

SUMMARY
Replication-dependent histone genes often reside in tandemly arrayed gene clusters, hindering systematic loss-of-function analyses. Here, we used CRISPR/Cas9 and the attP/attB double-integration system to alter numbers and sequences of histone genes in their original genomic context in Drosophila melanogaster. As few as eight copies of the histone gene unit supported embryo development and adult viability, whereas flies with 20 copies were indistinguishable from wild types. By hierarchical assembly, 40 alanine-substitution mutations (covering all known modified residues in histones H3 and H4) were introduced and characterized. Mutations at multiple residues compromised viability, fertility, and DNA-damage responses. In particular, H4K16 was necessary for expression of male X-linked genes, male viability and maintenance of ovarian germline stem cells, whereas H3K27 was essential for late embryogenesis. Simplified mosaic analysis showed that H3R26 is required for H3K27 trimethylation. We have developed a powerful strategy and valuable reagents to systematically probe histone functions in D. melanogaster.
Keywords: Drosophila, CRISPR/Cas9, FLP-FRT, attB-attP, dosage effects, histone mutant library, H4K16, mosaic system
INTRODUCTION
Chromatin is essential for genome packaging and regulation. The basic unit of chromatin is the nucleosome, consisting of 147 base pairs of DNA wrapped around a histone octamer comprised of two copies each of histone proteins H3, H4, H2A, and H2B (Luger et al., 1997). A fifth “linker histone”, H1, dynamically binds DNA residing between histone octamers at a subset of nucleosomes. Histones do not merely provide a binding platform for DNA; they also actively participate in DNA-related processes, such as transcription (Kouzarides, 2007). One mechanism for histones to carry out these functions is though post-translational modifications (PTMs) (Rothbart and Strahl, 2014).
In the past two decades, over 20 types of PTMs have been identified on histones, including acetylation, methylation, phosphorylation, ubiquitination, and crotonylation (Kouzarides, 2007; Tan et al., 2011; Tessarz and Kouzarides, 2014). Among these PTMs, 12 are added to lysine residues (Huang et al., 2014a). The N-terminal, flexible “tail” domains are the most heavily modified portions of histones, presumably because they are more easily accessible to histone-modifying enzymes than other domains. However, PTMs have also been detected within the globular core domains of histones (Xu et al., 2005; Ye et al., 2005). Histone PTMs are thought to modulate chromatin structure and gene expression either directly or via recruitment of specific chromatin-associated proteins.
Whether PTMs are always involved in chromatin structure remains controversial (Henikoff and Shilatifard, 2011). Studies involving genetic or chemical interventions targeting histone-modifying enzymes have provided substantial evidence for biological functions of specific PTMs. For example, H3K27 methylation by the polycomb repressive complex 2 (PRC2) is involved in maintenance of cellular identity (Conway et al., 2015). Unfortunately, because these modifying enzymes generally have other protein substrates in addition to histones (Glozak et al., 2005), and chromatin-regulating enzymes might also have functions unrelated to their enzymatic activities (Chaiyachati et al., 2013), these experimental data must be interpreted cautiously.
The roles of PTMs can be directly queried by systematic mutation of histone residues. Such studies have been carried out in Saccharomyces cerevisiae (Dai et al., 2008; Nakanishi et al., 2008), but experiments in higher organisms pose additional challenges. For example, there are 64 histone genes within the human genome, distributed at three major loci on different chromosomes (Marzluff et al., 2002), making it difficult to substantially alter levels of particular histone proteins inside human cells.
Currently, the only multicellular organism in which histone mutagenesis has been performed is Drosophila melanogaster, in which all core-histone genes reside at a single locus on the left arm of chromosome 2, with ~100 copies of histone gene-repeat units (His-GUs) per chromosome (McKay et al., 2015). Each His-GU (~5 kb in length) contains the four core-histone genes in two pairs (His2A–His2B and His3–His4), each under the control of a divergent promoter, plus the linker-histone gene, His1, which is regulated independently (Figure S1A).
Histone residue function in D. melanogaster has been explored by removing the His-GU cluster (Df(2L)HisC, referred to as HisC hereafter) and complementing it with transgenes from plasmids or bacterial artificial chromosomes (BACs) (Graves et al., 2016; Gunesdogan et al., 2010). These methods are labor intensive partly because four plasmids are needed for transgenic complementation and complex crossing procedures. Therefore, only limited sites within histone H3 and H4 have been analyzed (Coleman and Struhl, 2017; Graves et al., 2016; McKay et al., 2015; Pengelly et al., 2013; Yung et al., 2015). In addition, since the transgenes are randomly integrated, positional effects could confound data interpretation.
In this study, we generated an efficient histone-mutagenesis platform, enabling the functional study of each residue in all five histones with much higher throughput than with previous techniques. As a proof-of-concept study, we targeted H3 and H4, revealing several interesting insights that would have been difficult to obtain by other means.
RESULTS
Generation of a histone-deletion line
To generate a fly with deletion of the entire His-GU cluster, and to simultaneously introduce targeted integration sites to facilitate in situ complementation, we designed a strategy to knock-in two loxP donor sequences flanking the histone locus by homologous recombination (HR) with the ends-out targeting method (Xie and Golic, 2004). However, this strategy did not generate any suitable flies, possibly because the repetitive sequences near the His-GU cluster interfered with the process. As an alternative approach, we used CRISPR/Cas9-mediated knock-in technology to modify two fly lines simultaneously (Figure 1A) (Xue et al., 2014; Yu et al., 2013). In the first line, a flippase-recognition-target (FRT) site and PhiC31-mediated recombination site (attP) were inserted at the left side of the His-GU locus (Figures S1B–S1D). To avoid repetitive sequences and disruption of flanking genes, we chose a target site adjacent to CG8663 (2L: 21,394,202) (Figure S1A). This line also encoded flippase under the control of a heat-shock promoter (hs-FLP). In the second line, a donor plasmid containing the FRT site, attP site, and a red fluorescent protein (RFP) marker gene was targeted to integrate near CG3305 (2L: 21,559,013) to the right of the His-GU locus (Xue et al., 2014). An ends-in integration line (Rong and Golic, 2000) was recovered in this experiment (Figure S1E). PCR demonstrated that this founder line contained a duplicated FRT-attP-RFP cassette, as predicted (Figures 1A and S1F). The two founder lines were crossed and flippase-mediated recombination was induced by heat shock, resulting in production of heterozygous flies with the entire His-GU cluster deleted and replaced with duplicated attP-FRT-RFP sequences at the original chromosomal locus, enabling in situ integration of modified-histone arrays (Figure 1A).
Figure 1. Schematic overview of histone cluster deletion and molecular verification.
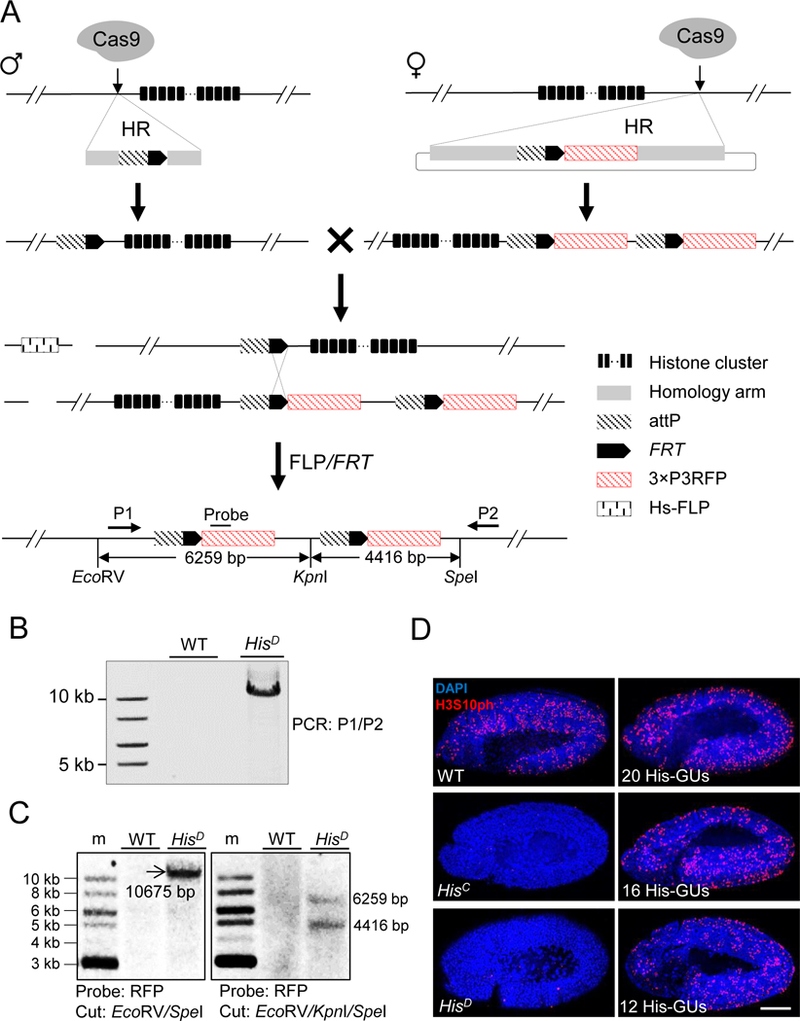
(A) Strategy for knocking out the entire histone cluster. CRISPR/Cas9-mediated homologous recombination (HR) pathway was used to knock-in attP-FRT cassette on both sides of the histone cluster. On the left side, single-stranded oligonucleotide DNA was used as the HR donor. On the right side, a plasmid with homologous arms was used. Flies with ‘ends-in’ HR events were identified during knocking-in at the right side, creating an attP-FRT duplication (top). The two fly lines were crossed and flippase activity was induced. Through HR between FRT sequences (middle), a histone null mutant was generated (bottom). Primers (P1 and P2) for long-range PCR, probe and restriction-enzyme sites for Southern blotting are indicated. The distances between these restriction sites are labeled.
(B) PCR analysis with P1 and P2 primers was used to validate histone cluster deletion (HisD). w1118 wild-type (WT) flies were used as a control.
(C) Southern-blot analysis of HisD. Size markers (m). Genomic DNA from either WT (w1118) or HisD was digested with EcoRV/SpeI (Left) or EcoRV/KpnI/SpeI (Right), separated and blotted with the probe indicated in Figure 1A.
(D) Confocal images of cycle 15 embryos immunostained with H3S10ph antibody (red) and DAPI (blue). WT: w1118; HisC: homozygous histone deletion (Bloomington Stock Center 8670); HisD: homozygous histone cluster deletion (generated in this study); 20His-GUs, 16 His-GUs, and 12His-GUs indicate homozygous histone cluster deletions with different copy numbers of histone gene units (His-GUs). No H3S10ph signal was detected in HisD and HisC embryos. Scale bar: 100 µm.
See also Figure S1.
We designated this His-GU-deletion fly line HisD, and confirmed its composition by PCR and Southern blotting (Figures 1B and 1C). Sequencing confirmed complete deletion of all canonical histone genes and precise insertion of duplicated attP-FRT-RFP. Maternal histone proteins and mRNAs support the development of homozygous HisC mutant embryos in the first 14 cell cycles of embryogenesis (Gunesdogan et al., 2014). We therefore cytologically assessed the expression of zygotic histone genes at cycle 15, and found phospho-histone H3 immunostaining in wild-type zygotes, but not in HisD or HisC (Figure 1D). These results demonstrated the generation of a core-histone deletion line enabling integration of two-copy of histone-donor simultaneously at the endogenous-histone locus on each copy of chromosome 2 via the PhiC31-mediated attP/attB integration system (Bischof et al., 2007).
Replacing 20 copies of His-GU in HisD results in a wild-type phenotype
Various numbers of His-GUs were introduced into the HisD background in situ via the attP/attB integration system. Viable flies were first recovered with introduction of eight His-GUs in the diploid genome, albeit with a low rescue ratio (<0.1) (Figure 2A). The adult rescue ratio improved with increasing copy number of His-GUs, to ~0.8 with 12 copies, ~0.95 with 16 copies, and ~1.0 with 20 copies.
Figure 2. Low histone dosage affects the fertility of rescued adults.

(A) Results of rescue tests for adult flies with different copy numbers of wild-type (WT) histone gene units (His-GUs; 4, 6, 8, 10, 12, 16, and 20 units) that were reintroduced into histone null mutants (HisD) to rescue the lethal phenotype. Results are means ± SD from triplicate determinations.
(B) Western blot analysis of levels of H3, H4, H2A, H2B, and H1 histones in virgin adult flies with 12, 16, or 20 His-GUs, HisD, and WT (w1118).
(C, D) Results of fertility tests of male (C) and female (D) adult flies with 12, 16, or 20 His-GUs, HisD, and WT (w1118). The fertility test is done by counting the number of surviving adult progeny produced by male or female flies of the given genotype crossed with w1118. Each point represents a vial of flies. The horizontal bar indicates the mean number of adult progeny produced. Error bars represent standard error of the mean (± SEM).
(E, F) Testes (E) and ovaries (F) from flies with different histone copy numbers were classified by their morphology into three categories, as show in the top panels: WT (w1118), moderate defect, and severe defect. Below, the numbers of adults with the given testis (E) and ovary (F) morphology in flies with 12, 16, or 20 His-GUs, HisD, and WT are shown. Total number of dissected testes and ovaries is listed on the right.
See also Figure S2.
Transgenic flies with 24 His-GUs in the diploid genome show no significant differences of histone protein or mRNA expression levels compared with wild-type flies, because of a histone gene-dosage compensation mechanism (McKay et al., 2015). We tested whether dosage compensation could also help to keep a steady level of histone proteins when the genes were integrated at their original locus by monitoring levels of histones H3, H4, H2B, and H2A, and linker histone H1, in HisD flies with different numbers of His-GUs. These histone proteins were expressed similarly in adults, embryos, and larvae, regardless of copy numbers of His-GUs from 12 to 20 in the diploid genome (Figures 2B, S2A and S2B). In addition, compared with the wild type, flies with 12 or 20 His-GUs did not show significant decreases in histone mRNA levels (Figure S2C).
Endogenous-histone mRNA transcription and processing in cells involves histone-locus body (HLB) nuclear subcompartments (Liu et al., 2006). The presence of HLBs in His-GU-rescued HisD flies was assessed by immunofluorescence in diploid cells on post-blastoderm-stage embryos. Binding of antibody against the HLB-specific-protein Lsm10 (Liu et al., 2006) demonstrated foci in 100% of nuclei in S phase in all rescued embryos (with 12, 16, or 20 His-GUs) and wild-type embryos, and no obvious foci in telophase cells (Figure S2D). The sizes of HLBs from HisD flies with 12 His-GUs or 20 His-GUs were similar to those of wild-type flies in S phase (Figure S2D). Immunostaining of salivary gland polytene chromosome spreads from third-instar larvae with anti-Lsm10 antisera confirmed these results: a single HLB spot was detected in larvae of rescued flies with 12, 16, or 20 His-GUs and in wild-type larvae. The HLBs in wild-type larvae were slightly larger than in the histone-rescued larvae, suggesting incomplete complementation of HisD (Figure S2E).
To test whether chromatin structure is affected by the number of His-GUs, we analyzed the polytene chromosomes from both wild-type flies and HisD flies with 12, 16, or 20 copies of His-GUs (Piacentini et al., 2009), which had no detectable abnormalities (Figures S2F and S2G). This result suggests that 12 or more copies of His-GUs give similar levels of histone expression in adult flies to the wild-type copy number.
It has been suggested that flies with 12 transgenic copies of His-GUs have a defect in female fertility (McKay et al., 2015). We counted how many adult progenies were produced by crossing either His-GU-rescued HisD males with wild-type females, or rescued HisD females with wild-type males. Fertility was normal in crosses involving males with ≥12 His-GUs (Figure 2C), but it was severely reduced in crosses involving females with 12 His-GUs, and slightly reduced with 16-His-GU females; with 20 His-GUs, fertility was similar to that of wild-type females (Figure 2D). Therefore, the number of His-GUs affected fertility in female flies, but not males.
To further characterize the female fertility defects, and to look for male defects that may have altered testis morphology but not fertility, we characterized the phenotypes of testes and ovaries in flies containing different numbers of His-GUs. Testes and ovaries were classified into three categories on the basis of morphological defects (Figures 2E and 2F). In male flies with ≥12 His-GUs, ≥85% of testes were phenotypically indistinguishable from wild type (Figure 2E). In female flies with 12 His-GUs, ovarian development was moderately affected in 28%, and severely disrupted in 35% (Figure 2F). Notably, however, histone expression levels were not significantly different in ovaries of flies with different His-GU copy numbers (Figure S2H).
Histone gene dosage affects oogenesis and spermatogenesis
To assess the effects of histone gene dosage on oogenesis and spermatogenesis, we immunostained ovaries and testes from flies and analyzed their morphologies. The D. melanogaster ovary consists of interconnected chains of egg chambers (ovarioles), with germline stem-cell niches located at the tip of each ovariole in a structure called the germarium (Figure 3A).
Figure 3. Low histone dosage affects testis and ovary development.
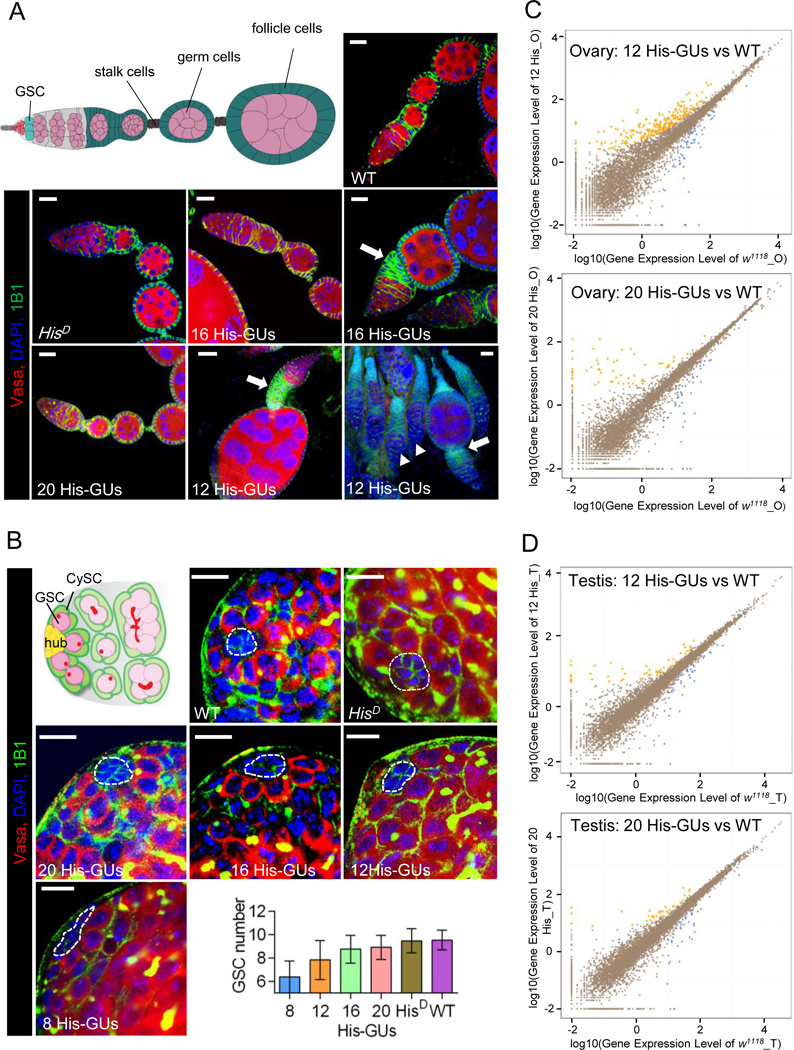
(A) Low copy numbers of histone gene units (His-GUs) caused severe budding defect in egg-chamber development. Illustration (top left) of a wild-type (WT) Drosophila germarium and egg chamber. Germline stem cells (GSCs; dark red) reside in a germline niche and divide asymmetrically and form cystoblasts, which develop into 16-cell cysts surrounded by follicle cells (green). The cyst cells then bud from the germarium as individual egg chambers. As the egg chambers continue to grow, they move further to the posterior and form a chain of egg chambers connected by stalk cells (blue). Confocal images of ovaries from adult flies carrying different copy numbers of His-GUs stained with anti-Vasa (red), DAPI (blue), anti-1B1 (green). Scale bar, 20 µm. The majority of ovaries in flies with 12 His-GUs had severe budding problems and did not have mid-stage egg chambers (solid arrow head).
(B) Low copy numbers of His-GUs caused GSC loss in the testis. Illustration of WT Drosophila testis (top left). Non-dividing hub cells (yellow) are surrounded by GSCs (dark red) and cyst stem cells (CySCs, dark green). GSCs with round fusome (red) divide asymmetrically and produce differentiated spermatogonia (green) with branched fusome. Low copy number (≤12 His-GUs) testes have fewer GSCs (P-value <0.00005). Testes from adult flies with different copy numbers of His-GUs were stained with anti-Vasa (red), DAPI (blue), anti-1B1 (green). Scale bar: 10 µm. The GSC number of testes was counted and results are means ± SEM (bottom right).
(C, D) Transcriptome comparison of ovaries (C) and testes (D) in flies with 12 or 20 His-GUs and WT (w1118). The scatter plots (yellow or blue) indicate differentially expressed genes. The RNA-seq experiments were performed with two biological replicates.
See also Figure S3.
We found that ovaries from female flies carrying 20 copies of His-GUs were phenotypically indistinguishable from wild-type ovaries. In female flies with 16 His-GUs, ~90% of ovaries had normal appearance, whereas ~10% had mild budding defects, in which early stages of oogenesis failed to separate from the germarium (Figures 3A and S3A). In female flies with 12 His-GUs, 36% of ovaries had severe defects in oogenesis, in which late-stage egg chambers were undetectable. All early-to-mid stages of egg chambers were completely missing from most of the ovaries in flies with eight His-GUs (Figure S3A). The budding defects might arise from reductions in the division rate of germ cells associated with reduced histone levels. High histone gene dosage (≥16 copies) may be required to maintain normal division of most germ cells in order to produce eggs. Notably, the germline stem-cell niche in the defective ovarioles looked quite normal, containing both stem cells and their differentiating progeny (Figure S3A), suggesting that mid-levels of histone gene dosage (12 copies) are sufficient to support the normal divisions of GSCs in the ovary.
As in the ovary, the Drosophila testis stem-cell niche is located at the testis apex, where a group of ~10–15 non-dividing somatic cells called the hub produce signals for the surrounding GSCs and cyst stem cells (Figure 3B) (Leatherman and Dinardo, 2010). The differentiating daughter of the GSC division (the gonialblast) undergoes four rounds of synchronous mitotic division and further differentiation into two-cell, four-cell, eight-cell, and 16-cell spermatogonial cysts that finally undergo meiosis and spermatogenesis to become sperm. With the decrease of histone copy numbers in male flies, the number of GSCs per testis niche also decreased slightly (Figure 3B). Male flies with 20 His-GUs resembled the wild type, with an average of 8.9 GSCs, whereas flies with 12 His-GUs had an average of 7.8 GSCs, and flies with eight His-GUs had an average of 6.0 GSCs. This result suggests that histone gene dosage might affect GSC division in the testis. However, even flies with only eight His-GUs still had 6.0 GSCs per testis niche and thus might still have been able to produce enough sperm to maintain fertility, agreeing with the finding that fertility was only mildly defective in the male flies with fewer copies of His-GUs. Microscopic examination of sperm differentiation processes also suggested there was no obvious defect even in male flies with eight His-GUs, except in the reduced number of GSCs (Figure S3B).
To investigate how histone gene dosage affected gene expression in reproductive systems, we performed transcriptomic analysis for ovaries of flies with 12 His-GUs and 20 His-GUs (Figures 3C,S3C and S3D). RNA sequencing (RNA-seq) revealed that 243 genes were up-regulated and 94 genes were down-regulated (|log2 ratio| ≥1) in ovaries of flies with 12 His-GUs, in comparison with those from wild-type flies (Figure S3E). Some ovarian-development-associated genes such as Asator, Disp, and Obst-B were drastically down-regulated in flies with 12 His-GUs (Figure S3F. However, in ovaries from flies with 20 His-GUs, only 1% of genes were differentially expressed compared with wild type, with 62 genes up-regulated and 67 genes down-regulated (Figures S3C and S3E). In testes of flies with 12 or 20 His-GUs, fewer than 1% of genes were differentially expressed compared with wild type (Figures 3D, S3D, and S3G). All these results indicated that the phenotypic defects in oogenesis were much more severe than those of spermatogenesis, and 20-His-GU male and female flies had normal organ development.
We performed immunochemistry with an antibody against Lsm10 to assess nuclear organization in nurse cells (Figure S3H). Nurse-cell chromosomes in stage-6 egg chambers were well dispersed in wild-type female flies, but persistently undispersed in 12-His-GU flies, forming large globules. By contrast, nurse-cell chromosomes in 20-His-GU flies had similar dispersal to those in wild-type flies. In summary, low histone dosage affects the transition from polytene to polyploid chromosomes in nurse cells.
A hierarchical assembly method for high-throughput, systematic construction of histone mutant arrays
Because 20 copies of His-GU were sufficient to give flies a wild-type phenotype, we developed a method to systematically construct plasmids containing five copies of His-GU, each with the same mutation, so that recombination into two sites on one chromosome would introduce 20 identical mutations in diploid genome into a histone null background.
Firstly, a histone unit (containing His1, His2A, His2B, His3, and His4) amplified from the D. melanogaster genome was cloned into a modified Gateway entry plasmid and a SalI restriction site was introduced between His2A and His4, resulting in the founder vector pSMART-attL (Figures 4A and S4A). Individual histone gene mutations were constructed in the founder vector. Through predesigned isocaudomer sites in the founder vector, two or three copies of mutated/wild-type histone genes were cloned in the same vector to give 2-His-GU and 3-His-GU constructs. Two copies of mutated His-GU were released and cloned into modified pUAST-attB-gw (Figure S4B). Finally, a construct with three mutated His-GUs was recombined with pUAST-attB-gw containing two mutated His-GUs via the Gateway system to yield pUAST-attB containing five copies of the mutated His-GU, and this vector was used in embryo injection.
Figure 4. Systematic mutagenesis of Drosophila melanogaster histone H3 and H4 residues.
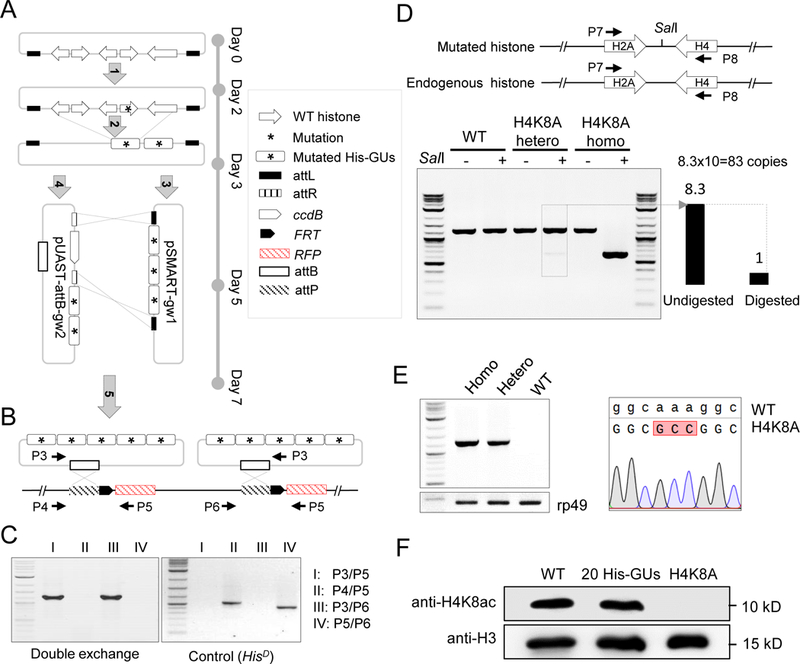
(A) Schematic representation of histone-mutagenesis procedures with Gateway assembly system (see details in Methods). ‘Five-step’ assembly was performed to insert five mutated-histone gene units (His-GUs) into the pUAST-attB integration vector.
(B) PhiC31-mediated ‘attB/attP’ exchange system for integration of two mutated 5 His-GUs into the genome in the HisD fly. Primer pairs for PCR verification are indicated.
(C) PCR verification of double attB/attP recombination.
(D) Molecular characterization of H4K8A mutant flies. The top panel is the schematic representation of the synthetic and native histone genes. Primers to amplify part of the histone genes are indicated. The SalI site introduced in the synthetic construct is shown. The bottom panel shows the gel image of amplified PCR fragments with or without SalI digestion. The copy number of His-GU was estimated as the ratio of band intensity of digested and undigested fragments, and is shown at the bottom left.
(E) Sequence confirmation of the H4K8A mutant. The left panel shows the PCR result using primers specifically to amplify the mutated DNA. The rp49 gene was used as a loading control. The sequencing trace is shown on the right.
(F) Analysis of H4K8ac (acetylated H4K8) in different fly lines by western blot.
See also Figure S4.
This approach enabled construction of many histone mutations, and their assembly into 5-His-GU vectors within a few days (Figure 4A). Through PhiC31-mediated double attP/attB integration at the locus of the attP sites in the HisD line, a single injection of such a vector enabled incorporation of 10 copies of a mutation on one chromosome (Figure 4B). A single round of crossing was then required to generate homozygous flies containing 20 copies of the same mutation. This system enabled testing of numerous histone mutations in a short time, greatly enhancing our ability to investigate histone function and the impact of histone modifications in this metazoan.
Validation of flies containing arrays of mutated histones
Given the possibility that the two attP sites on one chromosome in HisD flies could induce double attP/attB integration machinery, we used PCR to verify the relevant genotypes of recovered flies. As shown in Figures 4B and 4C, the precise double-exchange events that we sought indeed occurred. Some mutant flies with just one of the two attP/attB integrations (left or right) were also obtained (Figure S4F), as expected.
To rule out the possibility of potential recombination occurring between the mutated-histone locus and the adjacent endogenous-histone locus, we performed several assays to validate each mutant line. Because a SalI recognition site was inserted between His2A and His4 in all His-GU mutations, the amplified PCR product was digested with SalI and the size of the fragments was analyzed. As shown in Figure 4D, none of the DNA was cut by SalI in the wild-type line. However, the amplified DNA was completely digested in the homozygous H4K8A mutant line. In the heterozygous H4K8A mutant line, only a small proportion of amplified DNA could be digested, indicating the presence of both wild-type and mutated genes. This assay enabled us to estimate the copy number of histone genes. By quantifying the amounts of digested and undigested DNA, we estimated the copy number to be about 83 (Figure 4D).
Next, we designed a strategy to quickly identify the presence of mutated-histone genes by PCR with primers spanning the SalI site, which would only amplify fragments carrying mutations (Figure 4E). These fragments were sequenced to verify the presence of specific mutations (Figure 4E). Finally, a PTM-specific antibody, when available, was used for detection of the presence of a PTM in wild-type flies but not in mutants, as exemplified in Figure 4F with anti-H4K8ac antiserum.
Establishment of an alanine-substituted histone mutation library
Using the strategy described above, we targeted 44 amino acid residues from histones H3 and H4 with known PTMs, generating a library of 40 alanine-substitution mutation lines (Table 1). Each line was individually verified through attP/attB double-exchange PCR assays (Figure S4G), SalI digestion assays (Figure S4H), and DNA sequencing.
Table 1.
Phenotypic analysis of flies with mutations on modified histone H3 and H4 residues
| Mutants | Viabilitya | Fertilityb | DNA-damage sensitivityc | Gene-silencing defectsd | ||
|---|---|---|---|---|---|---|
| ♂ | ♀ | UV | X-Ray | |||
| H3T3A | ++ | ++ | sterile | + | +++ | + |
| H3T11A | + | + | sterile | nt | nt | - |
| H3R17A | ++ | + | sterile | nt | nt | - |
| H3K18A | +++ | ++ | ++ | ++ | ++ | - |
| H3K23A | ++ | ++ | + | + | + | +++ |
| H3K56A | ++ | ++ | sterile | +++ | +++ | +++ |
| H3K64A | +++ | ++ | ++ | + | + | - |
| H3T107A | ++ | ++ | + | + | +++ | - |
| H3K115A | +++ | +++ | ++ | + | +++ | - |
| H3K122A | +++ | ++ | ++ | + | +++ | - |
| H4T1A | ++ | ++ | ++ | + | +++ | ++ |
| H4R3A | +++ | ++ | + | + | ++ | - |
| H4K5A | +++ | ++ | +++ | + | + | - |
| H4K8A | ++ | +++ | +++ | ++ | ++ | - |
| H4K16A | ++* | ++ | + | + | +++ | - |
| H4K20A | + | nt | nt | nt | nt | - |
| H4R23A | ++ | +++ | + | + | +++ | +++ |
| H4K31A | +++ | ++ | ++ | + | ++ | + |
| H4Y51A | ++ | ++ | +++ | + | ++ | - |
| H4K77A | +++ | +++ | ++ | ++ | ++ | - |
| H4R92A | +++ | ++ | + | + | ++ | + |
| Lethal stage | Mutants | |||||
| Embryo | H3K4A, H3K14A, H3R26A, H3K27A, H3Y41A, H3T80A, H4Y88A, H4K91A | |||||
| Larva | H3K9A, H3T45A, H3K37A | |||||
| Pupa | H3R2A, H3T6A, H3R8A, H3S10A, H3S28A, H3K36A, H3K79A, H4K12A | |||||
Viability was represented by the rescue ratios of mutants, which was calculated as the following: ratio = (number of homozygous adult progenies)/(one-third of the number of total adult progenies) × 100. +++ >80%; ++ 30%–80%; + ≤30%.
Fertility was represented by the number of progeny of mutants when crossing with a wild-type counterpart. +++ >80; ++ 30–80; + ≤30; nt not tested.
DNA damage sensitivity was represented by the eclosion ratio, which was calculated as the following: ratio = [E (eclosed pharate number in UV or X-ray group)]/[C (eclosed pharate number in control group)] × 100. Third instar larvae were treated with 100 J/cm2 of UV or 30 Gy of X-ray irradiation. +++ <50%; ++ 50–75%; + ≥75%; nt not tested.
Gene silencing was measured by RT-PCR with primers specific to copia retrotransposon. – no changes compared with wild type. + Low expression; ++ Medium expression; +++ Strong expression.
Limited male (<1/1000 viable).
See also Figure S5.
Many histone alanine-substitution mutations impair viability
Each mutation was introduced in homozygous form to generate flies with 20 copies of the mutated His-GUs. Among the 40 lines, 19 (47.5%) represented essential residues, as the desired homozygous flies were not obtained despite screening hundreds of individuals (Table 1). By comparison, in yeast ~90% of such mutants are dispensable for cell survival (Dai et al., 2008; Nakanishi et al., 2008), suggesting that there are distinct requirements for histone modifications in single-cell yeast and in multicellular organisms. By maintaining the mutant lines over GFP-marked balancer chromosome, we performed survival tracing tests of homozygous mutant embryos and larvae, by monitoring the loss of GFP signal. This analysis enabled us to identify the stages at which the histone mutants ceased to develop, and this information potentially instructs further experiments. It also provided a platform to study the functional effects of histone mutations before the lethal stages. Among the 19 mutants, eight were embryonically lethal, three were lethal in larvae, and eight were lethal in pupae (Table 1; Figures S5A and S5B). Moreover, some of the mutants, which were not fully lethal, had an obvious reduction in viability, including H3T3A, H3T11A, and H4K16A, which produced 31.9%, 28.1%, and 38.2%, respectively, of the expected number of progeny compared with 20-His-GU (Figure S5D). These results suggest the existence of unknown requirements for particular histone residues or modifications during different developmental stages in multicellular organisms.
Histone mutants affect fertility more than adult morphology
For all viable mutants, we examined the visible morphology of adult flies, including the mouth parts, antennae, eyes, legs, sex combs, and wings. Minor defects in the wings were only observed in association with H3R17A and H4K20A, suggesting the majority of the viable mutations had minor effects during development. We measured the fertility of the flies by calculating ratios of adult progeny when mutants and wild types were backcrossed with wild type flies. The fertility of female flies was generally more sensitive to perturbation by histone mutations than that of males (Table 1). Male fertility was low for H3T11A (13.9%) and H3R17A (27.3%), but fertility ratios for males with other histone mutations were >50% (Figure S5E). However, female flies with H3T3A, H3T11A, H3R17A, and H3K56A mutations were completely sterile, and those with H4R23A, H4K16A, and H4R92A mutants were nearly sterile, with average ratios <10% (Figure S5F).
Because histone dosage affects fertility by interfering with oogenesis and spermatogenesis, we examined the testes and ovaries of mutant flies at 5 days old, classifying them into three categories as defined above (Figures 2E and 2F). Consistent with our observations in fertility tests, most males with alanine-substitution mutations had phenotypically wild-type testes (>90%, Figure S5G). Only four mutations (H3T3A, H3T11A, H3R17A, and H4K31A) resulted in notable defects in >20% of testes (Figure S5G). However, 13 of 20 histone mutations produced defects during oogenesis (Figure S5H). With some mutations, such as H3R17A, H4R23A, and H3K56A, none of the ovaries examined had wild-type morphology, consistent with the female sterile phenotype. Although a weak homozygote stock of H4K16A could be generated experimentally, its 90% decrease in fertility (compared with wild type) probably resulted from severely affected oogenesis (Figure S5H).
Histone mutations and DNA damage
Various types of histone modifications are functionally involved in the DNA-damage response (DDR) in different model systems. For example, in S. cerevisiae, both acetylation and deacetylation of histone H3K56 promote cell survival in response to genotoxic stress (Celic et al., 2006; Wurtele et al., 2012). However, the functions of histone mutations in relation to genotoxic-agent-induced DNA lesions are poorly understood in multicellular organisms. Therefore, we carried out a damage-sensitivity screen using both UV and X-ray irradiation (Figures S5I and S5J).
Consistent with previous reports in flies lacking the histone-modifying enzyme CBP/p300 (Das et al., 2009), the H3K56A mutation, which prevented PTM of this residue, resulted in a dramatically increased sensitivity to both UV and X-rays. The H4T1A mutation caused severe sensitivity to X-rays and minor sensitivity to UV irradiation, in agreement with a previous observation that phosphorylation of H4S1 is induced at DNA double-stranded breaks (DSBs) in S. cerevisiae (Cheung et al., 2005). Notably, several mutants had differential responses to UV light and X-rays, including H3T3A, H4R23A, and H4R92A (Figures S5I and S5J). These mutants had similar responses to wild type upon UV exposure, but were significantly more sensitive to X-rays, particularly H3T3A and H4R23A (Figures S5I and S5J). These residues are presumably specifically involved in DSB repair. Taken together, these histone mutations enabled us to reproduce the DNA-damage phenotypes known to occur on mutation of histone-modifying enzymes, and also to identify other histone residues that might be involved in regulating DDR in D. melanogaster.
Selective activation of retrotransposons is associated with specific histone mutations
Silencing of transposable elements in eukaryotic genomes prevents deleterious mutagenic effects. However, the regulatory mechanisms of silencing are poorly understood. To evaluate the effects of epigenetic regulation during this process in D. melanogaster, we analyzed the expression of copia, a heterochromatic long-terminal-repeat retrotransposon, which is usually silenced (de Setta et al., 2011). Several histone mutations (H3K9A, H3K23A, H3K56A, and H4R23A) caused strongly increased expression of copia in third-instar larvae, indicating de-repression of this retrotransposon (Figure S5K; Table 1). In Schizosaccharomyces pombe, localization of Swi6 (a homolog of D. melanogaster Heterochromatin Protein 1 (HP1)) to heterochromatic regions requires methylation on H3K9 (Nakayama et al., 2001). Our results suggested that several other histone PTMs (Figure S5K) may also be involved in retrotransposon silencing.
We selected two histone mutations (H3K9A and H3K56A) to investigate whether expression of other transposable elements, including Het-A, I-element, mdg, Mst40, and ZAM, was also up-regulated. In addition to copia (which is one of the most highly transcribed transposable elements in D. melanogaster), Mst40 was up-regulated in flies with the H3K9A mutation and mdg was up-regulated with H3K56A (Figures S5L and S5M). Further studies are required to dissect the underlying mechanisms involved in this specific regulation.
Histone H4 lysine 16 is required for ovarian GSC maintenance
Acetylation at H4K16 is evolutionarily conserved and functions to regulate the expansion of silent chromatin (Kimura et al., 2002), to modulate higher-order chromatin structure (Shogren-Knaak et al., 2006), to interfere with the activity of chromatin remodelers (Corona et al., 2002), and to control the replicative lifespan in budding yeast (Dang et al., 2009). In D. melanogaster, H4K16 acetylation is carried out by the acetyltransferase males-absent on the first protein (MOF) (Smith et al., 2000).
In our experiments, all homozygous H4K16A female flies displayed normal adult morphology. Western blotting and immunostaining with H4K16ac-specific antisera confirmed the absence of H4K16 acetylation in H4K16A homozygous flies (Figures 5A and S6A). Although previous results showed that MOF is important for female survival and lifespan (Conrad et al., 2012a), the H4K16A female flies in our study did not show a significant decrease in viability or lifespan (Figure S5D; Table 1). These results suggested that MOF-mediated H4K16 acetylation is not essential for female viability, and that MOF has additional roles during female survival, probably involving the male-specific lethal (MSL) complex. However, in H4K16A mutants, all adult females had a delayed development period and abnormal oogenesis, resulting in 90% less fertility than the wild type.
Figure 5. H4K16 was required for ovarian germline stem cell (GSC) maintenance and male viability.
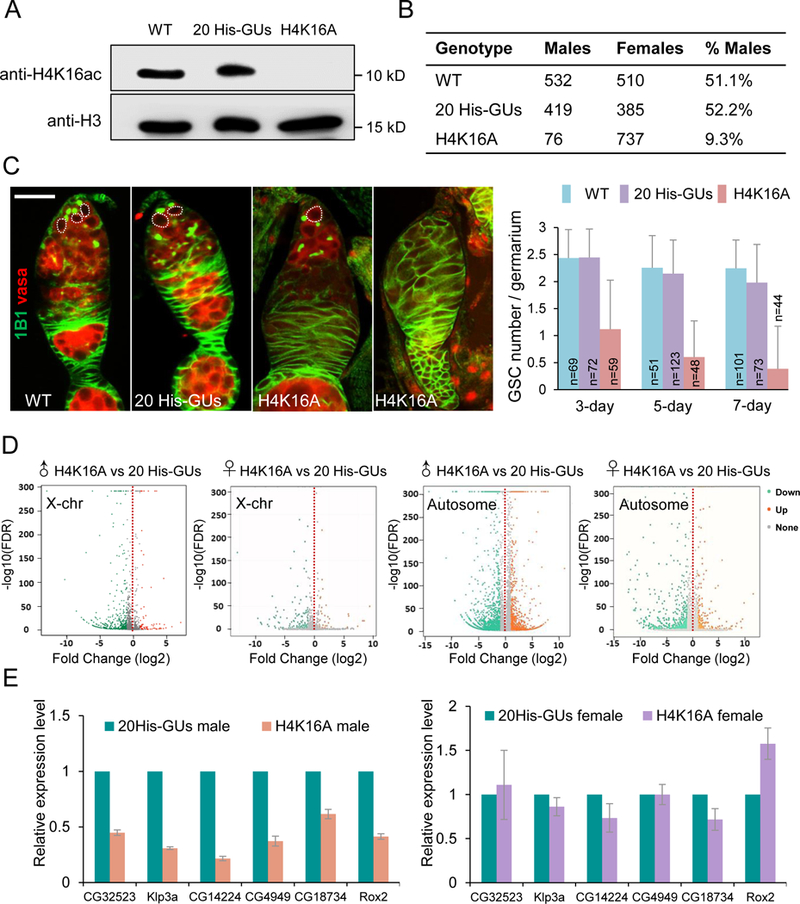
(A) Western blot of adult fly extract from wild type (WT; w1118), 20 His-GUs (20 copies of wild-type histone gene units), and H4K16A genotypes.
(B) The number of male and female progeny from wild type, 20 His-GUs and H4K16A parents.
(C) Confocal images of ovaries from w1118, 20 His-GUs and H4K16A flies. GSCs (dotted line) stained with 1B1 (green) and Vasa (red). Histogram shows the number of GSCs per germarium at days 3, 5, and 7 adulthood. The number of germaria examined per genotype is shown on each column. Values are means ± SEM. Scale bar: 20 µm.
(D) Transcriptome comparisons of third larvae salary glands between H4K16A mutants and 20His-GUs.
(E) RT-PCR verification of effects of H4K16A mutation. Six-linked genes (CG32523, Klp3a, CG14224, CG4949, CG18734, and Rox2) were chosen as the targets. Values are means ± SEM of three biological replicates (rp49 was the reference gene for normalization, RNA was extracted from salivary gland).
See also Figure S6.
By tracking ovarian morphology microscopically in females from 0 to 7 days old, we uncovered severe developmental defects in ovaries of flies with H4K16A mutations compared with wild-type controls (Figure 5C). Specifically, ovaries were examined for the maintenance of GSCs and their early progeny in the germaria by immunostaining with 1B1 antibody to the Hu-li tai shao Adducin homolog, which labels spectrosomes and fusomes, enabling the identification of GSCs, cystoblasts and differentiated cysts (Lin et al., 1994). In contrast to wild-type or 20-His-GU flies, which normally had two or three GSCs in each germarium during the first week of adulthood, most of the germaria from H4K16A flies had one or no GSCs (Figure 5C). To determine if this phenotype changed with age, GSC numbers were determined in ovaries isolated from 3 day, 5 day, and 7 day old H4K16A adult females. In contrast to control (wild-type or 20-His-GU) germaria, which had two or three GSCs at 3 days, 5 days, and 7 days, H4K16A mutants showed GSC loss, and on average contained only 1.2 GSCs at 3 days, 0.6 GSCs at 5 days, and 0.4 GSCs at 7 days (Figure 5C). Our results indicated that the H4K16 amino acid residue is required for ovarian GSC maintenance, but not for female adult viability.
Histone H4 lysine 16 is required for male viability
The genome-wide H4K16 acetylation mediated by MOF–MSL complexes contributes to the twofold transcriptional activation in X-chromosome dosage compensation (Conrad et al., 2012a). In our study, in flies with H4K16A mutations, about 90% of males did not survive to adulthood (Figure 5B). This finding was consistent with previously described male lethal or limited male (few male adults produced) phenotypes of mof conditional mutations (Raja et al., 2010). However, in our study, disruption of mof with the CRISPR/Cas9 system or mof RNAi resulted in 100% male lethality (Figure S6). Notably, >80% of salivary glands from male third-instar larvae with H4K16A mutations contained polytene chromosomes with disordered structure (Figure S6A), with MSL2 and most of the MOF protein still localized on the X chromosome. These results were consistent with the cytological observation that mof RNAi male larvae had 60% disruption in polytene X chromosomes (Figure S6B). Although MOF is required for genome-wide H4K16 acetylation in both sexes (Conrad et al., 2012a), chromatin structure was normal in most salivary gland polytene chromosomes of females with H4K16A mutations when compared with wild type(Figure S6A), and comparable to that in mof RNAi treated females (Figure S6B). These results suggested that MOF-mediated H4K16 acetylation may have cause more severe effects on higher-order chromatin structure in males than in females.
To directly test whether the H4K16A mutation impaired transcriptional activation of X chromosome-linked genes, genome-wide transcription analysis was performed in H4K16A male and female third-instar larvae salivary glands. In H4K16A males, 23% of X-linked genes (393 genes) were significantly down-regulated twofold or more compared with wild-type controls. By contrast, only 7% of X-linked genes were down-regulated in H4K16A females (Figures 5D and S6C). These results were consistent with previous findings that the MSL complex can induce twofold hypertranscription of hundreds of genes on the male X chromosome through specific histone H4K16 acetylation (Larschan et al., 2011). We confirmed the down-regulation of six known X-linked genes (Conrad et al., 2012a) in H4K16A male flies relative to wild-type controls by qRT-PCR (Figure 5E). For example, male expression levels of roX2 (long non-coding RNA on X) RNA, which is required for male sex-dosage compensation, were reduced by >50% in flies with H4K16A mutations relative to wild type. H4K16A was also associated with significant down-regulation of 19.9% of autosomal genes in males (compared with 6.9% in females) (Figure S6C), which was consistent with our observation that the effects of K16 mutation on global chromatin architecture in the male were more severe than in females. These data suggested that the H4K16 site not only contributes to male X-linked gene dosage compensation, but also globally regulates autosomal gene transcriptional activation in males. We conclude that modifications on H4K16 are required for male-specific viability.
A mosaic system enables study of the function of essential histone mutations
More than 45% of mutants in the histone mutation library did not produce homozygous viable adults, hindering any additional characterization (Table 1). We took H3K27A as an example, and examined its embryonic lethality through immunostaining at different time points with anti-H3K27me3 and anti-cyclin B. H3K27me3 staining in 3.5–4 h H3K27A embryos had a similar pattern to staining in 20-His-GU embryos (Figure 6A), presumably resulting from maternal histone supply (Gunesdogan et al., 2010). However, H3K27A embryos demonstrated failure of embryonic cell cycling at ~10 h, as shown by uniformly epidermal cell division after egg laying at 11.5–12 h (Figure 6A).
Figure 6. A rapid screen of mosaic analysis identifies histone modification sites involved in polycomb-group repressin.

(A) Pattern of cell division in epidermal cells from flies with 20 His-GUs (20 copies of wild-type histone gene units) and H3K27A mutations. Panels show time-matched individual embryos labeled for Cyclin B (white), H3K27me3 antibody (red), and DAPI (blue). Scale bar: 100 µm.
(B) Clonal analysis demonstrates H3K27 is required for polycomb target-gene repression. Wing imaginal disc clones of HisD homozygous cells in animals with transgenic His-GUsWT (left) or His-GUsH3K27A (right) were immunostained with anti-Ubx and anti-H3K27me3. HisD homozygous cells with transgene are marked by the absence of green fluorescent protein (GFP). The yellow box shows the clone cells. Scale bar: 50 µm.
(C) H3R26 is essential for efficient polycomb-mediated gene repression in Drosophila. Wing imaginal discs clones of HisD homozygous cells in animals with transgenic His-GUsH3R26A were immunostained with anti-Ubx or anti-Abd-B, anti-H3K27me3 or anti-H3K9me3. HisD homozygous cells with His-GUsH3R26A are marked by the absence of green fluorescent protein (GFP). Both Ubx and Abd-B were mis-expressed in homozygous H3R26A mutant cells (left column) and H3K27me3 was strongly reduced (top right corner). By contrast, H3K9me3 was unaffected in H3R26A clones (lower right corner).
(D) H3K27 tri-methylation is decreased in H3R26A Drosophila embryos. Western blot assay on lysate from 20 His-GUs, H3K27A, H3R26A, H3S28A, and H3K9A flies immunoblotted using anti-H3K27me3 antibody.
(E) Quantification of Figure 6D western blots. Densitometry of western blot bands was quantified with Image J software. Values are means ± SEM of three biological replicates.
See also Figure S7.
To develop a platform for systematic study of the effects of histone mutations in any tissue, we generated a FLP/FRT transgenic fly, which would specifically induce histone gene mitotic recombination. First, a Promoter/ubi-GFP-FRT DNA fragment was precisely integrated into the original Cas9/gRNA-targeted locus (as in Figure S1B) at the right side of the histone cluster by CRISPR-mediated HR (Figure S7A). This fly was crossed with a hsp70-Flp transgenic fly expressing Flp recombinase on the X chromosome, then crossed to give a homozygous stable fly stock for each mutated-histone mosaic analysis (Figure S7B). Subsequently, we performed mosaic analysis using FLP-FRT-mediated mitotic recombination (Xu and Rubin, 1993), after which we distinguished the desired clones by the absence of GFP signal. For example, to investigate whether the H3K27A mutation displayed homeotic transformation and Hox cluster mis-regulation, we observed defective phenotypes of wing morphogenesis in H3K27A/+ transgenic flies (occurring at a rate of ~2.9% (5/173)), consistent with previous results (McKay et al., 2015). The posterior-wing phenotype was reminiscent of gain-of-function of the Hox cluster gene ubx (Lewis 1978). Consistent with previous reports (McKay et al., 2015; Pengelly et al., 2013), we found that H3K27A mutation was associated with de-repression of ubx in the wing disc (Figure 6B).
To identify other histone residues involved in the polycomb pathway, we screened all lethal histone mutants with observation of ubx gene expression in clone cells. H3R26 and H3S28 were essential for repression of polycomb-targeted ubx gene (Figures 6C, S7C and S7D). H3R26A also exhibited de-repression of another Hox gene, abdominal B (abd-B), when expressed clonally in the imaginal wing disc (Figure 6C).
H3R26 and H3K27 are adjacent amino acid residues, we found that H3K27 methylation was considerably reduced in H3R26A clones (Figure 6C) compared with controls containing wild-type histones (Figure 6B). Notably, H3R26A clones had normal H3K9 methylation, indicating that not all modifications in H3 were affected by H3R26 alanine substitution (Figure 6C). Together, our results suggested that H3R26A causes de-repression of Hox genes by reducing H3K27 trimethylation.
Western blotting was performed on protein extracts from mutant embryos homozygous for histone mutations (Figure 6D). H3K27 trimethylation was diminished by 75% in extracts from embryos with the H3R26A mutation (compared with wild type). By contrast, a nearly wild-type level of H3K27 trimethylation was detected in embryos with the H3K9A mutation, and H3K27 trimethylation was reduced by 25% in H3S28A embryos (Figure 6E).
DISCUSSION
In this study, we have developed an efficient histone-mutagenesis system with several advantages over previous approaches. Our histone-deletion line facilitates histone rescue in situ. A single plasmid is sufficient for complementation, and the plasmid is targeted to the original histone locus, which eliminates consideration of positional effects associated with random integration of plasmids and BACs. Our high-throughput strategy to assemble multiple copies of His-GUs is fast and efficient and enables introduction of not only singular but also compound histone mutations. Below, we discuss some biological insights obtained with this method.
Histone dosage is critical for development of reproductive systems
Our results demonstrated that a low His-GU copy number causes developmental defects in both testes and ovaries, with more severe effects in ovary development (Figures 2D and 2F). The ovarian defect was not the result of a loss of GSCs, and instead the budding processes were impaired (Figure 3A), which leads to reduced fecundity or to sterility (Ruohola et al., 1991), and which explains the severe fertility defects in females. The number of GSCs was only slightly reduced in testes from adult males with low histone copy numbers (compared with wild type). Because histone copy numbers are altered globally in these flies, mosaic analysis could reveal whether reduced histone copies reflects an autonomous or non-autonomous effect on GSCs.
H4K16 is essential for male viability and female oogenesis
Our finding that H4K16 was critical for sex-dosage compensation and male development is consistent with the fact that MOF–MSL, which acetylates H4K16, contributes to male X-linked transcriptional activation (Conrad et al., 2012a). Notably, we recovered some H4K16A male adults and generated a weak homozygous mutant stock under normal culture conditions, whereas mof RNAi and mutant each lead to 100% male lethality. We propose that MOF has functions in male development beyond H4K16 acetylation.
H4K16A mutation caused a severe sex bias (1:10 male:female) in homozygotes, reminiscent of that resulting from inactivation of the non-coding roX gene (another dosage compensation component) in D. melanogaster (Ghosh et al., 2016). Given that MOF–MSL-mediated H4K16 acetylation is roX-dependent (Park et al., 2008), roX might act by stimulating H4K16 acetylation, directly or indirectly, which merits further exploration.
H4K16A mutation severely depleted GSCs in the ovary, which presumably contributed to the infertility in the mutants. This finding is not surprising, given that MOF is involved in maintaining pluripotency and self-renewal of embryonic stem cells, and mof mutations lead to failure in the reprogramming of stem cells (Mu et al., 2015). The H4K16A mutation might additionally compromise follicle-cell development, as suggested by the fact that Chameau, another H4K16 acetyltransferase, regulates the developmental transition of follicle cells into the amplification stages of oogenesis (McConnell et al., 2012).
Our mosaic analysis platform facilitates investigation of histone involvement in polycomb repression
H3K27me3 is essential for gene repression involving polycomb-group (PcG) proteins, but it is not clear which other histone residues are also involved. Traditional mosaic cloning analysis has identified H3S28 as one such residue (Yung et al., 2015). This method requires the generation of fly mutants with a complex genotype, which is laborious as it involves multistep crosses. Our strategy for mosaic analysis is much faster and simpler, enabling us to readily screen mutations of 19 essential histone residues. We confirmed the previous finding about H3S28, and further demonstrated that H3R26 is also essential for PcG function, thereby validating this strategy.
We showed that H3R26 is required for H3K27 trimethylation, which contributes to PcG-mediated gene repression. Additionally, H3R26 might stimulate PRC2 catalytic activity, as suggested by in vitro data showing that human PRC2 catalytic activity is partially dependent on H3R26 (Jiao and Liu, 2015). H3R26 may also facilitate PcG protein recognition, with the positive side chain of H3R26 contacting the SET domain of the E(z) methyltransferase. Whether H3R26 is modified remains unclear, although H3R26 methylation has been reported in mouse embryos (Torres-Padilla et al., 2007). Further studies are needed to clarify these issues.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Guanjun Gao (gaogj@shanghaitech.edu.cn).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Fly strains w1118, φC31, heterozygous histone deletion line Df(2L)HisC (Günesdogan et al., 2010) and all balancers were obtained from the Bloomington Stock Center (Indiana, USA). Fly strain yw hs-flp122; Ubi-GFP FRT was a gift from Renjie Jiao. All flies were cultured at 25°C on standard cornmeal/sugar/agar media (Fly food recipes: Agar 30g, Cornmeal 250g, Yeast 125g, Sucrose 360g, Water 5L).
METHOD DETAILS
CRISPR/Cas9-mediated HisD fly mutant construction
The CRISPR/Cas9 system was established and applied to gene targeting in Drosophila. Two guide RNAs (gRNAs), whose target sites flank the histone gene cluster in chromosome 2, were used to introduce DNA double-strand breaks at each side of the histone gene cluster into two w1118 flies. To take advantage of homologous recombination (HR) in Drosophila, two FRT sites were integrated into each side of the histone gene cluster. HisD was generated by crossing and inducing the rearrangement of two FRT sites and droppin the entire histone cluster out from one chromosome.
Mof mutant construction
The Mof mutant was generated by the fly CRISPR/Cas9 system established in our lab. The gRNA sequence was 5′-GGAGGAGCATGAGCCGGAAC-3′. General primers (Forward 5′-ACATGTTGTGATAAATCAGGGTTGC-3′ and Reverse 5′-AGATTACCGTATCGGAGGAGGGC-3′) were used to amplify the DNA fragment to verify the disruption of mof-gene coding sequence. Based on the sequencing results, we identified a 136 bp deletion (from 80 to 215 bp) at the N-terminus of the mof-CDS, which causes a frameshift from amino acid 27.
GFP-FRT transgenic fly construction
A Pomoter/ubi-GFP-FRT DNA fragment was knocked in to the original Cas9/gRNA-targeted locus at the right side of the histone gene cluster by CRISPR-mediated homologous recombination. This gRNA was the same as the gRNA used for the attp-FRT site knocked in at the right side of the histone cluster in Figure 1A.
In vitro synthesis and microinjection of Cas9-coding mRNA and gRNA
In vitro transcription of Cas9 mRNA was performed using the Sp6 mMESSAGE mMACHINE Kit (Ambion [Thermo Fisher Scientific, Waltham, MA, USA]), as described previously (Yu et al., 2013). In vitro transcription of the designed gRNAs was performed using the RiboMAX Large Scale RNA Production Systems-T7 Kit (Promega, Madison, WI, USA). Purified Cas9 mRNA, gRNA, and donor (single-stranded oligonucleotide DNA or plasmid) were mixed at final concentrations of 1 μg/μL, 50 ng/μL, and 0.3 μg/μL, respectively, followed by injection into w1118 embryos according to standard procedures.
Plasmid construction and synthesis
The Gateway cloning system was modified and used to assemble multi-copy histone units into a vector suitable for transgenesis. The attL1 and attL2 sequences were amplified from the pCR8/GW vector (K2500–20; Invitrogen [Thermo Fisher Scientific]) and assembled into a pSMART vector. The pSMART vector contains a kanamycin resistance marker, into which KpnI and NotI restriction sites were added between two attL sequences. The newly generated vector was named pSMART-attL. The attR1 and attR2 sequences, together with a ccdB gene and chloromycetin resistance gene, were amplified from the pDEST22 vector (Invitrogen, Gateway destination vector) and assembled into a pUAST-attB-w+ vector by SbfI and NotI restriction sites. The newly generated vector was named pUAST-attB-w+-attR. A primer pair that specifically targeted both sides of one histone unit in the chromosome was used for wild-type histone unit assembly. The forward primer contained KpnI and AgeI restriction sites, and the reverse primer contained NotI and XmaI restriction sites. KpnI and NotI restriction sites were used for shuttling one histone unit assembly from the Drosophila genome to the pSMART-attL vector. A SalI restriction site was introduced between His4 and His2A genes to facilitate histone mutagenesis.
To introduce amino acid mutations into specific histone modification sites, the wild-type His-GU was divided into three parts: His1; His2A, and His2B; and His3 and His4. Each part was cloned into a single pUC19 vector using the restriction-enzyme combinations XmaI/BglII, BspHI/SalI, and SalI/KpnI, respectively, creating three plasmids called pUC19-H1, pUC19-H2A/B and pUC19-H¾. Mutations were introduced through site-directed mutagenesis using primers that changed the destination codon to GCC (encoding alanine).
To increase the His-GU copy numbers within the pSMART-attL vector, KpnI and XmaI were used to excise a single histone unit and insert it back into the same plasmid, which was then linearized with AgeI and KpnI. Because AgeI and XmaI are isocaudomers, the same strategy was used to increase the His-GU copy numbers to three in this vector. In the pUAST-attB-w+-attR vector, KpnI and NotI restriction sites were introduced upstream of the attR1-ccdB-cmR-attR2 cassette. Two His-GUs were inserted into the pUAST-attB-w+-attR vector using KpnI and NotI. The Gateway LR clonase kit (11791–020; Invitrogen) was used to insert three His-GUs into the pUAST-attB-w+ destination vector. The final construct contained a total of five His-GUs: three from the pSMART-attL vector and two from the pUAST-attB-w+-attR vector (Figure 4A).
To insert an attP-FRT cassette downstream of the histone cluster in the genome, a synthetic plasmid donor named pAV-R, which contains homologous arms (ArmC, left; ArmD, right), an attP-FRT cassette and an RFP selection marker, was synthesized by Wuxi Qinglan Biotechnology (Yixing City, China).
To construct the conditional histone-deletion system, the pAV-R donor plasmid was modified to include an enhanced GFP marker driven by a ubiquitin promoter that was inserted between the left homologous arm and attB sites via the Golden Gate cloning method.
Plasmid midi-preparation
The constructs containing five copies of mutated or wild-type His-GUs were transformed into DH5α competent cells and cultured at 30°C for 24 h. A single colony was inoculated into a 1 L flask containing 200 mL of LB medium, and cultured at 30°C, with shaking at 220 rpm for 24 h. Bacterial wet weight was normalized to 1 g after harvest. Plasmid was extracted using a Plasmid Midi kit (12143; QIAGEN, Hilden, Germany) following the recommended protocol, and the plasmid concentration was quantified using a 2000c NanoDrop (Thermo Fisher Scientific). The plasmid was further purified by phenol/chloroform/isoamyl alcohol (25:24:1) extraction, and normalized to 1.5 μg/μL. The normalized plasmid was centrifuged at 18000 rpm for 1 h at 4°C. The supernatant was then transferred carefully to a new tube and stored at −20°C.
Drosophila embryo microinjection
HisD male flies were mated with φC31 virgin flies. After 3 or 4 days, 0–1 h embryos were harvested for microinjection. About 1000 embryos were injected for each histone mutant construct.
Genomic DNA preparation and histone construct double-integration verification
Drosophila adults (n = 3–5) were homogenized in 100 μL lysis buffer (10 mM Tris-HCl pH 8.0, 0.5 mM NaCl, 20 mM EDTA, 50 μg/mL RNase A and 1% SDS) and incubated at 30°C for 1 h. The tube was then transferred to a 65°C heating block for 30 min. Next, 100 μL of phenol/chloroform/isoamyl was added and the tube was gently vortexed. The tube was then spun at 15000 rpm for 10 min and the supernatant was transferred into a new tube. The DNA was precipitated with an equal volume of isopropanol at −20°C for 30 min then centrifuged at 15000 rpm at 4°C for 10 min. The supernatant was discarded, and the pelleted DNA was washed with 500 μL of 70% ethanol and dried using a vacuum pump at room temperature for 5 min. The genomic DNA was resuspended in 15 μL of Tris-EDTA (TE) buffer and stored at −20°C.
Taq Master Mix (2×) was used for histone construct double-integration verification. The genomic DNA was normalized to 100 ng for each 10 μL PCR reaction. Both forward and reverse primers were diluted to 1 μM in 10 μL of the reaction volume. The PCR program was set to one cycle of 94°C for 5 min, 32 cycles of 94°C for 30 s, 60°C for 30 s, and 72°C for 80 s, one cycle of 72°C for 7 min and hold at 12°C. PCR products were mixed with loading buffer and loaded onto a 1.5% agarose gel with 0.5 µg/mL ethidium bromide for electrophoresis. The verification pattern is shown and discussed in Figures S4G and S4H.
Histone mutant sequencing validation
Drosophila histone mutants were verified by histone-specific sequencing from genomic DNA. Two different primer pairs were used for mutant gene amplification from homozygous or heterozygous adult flies. For homozygous mutants, general histone primers (WZO172 and WZO101) were used to amplify a 1584 bp fragment for sequencing. For heterozygous mutants, transgene-specific primers (WZO371 and WZO101) were used to amplify a 1320 bp fragment for sequencing.
Homozygous fly mutant verification assay
A SalI digestion assay was used to distinguish between the endogenous and transgenic histone loci. In the transgenic histone unit, a SalI restriction site was introduced between His4 and His2A. A pair of primers (WZO167 and WZO337) flanking the SalI site was used to amplify a 1232 bp fragment, which could then be digested with SalI into two equal-length fragments. DNA fragments were mixed with the appropriate 6× DNA loading buffer and loaded onto a 1.5% agarose gel with 0.5 μg/mL ethidium bromide for electrophoresis. Gel images were generated using a TANON 1600 instrument and the DNA signal intensity was quantified via ImageJ software.
Rescue ratio and viability statistics
Heterozygous flies balanced over CyO were self-crossed to create homozygous histone mutants that were identified through the absence of the CyO phenotype. The adult rescue ratio was determined by dividing the actual with the theoretical number of homozygous mutants. Three biological replicas were performed. The flies were counted after almost all eclosion.
Fertility test
Either male or virgin-female histone mutant adult flies were crossed with three w1118 virgins of the opposite sex. Parents were allowed to mate for 7 days and then discarded. The number of adult progeny in each vial was counted to assess the fertility of the various histone mutants.
Morphological observation
To screen for morphological defects in each of the viable histone mutants and flies with different copy numbers of His-GUs, the mouthparts, antennae, eyes, legs, sex combs, and wings were observed using a stereomicroscope, and compared with those of w1118 control flies. Adult flies of the 10 His-GUs, 12 His-GUs, 16 His-GUs, 20 His-GUs, and 24 His-GUs genotypes were analyzed.
To determine the phenotypes within the ovaries and testes, virgin males and females were aged for 5 days, then their testes or ovaries were dissected and observed under an optical microscope. Wing, ovary, and testis phenotypes were photographed with a Leica Mz10f microscope.
Identification of the lethal phases of histone mutants
Lethal histone mutants were balanced over CyO, P{ActGFP}MR1 to identify homozygous mutant embryos or larvae by the absence of green fluorescent protein (GFP) expression. 150 GFP-positive first-instar larvae and 150 GFP-negative first-instar larvae were collected on an apple-juice plate, and tracked until adulthood. For each histone mutant, the number of progeny at second-instar larval, third-instar larval, pupal, and adult stages were counted. Embryonic-lethal mutants were identified by the absence of GFP-negative first-instar larvae.
DNA-damage-sensitivity screen
Viable homozygous histone mutants, and heterozygous histone mutants balanced over CyO, P{ActGFP}MR1, were screened for their DNA-damage responses. GFP-positive and GFP-negative wandering third-instar larvae were selected and randomly divided into control and experimental groups. The experimental groups were exposed to either 10 mJ/cm2 of UV radiation using a Stratalinker 2400 (Stratagene, San Diego, CA, USA) or 30 Gy X-ray radiation using a RS 2000 (Rad Source Technologies, Buford, GA, USA). After irradiation, all larvae were maintained under standard conditions for 7 days until eclosion, when adults were collected and counted. The eclosion rate was calculated as (the number of eclosed flies)/(the number of input larvae). The eclosion rate of the experimental groups was then normalized to its paired control as follows: normalized eclosion ratio (%) = [Ratio (experimental group)]/[Ratio (control group)]×100.
RNA extraction and RT-qPCR
Total RNA was extracted from Drosophila larvae, adults or salivary glands using an RNA extraction kit according to the manufacturer’s instructions (DP431; TIANGEN, Beijing, China). The total RNA concentration was measured using a NanoDrop 2000c, and 1 µg of total RNA was then used for cDNA synthesis (KR106; TIANGEN). 2×SYBR green master mix (ABI, 4367659) was added to the RNA extract for quantitative real-time PCR analysis using a LightCycler 480 instrument and software (Roche, Basel, Switzerland).
RNA sequencing
Total RNA from testes, ovaries, and salivary glands were separately isolated with TRIzol. Sample quality was verified on an Agilent 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA), and RNA was quantitated on an ABI StepOnePlus Real-Time PCR System (ABI, Foster City, CA, USA). RNA was sequenced on an Illumina HiSeq 2000 at the BGI (Shenzhen, China; http://www.genomics.cn/index). At the data-processing step, raw reads were filtered by removal of adaptor sequences, low-quality reads in which >50% of bases were low quality (≤5), and reads in which the percentage of unknown bases (N) was >10%.
The cleaned data were aligned to the reference sequence using SOAPaligner/SOAP2 (http://soap.genomics.org.cn/soapaligner.html). For the purpose of alignment, a maximum of two mismatches was permitted. The D. melanogaster genome and gene datasets were downloaded from FlyBase (r6.14), which was used as a reference. To assess sequencing saturation, the number of genes identified was plotted against the number of cleaned reads to determine when no further genes could be detected by adding reads, implying full saturation. To evaluate the quality of the RNA-seq dataset, the distribution of gene coverage in each sample was analyzed. Gene-expression levels were calculated using the Reads Per Kb per Million reads (RPKM) method. Furthermore, R package NOISeq (version 2.14.1) was utilized to identify differentially expressed genes between two samples with biological replicates. In this approach, the P-value corresponds to a differential gene-expression test, and the FDR (False Discovery Rate) is a method for determining the P-value threshold for multiple tests. We used an FDR ≤0.001 and |log2Ratio ≥1| as the threshold of significance for differences in gene expression.
Data access
The RNA-seq data sets generated by this study have been deposited in the NCBI Sequence Read Archive (SRA; https://www.ncbi.nlm.nih.gov/sra/) under accession numbers SRP157191, SRP157300, and SRP158300.
Nucleoprotein extraction and western blotting
Drosophila larvae or adults were homogenized in buffer M (50 mM Tris-HCl pH 7.5, 50 mM KCl, 5% Glycerol, 1 mM DTT, 0.5 mM PMSF, and 0.5 mM EDTA). Nuclei were purified with Miracloth, transferred into 1.5 mL tubes, and centrifuged at 12000g for 15 min at 4°C. The supernatant was discarded and nuclei were resuspended with 100 μL buffer M and 10 μL 3 M KCl was added to increase the final KCl concentration to 100 mM. Following genomic DNA sonication, 30 μL of 4×protein loading buffer (Takara, Mountain View, CA, USA) was added and the mixture heated for 6 min at 95°C. Protein samples were loaded onto a 15% SDS-PAGE gel for electrophoresis. After transfer to PVDF membrane and blocking in 5% dried milk dissolved in TBST, protein samples were incubated with primary antisera at 4°C overnight. After three 10 min TBST washes, membranes were incubated with secondary antisera from rabbit or mouse at room temperature for 1 h. After three 10 min TBST washes, and addition of enhanced chemiluminescence substrate, western blot signals were detected with an ImageQuant LAS 4000 instrument.
Analysis of confocal images
Confocal images were obtained with a Zeiss LSM 5 Pascal or a Zeiss LSM 510 Meta microscope. GSCs were scored as Vasa-positive cells making contact with the hub and containing spherical fusomes. Statistical analysis of stem-cell number was performed using Prism 5. Unpaired ANOVA analysis was used to compare three or more populations.
Polytene immunostaining
Salivary glands from wandering third-instar larvae were dissected in 0.7 M NaCl. For anti-HP1 (DSHB), anti-MSL2, and anti-MOF (anti-MSL2 and anti-MOF were gifts from Peter B. Becker) immunostaining, salivary glands were fixed for 7 min in 1.85% formaldehyde and 55% glacial acetic acid diluted in double-distilled water, then frozen in liquid nitrogen. The slides were washed with cold and room temperature 1×PBS for 10 min, then washed three times for 15 min each in PBS containing 0.1% Triton X-100 (PBST). The slides were blocked in normal goat serum (CWBIO, Beijing, China) for 1–2 h at room temperature. After blocking, the slides were incubated in 40 µL of primary antibody diluted in 0.1%PBST at 4°C overnight. Slides were washed three times for 15 min in PBST, and goat anti-mouse (A11029; Invitrogen) or goat anti-rabbit (A11036; Invitrogen) secondary antibodies were added to the slides for 2 h at room temperature in the dark. Slides were then washed three times for 5 min in PBS and mounted in Vectashield (H-1200; Vector Laboratories, Burlingame, CA, USA) containing 4’, 6-diamidino-2-phenylindole (DAPI). Images were obtained with a Zeiss Imager Z2. Detection of H4K16 acetylation on polytene chromosomes was performed as described previously (Piacentini et al., 2009).
Ovary immunostaining
Female flies were kept for 3, 5, and 7 days with a few male flies and transferred to fresh vials of food every other day. Ovaries were dissected in 1×PBS and fixed in 4% paraformaldehyde in PBS, pH 6.9, at room temperature for 15–20 min. Ovaries were washed in PBST containing 0.5% Triton X-100 for 30 min, followed by three 15 min washes in PBST containing 0.1% Triton X-100 at room temperature. Ovaries were blocked with goat serum (CWBIO) for 90 min at room temperature and then incubated at 4°C overnight in mouse monoclonal anti-1B1 (DSHB) antisera and rat anti-vasa (DSHB) antisera diluted in PBST containing 0.1% Triton X-100. Ovaries were washed four times for 15 min with 0.1% PBST, then incubated in the dark with goat anti-rat (A-11007; Invitrogen) and goat anti-mouse (A-11029; Invitrogen) secondary antibodies for 2 h at room temperature. Ovaries were washed four times for 15 min with 0.1% PBST again, eggs were discarded, and the remaining ovaries were mounted in Vectashield containing DAPI. Images were obtained with a Zeiss780 Observer Z2.
Embryo immunostaining
Embryo collections were obtained by restricting egg deposition to 30 min with subsequent ageing at 25°C for the appropriate length of time. Embryos were dechorionated with 50% bleach, fixed in 1:1 heptane (24,665–4; Sigma, St Louis, MO, USA):3.7% formaldehyde (344198; Calbiochem, San Diego, CA, USA) mixture for 20 min and then devitellinized in a 1:1 heptane:methanol mixture, followed by washes and storage in methanol as described in Drosophila protocols. Embryos were re-hydrated in PBTA solution (1×PBS, 1% BSA, 0.1% Triton X-100), blocked for 30 min at room temperature and incubated with primary antibodies at 4°C overnight in PBTA. Following three washes in PBTA for 20 min, secondary antibody incubation was in PBTA for 1 h at room temperature. After incubation, embryos were washed for 20 min in PBTA three times, then mounted in Vectashield. Micrographs were acquired on a Zeiss LSM 780 upright confocal microscope.
Clone analysis and wing-disc staining
Embryos were collected for 24 h in vials, and aged 1 day at room temperature, then heat-shocked at 38°C for 1 h, and incubated at 28°C until the third larval instar (about 4 days after heat-shocking). Third-instar larvae were dissected in 1×PBS and fixed for 20 min in 4% paraformaldehyde (158127; Sigma) in 1×PBS. Samples were washed for 15 min three times with 0.1% Triton X-100 in PBS, then incubated in blocking solution (1% BSA, 0.1% Triton X-100 in PBS) at room temperature for 1 h, then with primary antibodies in 0.1% Triton X-100 in PBS overnight at 4°C. After they were washed three times for 15 min with 0.1% Triton X-100 in PBS, samples were incubated with secondary antibodies before DAPI (diluted 1:1000) staining. Samples were washed for 15 min, three times with 0.1% Triton X-100 in PBS. Imaginal discs were carefully removed from the cuticle and mounted in Vectashield. Wing discs were imaged using a Zeiss LSM780 upright confocal microscope.
Supplementary Material
Data S1. Sequence of the histone gene targeting vector. The histone gene targeting vector contains five copy numbers of His-GUs (each copy contains five canonical histone genes, His1, His2B, His2A, His4, and His3). Total size of the vector is 34,570 bp.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-MSL2 | LMU munich | N/A |
| Mouse anti-MOF | LMU munich | N/A |
| Mouse anti-HP1 | DSHB | C1A9 |
| Mouse anti-1B1 | DSHB | 1B1 |
| Rabbit anti-H4K16ac | Santa Cruz | SC8662-R |
| Mouse anti-H4K8ac | PTM Biolabs | PTM-164 |
| Rat anti-vasa | DSHB | Anti-vasa |
| Goat anti-Rat | Invitrogen | A11007 |
| Goat anti-mouse | Invitrogen | A11029 |
| Goat anti-rabbit | Invitrogen | A11036 |
| Mouse anti-CyclinB | DSHB | F2F4 |
| Mouse anti-H3K27me3 | PTM Biolabs | PTM-651 |
| Ubx | DSHB | FP3.38 |
| Abd-B | DSHB | 1A2E9 |
| Rabbit anti-GFP | Abcam | ab6556 |
| Mouse anti-H3K9me3 | Active motif | 39285 |
| Bacterial and Virus Strains | ||
| ccdB Survival™2 | Invitrogen | Cat#A10460 |
| DH5α | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: w1118 | Bloomington Drosophila Stock Center | 3605 |
| D. melanogaster: phiC31 | Bloomington Drosophila Stock Center | 40161 |
| D. melanogaster: Df(2L)HisC | Bloomington Drosophila Stock Center | 8670 |
| Mof RNAi fly | Tsinghua fly center | THU4573 |
| D. melanogaster: yw hs-flp122 | This study | N/A |
| D. melanogaster: Ubi-GFP FRT | This study | N/A |
| Mof mutant | This study | N/A |
| Oligonucleotides | ||
| Primers for generating HisD, see Table S1 | This study | N/A |
| Primers for verifying histone integration, see Table S1 | This study | N/A |
| Primers for RT-qPCR, see Table S1 | This study | N/A |
| Software and Algorithms | ||
| Snapgene | GSL Biotech | http://www.snapgene.com/ |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Illustrator | Adobe | https://www.adobe.com/products/illustrator.html |
| LightCycler 480 | Roche | https://lifescience.roche.com |
| Graphpad | GraphPad Software | https://www.graphpad.com/ |
ACKNOWLEDGMENTS
We thank T. Chi for critically reading the manuscript, and PTMbiolabs for kindly providing some antibodies.
Funding: This work was supported by the National Key Research and Development Program of China 2017YFA0505103, National Science Fund for Distinguished Young Scholars 31725002 and National Natural Science Foundation of China 31471254 to J.D., National Natural Science Foundation of China 31171278, 31271542, 31671333, 91740107 to G.G., and by R01HD040307 to E.M.
Footnotes
Competing interests: The authors declare no competing interests.
Data and materials availability: All data is available in the main text or the supplementary materials.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bischof J, Maeda RK, Hediger M, Karch F, and Basler K (2007). An optimized transgenesis system for Drosophila using germ-line-specific phi C31 integrases. P Natl Acad Sci USA 104, 3312–3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celic I, Masumoto H, Griffith WP, Meluh P, Cotter RJ, Boeke JD, and Verreault A (2006). The sirtuins hst3 and Hst4p preserve genome integrity by controlling histone h3 lysine 56 deacetylation. Curr Biol 16, 1280–1289. [DOI] [PubMed] [Google Scholar]
- Chaiyachati BH, Jani A, Wan YS, Huang HC, Flavell R, and Chi T (2013). BRG1-mediated immune tolerance: facilitation of Treg activation and partial independence of chromatin remodelling. Embo J 32, 395–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung WL, Turner FB, Krishnamoorthy T, Wolner B, Ahn SH, Foley M, Dorsey JA, Peterson CL, Berger SL, and Allis CD (2005). Phosphorylation of histone H4 serine 1 during DNA damage requires casein kinase II in S. cerevisiae. Curr Biol 15, 656–660. [DOI] [PubMed] [Google Scholar]
- Coleman RT, and Struhl G (2017). Causal role for inheritance of H3K27me3 in maintaining the OFF state of a Drosophila HOX gene. Science 356, 41–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad T, Cavalli FM, Holz H, Hallacli E, Kind J, Ilik I, Vaquerizas JM, Luscombe NM, and Akhtar A (2012a). The MOF chromobarrel domain controls genome-wide H4K16 acetylation and spreading of the MSL complex. Dev Cell 22, 610–624. [DOI] [PubMed] [Google Scholar]
- Conway E, Healy E, and Bracken AP (2015). PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol 37, 42–48. [DOI] [PubMed] [Google Scholar]
- Corona DF, Clapier CR, Becker PB, and Tamkun JW (2002). Modulation of ISWI function by site-specific histone acetylation. EMBO Rep 3, 242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Hyland EM, Yuan DS, Huang H, Bader JS, and Boeke JD (2008). Probing nucleosome function: a highly versatile library of synthetic histone H3 and H4 mutants. Cell 134, 1066–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang W, Steffen KK, Perry R, Dorsey JA, Johnson FB, Shilatifard A, Kaeberlein M, Kennedy BK, and Berger SL (2009). Histone H4 lysine 16 acetylation regulates cellular lifespan. Nature 459, 802–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das C, Lucia MS, Hansen KC, and Tyler JK (2009). CBP/p300-mediated acetylation of histone H3 on lysine 56. Nature 459, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Setta N, Van Sluys MA, Capy P, and Carareto CM (2011). Copia retrotransposon in the Zaprionus genus: another case of transposable element sharing with the Drosophila melanogaster subgroup. J Mol Evol 72, 326–338. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Tibbit C, and Liu JL (2016). Effective knockdown of Drosophila long non-coding RNAs by CRISPR interference. Nucleic acids research 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, and Seto E (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23. [DOI] [PubMed] [Google Scholar]
- Graves HK, Wang PP, Lagarde M, Chen ZH, and Tyler JK (2016). Mutations that prevent or mimic persistent post-translational modifications of the histone H3 globular domain cause lethality and growth defects in Drosophila. Epigenetics & chromatin 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunesdogan U, Jackle H, and Herzig A (2010). A genetic system to assess in vivo the functions of histones and histone modifications in higher eukaryotes. EMBO reports 11, 772–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunesdogan U, Jackle H, and Herzig A (2014). Histone supply regulates S phase timing and cell cycle progression. eLife 3, e02443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S, and Shilatifard A (2011). Histone modification: cause or cog? Trends in genetics : TIG 27, 389–396. [DOI] [PubMed] [Google Scholar]
- Huang H, Sabari BR, Garcia BA, Allis CD, and Zhao Y (2014a). SnapShot: histone modifications. Cell 159, 458–458 e451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao LY, and Liu X (2015). Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Science 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Umehara T, and Horikoshi M (2002). Chromosomal gradient of histone acetylation established by Sas2p and Sir2p functions as a shield against gene silencing. Nat Genet 32, 370–377. [DOI] [PubMed] [Google Scholar]
- Kouzarides T (2007). Chromatin modifications and their function. Cell 128, 693–705. [DOI] [PubMed] [Google Scholar]
- Larschan E, Bishop EP, Kharchenko PV, Core LJ, Lis JT, Park PJ, and Kuroda MI (2011). X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature 471, 115–U139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherman JL, and Dinardo S (2010). Germline self-renewal requires cyst stem cells and stat regulates niche adhesion in Drosophila testes. Nature cell biology 12, 806–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis EB (1978). “A gene complex controlling segmentation in Drosophila.” Nature 276(5688): 565–570. [DOI] [PubMed] [Google Scholar]
- Lin H, Yue L, and Spradling AC (1994). The Drosophila fusome, a germline-specific organelle, contains membrane skeletal proteins and functions in cyst formation. Development 120, 947–956. [DOI] [PubMed] [Google Scholar]
- Liu J-L, Murphy C, Buszczak M, Clatterbuck S, Goodman R, and Gall JG (2006). The drosophila melanogaster cajal body. J Cell Biol 172, 875–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luger K, Mader AW, Richmond RK, Sargent DF, and Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- Marzluff WF, Gongidi P, Woods KR, Jin J, and Maltais LJ (2002). The human and mouse replication-dependent histone genes. Genomics 80, 487–498. [PubMed] [Google Scholar]
- McConnell KH, Dixon M, and Calvi BR (2012). The histone acetyltransferases CBP and Chameau integrate developmental and DNA replication programs in Drosophila ovarian follicle cells. Development 139, 3880–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay DJ, Klusza S, Penke TJR, Meers MP, Curry KP, McDaniel SL, Malek PY, Cooper SW, Tatomer DC, Lieb JD, et al. (2015). Interrogating the Function of Metazoan Histones using Engineered Gene Clusters. Developmental cell 32, 373–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mu X, Yan S, Fu C, and Wei A (2015). The Histone Acetyltransferase MOF Promotes Induces Generation of Pluripotent Stem Cells. Cell Reprogram [DOI] [PubMed]
- Nakanishi S, Sanderson BW, Delventhal KM, Bradford WD, Staehling-Hampton K, and Shilatifard A (2008). A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat Struct Mol Biol 15, 881–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama J, Rice JC, Strahl BD, Allis CD, and Grewal SI (2001). Role of histone H3 lysine 9 methylation in epigenetic control of heterochromatin assembly. Science 292, 110–113. [DOI] [PubMed] [Google Scholar]
- Park SW, Kuroda MI, and Park Y (2008). Regulation of histone H4 Lys16 acetylation by predicted alternative secondary structures in roX noncoding RNAs. Molecular and cellular biology 28, 4952–4962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly AR, Copur O, Jackle H, Herzig A, and Muller J (2013). A Histone Mutant Reproduces the Phenotype Caused by Loss of Histone-Modifying Factor Polycomb. Science 339, 698–699. [DOI] [PubMed] [Google Scholar]
- Piacentini L, Fanti L, Negri R, Del Vescovo V, Fatica A, Altieri F, and Pimpinelli S (2009). Heterochromatin Protein 1 (HP1a) Positively Regulates Euchromatic Gene Expression through RNA Transcript Association and Interaction with hnRNPs in Drosophila. Plos Genet 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raja SJ, Charapitsa I, Conrad T, Vaquerizas JM, Gebhardt P, Holz H, Kadlec J, Fraterman S, Luscombe NM, and Akhtar A (2010). The nonspecific lethal complex is a transcriptional regulator in Drosophila. Mol Cell 38, 827–841. [DOI] [PubMed] [Google Scholar]
- Rong YS, and Golic KG (2000). Gene targeting by homologous recombination in Drosophila. science 288, 2013–2018. [DOI] [PubMed] [Google Scholar]
- Rothbart SB, and Strahl BD (2014). Interpreting the language of histone and DNA modifications. Biochim Biophys Acta 1839, 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruohola H, Bremer KA, Baker D, Swedlow JR, Jan LY, and Jan YN (1991). Role of neurogenic genes in establishment of follicle cell fate and oocyte polarity during oogenesis in Drosophila. Cell 66, 433–449. [DOI] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, and Peterson CL (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. [DOI] [PubMed] [Google Scholar]
- Smith ER, Pannuti A, Gu W, Steurnagel A, Cook RG, Allis CD, and Lucchesi JC (2000). The drosophila MSL complex acetylates histone H4 at lysine 16, a chromatin modification linked to dosage compensation. Mol Cell Biol 20, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, and Rajagopal N (2011). Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarz P, and Kouzarides T (2014). Histone core modifications regulating nucleosome structure and dynamics. Nat Rev Mol Cell Biol 15, 703–708. [DOI] [PubMed] [Google Scholar]
- Torres-Padilla ME, Parfitt DE, Kouzarides T, and Zernicka-Goetz M (2007). Histone arginine methylation regulates pluripotency in the early mouse embryo. Nature 445, 214–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtele H, Kaiser GS, Bacal J, St-Hilaire E, Lee EH, Tsao S, Dorn J, Maddox P, Lisby M, Pasero P, et al. (2012). Histone H3 lysine 56 acetylation and the response to DNA replication fork damage. Mol Cell Biol 32, 154–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie HB, and Golic KG (2004). Gene deletions by ends-in targeting in Drosophila melanogaster. Genetics 168, 1477–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu F, Zhang K, and Grunstein M (2005). Acetylation in histone H3 globular domain regulates gene expression in yeast. Cell 121, 375–385. [DOI] [PubMed] [Google Scholar]
- Xu T, and Rubin GM (1993). Analysis of genetic mosaics in developing and adult Drosophila tissues. Development 117, 1223–1237. [DOI] [PubMed] [Google Scholar]
- Xue Z, Ren M, Wu M, Dai J, Rong YS, and Gao G (2014). Efficient gene knock-out and knock-in with transgenic Cas9 in Drosophila. G3 (Bethesda) 4, 925–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Ai X, Eugeni EE, Zhang L, Carpenter LR, Jelinek MA, Freitas MA, and Parthun MR (2005). Histone H4 lysine 91 acetylation a core domain modification associated with chromatin assembly. Molecular cell 18, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z, Ren M, Wang Z, Zhang B, Rong YS, Jiao R, and Gao G (2013). Highly efficient genome modifications mediated by CRISPR/Cas9 in Drosophila. Genetics 195, 289–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung PYK, Stuetzer A, Fischle W, Martinez AM, and Cavalli G (2015). Histone H3 Serine 28 Is Essential for Efficient Polycomb-Mediated Gene Repression in Drosophila. Cell reports 11, 1437–1445. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Sequence of the histone gene targeting vector. The histone gene targeting vector contains five copy numbers of His-GUs (each copy contains five canonical histone genes, His1, His2B, His2A, His4, and His3). Total size of the vector is 34,570 bp.