

To the Editor: Distal myopathy is a heterogenetic disorder characterized by early distal lower limb involvement, which has been linked to 18 disease-causing genes. The gene responsible for Laing distal myopathy (LDM, also called distal myopathy 1; OMIM 160500) was determined to be the myosin heavy chain 7 gene (MYH7) located on chromosome 14q11.[1]MYH7 encodes the myosin heavy chain beta isoform (MyHC-β) and is expressed predominantly in the cardiac ventricle and in type 1 skeletal muscle fibers. The mutant MyHC-β protein is also linked to familial hypertrophic cardiomyopathy (OMIM 192600), dilated cardiomyopathy (OMIM 115200), left ventricular non-compaction and myosin storage myopathy (OMIM 608358). Only deletion or missense mutations in exon 32 to 36 of the MYH7 gene are linked to LDM; missense mutations of exon 37 to 40 are linked to myosin storage myopathy.[2] The mutations of the MYH7 gene exhibit complete penetrance.
The typical symptoms of LDM begin in early childhood with foot drop and big toe weakness, leading to the “hanging big toe” sign. Then, the weakness progresses from the distal to proximal lower limb and upper limb, presenting with scapular winging, scoliosis, and finger extensor weakness. However, the extensor digitorum communis is always preserved. The clinical spectrum of symptoms caused by mutations of the MYH7 gene associated with LDM has been broadened and now includes proximal myopathy and asymptomatic hyperCKemia; some mutations may also have an intermediate clinical phenotype. Here, we describe a novel mutation of the MYH7 gene identified in a three-generation Chinese pedigree of LDM, presenting with pronounced type 1 fiber predominance and normal cardiac muscle.
The institutional ethics committee of Peking University Third Hospital approved this study (No. 2014155). Written informed consent was obtained from each participant. Four patients over three generations in a non-consanguineous Chinese family were included. The index case was a 27-year-old female who suffered from bilateral foot drop for more than 20 years. The medical histories of four affected family members were reviewed, and three of them underwent a complete clinical neurologic examination. Lower limb magnetic resonance imaging (MRI), a laboratory study including serum creatine kinase (CK), CK-MB, 12-lead electrocardiography, and echocardiogram were assessed. Electromyography for upper and lower extremities was evaluated in the index patient. A tibialis anterior muscle biopsy was available from the index patient. Staining for hemotoxylin and eosin (H&E), Gomori trichrome, ATPase, PAS, Oil red O, and NADH-TR was performed. Additionally, immunohistochemistry was performed to identify dystrophin, dysferlin, sarcoglycans, and myosin. DNA samples were acquired from three affected and six unaffected individuals in the family. Genomic DNA was extracted from peripheral blood leukocytes, and a panel of 168 distinct genes related to myopathy were sequenced using exome capture sequencing technology. Sequence data from the NCBI single nucleotide polymorphism (SNP) database and the 1000 Genomes Project database were used to exclude mutations with a frequency higher than 1% in healthy populations. The remaining mutations were considered to be candidates for disease-causing mutations. Sanger sequencing were performed to confirm the detected variants. The PolyPhen-2 (http://genetics. bwh.harvard.edu/pph2), MutationTaster (http://www.mutationtaster.org), and SIFT (http://sift.jcvi.org/) programs were used to predict the pathogenicity of the altered protein. The variants were identified using the Short Genetic Variations Database (dbSNP), the NHLBI Exome Variant Server (ESP 6500), the 1000 Genomes Project and the Exome Aggregation Consortium (ExAC) database. The American College of Medical Genetics and Genomics (ACMG) guidelines for the interpretation of sequence variants were applied for the confirmation of pathogenicity.
The pedigree analysis demonstrates an autosomal dominant inheritance pattern [Figure 1A]. The medical history and neurologic examinations of the index patient (IV.7), her sister (IV.8), and her mother (III.9) were reviewed. The proband (IV.7) was a 27-year-old female. In this individual, foot paralysis was first noticed at the age of 7 years, making it difficult for her to walk on her toes. She then developed bilateral foot drop one year later, and the weakness of the lower limbs slowly progressed. In her early 20s, she had difficulty using her fourth and fifth fingers but could still write. She experienced sore knees after walking long distances and had difficulty getting up from a chair without using both hands. Neck weakness began at age 25 years old. On examination, she presented with neck weakness, bilateral scapular winging, and weakness predominately in the distal upper and lower limbs. The deltoid, iliopsoas, wrist, and foot extensor muscles were paralyzed. Remarkable difficulty extending the big toes and the fourth and fifth fingers of the hands was observed. The deep tendon reflex was slightly decreased. She exhibited bilateral ankle contractures, but no pes cavus, scoliosis, or high-arched palate was noticed. She felt unsteady while walking and was easily tripped by big toes. The “hanging big toe” sign is shown in Figure 1. The mother (III.9) showed the typical “hanging big toe” sign, and extensor paralysis was present in the third, fourth, and fifth fingers. Although she suffered from weakness of the lower limbs, she can still walk slowly without assistance in her 50's. Scoliosis was observed on examination without pes cavus or high-arched palate. The sister (IV.8) had difficulty walking and was unable to run by age 10 years old. On examination, she exhibited a pattern of weakness similar to the index patient. The grandmother (II.3) was bed-ridden after a fall at age 60 years old and passed away in her 70's due to chronic renal failure caused by type 2 diabetes mellitus.
Figure 1.
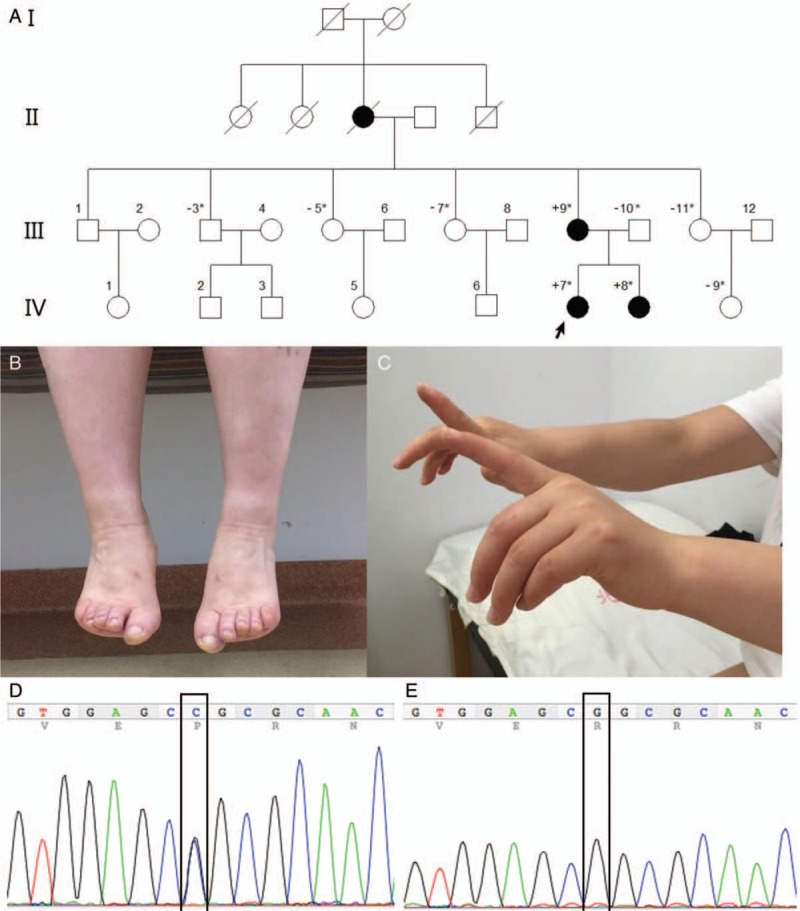
Pedigree and clinical characteristics of the index patient. (A) Pedigree of the family. Black filled circles are affected individuals. ∗ means genetic information for this family member was available. “+” indicates that this individual carries the MYH7 mutation. “−” indicates this individual does not carry the MYH7 mutation. “/” indicates that the individual is deceased. (B) Weakness for extension of the big toes in patient IV.7 (Hanging big toe sign). (C) Extended hands demonstrate a pronounced extensor paralysis of the third to fifth fingers bilaterally for the patient IV.7. Sequence chromatograms of MYH7 gene c.5027G>C (p.Arg1676Pro) heterozygous missense mutation in patient IV.7 (D) compare with wild type control in IV.9 (E).
The serum CK level for the index patient was within normal range. Cardiac ultrasound was normal with an ejection fraction of 56%. No premature contraction was observed on the electrocardiogram. The lower limb MRI showed mild fatty infiltration and atrophy of the tibialis anterior muscle. Calf hypertrophies was pronounced bilaterally. Nerve conduction studies were normal. The EMG showed a small amplitude, abundant polyphasic motor unit potential (MUP) pattern in the right deltoid, left rectus femoris, and right gastrocnemius, but the MUP of the left extensor digitorum communis was normal. We performed a left tibialis anterior muscle biopsy in the index patient. H&E staining revealed fiber size variability with atrophic rounded fibers and occasional angular fibers. A few fibers contained small- to medium-sized vacuoles; these vacuoles were blue-rimmed on H&E staining and red rimmed on Gomori trichrome staining. ATPase at pH 4.4, 4.6, 10.6 showed pronounced type 1 fiber predominance (nearly all fibers are type 1). NADH-tetrazoliumreductase stains showed no core or “moth-eaten” changes [Figure 2A and 2D]. Muscle fibers were positive for dystrophin, sarcoglycans, and dysferlin. The electron microscope examination showed no specific abnormalities except mild irregular Z bands.
Figure 2.

Hemotoxylin and eosin staining revealed increased in fiber size variability, without necrosis (A; scale bar 100 μm); NADH-tetrazoliumreductase staining (B; scale bar 100 μm); ATPase 4.6 (C) and 10.6 (D) show pronounced predominance of type I fibers with severe muscle grouping (scale bar 500 μm).
Genomic DNA was extracted from peripheral blood leukocytes, and a panel of 168 distinct genes related to myopathy were sequenced using exome capture sequencing technology. The DNA sequencing for the index patient revealed a c.5027G>C (p.Arg1676Pro) heterozygous missense mutation of the MYH7 gene, which was located in exon 35, the variant was validated by polymerase chain reaction [Figure 1]. The mutation was co-segregated with the phenotype in this family. The three affected individuals carried the novel mutation, while the six unaffected individuals did not. The c.5027G>C mutation was not present in the dbSNP, EVS, the 1000 Genomes Project and ExAC database. MutationTaster software suggested that the mutation was “disease-causing”. SIFT indicated the mutation was “damaging” with a score of 0. PolyPhen-2 indicated the mutation was “probably damaging,” with score of 0.995. The wild-type G-allele was highly conserved with a PhyloP score of 6.039.
In this family with distal myopathy, we used a targeted gene panel to test the coding exons of 168 myopathy-related genes in affected and unaffected individuals. The novel variant c.5027G > C (p.Arg1676Pro) of the MYH7 gene was identified from the panel, and the pathogenicity was supported by the following facts. The variant was not present in population-based polymorphism databases (EVS, dbSNP, 1000 Genomes Project, and ExAC, which include more than 65,000 exomes, of which more than 4300 are from an East Asian population). Furthermore, the variant was located in a highly conserved region. The pathogenicity is also supported by polymorphism prediction software (MutationTaster, PolyPhen-2, and SIFT), which suggested that p.Arg1676Pro was a deleterious mutation. Missense mutations in the MYH7 gene are a known mechanism of LDM, with an autosomal dominant inheritance. The clinical features of affected individuals were consistent with typical LDM. The variant was completely co-segregated with the phenotype: three affected individuals carried the novel mutation, while six healthy family members did not. Thus, the evidence indicated that the variant was likely to be pathogenic according to the American College of Medical Genetics and Genomics guidelines, which means that there is a greater than 90% certainty that the variant is disease causing.
The clinical presentation of the affected individuals in the present LDM family is homogeneous, including severe tibialis anterior and finger extensor muscle involvement, neck weakness, the “hanging big toe” sign and slow progression. The exceptional preservation of the index finger extensor was proved by a normal EMG study on the extensor digitorum communis, suggesting that selected preservation of certain extensor muscles is unique for LDM. Cardiac muscle is not commonly affected in LDM families with MYH7 rod domain mutations.[3] The patient carrying the p.Arg1676Pro mutation in this family showed no evidence of cardiac muscle involvement. Furthermore, the grandmother showed no cardiac issues, indicating that the cardiac muscle was spared until advanced age.
The muscle pathology showed fiber size variability and occasional angular atrophic fibers. A few possible rimmed vacuoles were observed, but it is nonspecific and common for hereditary distal myopathies. Pronounced type 1 fiber predominance was noticed, supported by ATPase 4.6 and 10.6 staining. Although the feature is common for LDM pathology in different degrees, but the extremely marked predominance (>95% of type 1 fiber) in this case is rarely presented in LDM cases. It has been reported that the differentia of fiber type and size is a key feature of LDM pathology, including fiber type disproportion (predominance of small type 1 fibers with type 2 fibers that are few in number but normal in size) and fiber type predominance.[1,4] But these features could also appear in other distal myopathies.
The MyHC-β protein consists of a globular head domain and a coiled-coil rod domain. Mutations in different domains of the MyHC-β protein result in distinct clinical phenotypes. Generally, disruption of the binding sites for the essential light chain and the regulatory light chain in the head and neck domain may cause cardiomyopathy, and structural change of the distal rod domain may cause skeletal myopathy.[2] The novel p.Arg1676Pro mutation found in this pedigree was located in exon 35 of rod domain, between the previously reported LDM phenotype mutations of p.Ala1663Pro and p.Leu1706Pro.[1] However, two adjacent mutations, p.Arg1677His and p.Ala1673Thr, in the rod domain both have a cardiomyopathy phenotype, including hypertrophic cardiomyopathy and dilated cardiomyopathy.[3] One possible explanation for this fact is the differences of polarity of the substituted amino acids. There are many mutations to proline that lead to LDM. Early publications suggested that the coiled-coil structure may be affected by an introduced proline. Studies of two mutations, p.Arg1500Pro and p.Leu1706Pro, revealed that the assembly of α-helices appears less stable with a proline in the myosin rod domain, but it can still be incorporated into thick filaments.[5] The proline is rare in the wild-type myosin rod domain and may affect hydrogen bonding. However, the residues of proline in the coiled-coil structure may also lead to a pronounced connection.[2] It is unclear whether proline mutations affect the stability of the coiled-coil structure or prevent the rod domain to form into thick filament. Because the clinical phenotype of the mutations in rod domain is different, the way they trigger dysfunction might be different. It is still possible that the phenotypic variability of MYH7 mutations is not fully derived from MyHC-β structure change. However, the underlying mechanism is still unclear.
In summary, we identified a family with LDM with a novel MYH7 gene p.Arg1676Pro mutation. We confirmed a benign phenotype without cardiomyopathy in this family, and a muscle biopsy revealed pronounced type 1 fiber predominance. Further research on the molecular structure of the mutations may increase the understanding of potential mechanisms of LDM.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patients have given their consent for their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Conflicts of interest
None.
Footnotes
How to cite this article: Liu XY, Zhang YS, Sun AP, Zhong YF, Zheng DF, Fan DS. A novel MYH7 mutation resulting in Laing distal myopathy in a Chinese family. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000148
References
- 1.Meredith C, Herrmann R, Parry C, Liyanage K, Dye DE, Durling HJ, et al. Mutations in the slow skeletal muscle fiber myosin heavy chain gene (MYH7) cause laing early-onset distal myopathy (MPD1). Am J Hum Genet 2004; 75:703–708. doi: 10.1086/424760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colegrave M, Peckham M. Structural implications of beta-cardiac myosin heavy chain mutations in human disease. Anat Rec (Hoboken) 2014; 297:1670–1680. doi: 10.1002/ar.22973. [DOI] [PubMed] [Google Scholar]
- 3.Walsh R, Rutland C, Thomas R, Loughna S. Cardiomyopathy: a systematic review of disease-causing mutations in myosin heavy chain 7 and their phenotypic manifestations. Cardiology 2010; 115:49–60. doi: 10.1159/000252808. [DOI] [PubMed] [Google Scholar]
- 4.Tajsharghi H, Oldfors A. Myosinopathies: pathology and mechanisms. Acta Neuropathol 2013; 125:3–18. doi: 10.1007/s00401-012-1024-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buvoli M, Buvoli A, Leinwand LA. Effects of pathogenic proline mutations on myosin assembly. J Mol Biol 2012; 415:807–818. doi: 10.1016/j.jmb.2011.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]