

Abstract
A palladium(II)-catalyzed enantioselective α-alkylation of azlactones with non-conjugated alkenes is described. The reaction employs a chiral BINOL-derived phosphoric acid as the source of stereoinduction and a cleavable bidentate directing group appended to the alkene to control the regioselectivity and stabilize the nucleopalladated alkylpalladium(II) intermediate in the catalytic cycle. A wide range of azlactones were found to be compatible under the optimal conditions to afford products bearing α,α-disubstituted α-amino acid derivatives with high yields and high enantioselectivity.
Keywords: palladium, directing group, chiral phosphoric acid (CPA), enantioselective, C–C bond formation
Graphical Abstract
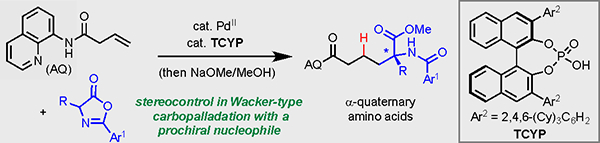
C–C Bond Formation. A method to achieve asymmetric α-alkylation of azlactones with non-conjugated alkenes through the dual catalytic action of Pd(II) and a chiral phosphoric acid. The reaction enables expedient access to quaternary α-amino acid products.
Palladium(II)-catalyzed Wacker-type nucleopalladation of alkenes is a synthetically enabling mode of reactivity to conjoin alkenes and various oxygen and nitrogen nucleophiles.[1,2] Stereocontrol in such reactions has been actively studied, most typically focused on stereodifferentiation of the two faces of the C=C bond (Scheme 1A).[2] Carbon (pro)nucleophiles, such as 1,3-dicarbonyls, also participate in nucleopalladation, though such reactions have been less extensively studied. In 1965, Tsuji described an early example of stoichiometric carbopalladation of 1,5-cyclooctadiene with sodium dimethyl malonate. The synthetic utility of stoichiometric carbopalladation was later demonstrated by Holton and Hegedus.[3] In the early 2000s, Widenhoefer reported a series of seminal studies on intramolecular redox-neutral cyclization of 1,3-dicarbonyl moieties and alkenes.[4] In 2016, our laboratory described substrate-directed hydrocarbofuntionalization of non-conjugated alkenes with various carbon (pro)nucleophiles.[5] He, Peng, and Chen recently identified a monodentate chiral oxazoline ligand to render this transformation enantioselective with internal alkenes.[6]
Scheme 1.

Background and Project Synopsis
With prochiral nucleophiles, carbopalladation presents the opportunity for stereocontrol in a manner that is distinct from cases with oxygen and nitrogen nucleophiles, namely establishing the absolute configuration of the nucleophilic carbon atom. α-Alkylation of carbonyl-based pronucleophiles is an extraordinarily enabling synthetic technology; however, non-conjugated alkenes have only been rarely employed as electrophiles despite their clear advantages over alkyl (pseudo)halides in terms of cost and availability.[7,8] The use of non-conjugated alkenes and carbonyl pronucleophiles as reaction partners in a Wacker-type manifold would represent a powerful approach towards enantioselective α-alkylation yet remains unknown to the best of our knowledge. The goal of the present study was to demonstrate this concept in the context of a synthetically significant problem, the α-alkylation of masked amino acids to form α-quaternary amino acid products (Scheme 1B).
We envisioned that such an approach could offer an appealing approach to access α-quaternary amino acids bearing a variety of substitution patterns. α-Quaternary amino acids are sought-after target compounds owing to their unusual conformational constraints in comparison to natural α-amino acids. Such compounds have unique range of biological activity, and are known to be enzyme inhibitors, ion-channel blockers, and antibiotics.[9] The widespread interest of α-quaternary amino acids has inspired the development of different synthetic methods, including (a) chiral-auxiliary-controlled electrophilic alkylation of amino acid enolate equivalents (b) asymmetric Strecker-type reactions, and (c) ring-opening reactions of chiral quaternary oxazolones.[10] Herein, we report a catalytic system to promote the stereocontrolled carbopalladation of a prochiral amino acid equivalent followed by protodepalladation of the resulting chelation stabilized alkylpalladium(II) intermediate to produce diverse α-quaternary amino acids.
To reduce this idea to practice, we elected to explore the combination of azlactones[11] as the masked amino acid pronucleophiles and chiral phosphoric acids (CPAs)[12] as the source of chiral information. Several important precedents informed this choice. Specifically, multiple groups have previously described CPA-catalyzed enantioselective α-functionalization of azlactones with a variety of electrophiles.[13] Additionally, stereocontrol in an assortment of mechanistically distinct palladium(II)-catalyzed reactions has been achieved using CPAs as chiral ligands/promoters.[14] In our proposed transformation, we envisioned the stereoinduction to occur through coordination of the CPA as an X-type ligand to palladium(II), and thus the enantioselectivity may be manifested in either the nucleopalladation by the azlactone or subsequent protodepalladation.
We initiated the investigation by examining the reaction between alkene 1 and azlactone 2 with different palladium precatalysts using (R)-TRIP (PA6) as chiral ligand. After an extensive optimization of reaction conditions (see SI), we found that by using 2 (3 equiv) in benzene (0.2 M) at 70 °C we were able to obtain the desired product 3 in 72% with 86:14 er. A notable finding from the optimization experiments was that the use of a non-polar solvent medium was critical for stereoinduction. We then explored a range of chiral BINOL-derived phosphoric acids (CPA) with different substituents at the 3 and 3’ positions, as shown in Table 1. Screening of various chiral phosphoric acids PA1–PA4 gave the product with moderate er (entries 1–4, Table 1). Upon modulating the steric bulk on the 3, 3’ substituents of the phosphoric acid promoter (PA5–PA7), we observed that TCYP (PA7), which contains cyclohexyl substituents, improved the er to 91:9 with 90% yield (Table 1, entry 8). Further increasing the level of steric encumbrance around the phosphoric acid by using PA8 gave 87:13 er and 79% yield (Table 1, entry 9). Additional optimization revealed that 55 °C temperature and 0.1 M concentration were optimal for this reaction, improving the er to 93:7 with 74% yield (Table 1, entry 11). Tuning the electronic properties of the CPA in order to perturb the pKa of the O–H bond did not significantly impact the reaction, with both electron-donating and -withdrawing groups showing similar er (Table 1, entries 12 and 13).
Table 1.
Optimization of the Reaction Conditions[a]
| Entry | Promoter (20%) | Yield[b](%) | Enantiomeric Ratio (er)[c] |
|---|---|---|---|
| 1 | - | 12 | 50:50 |
| 2 | PA1 | 74 | 83:17 |
| 3 | PA2 | 69 | 75:25 |
| 4 | PA3 | 29 | 55:45 |
| 5 | PA4 | 69 | 65:35 |
| 6 | PA5 | 73 | 80:20 |
| 7 | PA6 | 72 | 86:14 |
| 8 | PA7 | 90 | 91:9 |
| 9 | PA8 | 79 | 87:13 |
| 10[d] | PA7 | 75 | 92:8 |
| 11[d, e] | PA7 | 74 | 93:7 |
| 12 [d, e] | PA9 | 65 | 91:9 |
| 13 [d, e] | PA10 | 71 | 94:6 |
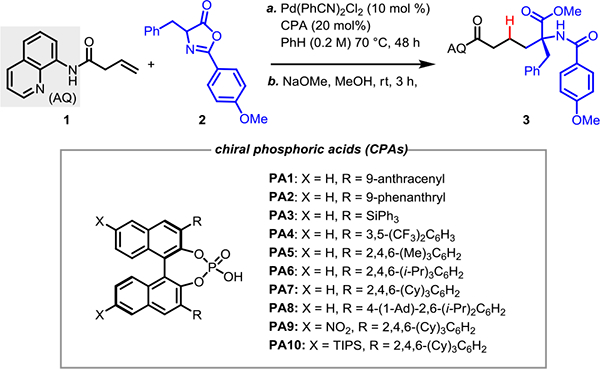
Reaction conditions: 1a (0.05 mmol), 2a (3.0 equiv), Pd(PhCN)2Cl2 (10 mol %), ligand (20 mol%), anhydrous benzene (0.2 M), 70 °C, 2 days.
Isolated yield.
Enantiomeric ratio determined by chiral SFC.
At 0.1 M concentration.
At 55 °C, 4 days.
We next sought to probe the effect of different substituent at the C-2 position under the optimized conditions, with the aim of identifying a generally useful azlactone protecting group that would provide high yield and er with various amino acid nucleophiles bearing different side chains.[15] In general, electron-donating groups on the para position of the aryl group provided the highest yield and enantiomeric ratio (3a–3g, Table 2; for additional examples, see Figure S2 in the SI). In particular, we identified the 4-phenoxybenzoyl group in 3a as highly effective, providing 95:5 er and 64% yield. We speculated that the 4-OPh group could be involved in π–π-stacking with the chiral phosphoric acid.[16] Based on this idea, we then tested the 4-methoxyphenoxyphenyl (PPMP) group, which led to further improvement in yield, while maintaining high enantioselectivity. Gratifyingly, using the PPMP-containing nucleophile, 3g was formed with 81% yield and 93:7 er. It is notable that the 3-OPh group also afforded product 3l with a high er of 93:7 and 55% yield. Substrates bearing electron-withdrawing groups on the para position of aryl group led to diminished yields (3i, Table 2 and Figure S2).
Table 2.
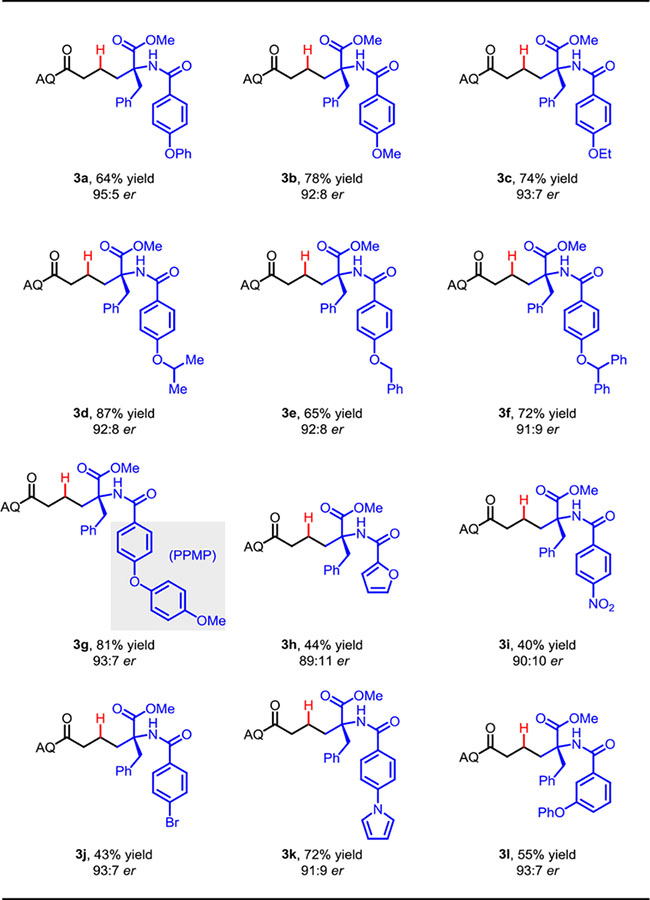
|
Reaction conditions: 1a (0.05 mmol), 2a (3.0 equiv), Pd(PhCN)2Cl2 (10 mol %), PA7 (TCYP) (20 mol%), anhydrous benzene (0.1 M), 55 °C, 4 days.
Isolated yield.
Enantiomeric ratio determined by chiral SFC.
We then explored the substrate scope by examining different C-4 substituents on the azlactone using the PPMP group. By starting with achiral α-amino acids, a wide range of azlactones were synthesized and tested in this reaction. Azlactones derived from phenylalanine containing electron-releasing groups (-CH3, -OCH3) or weak electron-withdrawing groups (-Br, -Cl and -F) on the aryl ring showed high yields and excellent er values (4a–4g, Table 3). Stronger electron-withdrawing groups (-CF3 and -NO2) gave products with slightly diminished yields and er values (4h and 4i), presumably due to attenuated nucleophilicity. Azlactones containing heterocyclic groups (indole, thiophene and furan) (4k–4l) and naphthyl groups (4m and 4n) also proved to be effective. Interestingly, with azlactones bearing α-substituents other than benzyl moieties, high yields were maintained (4p–4s), though enantioselectivity was more modest. When an α-substituted alkenyl AQ amide was tested, the reaction proceeded sluggishly, even at 70 °C, giving 4t in 34% yield with >20:1 dr and 82:18 er (major). Internal alkenes were unreactive in this system, which speaks to the sensitivity of the overall coordination assembly to subtle steric perturbations.
Table 3.
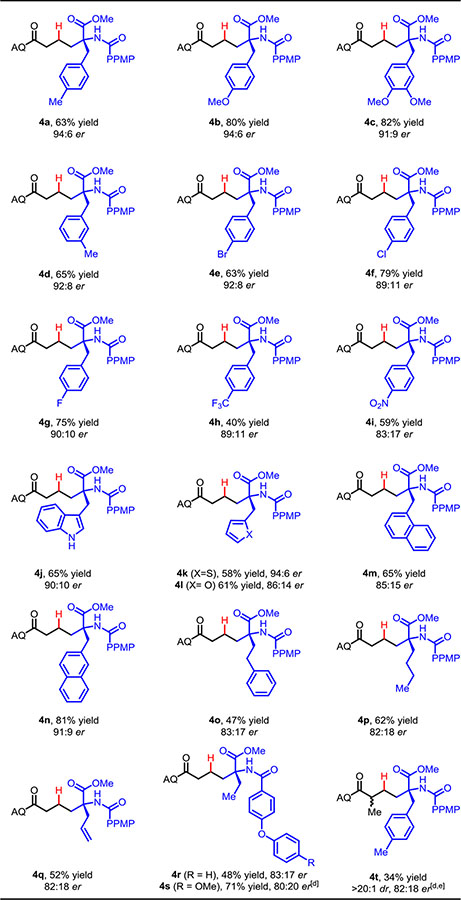
|
Reaction conditions as in Table 2.
Isolated yield.
Enantiomeric ratio determined by chiral SFC.
70 °C, 2 d.
The relative stereochemistry could not be assigned.
To demonstrate the synthetic utility of this method, we selected a representative enantioenriched product (3b) and carried out a series of diversification reactions that retained the stereochemical integrity of the newly formed chiral center (Scheme 2). Firstly, the 8-aminoquinoline directing group could be selectively cleaved using Ohshima’s alcoholysis protocol catalyzed by Ni(tmhd)2 (tmhd = 2,2,6,6,-tetramethyl-3,5-heptanedionate) to give product 5.[17] Additionally, the ester functional group could be reduced selectively to afford chiral aminoalcohol 6.[18] We also found that product 3b could undergo Daugulis-type alkynylation (7)[19] and arylation (8)[20] in 2:1 dr without erosion of stereochemical purity, thereby effecting a net two-step alkene 1,2-dicarbofunctionalization. The absolute configuration of the α-position for all of products was assigned as S by analogy to compound 8, the structure of which was confirmed by single-crystal X-ray crystallography (see SI). Lastly, we were also able to perform global deprotection with 6 M HCl to obtain free amino acid 9.[21]
Scheme 2.

Derivatization Reactions of 3b
Lastly, to elucidate the stereoinduction model in this unique enantioselective carbo-Wacker α-alkylation, we carried out a series of experimental and computational studies. We first sought to determine the number of CPA molecules involved in the enantiodetermining step. To this end, we examined how the ee of a representative reaction changed as a function of the ee of the CPA. Using TRIP as a model CPA, we found strong linear correlation between TRIP ee and product ee (Figure 1). The absence of a non-linear effect is consistent with 1:1 Pd:CPA ratio in the enantiodetermining step, which informed subsequent computational studies.
Figure 1.

Correlation between CPA ee and Product ee.
Armed with this information, we performed density functional theory (DFT) calculations to probe the reaction mechanisms and the origins of enantioselectivity using 1 as the model substrate, PA5 as the CPA ligand, and 2 as the azlactone (see SI: Figure S5 and Figure S6). For nucleopalladation, the activation free energy barrier for the (S)-nucleopalladation pathway (TS1_S, 10.3 kcal/mol) is lower than that of the (R)-pathway (TS1_R, 11.2 kcal/mol).[22] However, the subsequent protodepalladation step has a higher activation energy, indicating that the nucleopalladation step is reversible[5,23] and therefore, the reaction is under Curtin–Hammett control. The CPA-assisted protodepalladation step to form the (S)-enantiomer (TS2_S) has an activation free energy of 13.5 kcal/mol with respect to the π-alkene complex 10 in comparison to 14.9 kcal/mol for the (R)-enantiomer (TS2_R) indicating that the formation of the (S)-enantiomer is kinetically favored at the enantioselectivity-determining protodepalladation step, consistent with experimental selectivity.[24]
In conclusion, we have demonstrated a catalytic, enantioselective α-alkylation of azlactones with a non-conjugated alkene to access α-quaternary amino acids. The transformation involves a stereoselective Wacker-type carbopalladation across a 3-butenoic acid AQ amide followed by protodepalladation. The resulting products can be conveniently deprotected and further manipulated to access a variety of useful products.
Supplementary Material
Acknowledgements
This work was financially supported by Scripps Research, Pfizer, Inc., Bristol-Myers Squibb (Unrestricted Grant), the NIH (5R35GM125052–02 and 1R35GM128779) and the NSF (NSF-DBI 1759544, SURF fellowship to O.A.). Calculations were performed at the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF and the HPC Garibaldi cluster at Scripps Research. We thank Dr. Milan Gembicky and Dr. Curtis E. Moore (UCSD) for X-ray crystallographic analysis and Prof. Zachary K. Wickens (UW Madison) for helpful discussion.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].For reviews on Wacker oxidation, see:; a) Takacs JM, Jiang X, Curr. Org. Chem 2003, 7, 369; [Google Scholar]; b) Keith JA, Henry PM, Angew. Chem. Int. Ed 2009, 48, 9038; [DOI] [PubMed] [Google Scholar]; c) Dong JJ, Browne WR, Feringa BL, Angew. Chem. Int. Ed 2015, 54, 734. [DOI] [PubMed] [Google Scholar]
- [2].For representative reviews covering stereocontrol in nucleopalladation, see:; a) Jensen KH, Sigman MS, Org. Biomol. Chem 2008, 6, 4083; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) McDonald RI, Liu G, Stahl SS, Chem. Rev 2011, 111, 2981; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Garlets ZJ, White DR, Wolfe JP, Asian J Org. Chem 2017, 6, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].a) Tsuji J, Takahashi H, J. Am. Chem. Soc 1965, 87, 3275;14324316 [Google Scholar]; b) Holton RA, Kjonaas RA, J. Am. Chem. Soc 1977, 99, 4177; [Google Scholar]; c) Hegedus LS, Williams RE, McGuire MA, Hayashi T, J. Am. Chem. Soc 1980, 102, 4973. [Google Scholar]
- [4].a) Pei T, Widenhoefer RA, J. Am. Chem. Soc 2001, 123, 11290; [DOI] [PubMed] [Google Scholar]; b) Qian H, Widenhoefer RA, J. Am. Chem. Soc 2003, 125, 2056. [DOI] [PubMed] [Google Scholar]
- [5].Yang KS, J. A., Gurak Jr., Z. Liu, K. M. Engle, J. Am. Chem. Soc 2016, 138, 14705. [DOI] [PubMed] [Google Scholar]
- [6].Wang H, Bai Z, Jiao T, Deng Z, Tong H, He G, Peng Q, Chen G, J. Am. Chem. Soc 2018, 140, 3542. [DOI] [PubMed] [Google Scholar]
- [7].Mo F, Dong G, Science 2014, 345, 68. [DOI] [PubMed] [Google Scholar]
- [8].For a conceptually distinct enamine/photoredox/HAT catalysis approach to achieve enantioselective α-alkylation of aldehydes with non-conjugated alkenes, see:; Capacci AG, Malinowski JT, McAlpine NJ, Kuhne J, MacMillan DWC, Nat. Chem 2017, 9, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Ohfune Y, Shinada T, Eur. J. Org. Chem 2005, 5127; [DOI] [PubMed] [Google Scholar]; b) Cativiela C, Ordóñez M, Tetrahedron: Asymmetry 2009, 20, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Metz AE, Kozlowski MC, J. Org. Chem 2015, 80, 1; [DOI] [PubMed] [Google Scholar]; d) Pieczykolan M, Narczyk A, Stecko S, J. Org. Chem 2017, 82, 5636; [DOI] [PubMed] [Google Scholar]; e) Sato T, Izawa K, Aceña JL, Liu H, Soloshonok VA, Eur. J. Org. Chem 2016, 2757; [Google Scholar]; f) Ezquerra J, Pedregal C, Escribano A, Carman Carreno M, Garcia Ruano J, Tetrahedron Lett. 1995, 36, 3247. [Google Scholar]
- [10].For recent reviews, see:; a) Seebach D, Sting AR, Hoffmann M, Angew. Chem. Int. Ed. Engl 1996, 35, 2708; [Google Scholar]; b) Vogt H, Bräse S, Org. Biomol. Chem 2007, 5, 406; [DOI] [PubMed] [Google Scholar]; c) Hashimoto T, Maruoka K, Chem. Rev 2007, 107, 5656; [DOI] [PubMed] [Google Scholar]; d) Cativiela C, Díaz-de-Villegas MD, Tetrahedron: Asymmetry 2007, 18, 569; [Google Scholar]; e) Connon SJ, Angew. Chem. Int. Ed 2008, 47, 1176; [DOI] [PubMed] [Google Scholar]; f) De Castro PP, Carpanez AG, Amarante GW, Chem. Eur. J 2016, 22, 10294. [DOI] [PubMed] [Google Scholar]
- [11].For an example, of enantioselective conjugate addition of azlactones to Michael acceptors catalyzed by a bis-palladacycle complex, see:; Weber M, Jautze S, Frey W, Peters R, J. Am. Chem. Soc 2010, 132, 12222. [DOI] [PubMed] [Google Scholar]
- [12].For selected reviews, see:; a) Akiyama T, Chem. Rev 2007, 107, 5744; [DOI] [PubMed] [Google Scholar]; b) Terada M, Synthesis, 2010, 1929; [Google Scholar]; c) Kampen D, Reisinger CM, List B, Top. Curr. Chem 2010, 291, 395; [DOI] [PubMed] [Google Scholar]; d) Phipps RJ, Hamilton GL, Toste FD, Nat. Chem 2012, 4, 603; [DOI] [PubMed] [Google Scholar]; e) Parmar D, Sugiono E, Raja S, Rueping Chem M. Rev. 2014, 114, 9047. [DOI] [PubMed] [Google Scholar]
- [13].For representative reports, see:; a) Terada M, Tanaka H, Sorimachi K, J. Am. Chem. Soc 2009, 131, 3430; [DOI] [PubMed] [Google Scholar]; b) Jiang J, Qing J, Gong LZ, Chem. Eur. J 2009, 15, 7031; [DOI] [PubMed] [Google Scholar]; c) Terada M, Moriya K, Kanomata K, Sorimachi Angew K. Chem. Int. Ed 2011, 50, 12586; [DOI] [PubMed] [Google Scholar]; d) Han Z-Y, Guo R, Wang P-S, Chen D-F, Xiao H, Gong L-Z, Tetrahedron Lett. 2011, 52, 5963; [Google Scholar]; e) Zhang Z, Sun W, Zhu G, Yang J, Zhang M, Hong L, Wang R, Chem. Commun 2016, 52, 1377; [DOI] [PubMed] [Google Scholar]; f) Ávila EP, Justo RMS, Gonçalves VP, Pereira AA, Diniz R, Amarante GW, J. Org. Chem 2015, 80, 590; [DOI] [PubMed] [Google Scholar]; g) Zhang M, Yu C, Xie J, Xun X, Sun W, Hong L, Wang R, Angew. Chem. Int. Ed 2018, 57, 4921. [DOI] [PubMed] [Google Scholar]
- [14].Pd(II)/CPA-catalyzed enantioselective transformations:; a) Alper H, Hamel N, J. Am. Chem. Soc 1990, 112, 2803; [Google Scholar]; b) Chai Z, Rainey TJ, J. Am. Chem. Soc 2012, 134, 3615; [DOI] [PubMed] [Google Scholar]; c) Zhang D, Qiu H, Jiang L, Lv F, Ma C, Hu W, Angew. Chem. Int. Ed 2013, 52, 13356; [DOI] [PubMed] [Google Scholar]; d) Yu S-Y, Zhang H, Gao Y, Mo L, Wang S, Yao Z-J, J. Am. Chem. Soc 2013, 135, 11402; [DOI] [PubMed] [Google Scholar]; e) Jindal G, Sunoj RB, J. Am. Chem. Soc 2014, 136, 15998; [DOI] [PubMed] [Google Scholar]; f) Jiang T, Bartholomeyzik T, , Mazuela J Willersinn J, Bäckvall J-E, Angew. Chem. Int. Ed 2015, 54, 6024; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Wang H, Tong H-R, He G, Chen G, Angew. Chem. Int. Ed 2016, 55, 15387; [DOI] [PubMed] [Google Scholar]; h) Jain P, Verma P, Xia G, Yu J-Q, Nat. Chem 2017, 9, 140; [DOI] [PMC free article] [PubMed] [Google Scholar]; i) Smalley AP, Cuthbertson JD, Gaunt MJ, J. Am. Chem. Soc 2017, 139, 1412; [DOI] [PubMed] [Google Scholar]; j) Bao X, Wang Q, Zhu J, Angew. Chem. Int. Ed 2018, 57, 1995. [DOI] [PubMed] [Google Scholar]
- [15].Across several representative examples, we found that yields and er values were consistent in reaction performed on 0.05-mmol scale to 0.3-mmol scale. On larger scale (1.0 mmol), the reaction progressed more slowly, leading to slightly lower yields, while maintaining similar er. We elected to perform the examples in Tables 2 and 3 on 0.05-mmol scale due to the valuable nature of PA7 and because all of the reaction components have high formula weights, allowing for accurate measurement even on small scale.
- [16].The 4-OPh group appears to be relatively close to the CPA ligand in the calculated enantioselectivity-determining transition state, TS2_S, supporting this explanation.
- [17].Deguchi T, Xin H-L, Morimoto H, Ohshima T, ACS Catal. 2017, 7, 3157. [Google Scholar]
- [18].Joo J-E, Lee K-Y, Pham V-T, Tian Y-S, Ham W-H, Org. Lett 2007, 9, 3627. [DOI] [PubMed] [Google Scholar]
- [19].Ano Y, Tobisu M, Chatani N, J. Am. Chem. Soc 2011, 133, 12984. [DOI] [PubMed] [Google Scholar]
- [20].Zaitsev VG, Shabashov D, Daugulis O, J. Am. Chem. Soc 2005, 127, 13154. [DOI] [PubMed] [Google Scholar]
- [21].a) Liu Z, Wang Y, Wang Z, Zeng T, Liu P, Engle KM, J. Am. Chem. Soc 2017, 139, 11261; [DOI] [PubMed] [Google Scholar]; b) Thompson JF, Morris CJ, Gering RK, Anal. Chem 1959, 31, 1028. [Google Scholar]
- [22].For TS1_R, geometry optimization with B3LYP-D3 was unsuccessful after multiple attempts. Therefore, geometry optimization of TS1_R was performed with B3LYP instead. Single-point energy calculation for TS1_R was carried out with the same method as the rest of the structures (M06 and a mixed basis set of SDD for Pd and 6–311+G(d,p) with solvation energy corrections using the SMD model with C6H6). As nucleopalladation is not the enantioselectivity-determining step, using a different method for geometry optimization for TS1_R is not expected to affect the conclusions regarding the origin of enantioselectivity.
- [23].Tran V, Gurak JA Jr., Yang KS, Engle KM, Nat. Chem 2018, 10, 1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].The alternative diastereomeric palladium π-alkene intermediate and the corresponding nucleopalladation and protodepalladation transition states were also located computationally but were found to be higher in energy (see SI).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.