

Abstract
Recent approaches in the treatment of cancer focus on involving the immune system to control the tumor growth. The administration of immunotherapies, like checkpoint inhibitors, has shown impressive results in the long term survival of patients. Cancer vaccines are being investigated as further tools to prime tumor-specific immunity. Biomaterials show potential as adjuvants in the formulation of vaccines, and biomimetic elements derived from the membrane of tumor cells may widen the range of antigens contained in the vaccine. Here, we show how mice presenting an aggressive melanoma tumor model treated twice with the complete nanovaccine formulation showed control on the tumor progression, while in a less aggressive model, the animals showed remission and control on the tumor progression, with a modification in the immunological profile of the tumor microenvironment. We also prove that co-administration of the nanovaccine together with a checkpoint inhibitor increases the efficacy of the treatment (87.5% of the animals responding, with 2 remissions) compared to the checkpoint inhibitor alone in the B16.OVA model. Our platform thereby shows potential applications as a cancer nanovaccine in combination with the standard clinical care treatment for melanoma cancers.
Keywords: biohybrid, cancer vaccine, cell membrane, microfluidics, melanoma
The recent reports about the increase in the overall survival of cancer patients treated with immunotherapeutics, in particular checkpoint inhibitors, adoptive cell transfer, and chimeric antigen receptor T cells, rekindled the interest in the development of prophylactic and therapeutic cancer vaccines.1−3 Biomaterials are currently being investigated for the formulation of micro- and nanoparticulate vaccines that enable the co-delivery of antigens and adjuvants.4,5 Alternatively, biomaterials have been exploited in drug delivery systems for immune checkpoint inhibitors, including microneedles, scaffolds, micro- and nanoparticles, and platelets.6−10 Furthermore, the materials themselves may present immunogenic properties leading to the stimulation of toll-like receptors and danger associated molecular pathways, and consequently to the activation of antigen presenting cells (APCs).11 Among the biomaterials assessed, porous silicon (PSi) micro- and nanoparticles promote the maturation of immature dendritic cells (DCs) to a mature phenotype, with a shift in the cytokine profile toward a Th-1 biased profile, enabling the priming of cytotoxic T lymphocytes.12−14 The semi-synthetic modification of the biocompatible polymer dextran with acetal groups yields a pH-sensitive polymer characterized by immunostimulatory properties.15,16 Thermally oxidized PSi (TOPSi) nanoparticles and acetalated dextran (AcDEX) formulated into a nanocarrier by nanoprecipitation in glass capillary microfluidics induced the activation of peripheral blood monocytes in vitro, showing a potential application as a biomaterial-based platform for cancer vaccines.12
One of the reasons behind the failure of many vaccines in clinical trials is the choice of the antigenic component: Tumor associated antigens (TAAs) or cancer-testis antigens are expressed also by other healthy tissues, thus the likelihood of depletion of highly reactive T cells by central tolerance to avoid autoimmunity is high.2,17,18 On the contrary, tumor-specific antigens, or neoantigens, are derived from point mutations and are expressed only in cancer cells.19,20 The possibility of eliciting a stronger immune response is thereby higher upon stimulation with neoantigens compared to TAAs.21 Recently, vesicles derived from the membranes of cancer cells were proposed as an alternative source of antigens and neoantigens for the formulation of cancer vaccines.22 The advantage of using this antigenic source resides in the multiplicity and complexity of the antigens delivered to the APCs.23 We have previously investigated the feasibility of this antigenic source in vitro, in a multistage delivery system composed of a core made of adjuvant biomaterials, which was further coated with a layer of cell membranes (cancer cell membranes, CCM) by extrusion through a polymeric membrane.12
The immunosuppressive environment of the tumor microenvironment may reduce the potency of the immune response evoked by a therapeutic vaccination.24 To tackle this problem, we hyphothesized that a biohybrid vaccination platform (Scheme 1) should synergize with checkpoint inhibitors, such as CTLA-4 blocking antibodies. For this reason, we focused on the improvement of the immune infiltration and antigen-experience of T cells through an enhanced activation of APCs in the tumor surroundings. Furthermore, we hypothesize that a combinatorial therapy between our nanovaccine and an anti-CTLA4 antibody can enhance the efficacy of the vaccination due to the mechanism of action of the checkpoint inhibitor on the central regulation of the T cell activation. In this study, we assess the nanovaccine and the combination of anti-CTLA4 antibody administered concurrently with the vaccination on the efficacy of the treatment and on the priming of the immune response in vivo. We first evaluated the efficacy of the vaccination alone on the growth of an aggressive melanoma murine model, investigating also the changes in the immune profile at the tumor site. Subsequently, we assessed the synergistic potential of the combinatorial therapy with the immune checkpoint inhibitor on the growth of established tumors and on the infiltration of activated immune cells in the tumor microenvironment.
Scheme 1. Illustration of the Multistage Nanovaccine Platform.
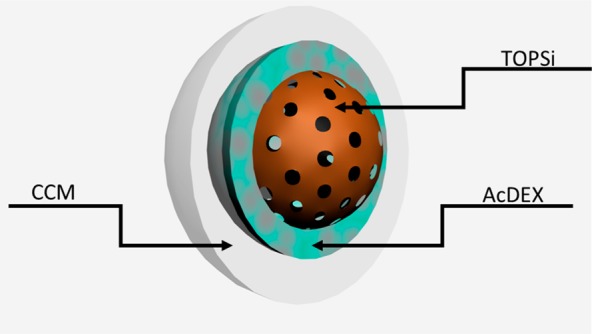
A PSi core (porous brown core) was encapsulated within a layer of AcDEX (light blue) using glass capillary microfluidics. Then, the nanoparticles were enveloped by a layer of cancer cell membrane (CCM; autologous with the tumor model evaluated, B16.F10 or B16.OVA; white layer).
Results
Tumor Membrane-Coated Nanoparticles: Cytocompatibility and Immunostimulatory Properties
We first evaluated the cytocompatibility of the system, tumor-membrane-coated (TOPSi@AcDEX@B16.OVA) nanoparticles (NanoCCM), onto an immortalized murine immune cell line, JAWS II, as such or in the presence of two different concentrations (1 and 10 μg/mL) of the murine anti-CTLA4 antibody. The formulation was assessed in a range of concentrations from 25 to 500 μg/mL for 24 h. As shown in Figure 1A, after 24 h, NanoCCM particles are highly cytocompatible over the whole range of concentrations assessed, inducing a slight cell proliferative effect, as reported also in previous works.5,12 The incubation of the nanovaccine together with the checkpoint inhibitor at the concentrations of 1 and 10 μg/mL did not alter the safety profile of the formulation. Thereby, we selected the concentration of 1 μg/mL of anti CTLA-4 antibody for further in vitro studies. After 48 h, the particles induce a dose-dependent decrease in the cellular viability for the highest concentrations assessed (250 and 500 μg/mL).
Figure 1.

Tumor-membrane coated TOPSi@AcDEX nanovaccines are cytocompatible and induce the maturation of murine APCs in vitro. (A) Cell viability (%) of JAWS II cells incubated with the tumor-membrane-coated TOPSi@AcDEX nanovaccine (NanoCCM) for 24 h (left) and 48 h (right), as such or in the presence of two different concentrations of a murine anti-CTLA4 antibody. Cells incubated in 10% medium and in Triton X-100 1% represent the negative and positive controls. (B) Percentage of CD80, CD86, and double positive JAWS II cells after 48 h (top) and 72 h (bottom). JAWS II cells were incubated with APC antimouse CD80+ antibody or PerCP-Cy 5.5 antimouse CD86+ antibodies, and the expression of the receptors was evaluated by flow cytometry. The cells were incubated with NanoCCM at the concentration of 100 μg/mL for 48 and 72 h. Cells incubated with medium and cells incubated with LPS represent the negative and positive controls, respectively. The results are expressed as mean ± SD (n > 3) and were analyzed with two-way ANOVA followed by Bonferroni post-test. *p < 0.05, **p < 0.01, and ***p < 0.001.
In the following assay, we assessed the immunostimulatory properties of the nanovaccine by incubating the nanoformulation, at the concentration of 100 μg/mL, with JAWS II and, subsequently, analyzing the activation profile of the cells from the expression of co-stimulatory signals (CD80 and CD86). As shown in Figure 1B, the nanovaccine, after 48 h of incubation, stimulated the expression of CD86 to levels comparable to those of lipopolysaccharide (LPS), proving the ability of the formulation to induce the maturation of APCs. The inclusion of the checkpoint inhibitor does not change the level of CD86 presented by the cells. The peak of the immunostimulatory effect of the formulation is reached at 48 h, and at 72 h, there is a decrease in the expression of the receptor, when compared to LPS, while still being significantly higher than the control in medium. As for the expression of CD80, at 48 h, there is no difference among all the samples, while for 72 h, the nanosystems present levels of expression even lower than the negative control. However, at 48 h, the incubation of the cells with NanoCCM resulted in a significant increase in the number of double positive cells, when compared to LPS. When the checkpoint inhibitor was added, the percentage of double positive cells decreased. After 72 h, the immunostimulating effect of the NanoCCM formulation fades, when compared to LPS. Interestingly, in the presence of the ICI, the cells displayed a higher percentage of double positive cells compared to the nanosystem alone. Moreover, we evaluated the mechanism of activation of APCs by determining the effect of the particles on the secretion of TNF-α by human peripheral blood monocytes; the effect of the cytokine was studied by co-culturing the medium of peripheral blood monocytes with Ramos Blue. As presented in Figure S1, only TOPSi NPs induce the secretion of TNF-α, while when the particles were encapsulated within the polymeric layer and then enveloped within the CCM, there was no secretion of TNF-α. These results are in agreement with what was previously reported elsewhere.12,13
Moreover, we evaluated the ability of the nanovaccine to mediate the cross-presentation of antigens to MHC-I. As presented in Figure S2, the incubation of JAWS-II cells with NanoCCM (wrapped with membrane derived from B16.OVA cells and spiked with SIINFEKL-cell penetrating peptide, CPP) induced the presentation of SIINFEKL on MHC-I. The cell membrane vesicles alone induced the cross-presentation, while the JAWS-II cells incubated with the polymer alone did not present any SIINFEKL peptide. Next, we investigated the possibility that the presence of the cell membrane wrapped around the particles could induce a partial cross-dressing with the APCs. For this, we prepared CCM and NanoCCM samples wrapped with the membrane of a BALB/c cell line (4T1). JAWS-II cells (C57BL/6 lineage) were pulsed with these formulations, and we assessed the percentage of the MHC-I H-2Kd molecule presented on the cells. As shown in Figure S3, the incubation with both CCM and NanoCCM results in a partial cross-dressing of the membranes. Finally, to clarify the vaccination mechanism of the biohybrid nanovaccine, splenocytes derived from OT-I mice were incubated with JAWS-II cells pulsed with the formulation (B16.OVA membrane spiked with SIINFEKL-CPP). The supernatant was collected and analyzed for the content in IFN-γ. As presented in Figure S4, APCs pulsed with the formulations presenting OVA and SIINFEKL (namely CCM and NanoCCM) activated OT-I cells with the secretion of IFN-γ. The nanosystem induced a statistically higher activation compared to CCM alone, despite the higher level of cross-presentation achieved with CCM.
Overall, we proved the cytocompatibility of the system and its ability to induce the maturation of APCs, with an increase in the expression of the co-stimulatory signals CD80 and CD86 to levels comparable to those of the positive control (cells activated by LPS). Moreover, the nanovaccine promotes the cross-presentation of antigens in APCs and the subsequent activation of T cells. Our system, thereby, has properties as adjuvant in a complete nanovaccine formulation.
Biohybrid Nanoparticles Increase the Response Against B16F10 Melanoma by Modulating the Infiltration of Dendritic Cells and Cytotoxic T-Lymphocytes
In these experiments, we tested our biohybrid platform by engrafting C57 mice with the highly immune suppressive B16F10 tumors. Hence, we treated the mice with TOPSi@AcDEX nanoparticles extruded with a homologous membrane (B16F10 cells) to promote an anti-tumor immune response. The percentage of mice responding to the therapy, defined as mice showing a tumor volume lower than 300 mm3, was 12.5, 28.6, and 33.3% in the mock, CCM, and AcDEX groups respectively (Figure 2A). Although modest, we registered an increase in the number of responding mice in the group treated with NanoCCM (44.4%). In a comparison between the tumor volume in different groups (Figure S2), no significant difference was found between the three treatment groups. We hypothesize that the intrinsic aggressiveness of the B16F10 tumor model prevents the possibility to fully evaluate the priming of a tumor-specific immune response. However, the efficacy of the treatment was limited to less than half of the cohort of animals, providing indications for future improvements of the formulation. Next, we investigated by flow cytometry whether or not the treatment with NanoCCM could affect the immune infiltration of cells into the tumor. Mice receiving NanoCCM showed a trend toward an increased infiltration of DCs (defined as CD11c+ cells; Figure 2B) with a large portion of those being activated and mature (CD80 and CD86 double positive cells; Figure 2C). In addition, we analyzed the infiltration by T-cells to understand if the effect of NanoCCM treatment on DCs would result also in an increased presence of tumor-infiltrating lymphocytes (TILs). Consistent with our hypothesis, we found that tumors of mice receiving the biohybrid nanoparticles were highly infiltrated by cytotoxic T-cells (defined as CD3+CD8+ cells; Figure 2D). Remarkably, only the treatment with NanoCCM was able to increase the presence of antigen-experienced TILs when compared to a mock (p < 0.05), as proven by the higher presence of PD-1+ T-cells (Figure 2E). However, the analysis of the PD-1+ T-cells provides only information on the level of the antigen experience of the T-cells, but not on the specific antigen the cells have been primed for.25−27
Figure 2.

Biohybrid nanovaccines increase the response to aggressive B16F10 established tumors and induce immunological changes into the tumor microenvironment. Female C57BL6/J mice (n = 7/8) were engrafted with 1.0 × 105 B16F10 melanoma cells on the right flank. After 6 days, the mice were randomized into 4 groups and treated subcutaneously with 5.4% isotonic glucose solution (mock group), bare TOPSi@AcDEX nanoparticles (AcDEX group), processed tumor-membrane vesicles (CCM group), or tumor-membrane-coated TOPSi@AcDEX nanovaccines (NanoCCM group). A second treatment injection was performed in the peri-tumoral region at day 13. (A) Single curves of each tumor for each group. In this tumor model, responders (green curves) are defined as mice that show an absolute tumor volume lower than 300 mm3. (B) Tumors were collected and stained for the presence of DCs (CD11c+). (C) The activation state of the DCs was characterized by using CD80 and CD86 surface markers. The presence of TILs was also assessed. (D) Flow cytometry was used to detect intratumoral cytotoxic T-cells (CD3+CD8+). (E) The activation state of the TILs was investigated by analyzing the presence of the PD-1 marker. All graphs represent mean ± SEM. Statistical analysis was performed with unpaired Student’s t-test or one-way ANOVA; the levels of significance were set at *p < 0.05 and **p < 0.01. The flow cytometry data for every mouse are normalized against the tumor volume of that mouse.
In line with our aim to increase the response to immunotherapy, we explored the correlation between immune infiltration of tumors and tumor size among all groups. Our analysis revealed that mice with smaller tumors showed significantly higher levels of TILs within the tumors (Figure 3A); in addition, PD-1+ TILs were enriched into the tumors of the mice with a low tumor burden (Figure 3B). The presence of DCs and their activation state also clearly distinguished nonresponders from responders (Figure 3C,D). Interestingly, the data from two responders scored higher than the responders in Figure 3A–C, leading us to hypothesize a trend with different levels of activation of the immune system within the responders court. However, the study was not powered enough to show statistical significance. Finally, we studied the correlation between tumor volume and immune infiltration, and we found that an exponential model would better describe the relationship between those two parameters. In fact, for all of the tested correlations, we achieved statistical significance (p < 0.05) and a relatively good fitting to an exponential model, suggesting that the correlation is unlikely to be linear, but it rather follows an exponential model since anti-tumor responses are likely the result of several co-operating factors (Figure 3E).
Figure 3.

Correlation between response to immune therapy and changes in the tumor microenvironment of melanoma tumors. The flow cytometry data presented in Figure 2 were used to perform an explorative analysis to correlate infiltration of immune cells to tumor size. Responding (red data points) and nonresponding (black data points) mice were grouped from all treatment groups and analyzed for the intratumoral presence of (A) cytotoxic T-cells, (B) antigen-experienced cytotoxic T-cells, (C) DCs, and (D) activated and mature DCs. The statistical analysis was performed with unpaired Student’s t-test, and the levels of significance were set at *p < 0.05 and **p < 0.01. The flow cytometry data for every mouse are normalized against the tumor volume of that mouse. (E) To evaluate the correlation between immunological features and tumor response, data from all mice were pooled, and the correlation was tested with Pearson’s correlation test (the p-value is indicated in each graph). The one phase exponential nonlinear models were used to fit the data and retrieve the R2 for each data set.
Biohybrid Nanoparticles Prime Tumor-Specific Immune Responses with Rejection of Established Melanomas
In this set of experiments, we investigated the in vivo efficacy of our cancer nanovaccine platform in a less aggressive setting. To this end, we engrafted C57BL6 immunocompetent mice with the B16.OVA tumor cells. Mice received treatments at 6 and 13 days after tumor engraftment. As shown in Figure 4A, treatment with only immunogenic TOPSi@AcDEX (i.e., AcDEX) nanoparticles or tumor membrane (i.e., CCM) resulted in modest responses in 50% and 66% of the treated mice, respectively. However, mice treated with the TOPSi@AcDEX particles coated with the tumor membrane (hereafter named NanoCCM) responded in 71% of the cases. Interestingly, this was the only group where we registered complete responses in 28.5% (2 out of 7) of the mice (Figure 4B). The average volume curves for all of the groups are presented in Figure S3, highlighting the synergistic effect of the core and CCM-layer over the single components. These results are consistent with our previous experiments and highlight the potential of our nanoplatform as a therapeutic cancer vaccine, since only two therapeutic injections of NanoCCM were necessary to achieve significantly better results compared to the mice in the control groups.
Figure 4.

Biohybrid nanoparticles increase the anti-tumor response and rejection rate against established B16.OVA tumors. Female C57BL6/J mice were engrafted with 2.5 × 105 B16.OVA cells on the right flank. After 6 days, mice were randomized into 4 groups and treated subcutaneously with 5.4% isotonic glucose solution (Mock group), bare TOPSi@AcDEX nanoparticles (AcDEX group), processed tumor-membrane (CCM group), or tumor-membrane-coated TOPSi@AcDEX nanoparticles (NanoCCM group). A second treatment injection was performed in the peri-tumoral region at day 13. (A) The single curves of each tumor for each group. Responders (green curves) are defined as mice that show an absolute tumor volume lower than 400 mm3. (B) The percentage of complete responses (i.e., cured mice) observed in each group.
Next, we investigated if NanoCCM could modulate the activation of DCs in this melanoma model. Consistent with the in vitro studies on APCs and with the results from the study in the more aggressive tumor model, the tumor tissues of mice receiving any of the immunotherapy treatment featured an increased number of tumor infiltrating DCs (tDCs defined as CD11c+ CD11b+; Figure 5C). However, we found substantial differences in the quality and activation state of DCs (Figure 5D). The AcDEX nanovaccine formulation proved to be the best in inducing the expression of the activation markers on the surface of DCs.
Figure 5.

In vivo immunological effects of TOPSi@AcDEX-based cancer vaccines. Individual tumor samples were collected at the end point for each mouse, and the tumor-infiltrating cells were analyzed. (A) The ratio of CD8+ to CD4+ TILs is reported for every group. (B) The percentage of OVA-specific CD8+ TILs was measured by pentamer staining of tumor samples. The percentages were normalized by the tumor volume of each mouse. Then, each data set was normalized against a mock to measure the fold-increase over the mock group. By definition, the mock group is 1. (C) DCs were defined as CD11c+ and CD11b+ double positive cells within the tumor microenvironment. Data were normalized to the tumor volume of every mouse to take into account the different sizes of tumor masses at the moment of tissue collection. (D) Total percentages of activated and mature DCs, defined as either CD86+ (gray) or CD80+ (black) single positive or CD86+ CD80+ double positive cells (red). (E) Relative distribution and polarization of the DCs subsets for each group. All graphs represent the mean ± SEM. Statistical analysis done with unpaired Student’s t test; *p < 0.05 and **p < 0.01.
Flow cytometry analysis revealed an increase in the percentage of the total activated DCs (either CD80+, CD86+, or double positive CD80+CD86+ cells), thereby proving AcDEX to be an excellent adjuvant. Nevertheless, when we studied the distribution of the activated population into the whole DCs population, the tumors of mice treated with NanoCCM presented the highest percentage of double positive CD80+CD86+DCs (39.36%), which represent the most efficient maturation status of an APC. We conclude that while all of the treatments induced a degree of maturation of DCs, only NanoCCM treatment was able to reshape the distribution between different maturation statuses, resulting in a lower percentage of early activated DCs (6.38% of CD86+ cells) and a higher percentage of fully activated DCs (39.36% of CD80+CD86+ cells) compared to the other groups (Figure 5E). It is noteworthy to stress that there was only a limited amount of tissue recovered in the animals cured after treatment with NanoCCM.
Biohybrid Nanoparticles Increase the Efficacy of Immune Checkpoint Blockade against Established Melanomas
In line with the efforts to convert “cold tumors” into “hot tumors”, we hypothesized that the biohybrid nanovaccine platform developed here would increase the response rate to checkpoint inhibition, especially increasing the number of the overall responders. To this end, we investigated how the co-administration of anti-CTLA4 antibody, known to potentiate the priming of T-cells, would increase the efficacy of our cancer nanovaccine platform.
Established murine melanoma tumors (B16.OVA) were treated at multiple time points (Figure 6A, black arrows) to prime and boost an antigen-specific response. The NanoCCM treatment was combined with intraperitoneal administration of aCTLA4 antibody (Nano-CCM + aCTLA4 group) and compared with aCTLA4 monotherapy. The aCTLA antibody was selected for the evaluation of the combinatorial therapy due to its mechanism of action that stimulated the priming of T cells at a systemic level, different for aPD-L1 antibodies.28 We hypothesize that our nanovaccine is priming an immune response against neoantigens, thereby a combo therapy with an aCTLA4 antibody is most likely to result in a synergistic effect. As expected, blocking the aCTLA4 inhibitory pathway slowed the growth of tumors compared to treatment with a 5.4% glucose isotonic solution (mock group). Mice receiving the combination treatment registered a significant reduction in tumor volumes compared to both mock and aCTLA monotherapies, as shown by the growth curves (Figure 6A) and their area under the curve (Figure 6B). One mouse using the aCTLA4 monotherapy experienced a complete response, however the combo therapy NanoCCM + aCTLA4 was able to induce complete responses in 25% of treated mice (Figure 6C). Consistent with the clinical data, while checkpoint inhibition slowed the growth of tumors, the overall benefit was inconsistent with few significant responses (Figure 6D, central panel). On the contrary, we recorded an increase in the number of responders among mice receiving the combination of aCTLA4 and NanoCCM cancer vaccines (87.5% of mice responded to the therapy).
Figure 6.

The efficacy of the anti-tumor effect of the vaccination with biohybrid nanoparticles is increased by co-administration of CTLA-4 checkpoint inhibitor. Female C57BL6/J mice were engrafted with 2.5 × 105 B16.OVA cells on the right flank. After 6 days, mice were randomized into 3 groups and treated subcutaneously with 5.4% isotonic glucose solution (mock group), intraperitoneally with 100 μg of anti-CTLA-4 monoclonal antibody, or with tumor-membrane coated TOPSi@AcDEX nanoparticles (NanoCCM) + anti-CTLA4 antibody. Two more rounds of treatment were performed at days 13 and 15 post-tumor engraftment. (A) Tumor volumes at each time point were normalized against the initial tumor volume. Then tumor growth curves were built by plotting the mean ± SEM of tumor volumes for each group. (B) The area under the curve (AUC) of the growth curve of each mouse is plotted in the box and whiskers graphs (Tukey’s representation). (C) The number of complete responses (i.e., cured mice) for each group is reported. (D) Single growth curves are reported for each group. Responders (green curves) are defined as mice which show an absolute tumor volume lower than 400 mm3. The percentage of responders in the group is reported next to each graph. Statistical analysis of tumor growth was done by two-way ANOVA with Tukey’s multiple comparison correction. The AUCs were analyzed by unpaired Student’s t test; *p < 0.05; **p < 0.01; ****p < 0.0001.
Immunological analysis showed that aCTLA4 monotherapy was not able to induce infiltration and expansion of CD8+ T cells into tumors of mice. However, the combination of cancer nanovaccine and the checkpoint inhibition induced a considerable increase in the number of tumor infiltrating cells (Figure 7A), supported by an increase in myeloid APCs (Figure 7B,C) into the tumors. When compared to a mock, both immunotherapies were able to increase the activation of DCs (Figure 7D). As shown in Figure 7E, the combination of aCTLA4 and NanoCCM reduced the fraction of early activated DCs (35.80%) compared to a mock (62.98%) and aCTLA monotherapy (47.68%). The combination group also increased the percentage of CD80+CD86+ double positive DCs (red portion of cake graphs in Figure 7E).
Figure 7.

Biohybrid nanoparticles favor the efficacy of the anti-CTLA-4 therapy by reshaping the tumor microenvironment. Individual tumor samples were collected at the end point for each mouse, and the tumor-infiltrating cells were analyzed. (A) The percentage of CD8+ TILs was normalized against the tumor volumes of each mouse to account for the size of tumor masses. (B) Infiltration of myeloid cells (defined as CD11b+ cells) into the tumor microenvironment. (C) DCs were defined as CD11c+ and CD11b+ double positive cells within the tumor microenvironment. Data were normalized against the tumor. (D) Total percentages of activated and mature DCs, defined as either CD86+ (gray), CD80+ (black) single positive, or CD86+ CD80+ double positive cells (red). (E) Relative distribution and polarization of the DCs subsets for each group. All graphs represent the mean ± SEM. Statistical analysis done with unpaired Student’s t test; *p < 0.05 and **p < 0.01.
Discussion
Tumor growth results in a continuous apoptosis/necrosis of tumor cells followed by their replacement during the invasion process.29 For this reason, the immune system is exposed to tumor antigens constantly.18,30 However, this process lacks a proper co-stimulation and pro-inflammatory signals, as the tumor microenvironment is heavily immunosuppressive.31,32 Therefore, antigen presentation is usually ineffective, and it rarely results in spontaneous and beneficial anti-tumor immunity.33 Therefore, it is crucial to provide the immune system with adjuvants capable of optimizing the maturation of APCs, which are at the center of every cancer vaccine approach.34 For this reason, cancer nanovaccines represent an attractive choice to stimulate anti-tumor responses in a specific and efficient manner.35 In this work, we describe the anti-tumor efficacy and immunological effects of our cancer nanovaccine platform based on biohybrid (TOPSi@AcDEX) nanoparticles coated with membranes derived from tumors cells. The nanovaccine promotes the activation of APCs (JAWS II cells) due to the biomaterials employed in the formulation of the system. TOPSi nanoparticles promote the maturation of immature monocyte-derived DCs due to their fast biodegradation in physiological fluids, with the release of silicic acid.13 AcDEX increases the expression of major histocompatibility complexes due to its rapid degradation in acidic conditions.15 The combination of the two biomaterials into one nanovaccine formulation primed a Th-1 biased immune response in vitro over human peripheral blood monocytes.12 The biohybrid nanovaccine, as result of the adjuvant properties of the biomaterials, can promote cross-presentation of the antigens presented on the cell membrane.36 The antigens and maturation cues provided to the APCs result in a complete immune response, with the activation of CD8 T cells and the secretion of IFN-γ.
Moreover, an alternative mechanism of antigen presentation to memory T cells takes place through the cross-dressing of membranes.37,38 The pulsing of APCs with the nanovaccine results in the cross-dressing of the membrane used to wrap the particles with the membrane of the APCs.
One of the main advantages of our nanoplatform is the possibility to induce immune responses against multiple tumor antigens, without the need to select specific peptides. Hence, tumor-membrane-coated nanoparticles share some similarities with whole tumor vaccines. Whole tumor vaccines have been studied in preclinical and clinical settings,39,40 and the main advantage of this approach is the ability to induce responses against a wide variety of antigens, thus overcoming the difficult selection of specific peptides.41,42 These classes of nanovaccines can be combined with cytokines to support the priming of cytotoxic T-lymphocytes (CTLs).43 For instance, interleukin (IL) 2 has been used in co-administration with tumor cells to sustain the proliferation of CTLs, resulting in responses in 72% of treated patients.44
We were able to induce a higher number of responses in a poorly immunogenic tumor model, observing a change in the infiltration of both DCs and T-cells (Figure 2). The modulation of the tumor microenvironment is a key aspect for the success of immunotherapy, and immune profiling is gaining momentum since it is often able to define patients who would respond to the therapy. While some studies point to the mutational load of tumors, which would help immune cells to recognize malignant ones,45 other studies point toward what has been defined as “immune-contexture”.46 The infiltration of tumor tissues by tumor cells and the presence of adaptive immunity (pre-existing or newly induced) are important prognostic factors when considering combination therapies.47 Our biohybrid nanoparticle (Figure 2) was able to increase the presence of TILs with an activated state (PD-1+), and this can potentially improve the response to the CTLA-4 blockade, as consistently described in a recent study elsewhere.48
We demonstrated that the combination of tumor membrane with proper immunologically active nanomaterial leads to better anti-tumor responses in B16.OVA melanoma models, with higher rejection rates among treated animals (Figure 4). We found an increased activation of APCs and, in particular, DCs after the treatment with NanoCCM (Figure 5D). The treatment with vesicles isolated from the tumor cells increases the number of the APCs in the tumor tissue, but is not sufficient to induce their activation without adding any adjuvant,22 while the nanoparticles alone induce the maturation of APCs that, however, are not tumor specific.15
In the last five years, the immunotherapy field has gained new attraction due to the success of immune checkpoint inhibitors (ICIs). This class of antibodies blocks inhibitory pathways that tumor cells exploit to inactivate cytotoxic CTLs.49−51 However, while very effective, the number of patients that benefit from the expensive treatment with ICIs remains limited. In fact, resistance mechanisms reduce the response rate,52 and the absence of antigen-specific T cells into the tumor microenvironment represents the biggest limitation. In addition, tumors with poor T cell infiltration, often defined as “cold tumors”, are resistant to checkpoint therapy, as ICIs do not create immune responses, but they simply protect primed T cells from inactivation.53−55 Recently, biomaterials have been developed for the local delivery of ICIs, in an effort to reduce side effects and to improve the efficacy.6−11 Therefore, in line with the efforts to convert “cold tumors” into “hot tumors”, we hypothesized that the biohybrid nanovaccine platform developed here would increase the response rate to checkpoint inhibition. We observed that blocking the inhibitory pathway with an anti-CTLA4 poorly improves the response to aggressive established tumors, as shown in Figure 6. However, an increase in the number of responders was achieved when the anti-CTLA-4 antibody was combined with the biohybrid nanovaccine platform. The nanovaccine platform provided a strong maturation signal to DCs that was converted into an increased presence of CD8+ T-lymphocytes into the tumor microenvironment, synergizing with anti-CTLA-4 treatment (Figure 7).
We would like to emphasize that the results herein presented are limited to murine melanoma models, implying that the efficacy of the treatment may be different in less immunogenic tumor models. Moreover, the results of the immunological profiling can be biased by the reduced tumor volume or by the rejection (no tumor retrievable) in the group treated with the complete formulation. We would also like to stress the absence of a direct causative relationship between the phenotypical changes recorded in the tumor microenvironment and the efficacy of the treatments. The analysis of the presentation of co-stimulatory signals by DCs does not represent per se a justification to affirm the presence of an immune response induced by the nanovaccine. Further studies are needed to evaluate the in vivo mode of action of the nanovaccine, analyzing in particular the molecular and functional changes induced in APCs.
Conclusion
In summary, the biohybrid nanovaccine is able to elicit an anti-tumor immune response in aggressive melanoma models and to modify also the immunological profile within the tumor microenvironment. The administration of the vaccine enhances the activation of APCs, leading to an increased priming of CD8+ T cells. Moreover, the combination therapy with the administration of the nanovaccine platform and a checkpoint inhibitor improves the anti-tumor efficacy of the checkpoint inhibitor alone. The versatility of the biohybrid (tumor-membrane-coated) nanoparticle platform represents an advantage in the personalized medicine field, where tumor membranes can be obtained from patient’s biopsies in order to eliminate histocompatibility problems and target patient-specific neo-antigens present in the tumor cells. The production of the nanovaccine is faced with the scale up from a laboratory to clinical batches. However, the scaling up is facilitated by the production of the particles by microfluidics, in single devices that may produce up to 700 g/day.56 In addition, this class of cancer nanovaccines can be easily combined with checkpoint inhibitors in order to protect primed T cells from inactivation, increasing the rate of responders to standard care immunotherapies available on the market.
Materials and Methods
Cell Lines and Reagents
The murine melanoma cell line B16.OVA, a mouse melanoma cell line expressing chicken ovalbumin (OVA), was kindly provided by Prof. Richard Vile (Mayo Clinic, Rochester, MN, USA). This cell line was cultured in Roswell Park Memorial Institute (RPMI) 1640 media (Gibco, Thermo Fisher) supplemented with 10% of fetal bovine serum (FBS, Gibco, Thermo Fisher), 1% of l-glutamine (Glutamax, Gibco, Thermo Fisher), and 1% of penicillin/streptomycin (Gibco, Thermo Fisher). To ensure the expression of OVA protein and the selection of OVA-positive clones, cells were cultured with geneticin antibiotic at a 10% concentration (G418, Gibco, Thermo Fisher). B16F10 cells were cultured as B16.OVA, without the addiction of geneticin to the flasks. B16.OVA and B16F10 cells were used both as source of the membranes to coat the system and as tumor models implanted in the animals. JAWS II (ATCC CRL-11904) was used to evaluate the cytocompatibility and the immunostimulative properties of the system in the presence of the check-point inhibitor. JAWS II cells were cultured in 20% α-modified Eagle’s medium (aMEM, HyClone, USA) supplemented with 5 ng/mL of murine GM-CSF. OT-1 splenocytes were isolated from 4–8 week old C57BL/6-Tg(TcraTcrb)1100Mjb/J (OT-1) mice (The Jackon Laboratories) and frozen at −80 °C until use. The splenocytes were then thawed in lymphocyte media (RPMI supplemented with 10% of FBS, 20 mmol/L l-glutamine, 1% of penicillin/streptomycin, 15 mmol/L HEPES, 50 μmol/L β-mercaptoethanol, 1 mmol/L Na pyruvate, 160 ng/mL murine IL-2, and 0.3 μg/mL anti-CD3 antibody clone 145–2C11).57In vivo Mab anti-CTLA-4 antibody was purchased form Bio X Cell (West Lebanon, NH, USA).
Animal Experiments
C57BL/6J mice were obtained from Scanbur (Denmark) at 4–6 weeks of age. Mice were kept in air-isolated cages with unlimited access to food. All procedures were carried out under sterile conditions. Mice were anesthetized using isofluorane vaporizers. Subcutaneous tumor models were developed by injecting either 1 × 105 or 2.5 × 105 B16.OVA tumor cells (when 80% confluent in T175 flasks) in the right flank of each mouse in 100 μL of nonsupplemented RPMI-1640 medium. Details about the treatment schedule are given in the figure legends. During the experiments, the tumor volume was recorded every 2 days by using a digital caliper. Maximum (L) and minimum (l) tumor diameters were recorded, and tumor volumes were calculated according to the formula: (L × l2)/2.
All animal experiments were reviewed and approved by the Experimental Animal Committee of the University of Helsinki and the Provincial Government of Southern Finland.
Top-Down Production of Thermally Oxidized Porous Silicon (TOPSi) Particles
The detailed protocol for the preparation of TOPSi nanoparticles can be found elsewhere.58,59 The anodization of silicon wafers into a solution of hydrofluoric acid:ethanol (1:1) introduces pores within the silicon wafer. The porous layer is then detached, oxidized for 2 h at 300 °C, and milled in ethanol with a high-energy ball mill to yield nanoparticles. The particles are then segregated into the different size ranges by centrifugation and kept at +4 °C in ethanol.
Synthesis of Acetalated Dextran (AcDEX)
Acetalated dextran was synthesized according to the reaction reported elsewhere.12,60,61 Briefly, we added dextran (1 g, MW 9000–11,000 kDa; Sigma-Aldrich, USA) to a two-neck flask, previously dried, and we purged the flask with dry N2. Dextran powder was dissolved in anhydrous dimethyl sulfoxide (10 mL, Sigma-Aldrich, USA) before adding pyridinium-p-toluenesulfonate (15.6 mg; Sigma-Aldrich, USA) and 2-methoxypropene (3.4 mL; Sigma-Aldrich, USA). We quenched the reaction with trimethylamine (1 mL; Sigma-Aldrich, USA) after 1 h, and we employed H2O (200 mL) to precipitate the modified dextran. The pellet was centrifuged (10 min, 20,000g) and washed twice with trimethylamine solution (100 mL; 0.01% v/v; pH 8). Finally, to remove the residual trimethylamine solution, the powder was vacuum-dried at 40 °C for 48 h, producing a fine white powder of acetalated dextran (1.10 g).15
Isolation of Cancer Cell Membranes (CCM) from B16F10 and B16.OVA Cells
We isolated the cell membranes following the protocol reported elsewhere.12,22 We cultured the cells as described above. Upon reaching 80% of confluence, we removed the medium, washed the cells with 1 × phosphate buffer solution-ethylenediaminetetraacetic acid (PBS-EDTA; pH 7.4) solution, and detached them using a 0.25% trypsin-PBS EDTA solution (HyClone, USA). The cells were centrifuged at 409g for 5 min, and the cells were washed 3 times with 1 × PBS (pH 7.4). Then, we resuspended the pellet of cells into lysing buffer (20 mM of TRIS HCl; Sigma-Aldrich, USA; 10 mM of KCl; Sigma-Aldrich, USA; 2 mM of MgCl2; Sigma-Aldrich, USA; 1 protease inhibitor mini tablet, EDTA free; Pierce, Thermo Fisher, USA) and pipetted them thoroughly. The cells were then centrifuged at 3200g for 5 min, the supernatant collected, and the pellet resuspended again in lysing buffer and pipetted. The cells were centrifuged a second time at 3200g for 6 min. We pooled the supernatant and centrifuged it at 20,000g with a TLA 120.2 rotor in a ultracentrifuge (Optima MAX, Beckmann Coulter, USA) for 20 min. We then collected the supernatant and centrifuged it at 45,000g for 5 min. The supernatant was then discarded, and we resuspended the membranes in Milli Q water.
Production of the Nanovaccine Components by Microfluidics Nanoprecipitation and Encapsulation with CCM by Film Extrusion
The core structure of the formulation was prepared by nanoprecipitation in a glass capillary microfluidics device, as reported elsewhere.12 We employed a device presenting a 3D co-flow geometry for nanoprecipitation. The assembly of the device has been described elsewhere.62 The inner capillary (ID 580 μm, OD 1 mm; World Precision Instruments Inc., USA) was pointed with a micropipette puller (P-97, Sutter Instrument Co., USA) to a diameter of approximately 20 μm. We then tapered the capillary to enlarge the diameter to approximately 100 μm. This capillary was then inserted and aligned coaxially into the outer capillary (ID 1.10 mm, Vitrocom, USA), and we assembled them in the microfluidics platform. In the nanoprecipitation technique, the inner and outer solutions are miscible and are pumped, keeping the flow rates constant, in the microfluidics device in the same direction. The flow rate of the two phases was controlled by two pumps (PHD 2000, Harvard Apparatus, USA), and the liquids were pumped from syringes into the capillaries through polyethylene tubes. To prepare TOPSi@AcDEX particles, we resuspend TOPSi particles in an ethanol solution of AcDEX for the inner phase, while we employed a 1% (w/v) poly(vinyl alcohol) aqueous solution for the outer phase. We fixed the inner flow rate to 2 mL/h and the other to 40 mL/h. The collection vial was kept under stirring (approximately 300g), and the collected particles were washed once with Milli-Q water (12959g, 5 min). Finally, the cell membrane layer was added on the surface of the particles by film extrusion through a 0.8 μm filter (Nucleopore Track-Etch Membrane, Whatman, UK) with an Avanti extruder (Avanti Lipids, USA). The cell membrane used for the extrusion of the samples was homologous to the cells used to establish the tumor models, B16.F10 in experiment 1 and B16.OVA in experiments 2 and 3.
Characterization of the Nanovaccine by Dynamic Light Scattering (DLS) and Electrophoretic Light Scattering (ELS)
The hydrodynamic radius and the surface charge of the nanovaccine were evaluated by DLS and ELS with a Zetasizer NanoZS (Malvern Instruments Ltd., UK). Each sample was diluted 1:50, and 1 mL of the dilution was pipetted in a disposable polystyrene cuvette (Sarstedt AG&Co., Germany) to determine the size. The temperature of each sample was equilibrated to 25 °C before each measurement. About 750 μL of the particle suspension in Milli Q-water (pH 7.4) was pipetted into a disposable folded capillary cell (DTS1070, Malvern Ltd., UK).
Cytocompatibility Studies
We assessed the cytocompatibility of TOPSi@AcDEX@CCM alone or in the presence of two different concentrations of murine anti-CTLA-4 antibody (In vivo Mab antimouse CTLA-4, CD152, clone 9D9, Bio X Cell, USA). Briefly, 50 μL of a 4 × 105 cell/mL suspension of JAWS II cells was seeded into 96-well plates (Corning Inc., USA) and left attaching overnight. We added 50 μL of the appropriate sample, redispersed in medium, in each well. The plates were then incubated at 37 °C with 5% CO2. At the time point, we then added 100 μL of Cell Titer Glo (Promega, USA) solution to each well. A Varioskan Lux reader (Thermo Fisher Scientific Inc., USA) was employed to read the resulting luminescence. Triton X-100 (1%) and 20% of FBS aMEM were used as positive and negative controls, respectively.
Immunostimulation Assay
We evaluated the immunostimulative properties of the nanovaccine and the effect of the combination with a check-point inhibitor in JAWS II cells. About 0.7 mL of a 4 × 105 cell/mL suspension was seeded in 12-well plates (Corning Inc., USA) and left attaching overnight. 0.7 mL of the appropriate sample was added to each well. The plates were then incubated at 37 °C with 5% CO2. Upon each time point, the medium was removed and centrifuged to recover the cells in suspension, and the adherent cells were detached with cold PBS-EDTA. The cells were then centrifuged and suspended in 90 μL of cold PBS. Five μL of APC antimouse CD80 (BD Biosciences, USA) and 5 μL of PerCP-Cy 5.5 antimouse CD86 (BioLegend, USA) antibodies were added to each sample. The samples were incubated for 20 min in the dark at +4 °C. To remove the unbound antibody, the cells were centrifuged, the supernatant discarded, and the cell pellet washed twice with cold PBS. We then suspend the cells in 700 μL of cold PBS and analyzed by flow cytometry (FACS) on a LSR II (BD Biosciences, USA). A compensation of the signal from each fluorochrome in the multistaining analysis was run. Lipopolysaccharide (LPS) and 20% of FBS aMEM were used as positive and negative controls, respectively.
Flow Cytometry and Analysis of Tumor-Infiltrating Immune Cells
Tumors were excised from the euthanized animals, and single cell suspensions were obtained by gently disrupting the tissue samples on a cell strainer (70 μm net-size) with a syringe plunger. Samples were then cryopreserved at −80 °C, by adding 10% of DMSO to cell suspension, until the day of analysis. Samples were quickly thawed into a water bath (+37 °C) and washed once in PBS. Anti-CD16/32 antibody was used to block unspecific staining from Fc receptors. Cells were then stained for 30 min with antibody cocktails on ice. Next, cells were washed to eliminate excess antibodies and fixed in 4% of formalin for 10 min on ice. Samples were washed two times and analyzed by flow cytometry. A Gallios flow cytometer (Beckman Coulter) was used to acquire the data, and FlowJo (Threestar) software was used for data analysis. Antibodies from BD-biosciences were used to stain cells for T-cell (CD8, CD4 and CD3) or DCs (CD11b, CD11c, CD86 and CD80) specific markers.
Statistical Analysis
Statistical significance was determined using GraphPad Prism 6 (GraphPad Software, Inc., La Jolla, CA, USA). A detailed description of the statistical methods used to analyze the data from each experiment can be found in each figure legend.
Acknowledgments
We thank Ms. Nazanin Zanjanizadeh Ezazi for assistance in the preparation of Scheme 1. F.F. acknowledges the Faculty of Pharmacy of the University of Helsinki for an assistant research grant. M.F. and C.C. acknowledge the Drug Research Doctoral Program of the University of Helsinki for a Ph.D. grant. C.G. would like to acknowledge financial support from the ULLA Consortium, the Jo Kolk Studiefonds, and the LUF International Study Fund. V.C. acknowledges financial support from the HiLIFE Research Funds, Jane and Aatos Erkko Foundation, and the European Research Council under the European Union’s Horizon 2020 Program (grant no. 681219). H.A.S. acknowledges financial support from the University of Helsinki Research Funds, the Sigrid Jusélius Foundation (decision no. 4704580), the HiLIFE Research Funds, and the European Research Council proof-of-concept grant (decision no. 825020).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b09613.
Figures analyzing the in vitro mechanism of activation of APCs and the average tumor volumes for each group (PDF)
Author Contributions
# These authors contributed equally to this work.
Author Contributions
F.F. and M.F. designed, planned, and executed the experiments. C.G, C.C., J.C., and S.F. contributed to the executing of the animal experiments. V.C. and H.A.S. supervised the experimental design and its execution. Z.L. synthesized the AcDEX polymer. E.M. and J.S. fabricated the PSi nanoparticles. F.F., C.C., J.H., V.C., and H.A.S. wrote and edited the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Kapadia C. H.; Perry J. L.; Tian S.; Luft J. C.; DeSimone J. M. Nanoparticulate Immunotherapy for Cancer. J. Controlled Release 2015, 219, 167–180. 10.1016/j.jconrel.2015.09.062. [DOI] [PubMed] [Google Scholar]
- van der Burg S. H.; Arens R.; Ossendorp F.; van Hall T.; Melief C. J. Vaccines for Established Cancer: Overcoming the Challenges Posed by Immune Evasion. Nat. Rev. Cancer 2016, 16, 219–233. 10.1038/nrc.2016.16. [DOI] [PubMed] [Google Scholar]
- Branca M. A. Rekindling Cancer Vaccines. Nat. Biotechnol. 2016, 34, 1019–1024. 10.1038/nbt.3690. [DOI] [PubMed] [Google Scholar]
- Koshy S. T.; Mooney D. J. Biomaterials for Enhancing Anti-Cancer Immunity. Curr. Opin. Biotechnol. 2016, 40, 1–8. 10.1016/j.copbio.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauleth-Ramos T.; Shahbazi M.-A.; Liu D.; Fontana F.; Correia A.; Figueiredo P.; Zhang H.; Martins J. P.; Hirvonen J. T.; Granja P.; Sarmento B.; Santos H. A. Nutlin-3a and Cytokine Co-loaded Spermine-Modified Acetalated Dextran Nanoparticles for Cancer Chemo-Immunotherapy. Adv. Funct. Mater. 2017, 27, 1703303. 10.1002/adfm.201703303. [DOI] [Google Scholar]
- Chen Q.; Wang C.; Chen G.; Hu Q.; Gu Z. Delivery Strategies for Immune Checkpoint Blockade. Adv. Healthcare Mater. 2018, 7, 1800424. 10.1002/adhm.201800424. [DOI] [PubMed] [Google Scholar]
- Fan Q.; Chen Z.; Wang C.; Liu Z. Toward Biomaterials for Enhancing Immune Checkpoint Blockade Therapy. Adv. Funct. Mater. 2018, 28, 1802540. 10.1002/adfm.201802540. [DOI] [Google Scholar]
- Francis D. M.; Thomas S. N. Progress and Opportunities for Enhancing the Delivery and Efficacy of Checkpoint Inhibitors for Cancer Immunotherapy. Adv. Drug Delivery Rev. 2017, 114, 33–42. 10.1016/j.addr.2017.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C.; Ye Y.; Hochu G. M.; Sadeghifar H.; Gu Z. Enhanced Cancer Immunotherapy by Microneedle Patch-Assisted Delivery of Anti-PD1 Antibody. Nano Lett. 2016, 16, 2334–2340. 10.1021/acs.nanolett.5b05030. [DOI] [PubMed] [Google Scholar]
- Wang C.; Sun W.; Ye Y.; Hu Q.; Bomba H. N.; Gu Z. In Situ Activation of Platelets with Checkpoint Inhibitors for Post-Surgical Cancer Immunotherapy. Nat. Biomed. Eng. 2017, 1, 0011. 10.1038/s41551-016-0011. [DOI] [Google Scholar]
- Wang C.; Ye Y.; Hu Q.; Bellotti A.; Gu Z. Tailoring Biomaterials for Cancer Immunotherapy: Emerging Trends and Future Outlook. Adv. Mater. 2017, 29, 1606036. 10.1002/adma.201606036. [DOI] [PubMed] [Google Scholar]
- Fontana F.; Shahbazi M. A.; Liu D.; Zhang H.; Mäkilä E.; Salonen J.; Hirvonen J. T.; Santos H. A. Multistaged Nanovaccines Based on Porous Silicon@Acetalated Dextran@Cancer Cell Membrane for Cancer Immunotherapy. Adv. Mater. 2017, 29, 1603239. 10.1002/adma.201603239. [DOI] [PubMed] [Google Scholar]
- Shahbazi M. A.; Fernandez T. D.; Mäkilä E. M.; Le Guevel X.; Mayorga C.; Kaasalainen M. H.; Salonen J. J.; Hirvonen J. T.; Santos H. A. Surface Chemistry Dependent Immunostimulative Potential of Porous Silicon Nanoplatforms. Biomaterials 2014, 35, 9224–9235. 10.1016/j.biomaterials.2014.07.050. [DOI] [PubMed] [Google Scholar]
- Xia X.; Mai J.; Xu R.; Perez J. E.; Guevara M. L.; Shen Q.; Mu C.; Tung H. Y.; Corry D. B.; Evans S. E.; Liu X.; Ferrari M.; Zhang Z.; Li X. C.; Wang R. F.; Shen H. Porous silicon Microparticle Potentiates Anti-Tumor Immunity by Enhancing Cross-Presentation and Inducing Type I Interferon Response. Cell Rep. 2015, 11, 957–966. 10.1016/j.celrep.2015.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broaders K. E.; Cohen J. A.; Beaudette T. T.; Bachelder E. M.; Fréchet J. M. Acetalated Dextran is a Chemically and Biologically Tunable Material for Particulate Immunotherapy. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 5497–5502. 10.1073/pnas.0901592106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachelder E. M.; Pino E. N.; Ainslie K. M. Acetalated Dextran: A Tunable and Acid-Labile Biopolymer with Facile Synthesis and a Range of Applications. Chem. Rev. 2017, 117, 1915–1926. 10.1021/acs.chemrev.6b00532. [DOI] [PubMed] [Google Scholar]
- Zhu G.; Zhang F.; Ni Q.; Niu G.; Chen X. Efficient Nanovaccine Delivery in Cancer Immunotherapy. ACS Nano 2017, 11, 2387–2392. 10.1021/acsnano.7b00978. [DOI] [PubMed] [Google Scholar]
- Gajewski T. F.; Schreiber H.; Fu Y. X. Innate and Adaptive Immune Cells in the Tumor Microenvironment. Nat. Immunol. 2013, 14, 1014–1022. 10.1038/ni.2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott P. A.; Hu Z.; Keskin D. B.; Shukla S. A.; Sun J.; Bozym D. J.; Zhang W.; Luoma A.; Giobbie-Hurder A.; Peter L.; Chen C.; Olive O.; Carter T. A.; Li S.; Lieb D. J.; Eisenhaure T.; Gjini E.; Stevens J.; Lane W. J.; Javeri I.; Nellaiappan K.; et al. An Immunogenic Personal Neoantigen Vaccine for Patients With Melanoma. Nature 2017, 547, 217–221. 10.1038/nature22991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin U.; Derhovanessian E.; Miller M.; Kloke B. P.; Simon P.; Lower M.; Bukur V.; Tadmor A. D.; Luxemburger U.; Schrors B.; Omokoko T.; Vormehr M.; Albrecht C.; Paruzynski A.; Kuhn A. N.; Buck J.; Heesch S.; Schreeb K. H.; Muller F.; Ortseifer I.; et al. Personalized RNA Mutanome Vaccines Mobilize Poly-specific Therapeutic Immunity Against Cancer. Nature 2017, 547, 222–226. 10.1038/nature23003. [DOI] [PubMed] [Google Scholar]
- Yarchoan M.; Johnson B. A. 3rd; Lutz E. R.; Laheru D. A.; Jaffee E. M. Targeting Neoantigens to Augment Antitumour Immunity. Nat. Rev. Cancer 2017, 17, 209–222. 10.1038/nrc.2016.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang R. H.; Hu C. M.; Luk B. T.; Gao W.; Copp J. A.; Tai Y.; O’Connor D. E.; Zhang L. Cancer Cell Membrane-Coated Nanoparticles for Anticancer Vaccination and Drug Delivery. Nano Lett. 2014, 14, 2181–2188. 10.1021/nl500618u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung A. S.; Koshy S. T.; Stafford A. G.; Bastings M. M.; Mooney D. J. Adjuvant-Loaded Subcellular Vesicles Derived From Disrupted Cancer Cells for Cancer Vaccination. Small 2016, 12, 2321–2333. 10.1002/smll.201600061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth M. J.; Ngiow S. F.; Ribas A.; Teng M. W. Combination Cancer Immunotherapies Tailored to the Tumour Microenvironment. Nat. Rev. Clin. Oncol. 2016, 13, 143–158. 10.1038/nrclinonc.2015.209. [DOI] [PubMed] [Google Scholar]
- Inozume T.; Hanada K.; Wang Q. J.; Ahmadzadeh M.; Wunderlich J. R.; Rosenberg S. A.; Yang J. C. Selection of CD8+PD-1+ Lymphocytes in Fresh Human Melanomas Enriches for Tumor-Reactive T Cells. J. Immunother. 2010, 33, 956–964. 10.1097/CJI.0b013e3181fad2b0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gros A.; Robbins P. F.; Yao X.; Li Y. F.; Turcotte S.; Tran E.; Wunderlich J. R.; Mixon A.; Farid S.; Dudley M. E.; Hanada K.; Almeida J. R.; Darko S.; Douek D. C.; Yang J. C.; Rosenberg S. A. PD-1 Identifies the Patient-Specific CD8+ Tumor-Reactive Repertoire Infiltrating Human Tumors. J. Clin. Invest. 2014, 124, 2246–2259. 10.1172/JCI73639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon S.; Labarriere N. PD-1 Expression on Tumor-Specific T Cells: Friend or Foe for Immunotherapy?. Oncoimmunology 2018, 7, e1364828 10.1080/2162402X.2017.1364828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll D. M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R. K.; Martin J. D.; Stylianopoulos T. The Role of Mechanical Forces in Tumor Growth and Therapy. Annu. Rev. Biomed. Eng. 2014, 16, 321–346. 10.1146/annurev-bioeng-071813-105259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive Networks in the Tumour Environment and Their Therapeutic Relevance. Nat. Rev. Cancer 2005, 5, 263–274. 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
- Crespo J.; Sun H.; Welling T. H.; Tian Z.; Zou W. T Cell Anergy, Exhaustion, Senescence and Stemness in the Tumor Microenvironment. Curr. Opin. Immunol. 2013, 25, 214–221. 10.1016/j.coi.2012.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce J. A.; Fearon D. T. T Cell Exclusion, Immune Privilege and the Tumor Microenvironment. Science 2015, 348, 74–80. 10.1126/science.aaa6204. [DOI] [PubMed] [Google Scholar]
- Schreiber R. D.; Old L. J.; Smyth M. J. Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 2011, 331, 1565–1570. 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- Irvine D. J.; Hanson M. C.; Rakhra K.; Tokatlian T. Synthetic Nanoparticles for Vaccines and Immunotherapy. Chem. Rev. 2015, 115, 11109–11146. 10.1021/acs.chemrev.5b00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontana F.; Liu D.; Hirvonen J.; Santos H. A. Delivery of Therapeutics with Nanoparticles: What’s New in Cancer Immunotherapy?. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1421 10.1002/wnan.1421. [DOI] [PubMed] [Google Scholar]
- Wang C.; Li P.; Liu L.; Pan H.; Li H.; Cai L.; Ma Y. Self-adjuvanted Nanovaccine for Cancer Immunotherapy: Role of Lysosomal Rupture-induced ROS in MHC Class I Antigen Presentation. Biomaterials 2016, 79, 88–100. 10.1016/j.biomaterials.2015.11.040. [DOI] [PubMed] [Google Scholar]
- Wakim L. M.; Bevan M. J. Cross-dressed Dendritic Cells Drive Memory CD8+ T-cell Activation after Viral Infection. Nature 2011, 471, 629–632. 10.1038/nature09863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campana S.; De Pasquale C.; Carrega P.; Ferlazzo G.; Bonaccorsi I. Cross-dressing: an Alternative Mechanism for Antigen Presentation. Immunol. Lett. 2015, 168, 349–354. 10.1016/j.imlet.2015.11.002. [DOI] [PubMed] [Google Scholar]
- Sloan A. E.; Dansey R.; Zamorano L.; Barger G.; Hamm C.; Diaz F.; Baynes R.; Wood G. Adoptive Immunotherapy in Patients with Recurrent Malignant Glioma: Preliminary Results of Using Autologous Whole-Tumor Vaccine Plus Granulocyte-Macrophage Colony-Stimulating Factor and Adoptive Transfer of Anti-CD3-Activated Lymphocytes. Neurosurg. Focus 2000, 9, 1–8. 10.3171/foc.2000.9.6.10. [DOI] [PubMed] [Google Scholar]
- Euhus D. M.; Gupta R. K.; Morton D. L. Induction of Antibodies to a Tumor-Associated Antigen by Immunization with a Whole Melanoma Cell Vaccine. Cancer Immunol. Immunother. 1989, 29, 247–254. 10.1007/BF00199212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W.; Guo C.; Liu Q. G.; Pan C. Experimental Study of Specific Immunotherapy Induced by H22 Autologous Tumor as Whole Tumor Cell Vaccine. Biomed. Pharmacother. 2009, 63, 404–408. 10.1016/j.biopha.2008.08.020. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Sun H. The Tumor Protection Effect of High-Frequency Administration of Whole Tumor Cell Vaccine and Enhanced Efficacy by the Protein Component from Agrocybe Aegerita. Int. J. Clin. Exp. Med. 2015, 8, 6914–6925. [PMC free article] [PubMed] [Google Scholar]
- Chiang C. L.-L.; Kandalaft L. E.; Coukos G. Adjuvants for Enhancing the Immunogenicity of Whole Tumor Cell Vaccines. Int. Rev. Immunol. 2011, 30, 150–182. 10.3109/08830185.2011.572210. [DOI] [PubMed] [Google Scholar]
- Fishman M.; Hunter T. B.; Soliman H.; Thompson P.; Dunn M.; Smilee R.; Farmelo M. J.; Noyes D. R.; Mahany J. J.; Lee J. H.; Cantor A.; Messina J.; Seigne J.; Pow-Sang J.; Janssen W.; Antonia S. J. Phase II Trial Of B7–1 (CD-86) Transduced, Cultured Autologous Tumor Cell Vaccine Plus Subcutaneous Interleukin-2 for Treatment of Stage IV Renal Cell Carcinoma. J. Immunother. 2008, 31, 72–80. 10.1097/CJI.0b013e31815ba792. [DOI] [PubMed] [Google Scholar]
- Roh W.; Chen P. L.; Reuben A.; Spencer C. N.; Prieto P. A.; Miller J. P.; Gopalakrishnan V.; Wang F.; Cooper Z. A.; Reddy S. M.; Gumbs C.; Little L.; Chang Q.; Chen W. S.; Wani K.; De Macedo M. P.; Chen E.; Austin-Breneman J. L.; Jiang H.; Roszik J.; et al. Integrated Molecular Analysis of Tumor Biopsies on Sequential CTLA-4 and PD-1 Blockade Reveals Markers of Response and Resistance. Sci. Transl. Med. 2017, 9, eaah3560 10.1126/scitranslmed.aah3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascierto P. A.; Capone M.; Urba W. J.; Bifulco C. B.; Botti G.; Lugli A.; Marincola F. M.; Ciliberto G.; Galon J.; Fox B. A. The Additional Facet of Immunoscore: Immunoprofiling as a Possible Predictive Tool for Cancer Treatment. J. Transl. Med. 2013, 11, 54. 10.1186/1479-5876-11-54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman W. H.; Zitvogel L.; Sautes-Fridman C.; Kroemer G. The Immune Contexture in Cancer Prognosis and Treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
- Rashidian M.; Ingram J. R.; Dougan M.; Dongre A.; Whang K. A.; LeGall C.; Cragnolini J. J.; Bierie B.; Gostissa M.; Gorman J.; Grotenbreg G. M.; Bhan A.; Weinberg R. A.; Ploegh H. L. Predicting the Response to CTLA-4 Blockade by Longitudinal Noninvasive Monitoring of CD8 T Cells. J. Exp. Med. 2017, 214, 2243–2255. 10.1084/jem.20161950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engeland C. E.; Grossardt C.; Veinalde R.; Bossow S.; Lutz D.; Kaufmann J. K.; Shevchenko I.; Umansky V.; Nettelbeck D. M.; Weichert W.; Jager D.; von Kalle C.; Ungerechts G. CTLA-4 and PD-L1 Checkpoint Blockade Enhances Oncolytic Measles Virus Therapy. Mol. Ther. 2014, 22, 1949–1959. 10.1038/mt.2014.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdeil G.; Fuertes Marraco S. A.; Murray T.; Speiser D. E. From T cell “Exhaustion” to Anti-Cancer Immunity. Biochim. Biophys. Acta, Rev. Cancer 2016, 1865, 49–57. 10.1016/j.bbcan.2015.06.007. [DOI] [PubMed] [Google Scholar]
- Zitvogel L.; Kroemer G. Targeting PD-1/PD-L1 Interactions for Cancer Immunotherapy. Oncoimmunology 2012, 1, 1223–1225. 10.4161/onci.21335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S.; Akbay E. A.; Li Y. Y.; Herter-Sprie G. S.; Buczkowski K. A.; Richards W. G.; Gandhi L.; Redig A. J.; Rodig S. J.; Asahina H.; Jones R. E.; Kulkarni M. M.; Kuraguchi M.; Palakurthi S.; Fecci P. E.; Johnson B. E.; Janne P. A.; Engelman J. A.; Gangadharan S. P.; Costa D. B.; et al. Adaptive Resistance to Therapeutic PD-1 Blockade is Associated with Upregulation of Alternative Immune Checkpoints. Nat. Commun. 2016, 7, 10501. 10.1038/ncomms10501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali O. A.; Lewin S. A.; Dranoff G.; Mooney D. J. Vaccines Combined with Immune Checkpoint Antibodies Promote Cytotoxic T-cell Activity and Tumor Eradication. Cancer Immunol. Res. 2016, 4, 95–100. 10.1158/2326-6066.CIR-14-0126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Zhang S.; Hu Y.; Yang Z.; Li J.; Liu X.; Deng L.; Wang Y.; Zhang X.; Jiang T.; Lu X. Targeting PD-1 and Tim-3 Pathways to Reverse CD8 T-Cell Exhaustion and Enhance Ex Vivo T-Cell Responses to Autologous Dendritic/Tumor Vaccines. J. Immunother. 2016, 39, 171–180. 10.1097/CJI.0000000000000122. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Skowron K. B.; Namm J. P.; Burnette B.; Fernandez C.; Arina A.; Liang H.; Spiotto M. T.; Posner M. C.; Fu Y. X.; Weichselbaum R. R. Combination of Radiotherapy and Vaccination Overcomes Checkpoint Blockade Resistance. Oncotarget 2016, 7, 43039–43051. 10.18632/oncotarget.9915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D.; Zhang H.; Cito S.; Fan J.; Mäkilä E.; Salonen J.; Hirvonen J.; Sikanen T. M.; Weitz D. A.; Santos H. A. Core/Shell Nanocomposites Produced by Superfast Sequential Microfluidic Nanoprecipitation. Nano Lett. 2017, 17, 606–614. 10.1021/acs.nanolett.6b03251. [DOI] [PubMed] [Google Scholar]
- Tähtinen S.; Grönberg-Vähä-Koskela S.; Lumen D.; Merisalo-Soikkeli M.; Siurala M.; Airaksinen A. J.; Vähä-Koskela M.; Hemminki A. Adenovirus Improves the Efficacy of Adoptive T-cell Therapy by Recruiting Immune Cells to and Promoting Their Activity at the Tumor. Cancer Immunol. Res. 2015, 3, 915–925. 10.1158/2326-6066.CIR-14-0220-T. [DOI] [PubMed] [Google Scholar]
- Bimbo L. M.; Mäkilä E.; Laaksonen T.; Lehto V.-P.; Salonen J.; Hirvonen J.; Santos H. A. Drug permeation Across Intestinal Epithelial Cells Using Porous Silicon Nanoparticles. Biomaterials 2011, 32, 2625–2633. 10.1016/j.biomaterials.2010.12.011. [DOI] [PubMed] [Google Scholar]
- Santos H. A.; Riikonen J.; Salonen J.; Mäkilä E.; Heikkilä T.; Laaksonen T.; Peltonen L.; Lehto V. P.; Hirvonen J. In Vitro Cytotoxicity of Porous Silicon Microparticles: Effect of the Particle Concentration, Surface Chemistry and Size. Acta Biomater. 2010, 6, 2721–2731. 10.1016/j.actbio.2009.12.043. [DOI] [PubMed] [Google Scholar]
- Liu D.; Cito S.; Zhang Y.; Wang C. F.; Sikanen T. M.; Santos H. A. A Versatile and Robust Microfluidic Platform Toward High Throughput Synthesis of Homogeneous Nanoparticles With Tunable Properties. Adv. Mater. 2015, 27, 2298–2304. 10.1002/adma.201405408. [DOI] [PubMed] [Google Scholar]
- Liu D.; Zhang H.; Mäkilä E.; Fan J.; Herranz-Blanco B.; Wang C. F.; Rosa R.; Ribeiro A. J.; Salonen J.; Hirvonen J.; Santos H. A. Microfluidic Assisted One-Step Fabrication of Porous Silicon@Acetalated Dextran Nanocomposites for Precisely Controlled Combination Chemotherapy. Biomaterials 2015, 39, 249–259. 10.1016/j.biomaterials.2014.10.079. [DOI] [PubMed] [Google Scholar]
- Herranz-Blanco B.; Ginestar E.; Zhang H.; Hirvonen J.; Santos H. A. Microfluidics Platform for Glass Capillaries and its Application in Droplet and Nanoparticle Fabrication. Int. J. Pharm. 2017, 516, 100–105. 10.1016/j.ijpharm.2016.11.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.