Kehl T, et al. In vitro metabolic fate of nine LSD‐based new psychoactive substances and their analytical detectability in different urinary screening procedures. Anal Bioanal Chem. 2019. 10.1007/s00216-018-1558-9 [DOI] [PubMed] [Google Scholar]
- 24. Martin R, Schurenkamp J, Gasse A, Pfeiffer H, Kohler H. Determination of psilocin, bufotenine, LSD and its metabolites in serum, plasma and urine by SPE‐LC–MS/MS. Int J Leg Med. 2013;127(3):593‐601. [DOI] [PubMed] [Google Scholar]
- 25. Tagliazucchi E, Roseman L, Kaelen M, et al. Increased global functional connectivity correlates with LSD‐induced ego dissolution. Curr Biol. 2016;26(8):1043‐1050. [DOI] [PubMed] [Google Scholar]
- 26. Sheiner LB, Stanski DR, Vozeh S, Miller RD, Ham J. Simultaneous modeling of pharmacokinetics and pharmacodynamics: application to d‐tubocurarine. Clin Pharmacol Ther. 1979;25(3):358‐371. [DOI] [PubMed] [Google Scholar]
- 27. Harding SD, Sharman JL, Faccenda E, et al. The IUPHAR/BPS guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucl Acid Res. 2018;46:D1091‐D1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sokoloff L, Perlin S, Kornetsky C, Kety SS. The effects of D‐lysergic acid diethylamide on cerebral circulation and overall metabolism. Ann N Y Acad Sci. 1957;66(3):468‐477. [DOI] [PubMed] [Google Scholar]
- 29. Wagner JG, Aghajanian GK, Bing OH. Correlation of performance test scores with “tissue concentration” of lysergic acid diethylamide in human subjects. Clin Pharmacol Ther. 1968;9(5):635‐638. [DOI] [PubMed] [Google Scholar]
- 30. Hoch PH. Studies in routes of administration and counteracting drugs In: Cholden L, ed. Lysergic acid diethylamide and mescaline in experimental psychiatry. New York: Grune and Stratton; 1956:8‐12. [Google Scholar]
- 31. Kaelen M, Barrett FS, Roseman L, et al. LSD enhances the emotional response to music. Psychopharmacology (Berl). 2015;232(19):3607‐3614. [DOI] [PubMed] [Google Scholar]
- 32. Mueller F, Dolder PC, Schmidt A, Liechti ME, Borgwardt S. Altered network hub connectivity after acute LSD administration. Neuroimage Clin. 2018;18:694‐701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mueller F, Lenz C, Dolder PC, et al. Acute effects of LSD on amygdala activity during processing of fearful stimuli in healthy subjects. Transl Psychiatry. 2017;7(4):e1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mueller F, Lenz C, Dolder PC, et al. Increased thalamic resting state connectivity as a core driver of LSD‐induced hallucinations. Acta Psychiatr Scand. 2017;136(6):648‐657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schmidt A, Mueller F, Lenz C, et al. Acute LSD effects on response inhibition neuronal networks. Psychol Med. 2017;48:1464‐1473. [DOI] [PubMed] [Google Scholar]
- 36. Kraehenmann R, Pokorny D, Vollenweider L, et al. Dreamlike effects of LSD on waking imagery in humans depend on serotonin 2A receptor activation. Psychopharmacology (Berl). 2017;234(13):2031‐2046. [DOI] [PubMed] [Google Scholar]
- 37. Carhart‐Harris RL, Kaelen M, Bolstridge M, et al. The paradoxical psychological effects of lysergic acid diethylamide (LSD). Psychol Med. 2016;46(7):1379‐1390. [DOI] [PubMed] [Google Scholar]
- 38. Carhart‐Harris RL, Kaelen M, Whalley MG, Bolstridge M, Feilding A, Nutt DJ. LSD enhances suggestibility in healthy volunteers. Psychopharmacology (Berl). 2015;232(4):785‐794. [DOI] [PubMed] [Google Scholar]
- 39. Dolder PC, Liechti ME, Rentsch KM. Development and validation of an LC‐MS/MS method to quantify lysergic acid diethylamide (LSD), iso‐LSD, 2‐oxo‐3‐hydroxy‐LSD, and nor‐LSD and identify novel metabolites in plasma samples in a controlled clinical trial. J Clin Lab Anal. 2018;32(2):e22265. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Diagnostic plots for a representative subject. Upper left panel: Observed and predicted concentrations of LSD vs. time. Upper middle panel: Observed vs. predicted concentrations of LSD. Upper right panel: Residual vs. predicted concentrations of LSD. Lower left panel: Observed and predicted effects of LSD vs. time. Lower middle panel: Observed vs. predicted effects of LSD. Lower right panel: Residual vs. predicted effects of LSD.
Figure S2. LSD plasma concentration–time curves. LSD was orally administered as a solution in ethanol at a dose of 100 μg at t = 0. The data represent individual observed LSD plasma concentrations as measured at the different time points (●) and the LSD concentrations predicted by the one‐compartment pharmacokinetic model (black lines).
Figure S3. Ego dissolution induced by LSD. (A) “Ego dissolution” was induced to variable extents. The data are expressed as the mean ± SEM in 27 subjects after administration of 100 μg LSD or placebo at t = 0 h. The line represents the mean of the individual predictions based on the pharmacokinetic/pharmacodynamic model. (B) Individual LSD effect‐time curves for Visual Analog Scale ratings (0–100%) of “ego dissolution.” Curves represent the individual pharmacokinetic/pharmacodynamic model predictions in 27 subjects with the mean marked in bold.
Figure S4. Subjective responses to LSD. LSD was orally administered as a solution in ethanol at a dose of 100 μg at t = 0 h. The data represent individual observed LSD responses on the “any drug effect” Visual Analog Scale (rated 0–100%) at the different time points (●) and the pharmacokinetic/pharmacodynamic model‐predicted effect (black lines).
Figure S5. Subjective responses to LSD. LSD was orally administered as a solution in ethanol at a dose of 100 μg at t = 0 h. The data represent individual observed LSD responses on the “good drug effect” Visual Analog Scale (rated 0–100%) at the different time points (●) and the pharmacokinetic/pharmacodynamic model‐predicted effect (black lines).
Figure S6. Subjective responses to LSD. LSD was orally administered as a solution in ethanol at a dose of 100 μg at t = 0 h. The data represent individual observed LSD responses on the “bad drug effect” Visual Analog Scale (rated 0–100%) at the different time points (●) and the pharmacokinetic‐pharmacodynamic model‐predicted effect (black lines).
Figure S7. Individual LSD plasma concentration–time curves in (A) 27 healthy subjects after 100 μg of LSD formulated as oral solution in ethanol (present study) and in (B) 24 healthy subjects after administration of 100 μg LSD formulated in gelatine capsules (Dolder et al., 2017). Higher average plasma concentrations were reached after the oral solution compared with the capsules. The geometric mean (range) C max of the LSD solution was 1.7 (1.0–2.9) ng/mL compared to 1.3 (0.3–3.7) ng/mL for the LSD capsule tested previously in different subjects (Dolder et al., 2017). The geometric mean (range) AUC∞ values were 13 (7.1–28) and 8.1 (1–19) ng·h/mL for the solution and capsule, respectively. There was less variation in the LSD concentrations between the subjects after the solution compared to the capsules. Each line indicates the LSD concentration in one subject predicted using an identical one‐compartment pharmacokinetic model for each subject and formulation. The bold line indicates the mean of all curves. LSD was administered at t = 0 h.
Figure S8. Individual LSD subjective effect–time curves (VAS any drug effect) in (A) 27 healthy subjects after 100 μg of LSD formulated as oral solution in ethanol (present study) and in (B) 24 healthy subjects after administration of 100 μg LSD formulated in gelatine capsules (Dolder et al., 2017). Times of onset and offset, effect duration and peak drug effects were comparable after the oral solution compared with the capsules. Time to onset was more variable after the capsule administration compared with the solution. Each line indicates the LSD effect in one subject predicted using an identical pharmacokinetic‐pharmacodynamic model for each subject and formulation. The bold line indicates the mean of all curves. LSD was administered at t = 0 h.
Table S1. Analyte specific settings used for the analysis of O‐H‐LSD, nor‐LSD, and LSD
Table S2. Intra‐assay accuracy and precision data of O‐H‐LSD, nor‐LSD, and LSD.
Table S3. Pharmacokinetic parameters for LSD and O‐H‐LSD based on non‐compartmental analysis
Table S4. Pharmacodynamic parameter estimates (PK‐PD link model)
Table S5. Pharmacokinetic parameters for LSD 100 μg capsules based on non‐compartmental analysis
Table S6. Characteristics of the subjective response (any drug effect) in response to the solution or capsule