

To the Editor: Diabetic kidney disease (DKD) is one of the major causes of end-stage renal failure. Progressive mesangial expansion in the glomerulus is widely recognized. More and more evidence shows that tubulointerstitial fibrosis is also regarded as a prominent feature of DKD, in which tubular epithelial-mesenchymal transition (EMT) may play an important role.[1] During EMT, tubular epithelial cells lose their characteristic and produce high levels of myofibroblast makers such as α-smooth actin (α-SMA) which is the signature protein of EMT.[2] However, renal fibrogenesis is a complicated process in which the contribution of DKD is unknown.
The endoplasmic reticulum (ER) is a key player in maintenance of protein homeostasis. Disturbances such as ischemia, oxidative stress and hypoxia can cause accumulation of misfolded proteins in the ER lumen, which induce endoplasmic reticulum stress (ERS).[3] ERS activates the highly conserved unfolded protein response which is initiated by three kinds of ER transmembrane signal protein pathways. PERK is one of them, which is combined with GRP78 to make it inactive in the physiological state. However, in the case of stress, PERK is dissociated with GRP78 and activated by its own phosphorylation, which serves as the central regulator of translational control during ERS. In the meantime, PERK has protein-kinase activity to phosphorylate the α-submit of eIF2α.
Several mechanisms including hyperglycemia can cause ERS. On the other hand, ERS greatly affects the secretion and action of insulin, which leads to elevated blood glucose level.[4] Mice lacking PERK exhibit several defects including small size, bone abnormalities and type 1 diabetes.[5] Furthermore, there is evidence that ERS can lead to diabetic vascular complications.[6] ER stress has been demonstrated to contribute to the onset and progression of diabetic retinopathy by induction of multiple inflammatory signaling pathways.[7] Liu et al[8] has proved that ER stress contributes to premature senescence of renal tubular epithelial cells in patients with diabetes. But it is not very clear how ERS participates in the development of DKD. The purpose of the present study was to get a comprehensive understanding of how ERS participates in tubulointerstitial fibrosis in DKD and the mechanisms involved.
The Ethics Committee of Renmin Hospital of Wuhan University confirmed that the ethics approval of this study was not needed. Anti-PERK, phosphor-PERK, GRP78, eIF2α antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-phosphor-eIF2α and α-SMA antibody were obtained from Abcam Biotechnology (Cambridge, UK). Thapsigargin was purchased from Sigma (St. Louis, MO, USA). GSK2606414 was purchased from Selleck Chemicals (Houston, TX, USA). Rat renal proximal tubule epithelial cells (NRK-52E) were presented from the laboratory of nephrology department, Renmin Hospital of Wuhan University (obtained from American Type Culture Collection). NRK-52E cells were cultured (at 37°C, in a 5% CO2 atmosphere) in Dulbecco Modified Eagle's Medium (DMEM) containing glucose (5.6 mmol/L), 10% fetal bovine serum, 100 mg/mL streptomycin, 100 U/mL penicillin. Cells were seeded at 1 × 105 cells/well in 6-well dishes and were quiescent with serum-free medium for 24 h before use in all experiments.
After finishing experiments, the media was removed and the cells were washed twice with ice-cold PBS. Cells were lysed with RIPA lysates (Beyotime Biotechnology, Shanghai, China), containing 1 mmol/L PMSF and quantified using bicinchoninic acid protein assay kit (Beyotime Biotechnology, Shanghai, China). Cell homogenates were separated by 10% SDS-PAGE and transferred to polyvinylidene fluoride membranes, which were then blocked for 1 h at room temperature with 5% skim milk powder in TBST. The blots were incubated with one of the following primary polyclonal antibodies: α-SMA, GRP78, PERK, phosphor-PERK, eIF2α, or phosphor-eIF2α. Horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G was used as the secondary antibody. After the chemiluminescence reaction, bands were detected by exposing the blots to X-ray film. The same membrane was reused to detect β-actin by incubating it with monoclonal anti-β-actin antibody. For a quantitative analysis, the bands were detected and evaluated densitometrically with Bandscan software (Glyko Inc., Novato, CA, USA), and normalized to corresponding density of β-actin.
All data were expressed as mean ± standard deviation (SD). Each experiment was repeated three times independently. Data were compared among groups using one-way analysis of variance (ANOVA), while between-group comparisons of means were analyzed by Least Significant Difference (LSD) t test. All statistical analyses were performed by the SPSS Statistical Software version 21.0 (SPSS Inc., Chicago, USA). A value of P < 0.05 was considered statistically significant.
As shown in Figure 1A, NRK-52E cells were cultured in mannitol (5.6 mmol/L D-glucose + 19.4 mmol/L D-mannitol; Mann group), or medium containing different level of glucose (NC group: 5.6 mmol/L; H1 group: 15 mmol/L; H2 group: 25 mmol/L; H3 group: 50 mmol/L) for 48 h, or medium containing 25 mmol/L glucose for 24 h (H/24 h group) or 48 h (H/48 h group). High glucose increased the protein of α-SMA in NRK-52E cells in a dose-dependent (NC: 0.21 ± 0.01 vs. H1: 0.40 ± 0.02, P < 0.01; NC: 0.21 ± 0.01 vs. H2: 0.45 ± 0.04, P < 0.01; NC: 0.21 ± 0.01 vs. H3: 0.53 ± 0.03, P < 0.01; F = 94.13, P < 0.01) and time-dependent manner (NC/24 h: 0.15 ± 0.01 vs. H/24 h: 0.25 ± 0.01, P < 0.01; NC/48 h: 0.15 ± 0 vs. H/48 h: 0.38 ± 0.02, P < 0.01; F = 199.67, P < 0.01). The addition of mannitol had no significant effect on α-SMA (NC: 0.21 ± 0.01 vs. Mann: 0.23 ± 0.02, P = 0.36).
Figure 1.
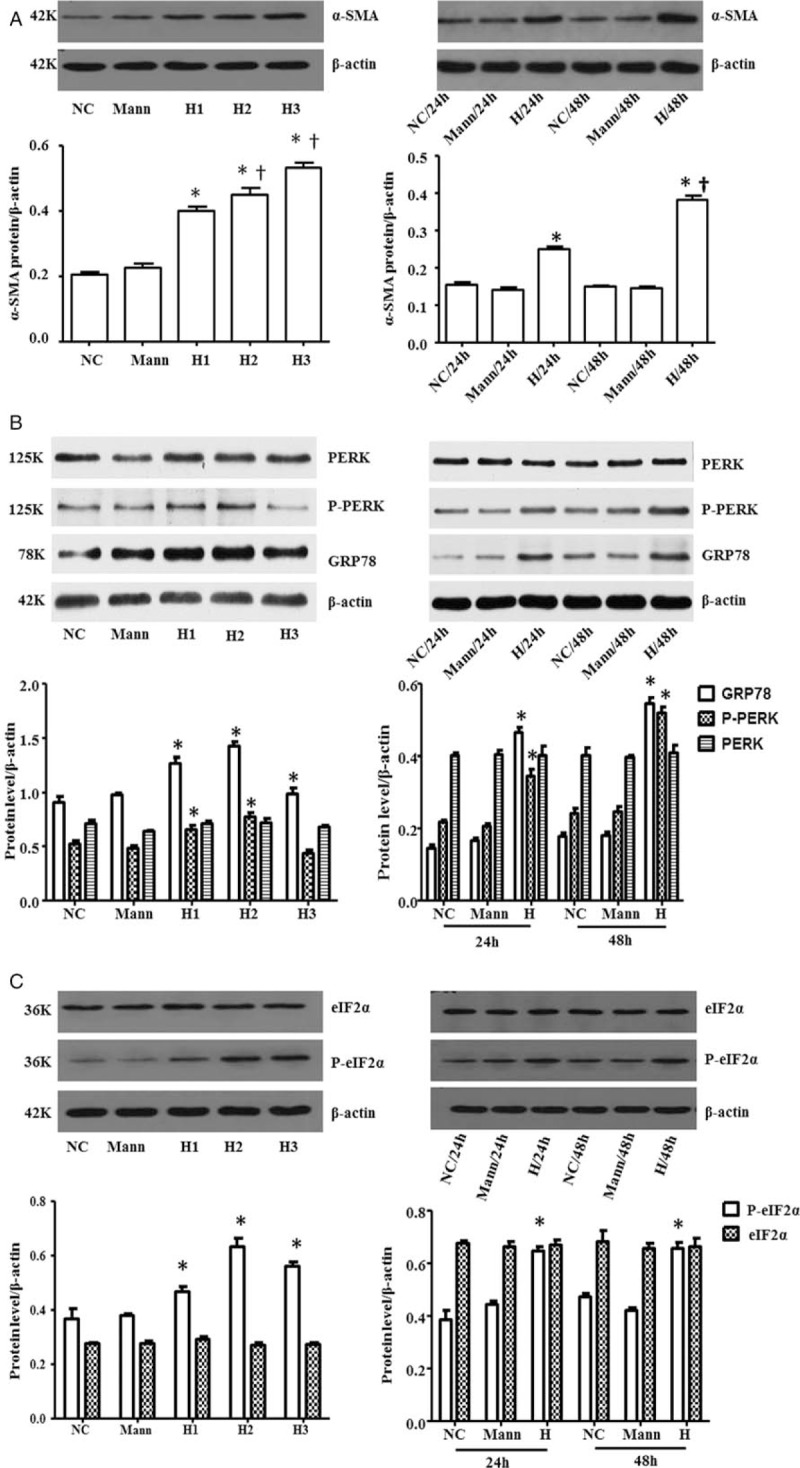
High concentration of glucose up-regulated α-SMA expression and triggered ERS in NRK-52E cells. Cells were cultured in medium with different level glucose (NC group: 5.6 mmol/L; H1 group: 15 mmol/L; H2 group: 25 mmol/L; H3 group: 50 mmol/L), or medium containing 25 mmol/L glucose in different time for 24 h (H/24 h group) or 48 h (H/48 h group). The results showed that high glucose induced the overexpression of α-SMA protein (A). The expression of GRP78 protein and phosphorylation of PERK and eIF2α were increased in high glucose medium, while the express of PERK and eIF2α protein did not change (B and C). ∗P < 0.05 vs. NC group, †P < 0.05 vs. H1 group and H/24 h group. α-SMA:α-smooth actin; ERS: Endoplasmic reticulum stress; eIF2α: Eukaryotic translation-initiation factor 2α; GRP78: 78kd-glucose-reglulated protein; Mann: Mannitol; NC: Normal control; PERK: Protein kinase R-like ER kinase.
And as shown in Figure 1B and 1C, compared with the levels of NC group, the expression of GRP78 protein (NC: 0.90 ± 0.09 vs. H1: 1.27 ± 0.09, P = 0.001; NC: 0.90 ± 0.09 vs. H2: 1.42 ± 0.07, P < 0.01; NC: 0.90 ± 0.09 vs. H3: 1.14 ± 0.06, P = 0.03; F = 25.54, P < 0.01) and phosphorylation of PERK (NC: 0.43 ± 0.06 vs. H1: 0.65 ± 0.02, P < 0.01; NC: 0.43 ± 0.06 vs. H2: 0.77 ± 0.07, P < 0.01; NC: 0.43 ± 0.06 vs. H3: 0.40 ± 0.01, P = 0.43; F = 37.45, P < 0.01), eIF2α (NC: 0.37 ± 0.06 vs. H1: 0.47 ± 0.04, P = 0.018; NC: 0.37 ± 0.06 vs. H2: 0.63 ± 0.05, P < 0.01; NC: 0.37 ± 0.06 vs. H3: 0.56 ± 0.03, P < 0.01; F = 22.07, P < 0.01) were increased with the concentration of glucose. The peak expression of these proteins appeared after cells being cultured 48 h in the 25 mmol/L glucose. However, the protein of PERK (NC: 0.71 ± 0.06 vs. H1: 0.72 ± 0.03, P = 0.86; NC: 0.71 ± 0.06 vs. H2: 0.72 ± 0.07, P = 0.89; NC: 0.71 ± 0.06 vs. H3: 0.67 ± 0.03, P = 0.37; F = 1.64, P = 0.24) and eIF2α (NC: 0.27 ± 0.01 vs. H1: 0.29 ± 0.02, P = 0.20; NC: 0.27 ± 0.01 vs. H2: 0.27 ± 0.02, P = 0.58; NC: 0.27 ± 0.01 vs. H3: 0.27 ± 0.01, P = 0.87; F = 1.09, P = 0.41) did not change. Meanwhile, mannitol (osmotic control) had no effect on all these proteins. These results suggested that high glucose-induced ERS-related protein overproduction was not due to high osmotic stress.
To examine whether ERS induced overexpression of α-SMA, NRK-52E cells were treated with the thapsigargin (Thaps), which was the inducing agent of ER-stress. Cells were incubated for 24 h or 48 h in Thaps concentration of 0.1 and 0.2 μmol/L respectively (Thaps 0.1/24 h group, Thaps 0.2/24 h group, Thaps 0.1/48 h group, Thaps 0.2/48 h group). The levels of α-SMA protein were all upregulated (NC: 0.35 ± 0.03 vs. Thaps 0.1/24 h: 0.80 ± 0.08, P < 0.01; NC: 0.35 ± 0.03 vs. Thaps 0.2/24 h: 0.63 ± 0.07, P < 0.01; NC: 0.35 ± 0.03 vs. Thaps 0.1/48 h: 0.72 ± 0.08, P < 0.01; NC: 0.35 ± 0.03 vs. Thaps 0.2/48 h: 0.52 ± 0.04, P < 0.01; F = 27.66, P < 0.01). The production of α-SMA was the highest when Thaps concentration was 0.1 μmol/L and the incubation time was 24 h (0.83 ± 0.08). To determine the mechanism of Thaps-induced α-SMA, we used GSK2606414 in subsequent intervene experiment. Cells were pre-treated with GSK2606414 (30 min), then cultured with Thaps. The overexpression of α-SMA was blocked greatly (0.91 ± 0.04 vs. 0.60 ± 0.03, P < 0.01), indicating that the PERK-e IF2α pathway was partly essential for ER-stress induced α-SMA activation [Figure 2A].
Figure 2.
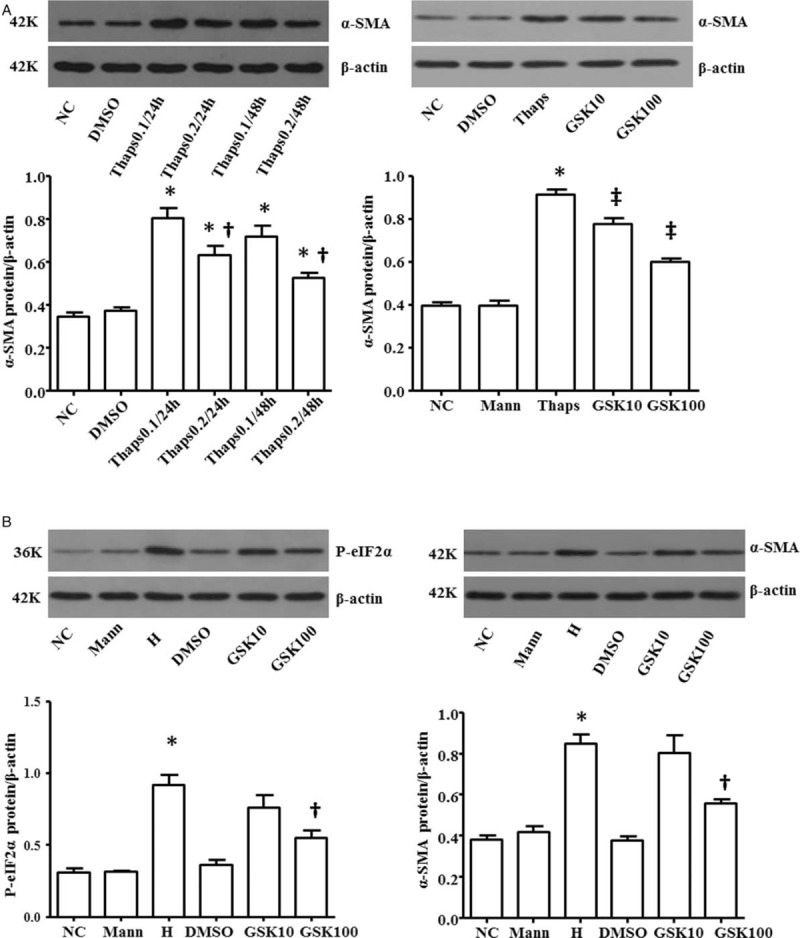
Role of PERK- eIF2α pathway on α-SMA overexpression induced by Thaps and high glucose. Cells were incubated for 24 h or 48 h in different concentration of Thaps (Thaps 0.1/24 h group; Thaps 0.2/24 h group; Thaps 0.2/24 h group; Thaps 0.2/48 h group). The production of α-SMA was the highest when Thaps concentration was 0.1 mol/L and the incubation time was 24 h. Cells were pre-treated by GSK2606414 followed by cultured with Thaps (Thaps group) or high glucose (H group), the overexpression of α-SMA induced by Thaps was blocked greatly. ∗P < 0.05 vs. NC group, †P < 0.05 vs. Thaps 0.1/24 h group, ‡P < 0.05 vs. Thaps group (A). GSK2606414 treatment inhibited high glucose-induced phosphorylation of eIF2α and α-SMA. ∗P < 0.05 vs. NC group, †P < 0.05 vs. H group (B). α-SMA:α-smooth actin; eIF2α: Eukaryotic translation-initiation factor 2α; PERK: Protein kinase R-like ER kinase; Thaps: Thapsigargin.
Cells were pre-treated with different concentration of GSK2606414 (10 nmol/L, 100 nmol/L) for 30 min, then cultured with high glucose for 24 h. We observed that GSK2606414 (100 nmol/L) treatment inhibited high glucose-induced phosphorylation of eIF2α (0.55 ± 0.09 vs. 0.92 ± 0.13, P = 0.001) and α-SMA (0.56 ± 0.04 vs. 0.85 ± 0.08, P = 0.001) expression. The data indicated that high glucose activated α-SMA expression through PERK-e IF2α pathway [Figure 2B].
DKD is currently the leading cause of irreversible ESRD (end-stage renal disease) requiring dialysis. Rather than a solely glomerular disease, tubulointerstitial injury is also a major characteristic of DKD and an important predictor of renal dysfunction. The severity of tubulointerstitial fibrosis correlates with progressive renal damage. Various studies have demonstrated that EMT is a direct contributor to the kidney myofibroblast accumulation in the development of renal fibrosis, including DKD. In this process of EMT, tubular cells lost their cell markers and expressed a high level of mesenchymal markers, such as α-SMA. The present study showed that α-SMA as an index of fibrosis was increased in NRK-52E cells exposed to high glucose, which was consistent with previous research.[9]
ER is a vast membranous network that is recognized as a protein-folding factory. A number of pathophysiological conditions are associated with ER stress, including diabetes and Alzheimer's disease. GRP78 is an important molecular indicator of ERS. The most immediate response to ER stress is the homodimerization and trans-phosphorylation of PERK, which is a Ser/Thr protein kinase and its activation results in the phosphorylate of eIF2α. Lakshmana et al[6] reported that the phosphorylation of myocardial PERK and eIF2α was shown to be significantly increased in the Spontaneous Diabetic Torill (SDT) rats, when compared with the SD rats. Loss of PERK in humans leads to Wallcot-Rallison syndrome (WRS), a disorder involving neonatal insulin-dependent diabetes resulting from destruction of pancreatic islet beta cells.[5] These researches indicate that PERK-eIF2α pathway is closely related to diabetes. In the present study, we demonstrated that high glucose increased the levels of GRP78 protein expression and PERK, eIF2α phosphorylation. Otherwise, mannitol had no effect on all these proteins, which suggests these effects were not induced by hyperosmosis.
Moderate ER stress can maintain cell homeostasis. When the stress is excessive or prolonged, it can initiate pathologic reactions. Recently, ERS has been revealed to be closely linked to inflammatory and stress signal networks, including the action of JNK-AP1, NF-κB pathways. Furthermore, the relationship between ERS and renal damage is also gradually concerned. It has been proved that ERS plays a vital role in the development of renal fibrosis. And inhibition of ERS can improve renal fibrosis progression.[10] Fang et al[11] reported ER stress seemed to play an important role in albuminuria-induced kidney epithelial cells injury. In order to clarify the relationship between ERS and EMT, we used ERS-inducer thapsigargin to incubate NRK-52E cells. The results showed that thapsigargin increased the level of α-SMA protein. Subsequently, we used a potent selective PERK inhibitor–GSK2606414 to pre-treat cells. After co-cultured with GSK2606414, the increased level of α-SMA induced by thapsigargin was blocked significantly. These results manifested that thapsigargin stimulated overexpression of α-SMA by inducing ERS, which is partially through the PERK-eIF2α pathway.
However, it has remained unclear how hyperglycemia induces EMT mediated by ER stress. In cancer cells, EMT can drive tumor metastasis, and disruption of PERK-eIF2α axis compromises the malignant phenotype of EMT cancer cells. Because of that, PERK pathway inhibitors warrant further exploration as potential cancer therapies.[12] Qi et al[13] proved that 4-PBA exerted a marked renoprotective effect through suppressing the expression of p-PERK in STZ-induced diabetic rats. Furthermore, as is well-known, P58IPK is an important component whose intracellular effect on ERS was achieved by inhibiting PERK activation. The ultimate result of a lack of P58IPK is deficiency of insulin, which mimics β-cell failure in type 1 and late-stage type 2 diabetes.[4] Yang et al[7] revealed the protecting role of P58IPK against ER stress-mediated diabetic retinopathy. In the present study, NRK-52E cells were cultured for 48 h with high glucose after they were pre-treated with GSK2606414. We observed that GSK2606414 inhibited high glucose-induced P-eIF2α and α-SMA expression. Based on these researches, we hypothesized that high glucose induced α-SMA overexpression in NRK-52E cells partly through the PERK-eIF2α pathway in the process of ERS. Our results provided some evidences to support this theory. Firstly, it had been proved that high concentration glucose triggered ERS in NRK-52E cells. Secondly, our results indicated that ERS and high glucose both induced renal proximal tubular cells to undergo EMT. Lastly, the study proved that GSK2606414 treatment reduced significantly overexpression of P-eIF2α and α-SMA protein stimulated by high glucose.
In conclusion, our findings confirmed that high glucose stimulated α-SMA overexpression and ER stress in NRK-52E cells. In addition, this study also showed that high glucose-induced EMT was at least partially through the PERK-eIF2α pathway. As is well-known, EMT contributes to the development of tubulointertial injury. Collectively, the finding suggests inhibiting ER stress is a potential novel therapy of alleviating DKD. This experiment was only done in cells. In the future, we expect that animal experiments will be conducted to further explore the corresponding mechanism.
Funding
This work was supported by grants from the Natural Science Foundation of Hubei Province (No. 2017CFB779) and the Fundamental Research Funds for the Central Universities (No. 2042017kf0133).
Conflicts of interest
None.
Footnotes
How to cite this article: Bao Y, Ao Y, Yi B, Batubayier J. High levels of glucose induce epithelial-mesenchymal transition in renal proximal tubular cells through PERK-eIF2α pathway. Chin Med J 2019;00:00–00. doi: 10.1097/CM9.0000000000000157
References
- 1.Hills C, Price GW, Wall MJ, Kaufmann TJ, Chi-Wai TS, Yiu WH, et al. Transforming growth factor Beta 1 drives a switch in connexin mediated cell-to-cell communication in tubular cells of the diabetic kidney. Cell Physiol Biochem 2018; 45:2369–2388. doi: 10.1159/000488185. [DOI] [PubMed] [Google Scholar]
- 2.Lovisa S, LeBleu VS, Tampe B, Sugimoto H, Vadnagara K, Carstens JL, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med 2015; 21:998–1009. doi: 10.1038/nm.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xu FF, Liu XH. Calreticulin translocation aggravates endoplasmic reticulum stress-associated apoptosis during cardiomyocyte hypoxia/reoxygenation. Chin Med J 2015; 128:353–360. doi: 10.4103/0366-6999.150103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ladiges WC, Knoblaugh SE, Morton JF, Korth MJ, Sopher BL, Baskin CR, et al. Pancreatic beta-cell failure and diabetes in mice with a deletion mutation of the endoplasmic reticulum molecular chaperone gene P58IPK. Diabetes 2005; 54:1074–1081. [DOI] [PubMed] [Google Scholar]
- 5.Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, et al. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol 2002; 22:3864–3874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lakshmanan AP, Harima M, Suzuki K, Soetikno V, Nagata M, Nakamura T, et al. The hyperglycemia stimulated myocardial endoplasmic reticulum (ER) stress contributes to diabetic cardiomyopathy in the transgenic non-obese type 2 diabetic rats: a differential role of unfolded protein response (UPR) signaling proteins. Int J Biochem Cell Biol 2013; 45:438–447. doi: 10.1016/j.biocel.2012.09.017. [DOI] [PubMed] [Google Scholar]
- 7.Yang H, Liu R, Cui Z, Chen ZQ, Yan S, Pei H, et al. Functional characterization of 58-kilodalton inhibitor of protein kinase in protecting against diabetic retinopathy via the endoplasmic reticulum stress pathway. Mol Vis 2011; 17:78–84. [PMC free article] [PubMed] [Google Scholar]
- 8.Liu J, Yang JR, Chen XM, Cai GY, Lin LR, He YN. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am J Physiol Cell Physiol 2015; 308:C621–C630. doi: 10.1152/ajpcell.00096.2014. [DOI] [PubMed] [Google Scholar]
- 9.Zhang W, Song S, Liu F, Liu Y, Zhang Y. Beta-casomorphin-7 prevents epithelial-mesenchymal transdifferentiation of NRK-52E cells at high glucose level: involvement of AngII-TGF-β1 pathway. Peptides 2015; 70:37–44. doi: 10.1016/j.peptides.2015.04.002. [DOI] [PubMed] [Google Scholar]
- 10.Liu QF, Ye JM, Deng ZY, Yu LX, Sun Q, Li SS. Ameliorating effect of Klotho on endoplasmic reticulum stress and renal fibrosis induced by unilateral ureteral obstruction. Iran J Kidney Dis 2015; 9:291–297. [PubMed] [Google Scholar]
- 11.Fang L, Xie D, Wu X, Cao H, Su W, Yang J. Involvement of endoplasmic reticulum stress in albuminuria induced inflammasome activation in renal proximal tubular cells. PLoS One 2013; 8:e72344.doi: 10.1371/journal.pone.0072344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Feng YX, Sokol ES, Del VCA, Sanduja S, Claessen JH, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK-eIF2α and sensitizes cells to endoplasmic reticulum stress. Cancer Discov 2014; 4:702–715. doi: 10.1158/2159-8290.CD-13-0945. [DOI] [PubMed] [Google Scholar]
- 13.Qi W, Mu J, Luo ZF, Zeng W, Guo YH, Pang Q, et al. Attenuation of diabetic nephropathy in diabetes rats induced by streptozotocin by regulating the endoplasmic reticulum stress inflammatory response. Metabolism 2011; 60:594–603. doi: 10.1016/j.metabol.2010.07.021. [DOI] [PubMed] [Google Scholar]