

Abstract
Down syndrome (DS) or Trisomy 21 is a developmental disorder leading to cognitive deficits, including disruption of hippocampus-dependent learning and memory. Enhanced inhibition has been suggested to underlie these deficits in DS based on studies using the Ts65Dn mouse model. Here we show that, in this mouse model, GABAergic synaptic inhibition onto dendrites of hippocampal pyramidal cells is increased. By contrast, somatic inhibition was not altered. In addition, synaptic NMDAR currents were reduced. Furthermore, dendritic inhibition was mediated via nonlinear α5-subunit containing GABAARs that closely matched the kinetics and voltage dependence of NMDARs. Thus, enhanced dendritic inhibition and reduced NMDA currents strongly decreased burst-induced NMDAR-mediated depolarization and impaired LTP induction. Finally, selective reduction of α5-GABAAR-mediated inhibition rescued both burst-induced synaptic NMDAR activation and synaptic plasticity. These results demonstrate that reduced synaptic NMDAR activation and synaptic plasticity in the Ts65Dn mouse model of DS can be corrected by specifically targeting nonlinear dendritic inhibition.
SIGNIFICANCE STATEMENT Mild to moderate intellectual disability is a prominent feature of Down syndrome. Previous studies in mouse models suggest that increased synaptic inhibition is a main factor for decreased synaptic plasticity, the cellular phenomenon underlying memory. The present study shows that increased inhibition specifically onto dendrites together with reduced NMDAR content in excitatory synapses may be the cause. Reducing a slow nonlinear component that is specific to dendritic inhibitory inputs and mediated by α5 subunit-containing GABAA receptors rescues both NMDAR activation and synaptic plasticity.
Keywords: alpha5 GABAA receptors, CA1 pyramidal cells, dendritic inhibition, Down syndrome, hippocampus, NMDA receptors
Introduction
Trisomy of chromosome 21, also known as Down syndrome (DS), is the most common genetic neurodevelopmental disorder associated with mild-to-moderate intellectual disability (Lott and Dierssen, 2010). While implicit learning is relatively preserved, there are strong deficits in explicit learning capabilities, including hippocampus-dependent tasks. Triplication of the human chromosome 21 might increase expression of developmental genes regulating size and fate of neural progenitor cell pools (Dierssen, 2012). In addition to smaller brain size and reduced neurogenesis, an imbalance between the cortical excitatory and inhibitory synaptic transmission (E/I balance) has been implicated as a major cause of cognitive deficits in DS (Kleschevnikov et al., 2004; Costa and Grybko, 2005; Fernandez et al., 2007; Dierssen, 2012; Bartesaghi et al., 2015) similar to other neurodevelopmental disorders, including autism and schizophrenia (Lewis, 2014; Braat and Kooy, 2015; Pocklington et al., 2015).
In the mouse, ∼50% of the ortholog genes of the human chromosome 21, including the DS critical region are located on the chromosome 16 (Reeves et al., 1995; Chakrabarti et al., 2007). Several mouse models of DS have been developed, including the widely used Ts65Dn mouse, which has an additional fragment of the murine chromosome 16 containing ∼100 ortholog genes (Chakrabarti et al., 2007; Gardiner, 2014) and recapitulates the hallmarks of the DS phenotype, including cognitive impairments (Reeves et al., 1995; Dierssen, 2012). Although the precise contributions of all triplicated genes have not been clearly identified, there is evidence that the number of medial ganglionic eminence-derived somatostatin (SOM) and parvalbumin (PV)-expressing GABAergic interneurons is increased due to enhanced expression of the transcription factors Olig1 and Olig2 in interneuron progenitors (Chakrabarti et al., 2010). In addition, there may be an increased number of NO-synthase (NOS)/NPY-positive neurogliaform cells, which derive from the same progenitor pool in the medial ganglionic eminence (Tricoire et al., 2010; Raveau et al., 2018). Similarly, the triplicated expression of the serine/threonine kinase DYRK1A affects interneuron differentiation and shifts the E/I balance toward more inhibition (Souchet et al., 2014).
On a functional level, it has been shown that hippocampal LTP is reduced in Ts65Dn mice in a GABAA-receptor-dependent manner (Kleschevnikov et al., 2004, 2012; Costa and Grybko, 2005; Fernandez et al., 2007). Furthermore, partial block of GABAA receptors did not only rescue hippocampal LTP, but also reversed learning deficits in Ts65Dn mice (Fernandez et al., 2007). However, it is largely unknown which functional properties of inhibitory transmission onto hippocampal and cortical pyramidal neurons are altered in Ts65Dn mice.
Pharmacological evidence indicates that specific subtypes of GABAA receptors may be involved in DS phenotypes. Behavioral studies have shown that pharmacological modulation of α5-containing GABAA receptors (α5-GABAARs) ameliorates cognitive deficits in Ts65Dn mice and rescues impaired LTP induction (Braudeau et al., 2011; Martínez-Cué et al., 2014; Rudolph and Möhler, 2014). Furthermore, it was shown that genetic ablation of the α5-subunit of GABAARs in TS mice partially rescues learning as well as LTP deficits (Vidal et al., 2018) pointing to a specific role of these GABA receptors in DS. Hence, selective modulation of α5-GABAAR by negative allosteric modulators (α5-NAM) may represent an attractive treatment strategy, as these drugs do not generate proconvulsant or anxiogenic side effects (Rudolph and Knoflach, 2011; Rudolph and Möhler, 2014).
Immunohistochemistry indicates that α5-GABAARs are localized in hippocampal pyramidal cell dendrites in the synaptic as well as in extrasynaptic membrane (Serwanski et al., 2006). We recently showed that these receptors mediate synaptic inhibition of pyramidal cell dendrites evoked by SOM- and NOS-positive interneurons (Schulz et al., 2018), in addition to the contribution to tonic inhibition (Caraiscos et al., 2004; Prenosil et al., 2006; Brickley and Mody, 2012). By contrast, PV-interneuron-mediated inhibition targeting pyramidal cell somata was independent of α5-GABAARs. Together with results from in vivo pharmacological studies in Ts65Dn mice, this suggests that SOM and/or NOS interneuron-mediated dendritic inhibition may contribute to cognitive deficits in DS. However, the role of dendritic inhibition in DS models has not been addressed.
In the current study, we show, for the first time, that reduced NMDAR-EPSPCs and increased dendritic inhibition converge toward reduced NMDAR-function during burst PSPs in Ts65Dn mice. We further elucidate the mechanisms by which an α5-NAM counteract the increased dendritic inhibition and rescue disturbed NMDAR-dependent LTP.
Materials and Methods
Animals.
Ts65Dn mice (B6EiC3Sn a/A-Ts(1716)65Dn) were obtained from The Jackson Laboratory and bred by backcrossing of Ts65Dn females with B6C3F1/OlaHsd males (F1 hybrid males from C57BL/6JOlaHsd ♀ × C3H/HeNHsd ♂ obtained from Harlan). Breeding was performed at the animal facilities of the University of Basel. The supernumerary chromosome 1716 in offspring was identified by standard PCR procedures as described previously (Reinholdt et al., 2011). Some Ts65Dn mice plus WT littermates were kindly provided by Carmen Martinez-Cue from the University of Cantabria for pilot experiments. For most experiments, mice were young adults of 8–15 weeks. For some experiments (see Fig. 3C–F), some younger C57BL/6 mice were also used (6–10 weeks of age).
Figure 3.

Excitability of dendrite-targeting interneurons is not increased in Ts65Dn mice. A, Membrane potential responses of an interneuron located in SR in a WT mouse to linear current steps. Inset, Relationship between membrane potential and injected current. The input resistance was derived from the slope of the regression line. B, The same for a representative SR interneuron in Ts65Dn mice. C, D, AP frequency versus current relationship (F-I curve) for current injections into the SR interneurons shown in A and B. The AP frequency was fitted by the logarithmic function, F = gain × ln(I/I0), where I0 denotes the threshold current. The estimated F-I gains from the fits are indicated. E, Scatter plots of the resting membrane potential for the group of pyramidal neurons (PN), SR interneurons (SR IN), and interneurons located in SO IN. F–H, Scatter plots of AP threshold, input resistance, and F-I gain. The statistically significant difference in the F-I gain of SR interneurons in WT versus Ts65Dn mice is indicated (*p = 0.008, unpaired two-sample t tests).
Mice were housed in groups of up to 5 animals in standard individually ventilated cages in standard laboratory conditions with a 12 h light/dark cycle, and access to food and water ad libitum. Wet chow was fed to weaned mice to ensure sufficient growth in Ts65Dn mice. All experiments were approved by the Basel Cantonal Committee on Animal Experimentation according to federal and cantonal regulations.
Slice preparation for patch-clamp recordings.
Mice were anesthetized with isoflurane (4% in O2, Vapor, Draeger) and killed by decapitation, in accordance with national and institutional guidelines. To increase cell viability for single-cell patch-clamp recordings, animals were exposed to oxygen-enriched atmosphere for 10 min before decapitation. Slices were cut as previously described (Geiger et al., 2002; Bischofberger et al., 2006). Briefly, the brain was dissected in ice-cold sucrose-based solution at ∼4°C. Horizontal 350-μm-thick hippocampal brain slices were cut at an angle of 20° to the dorsal surface of the brain along the dorsoventral axes of the hippocampus using a VT1200 vibratome (Leica Microsystems). For cutting and storage, a sucrose-based solution was used, containing the following (in mm): 87 NaCl, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 75 sucrose, 0.5 CaCl2, 7 MgCl2, and 10 glucose (equilibrated with 95% O2/5% CO2). Slices were kept at 35°C for 30 min after slicing and subsequently stored at room temperature until experiments were performed at 32°C–33°C.
Slice preparation for field potential recordings.
WT and Ts65Dn mice were anesthetized using a mixture of 2.5% isoflurane in O2 and decapitated as approved by the local institutional animal welfare committee. After removing the brain, the left side of the hippocampal formation was dissected out in a solution containing the following (in mm): 124 NaCl, 2.5 KCl, 1.25 KH2PO4, 2 MgSO4, 2.5 CaCl2, 26 NaHCO3, and 10 glucose at room temperature. Transverse slices (400-μm-thick) from the medium part of the hippocampus were cut with a TC-2 Sorvall tissue chopper (MTS), transferred to the recording chamber, and stored at room temperature for 1 h before the experiment.
Patch-clamp recordings.
CA1 pyramidal neurons were visually identified in the pyramidal cell layer close to the border of stratum radiatum (SR) using infrared differential interference contrast video microscopy. Slices were continuously superfused with ACSF at near physiological temperature (32°C-33°C). The ACSF contained the following (in mm): 125 NaCl, 25 NaHCO3, 25 glucose, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2 (equilibrated with 95% O2/5% CO2). Patch pipettes were pulled from borosilicate glass tubing with a 2.0 mm outer diameter and 0.5 mm wall thickness (Hilgenberg) on a Flaming-Brown P-97 puller (Sutter Instruments).
For most current-clamp recordings, patch pipettes (4–7 mΩ) were filled with a solution containing the following (in mm): 120 KMeSO4, 4 KCl, 10 EGTA, 10 HEPES, 2 MgCl, 10 Na2-phosphocreatine, 2 Na2ATP, 0.3 GTP, and 0.2% biocytin adjusted to pH 7.3 with KOH. For voltage-clamp recordings, patch pipettes (2–4 mΩ) were filled with a Cs gluconate-based solution containing the following (in mm): 135 CsGluc, 4 CsCl, 10 EGTA, 10 HEPES, 2 MgCl, 2 Na2ATP, 2 TEA-Cl, 5 and QX314 adjusted to pH 7.3 with CsOH.
Voltage and current signals were measured with a Multiclamp 700A amplifier (Molecular Devices), low-pass filtered with cutoff frequencies of 8 kHz, and digitized at 20 kHz using a CED Power 1401 interface (Cambridge Electronic Design). Recorded cells were only included if the initial seal exceeded a resistance of 1 GΩ, the initial membrane potential was well below −55 mV, and there were no signs of cellular deterioration (e.g., decreasing input resistance) or rundown of synaptic transmission during the recording. Bridge balance was used to compensate the series resistance (RS = 10–40 mΩ) in current-clamp recordings. Series resistance in voltage-clamp experiments (RS = 5–20 mΩ) was monitored online, and experiments were discarded if RS changed >20%. Data acquisition was controlled using IGOR Pro 6.31 (WaveMetrics) and the CFS library support from Cambridge Electronic Design.
For the assessment of cellular excitability in interneurons and pyramidal neurons (see Fig. 3), we used a potassium gluconate-based internal solution as follows (in mm): 135 potassium gluconate, 21 KCl, 2 MgCl2, 2 Na2ATP, 0.3 Na-GTP, 10 HEPES, 0.5 EGTA, adjusted to pH 7.3 with KOH. These experiments were performed in the presence of ionotropic receptor blockers 10 μm NBQX, 25 μm AP5, and 100 μm picrotoxin. Interneurons were identified by their location in SR or stratum oriens (SO). In SO, only interneurons with a horizontally elongated soma and without prominent apical dendrites were chosen according to properties of dendrite-targeting somatostatin-positive interneurons (Maccaferri et al., 2000; Lien et al., 2002). A few interneurons with a prominent fast-spiking phenotype indicative of PV+ interneurons, which mainly target axon initial segments and perisomatic compartments, were excluded from analysis.
To evaluate cellular properties, 1-s-long current steps of increasing amplitude (steps of 25 pA) were injected during current-clamp recordings. In all current-clamp protocols other than the initial assessment of cellular properties, the resting membrane potential was kept constant close to the initial potential of −67 to −70 mV by small constant current injections throughout the experiment. During pharmacological manipulations, the input resistance was regularly assessed by small current steps of 25 pA amplitude.
Extracellular synaptic stimulation.
For stimulation of synaptic inputs, 4–6 mΩ pipettes filled with HEPES-buffered Na2+-rich solution were used to apply brief negative current pulses (200 μs). To stimulate Schaffer collaterals (SCs), the pipettes were placed into SR close to the CA2-CA1 border at a distance of ≥500 μm (500–800 μm) from the recorded neuron, and stimulation was applied at intermediate intensity (50–100 μA). To record NMDA-AMPA ratios, the pipettes were placed close to the recorded cell at a distance of <200 μm in either SR or SO, and stimulation was applied at low intensity (30 μA). GABAergic IPSCs were evoked at a range of intensities (10–40 μA). To avoid the confounding influence of potentially altered GABAB receptor signaling in Ts65Dn mice (Kleschevnikov et al., 2012), 1 μm CGP54626 was added to block GABAB signaling, except for field potential recording experiments (see Fig. 6). Stimulus artifacts have been truncated in most figures for clarity.
Figure 6.
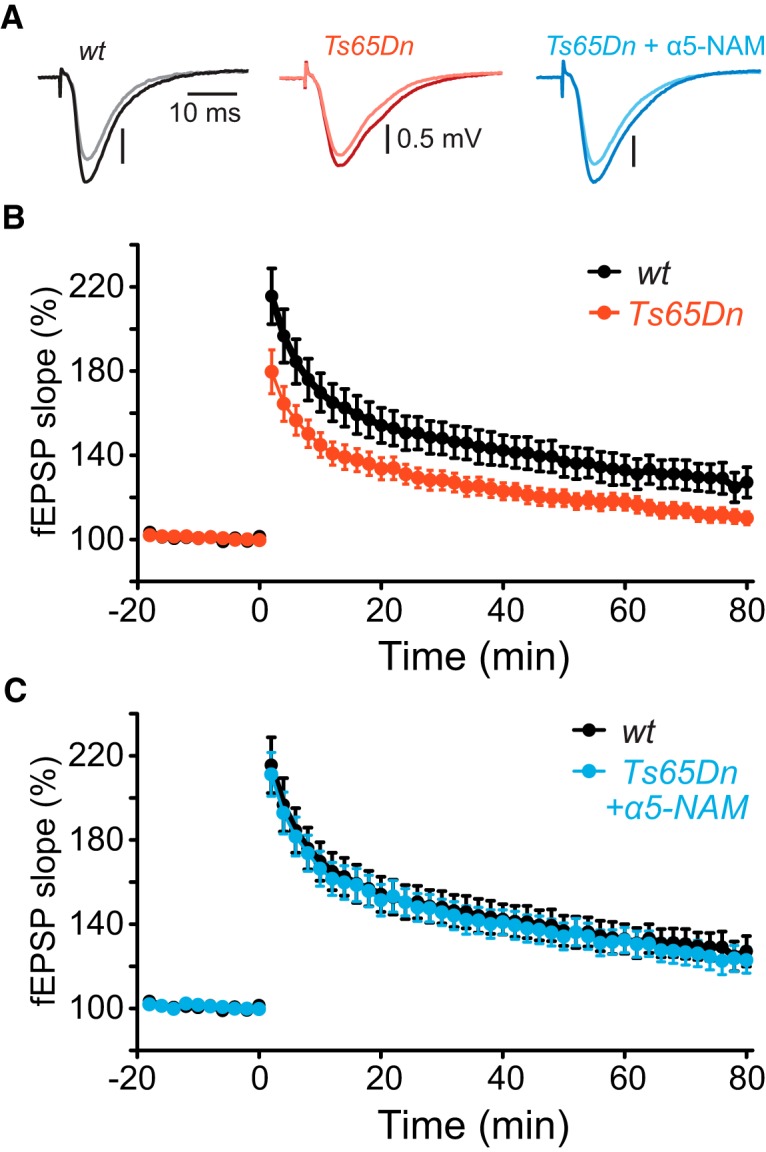
Acute application of a α5-NAM rescues NMDAR-dependent synaptic plasticity. A, The mean fEPSPs recorded in SR before (light color) and after TBS (10 × 5 pulses at 100 Hz every 200 ms; dark color) of the SC from a slice of a WT (gray), a Ts65Dn mouse (red), and a Ts65Dn mouse in the presence of the α5-NAM (blue). B, Time courses of the fEPSP slope. TBS was applied at time 0. The fEPSP slope was significantly less elevated in Ts65Dn slices (110.1 ± 3.1%, n = 19; red) compared with WT littermates (127.1 ± 7.2%, n = 14; p < 0.05, two-sample t test; black) at 80 min after induction. C, Application of the α5-NAM (0.3 μm) enhanced LTP in Ts65Dn mice up to normal levels. The potentiation of the fEPSP slope in Ts65Dn slices was increased in the presence of the α5-NAM (122.9 ± 6.2%, n = 11; blue) relative to control conditions (p < 0.05; two-sample t test), and comparable with WT slices in control condition (127.1 ± 7.2%, n = 14; black, same experiments as in B).
NMDA-AMPA ratios.
To measure the balance between NMDAR- and AMPAR-mediated currents, the neuron was clamped to −70 mV and stepped alternatingly to −90 mV and to 50 mV. Voltage steps were of 1 s duration, and a single stimulus was applied 750 ms after the voltage step onset. At −90 mV, AMPAR-mediated inward currents were recorded. At 50 mV, the NMDAR-mediated outward currents were recorded. The interstimulus interval was ≥50 s. Experiments were performed in the presence of 10 μm nimodipine to reduce dendritic Ca2+ channel activation, 100 μm picrotoxin to block GABAAR, and the NMDAR coagonist 10 μm glycine to avoid potential washout of endogenous glycine and ensure optimal synaptic NMDAR activation. PSCs were recorded in whole-cell configuration. The series resistance (5–12 mΩ) was compensated (50%–90%, 5 kHz bandwidth). The conductance was calculated as the PSC amplitude divided by the driving force (i.e., the difference between membrane potential and synaptic reversal potential). The synaptic reversal potential for glutamatergic synapses was assumed to be 0 mV.
Voltage dependence of NMDA-PSCs.
NMDAR-PSCs were recorded at increasing membrane potentials with an Axopatch 200B (see Fig. 3C). Series resistance was compensated at 90%–95%. For the analysis of the voltage dependence, only experiments with a maximal PSC amplitude of >0.5 nA were included. Membrane potentials were corrected offline by the calculated liquid junction potential of −15.7 mV (JPCalcWin) (Barry, 1994).
Measurement of α5-GABAAR-mediated IPSCs.
All IPSCs were recorded at 80% series resistance (5–12 mΩ) compensation. To assess the decay phase of outward currents, IPSCs were recorded at −20 mV before and after a wash-in phase of >5 min of RO4938581 (1 μm). Experiments were performed at 32°C–33°C, except for the assessment of the voltage dependence (see Fig. 3F), which was performed at room temperature (22°C–24°C) for greater recording stability.
Local field potential recordings and LTP induction.
fEPSPs were recorded from hippocampal slices of WT and Ts65Dn mice using glass micropipettes (Clark GC 120F; 1–4 mΩ) filled with 2 m NaCl and placed in the SR of the CA1 region. Slices were constantly superfused with an ACSF at 30°C containing the following (in mm): 120 NaCl, 3.5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1.25 NaH2PO4, 26 NaHCO3, and 10 glucose equilibrated with 95% O2/5% CO2. fEPSPs were evoked by stimulating the SR (0.033 Hz, 100 μs) with insulated bipolar platinium/iridium electrodes (RDM). The stimulus strength was adjusted to evoke fEPSPs equal to 30% of the relative maximum amplitude without superimposed population spike. This was determined from separate input–output experiments recorded for each slice. After stable baseline recordings, LTP was induced using a theta burst stimulation paradigm (TBS) consisting of a pattern of 5 stimuli at 100 Hz repeated 10 times at 200 ms intervals. The duration of the stimulation pulses was doubled during TBS without modifying the stimulation strength.
fEPSPs were amplified with a Cyberamp 380 amplifier (Molecular Devices), filtered at 2.4 kHz and digitized at 20 kHz with a Digidata 1322 acquisition board (Molecular Devices). The pClamp data acquisition software (Molecular Devices) was used to record and to analyze the signals.
Drugs.
Nimodipine (Sigma-Aldrich) was dissolved at 20 mm in DMSO on the day of the experiment. All other drugs were stored as aliquots at −20°C. d-AP5 (50 mm; Tocris Bioscience) was dissolved in water. Picrotoxin was dissolved at 50 mm in ethanol. CGP 54626 hydrochloride (10 mm; Tocris Bioscience), NBQX (20 mm; Tocris Bioscience), L-655,708 (1 mm; Tocris Bioscience), and the α5-NAM RO4938581 (10 mm; F. Hoffmann-La Roche) were dissolved in DMSO.
Data analysis.
Analysis of patch-clamp data was performed offline using the open source analysis software Stimfit (https://neurodroid.github.io/stimfit) (Guzman et al., 2014) and customized scripts written in Python. For calculations of the integral, traces were low-pass filtered (third-order Butterworth, 100 Hz cutoff) to determine the start and endpoint of the integral as the intersection of the smoothed trace with the baseline. The integral was then calculated as the sum of all values of the original trace minus the baseline in between start and endpoint. The analysis of voltage-clamp data was performed on mean waveforms. The waveform of the IPSCs mediated by α5-GABAARs was obtained by subtracting the mean IPSC waveform after the addition of an α5-NAM from the waveform recorded under control conditions. The analysis of current-clamp data was performed on single-trial data to avoid distortion by occasional action potential (AP) discharge. The analysis of the AP5-sensitive burst components after distant SC stimulation was restricted to the last PSP of the burst, where the strongest NMDAR contribution was expected. Before the calculation of the integral, occasional APs were cutoff at the AP threshold defined as the membrane potential at which the voltage slope reached 10 Vs−1. The median of all single episode measurements of a whole epoch (defined by the drug treatment) was then saved.
Standard electrophysiological parameters were determined from a family of 1 s steps of increasing amplitudes (25 pA step size). Input resistance was determined from the slope of a regression line fitted to four mean membrane potentials produced by a series of subthreshold current pulses (−25, 0, 25, 50 pA). The rheobase current was defined as the minimal current amplitude necessary to evoke an AP within 1 s pulses. AP frequency versus current relationship (F-I curve) for somatic current injections was fitted by the logarithmic function F = gain × ln(I/I0), where I0 denotes the threshold current (Engel et al., 1999). AP properties were measured on the first spike elicited by the rheobase current.
The slope of the fEPSP was measured by fitting a straight line from 40% to 70% of the peak amplitude using the Clampfit program.
Experimental design and statistical analysis.
Statistical analyses were performed in Prism 6 (GraphPad). Before statistical evaluation, data were always tested for normality by the Shapiro–Wilk normality test. In most instances, statistical estimations of significance of paired data were derived from paired two-tailed Student's t tests. For comparisons between groups, statistical tests were two-sample, two-tailed Student's t tests. Datasets that failed the Shapiro–Wilk normality test were subsequently analyzed with the nonparametric Wilcoxon Signed Rank and the Mann–Whitney tests for paired and unpaired data, respectively. The significance level was set to p = 0.05. The number of cells per group varied for different experiments typically ranging from n = 10–18 cells. Assuming an effect size of 1 SD, the statistical power ranged from 0.62 to 0.96 for comparison between genotypes, and from 0.58 to 0.99 for repeated-measures comparisons. Because of the phenotypical features of Ts65Dn mice (i.e., smaller head and brain size), a blinding of the experimenter to the genotype of mice was not possible. No randomization was performed. Ts65Dn mice and WT littermates were used alternately from the same litters. All data are shown as mean ± SEM. Unless stated otherwise, the number (n) of observations indicated reflects the number cells recorded from.
Results
Normal excitability of CA1 pyramidal neurons in Ts65Dn mice
CA1 pyramidal neurons were examined in hippocampal brain slices from adult Ts65Dn mice and WT littermates. Fundamental electrophysiological parameters, such as membrane potential, capacitance, input resistance, AP threshold, and other AP parameters (rheobase, AP amplitude, half-width, rise, and decay slope), were normal (Table 1). Therefore, it is unlikely that altered cellular excitability might contribute to the potential differences in hippocampal synaptic plasticity, learning, and memory.
Table 1.
Cellular properties of CA1 pyramidal neurons in Ts65Dn mice and WT littermatesa
| Ts65Dn (n = 27) | WT (n = 25) | |
|---|---|---|
| Response to current steps | ||
| Membrane potential (mV) | −67.7 ± 3.0 | −67.2 ± 1.9 |
| Capacitance (pF) | 231.0 ± 19.3 | 241.4 ± 12.5 |
| Input resistance (mΩ) | 95.2 ± 20.0 | 90.3 ± 22.6 |
| τmembrane (ms) | 20.9 ± 7.3 | 21.4 ± 6.5 |
| IH dependent sag (mV) | 2.3 ± 0.5 | 2.4 ± 0.8 |
| Rheobase current (pA) | 154.6 ± 45.0 | 168.0 ± 53.8 |
| Spike waveform | ||
| Firing threshold (mV) | −41.8 ± 2.5 | −41.1 ± 2.0 |
| Amplitude (mV) | 88.5 ± 8.7 | 88.4 ± 9.0 |
| Half-width (ms) | 0.83 ± 0.12 | 0.82 ± 0.12 |
aData are mean ± SD.
Reduced NMDAR activation in CA1 pyramidal neurons of Ts65Dn mice
Hippocampal LTP induced by TBS is decreased in Ts65Dn mice (Costa and Grybko, 2005; Martínez-Cué et al., 2013). This could be due to reduced activation of synaptic NMDARs or reduced downstream signaling during LTP expression. On the other hand, memantine has been shown to counteract cognitive deficits in Ts65Dn mice during fear conditioning indicating that NMDARs might be hyperactive in DS (Costa et al., 2008). To directly measure synaptic NMDAR and AMPAR activation, EPSCs were recorded after stimulation of SC inputs in SR (Fig. 1A,B) in the presence of GABAA-receptor antagonists. AMPAR-mediated PSCs evoked in SR were of similar amplitude in Ts65Dn and WT mice independent of stimulation intensity (Fig. 1C). In contrast, NMDAR-mediated PSCs tended to be smaller in Ts65Dn mice (Fig. 1D), although this effect was not statistically significant (p = 0.37, F(1,32) = 0.8224, two-way ANOVA; n1 = 16, n2 = 18) probably due to the large cell-to-cell variability of evoked current amplitudes. To increase statistical power, we normalized the NMDAR-mediated conductance by the AMPAR-mediated conductance recorded in the same cell. The NMDA-AMPA conductance ratio was 0.84 ± 0.04 (n = 27) in Ts65Dn mice and thus ∼30% smaller than in WT mice (1.18 ± 0.06, n = 31, p < 0.0001, two-sample t test; Fig. 1E). To test whether the decrease in the synaptic NMDAR content was synapse specific, we recorded also evoked synaptic inputs onto basal dendrites by stimulating in SO in a subset of neurons. The NMDA-AMPA ratio of SO inputs was not different between Ts65Dn and WT mice (1.24 ± 0.17, n = 9 vs 1.31 ± 0.08, n = 15; p = 0.7; Fig. 1F–H). Therefore, these results indicate that glutamatergic synapses show a subcellular compartment-specific reduction of the synaptic NMDAR complement.
Figure 1.

Impaired synaptic NMDAR activation in Ts65Dn mice. A, Experimental design. SCs were locally stimulated in SR in the presence of the GABAA antagonist picrotoxin (100 μm). B, AMPAR-mediated PSCs and NMDAR-mediated PSCs were measured in pyramidal neurons at −90 mV and 50 mV, respectively. C, Input–output relationship of AMPAR PSC amplitude evoked in SR versus stimulating current strength. D, Input–output relationship of NMDAR PSCs. E, The mean NMDA-AMPA conductance ratio of PSCs evoked in SR using 30 μA pulses was significantly smaller in Ts65Dn mice (red; n = 27) than in WT littermates (p < 0.0001; two-sample t test, n = 31). F, Scatter plot of the mean NMDA-AMPA conductance ratio measured during SR versus SO stimulation (30 μA) in the same WT neurons (n = 15 neurons). There was no positive correlation of the NMDA-AMPA conductance ratios between stimulation sites (R2 = 0.24, p = 0.06, slope = −0.48 ± 0.24), indicating that largely independent sets of inputs had been activated. Group means of NMDA-AMPA conductance ratios measured in SR and SO are indicated by horizontal and vertical dashed lines, respectively. G, The same scatter plot for NMDA-AMPA conductance ratios recorded in neurons from Ts65Dn brain slices (red; n = 9). For comparison, group means and values from individual WT neurons are shown in light gray. SO NMDA-AMPA conductance ratios in Ts65Dn mice scatter around the group mean measured in WT slices (vertical dashed line), whereas the SR NMDA-AMPA conductance ratios in Ts65Dn mice lie consistently below the group mean measured in WT slices (horizontal dashed line). H, There was no statistically significant difference of NMDA-AMPA conductance ratio of PSCs evoked in SO between neurons in Ts65Dn and WT littermates (p = 0.71; two-sample t test). I, Postsynaptic potentials evoked by burst stimulation (5 pulses, 50 Hz) in SR in control condition (black) and after the application of the NMDAR-antagonist AP5 (50 μm, green) reveal a NMDAR-mediated PSP component (shaded area) in WT (n = 10 neurons) but not in Ts65Dn mice (n = 13). Experiments were performed in the presence of 1 μm CGP54626. J, Group means of the NMDAR-mediated contribution to the amplitude and integral of the last PSP. Statistical significant contributions are indicated (*p < 0.05, one-sample t tests).
Next, we studied the contribution of NMDARs to burst EPSPs evoked by SC activation in SR in the absence of GABAAR antagonists. Burst stimulation (5 pulses at 50 Hz) revealed a significant NMDAR contribution in WT mice to both, the peak amplitude and integral of the fifth burst PSP of 6.2 ± 2.5% (p = 0.038; one-sample t test, n = 10) and 15.5 ± 2.5% (p < 0.001), respectively (Fig. 1I). However, in Ts65Dn mice, the contribution of NMDARs to amplitude and integral of burst PSPs was virtually absent (both: p > 0.6, n = 15; Fig. 1I,J). Together, these results show that NMDAR recruitment is deficient in response to inputs from CA3, the major input source of CA1 pyramidal neurons.
Increased dendritic inhibition in Ts65Dn mice
Increased inhibition has been proposed to be the main cause for impaired synaptic plasticity in Ts65Dn mice. Although there is substantial evidence showing that reduction of GABA signaling rescues LTP deficits in Ts65Dn mice (Kleschevnikov et al., 2004; Costa and Grybko, 2005; Fernandez et al., 2007; Martínez-Cué et al., 2013), GABAAR-mediated IPSCs have rarely been investigated in CA1 pyramidal neurons of Ts65Dn mice (Chakrabarti et al., 2010; Best et al., 2012). Therefore, we systematically analyzed GABAAR-mediated IPSCs in CA1 pyramidal cells, evoked either in dendritic layers (SR) or close to the soma (SP) with different stimulation intensities. As shown in Figure 2, dendritic IPSCs evoked in SR increased with increasing stimulation intensity (p < 0.0001, F(3,72) = 77.46, two-way ANOVA; Fig. 2C). In addition, there was a significant genotype × stimulation intensity interaction (p < 0.0001, F(3,72) = 8.316). Post hoc tests revealed that SR-IPSCs were larger in Ts65Dn mice compared with littermate controls at stimulation intensities of 30 μA (610.1 ± 75.5 pA, n = 12 vs 395.0 ± 60.8 pA, n = 14, p = 0.043) and 40 μA (713.1 ± 83.9 pA, n = 12 vs 464.3 ± 68.4 pA, n = 14, p = 0.016, Sidak's multiple-comparisons test). By contrast, for somatic IPSCs evoked in SP, there was neither a significant main effect of genotype (p = 0.35, F(1,21) = 0.9191, n1 = 9, n2 = 14), nor a significant genotype × stimulation intensity interaction (p = 0.74, F(3,63) = 0.4194; Fig. 2D). These results show that dendritic inhibition is significantly increased in Ts65Dn mice, whereas somatic inhibition is unchanged.
Figure 2.

Increased dendritic but not somatic inhibition in Ts65Dn mice. A, Left, Experimental design. Axon collaterals of local interneurons targeting the dendritic compartment were stimulated in in SR. IPSCs evoked at increasing stimulation strength were recorded at 0 mV. Right, Representative mean IPSCs recorded from pyramidal neurons in Ts65Dn and WT mice. B, The same for local stimulation of interneuron axon collaterals targeting the perisomatic compartment in stratum pyramidale (SP). C, Input–output relationship of IPSC amplitude evoked in SR versus stimulating current strength. A two-way ANOVA revealed a significant genotype × stimulation intensity interaction (*p < 0.0001, F(3,72) = 8.316). Statistically significant differences between neurons from Ts65Dn (n = 12) and WT mice (n = 14) are indicated for specific stimulation current intensities (p < 0.05; Sidak's multiple-comparisons test). D, Input–output relationship of IPSC amplitude evoked in SP versus stimulating current strength (WT, n = 14; Ts65Dn, n = 9).
Increased dendritic inhibition is not caused by increased excitability of dendrite-targeting interneurons Ts65Dn mice
Increased inhibition during local electric stimulation could be potentially the result of an enhanced recruitment of inhibitory fibers due to altered cellular excitability of interneurons in Ts65Dn mice. To test for this possibility, we recorded from interneurons located in SR in the presence of ionotropic receptor blockers (10 μm NBQX, 25 μm AP5, 100 μm picrotoxin; Fig. 3). There was no difference in resting membrane potential (Fig. 3A,B,E), AP voltage threshold (Fig. 3F), input resistance (Fig. 3G), or rheobase (100.0 ± 11.3 pA, n = 17 vs 92.5 ± 8.8 pA, n = 10; p = 0.65, unpaired two-sample t test) between SR interneurons in Ts65Dn and WT animals. Surprisingly, the analysis of the relationship of AP frequency versus current (F-I curve) for somatic current injections (Fig. 3C,D,H) showed a decreased frequency gain per e-fold current increase in SR interneurons in Ts65Dn mice (41.3 ± 2.2 Hz, n = 17 vs 52.6 ± 3.6 Hz, n = 10; p = 0.008, unpaired two-sample t test). This difference indicates that excitability of SR interneurons is not increased in Ts65Dn mice, but instead decreased during continuous current injection. Under the same recording conditions, we did not find any indication for increased intrinsic excitability in pyramidal neurons or dendrite-targeting interneurons located in SO (Fig. 3E–H), either. Together, these results indicate that increased dendritic inhibition in Ts65Dn mice is not due to increased cellular excitability and differential recruitment of dendrite-targeting interneurons.
Dendritic α5-GABAAR-mediated IPSCs match NMDAR properties
We have previously shown that dendritic inhibition in CA1 pyramidal cells is largely mediated via α5-GABAAR (Schulz et al., 2018). By contrast, perisomatic IPSCs evoked in SP were not affected by α5-GABAAR-selective modulators (Schulz et al., 2018). Therefore, we aimed to reduce enhanced dendritic inhibition in Ts65Dn mice by application of the α5-NAM RO4938581 (0.6 μm), resulting in a decrease of dendritic IPSCs by 19.7 ± 3.8% at a membrane potential of −70 mV (n = 9) (Ballard et al., 2009; Schulz et al., 2018). These receptors were shown to have slow-gating kinetics and nonlinear voltage dependence. Therefore, we compared the decay time course of NMDAR-mediated currents with the time course of α5-GABAAR-mediated dendritic inhibition. As shown in Figure 4, the decay time constant of NMDA EPSCs in Ts65Dn mice was not significantly different from WT littermates (46.2 ± 2.2 ms, n = 9, vs 46.9 ± 2.5 ms, n = 15, p = 0.85, two-sample t test; Fig. 4A,B). Dendritic IPSCs evoked in SR showed a faster time course (28.0 ± 0.8 ms, n = 5; Fig. 4D,E) than NMDARs. However, the α5-NAM-sensitive component was significantly slower (p = 0.001, paired t test) and showed a similar decay time constant (56.2 ± 3.1 ms, n = 5) to NMDAR-mediated currents (Fig. 4B, black vs Fig. 4E, light blue). We further analyzed the current-voltage relationship of GABAergic currents evoked in SR, revealing a pronounced nonlinear voltage dependence. The maximal slope of the IV-curve equivalent to the maximal conductance was reached at −50 mV, very similar to the voltage-dependent activation threshold of NMDARs (Fig. 4C vs Fig. 4F).
Figure 4.

α5-GABAR-mediated dendritic inhibition matches slow decay and voltage dependence of NMDARs. A, Mean NMDAR-mediated PSCs recorded from a WT and a Ts65Dn pyramidal neuron after stimulation in SR. Biexponential fits to the decay phase (lighter thick lines) and weighted decay taus are indicated. B, Group data show that there was no difference in the weighted decay tau between WT (n = 15) and Ts65Dn (p = 0.8, n = 9; two-sample t test). C, Current-voltage relationship for NMDAR-mediated PSCs recorded in the presence of picrotoxin (100 μm) and NBQX (10 μm). D, Mean IPSCs evoked in the outer third of SR before and after the addition of an α5-NAM (blue represents RO4938581, 0.6 μm) in the presence of NBQX (10 μm) and AP5 (25 μm). Biexponential fits to the decay phase (lighter thick lines) and weighted decay taus are indicated. Bottom, The difference between the mean IPSCs before and after addition of the α5-NAM reveals the slow dynamics of the component that is mediated by α5-GABARs. E, Group data show that the α5-NAM significantly reduced the decay tau (*p < 0.001; paired t test, n = 5). The α5-GABAR-mediated IPSCs had a much slower decay tau than the control IPSCs (*p = 0.001; paired t test). F, Current-voltage relationship for pharmacologically isolated α5-GABAR-IPSCs (blue represents 0.6 μm RO4938581, n = 6; turquoise represents 50 nm L-655,708, n = 3). A linear fit to the outward IPSCs above −50 mV is indicated (dashed line). There is a strong deviation of inward currents at more hyperpolarized membrane potentials, indicating outward rectification. Inset, The grand mean of α5-GABAR-IPSCs from experiments with application of 50 nm L-655,708. The membrane potential was corrected by the calculated liquid junction potential of −15.7 mV in C, F.
This suggests that the absence of NMDAR activation during burst PSPs in pyramidal cells in Ts65Dn mice (Fig. 1I,J) is due to a synergistic action of reduced synaptic NMDAR currents (Fig. 1D,E) and increased dendritic IPSCs (Fig. 2).
Rescue of impaired NMDAR activation in Ts65Dn mice by α5-NAM
To directly study whether dendritic inhibition interferes with NMDAR activation in Ts65Dn mice via slow α5-GABAAR, we tested the effect of 1 μm of the α5-NAM RO4938581 on SC-evoked burst PSPs, which was reported to block ∼50% of the α5-GABAR-mediated currents (Ballard et al., 2009; Schulz et al., 2018). Strikingly, application of the α5-NAM restored the NMDAR activation during burst PSPs in Ts65Dn mice up to the level found in WT littermates under control condition (Fig. 5A). The presence of the α5-NAM allowed NMDARs to contribute with 9.7 ± 3.7% (n = 8) significantly to the PSP amplitude in Ts65Dn mice (p = 0.035, one-sample t test; Fig. 5B). Likewise, the reduction of slow α5-GABAAR-mediated currents resulted in a significant NMDAR contribution to the integral in Ts65Dn mice (11.8 ± 4.8%, p = 0.008, Wilcoxon Signed Rank test), comparable with the one in WT mice under control conditions (15.5 ± 2.5%, n = 10). This shows that blocking 50% of the α5-GABARs in dendritic synapses is sufficient to compensate for increased dendritic inhibition as well as for the decreased NMDAR current density in SC synapses.
Figure 5.

Reduction of α5-GABAR-dependent inhibition increases synaptic NMDAR activation and spike output in Ts65Dn mice. A, Postsynaptic potentials evoked by burst stimulation (5 pulses, 50 Hz) of the SC in control condition (gray) were enhanced by the acute application of the α5-NAM (1 μm RO4938581; blue). Under this condition, application of the NMDAR-antagonist AP5 (50 μm, green) revealed an NMDAR-mediated component in both WT and Ts65Dn mice (blue shaded area). B, Group means of the NMDAR-mediated contribution to the amplitude and integral of the last PSP. All significant contributions by NMDAR to the PSP in WT (n = 7) and Ts65Dn mice (n = 8) are indicated: *p < 0.05 (one-sample t test). For comparison, the NMDAR-mediated component in the absence of the α5-NAM is indicated by the dashed lines (from Fig. 1E). C, Example voltage traces of theta burst-evoked AP firing in pyramidal cells during SC stimulation before and after the addition of the α5-NAM (1 μm) in Ts65Dn mice. D, Group means show a significantly increased spike rate (p = 0.022; paired t test, n = 5). E, The input resistance was not significantly increased by the α5-NAM (p = 0.76, n = 8; paired t test) in Ts65Dn mice. However, there was a small increase of the input resistance in WT pyramidal neurons (p < 0.005; paired t test, n = 17).
Next, we investigated the effect of the α5-NAM on spiking output evoked by TBS of SC fibers (5 stimuli at 50 Hz every 200 ms), a physiologically relevant activity pattern often used for induction of synaptic plasticity (Fig. 5C). Application of the α5-NAM effectively increased firing frequency by a factor of 2 (13.9 ± 2.5 Hz vs 6.7 ± 2.0 Hz, n = 5, p = 0.022, paired t test; Fig. 5D). As the input resistance in pyramidal cells of Ts65Dn mice was not significantly increased by the α5-NAM (p = 0.76, n = 8, paired t test; Fig. 5E), this indicates that decreased dendritic inhibition powerfully increase postsynaptic depolarization and output firing.
Finally, we measured TBS-induced LTP of synaptic field potentials in SR (Raymond and Redman, 2006) (Fig. 6). Consistent with previous studies (Costa and Grybko, 2005; Martínez-Cué et al., 2013), the relatively mild TBS paradigm induced LTP in slices from WT littermates (127.1 ± 7.2% at 80 min, n = 14), but failed to induce LTP in Ts65Dn mice (110.1 ± 3.1% of baseline control, n = 19, p < 0.05, two-tailed Student's t test; Fig. 6A,B). Reducing α5-GABAAR-mediated inhibition by acute application of the α5-NAM (0.3 μm) compensated for both dendritic overinhibition and smaller NMDAR-mediated currents, and rescued the deficit in LTP in Ts65Dn mice (122.9 ± 6.2%, n = 11; Fig. 6C). Together, these results demonstrate that synaptic plasticity in Ts65Dn mice can be effectively rescued by targeting α5-GABAARs to counterbalance increased dendritic inhibition as well as decreased synaptic NMDAR availability.
Discussion
In the present study, we found a reduced NMDAR complement in SC inputs and reduced NMDAR-mediated depolarization during afferent burst activity in hippocampal CA1 pyramidal cells of Ts65Dn mice. This was due to a combination of decreased NMDAR-EPSCs as well as increased dendritic inhibition mediated by nonlinear α5-GABAAR. Both NMDAR-mediated depolarization and NMDAR-dependent LTP deficits could be normalized by acute application of an α5-NAM, showing that dendritic inhibition is a major contributor to disturbed NMDAR recruitment during physiologically relevant activity patterns.
Control of NMDAR-dependent plasticity and learning by dendritic inhibition
The NMDAR component of the fEPSP recorded in the dentate gyrus during brief burst activation was previously shown to be reduced in Ts65Dn mice relative to control. Complete blockade of GABAARs with picrotoxin rescued the NMDAR component up to control levels (Kleschevnikov et al., 2004), suggesting that synaptic NMDAR expression is normal in dentate gyrus in Ts65Dn mice. Hence, different mechanisms can result in NMDAR hypoactivation. In addition to reduced NMDAR content (CA1 radiatum), a major cause is increased GABAergic inhibition (CA1 radiatum and DG).
The present study demonstrates that not all forms of inhibition are equally relevant. There was a specific increase of dendritic inhibition of pyramidal cells in Ts65Dn mice mediated via α5-GABAAR, which compromise postsynaptic integration of EPSPs (Fig. 7). By contrast, somatic inhibition was not significantly different in Ts65Dn mice. The identity of the presynaptic interneurons mediating the dendritic inhibition is unclear at present. However, it is likely that the increased number of dendrite targeting SOM and NOS interneurons contribute to enhanced inhibition in DS (Chakrabarti et al., 2010; Raveau et al., 2018). Dendritic depolarization is critically important for the activity-dependent opening of voltage-dependent NMDARs. Our results show that the reduced NMDAR currents together with the exaggerated dendritic inhibition in Ts65Dn mice powerfully interferes with the NMDAR-activation process via the nonlinear slow α5-GABARs (Fig. 7B,C). However, the disturbed E/I balance in DS animals can be effectively rectified by an α5-NAM via reduction of dendritic inhibition up to a level that enables relatively normal NMDAR activation during brief burst EPSPs (Fig. 7D). Therefore, our new results highlight the importance of dendritic inhibition in this DS model and may explain why a selective downmodulation of α5-GABARs by ∼50% rescues both NMDAR-dependent LTP in CA1 as well as learning behavior (Martínez-Cué et al., 2013).
Figure 7.

Mechanisms underlying the reduced NMDAR activation in Ts65Dn mice. A, Schematic drawing of SC-activated synapses onto CA1 pyramidal cells. B, Brief burst activation of SC inputs engages a positive feedback loop between membrane depolarization and voltage-dependent NMDAR activation in CA1-pyramidal cell dendrites of WT mice. This positive feedback is regulated by α5-GABAR-mediated voltage-dependent inhibition. C, Both increased dendritic inhibition and reduced NMDAR expression disrupt NMDAR activation in Ts65Dn mice. D, Application of the α5-NAM reduces dendritic inhibition and restores dendritic E/I balance to enable normal NMDAR activation in Ts65Dn mice.
Dendritic inhibition is crucially important for plasticity and learning. In CA1 pyramidal cells it was shown, that synchronous pairing of SC stimulation with distal entorhinal inputs generates NMDAR-dependent dendritic plateau potentials and burst firing, which can induce long-lasting changes in synaptic strength (Takahashi and Magee, 2009). Consequently, the generation of plateau potentials and burst firing during spatial exploration can induce the formation of new place fields in CA1 pyramidal cells (Bittner et al., 2015). Dendritic inhibition in particular is ideally placed to regulate the local membrane potentials in the dendrites. Using optogenetic silencing, it has been shown that PV-positive soma-targeting basket cells affect spike timing of CA1 pyramidal cells relative to extracellular theta oscillations without a large effect on firing frequency during spatial exploration. By contrast, silencing of dendrite-targeting SOM interneurons strongly increases NMDAR-dependent burst firing in pyramidal cells (Lovett-Barron et al., 2012; Royer et al., 2012). Similarly, it has been shown that neurogliaform cells, which also target distal dendrites (Bloss et al., 2016), prevent the activation of dendritic calcium spikes in CA1 pyramidal cells (Milstein et al., 2015). This demonstrates that dendrite-targeting interneurons effectively control NMDAR activation and synaptic plasticity.
Both SOM interneurons and neurogliaform cells control NMDAR activation via α5-GABARs (Zarnowska et al., 2009; Capogna and Pearce, 2011; Schulz et al., 2018). What are the mechanisms underlying the α5-GABAR-mediated control of NMDAR opening? The powerful inhibitory effect of α5-GABAAR containing synapses has been attributed to the slow time course of synaptic currents which is probably mediated by a slow GABA transient together with the slow gating kinetics of the high-affinity α5-GABAARs (Karayannis et al., 2010). Furthermore, the nonlinear voltage dependence provides large inhibitory conductance on demand, specifically when synaptic burst activity depolarizes distal dendrites (Schulz et al., 2018). Thus, α5-GABAAR-mediated inhibition in the hippocampus is in a powerful position to control the generation of dendritic calcium spikes, synaptic plasticity, and the formation of new spatial memories.
Using in vivo tetrode recordings in another mouse model of DS (Dp(16)1Yey), specific deficits in firing of CA1 pyramidal cells were recently reported (Raveau et al., 2018). Burst firing during spatial exploration and memory consolidation was reduced. By contrast, spike timing relative to the hippocampal theta cycle was normal. Furthermore, the firing rate within place fields was lower and the spatial information content of the pyramidal cell population was lower in DS mice compared with WT controls. As all these parameters are dependent on proper synaptic NMDAR activation, our results suggest that these deficits would be improved or rescued by targeting dendritic inhibition via modulators of α5-GABAARs. Further pharmacological studies in this mouse line will be necessary to confirm this hypothesis.
NMDAR deficits in DS mouse models
Deficits in NMDAR-dependent LTP have been reported previously in mouse models of DS. However, because these deficits could be corrected by blockade of GABAergic inhibition (Kleschevnikov et al., 2004; Costa and Grybko, 2005; Belichenko et al., 2009), they have been mainly ascribed to pathologically increased inhibition. The present study demonstrates, in addition, a reduction of NMDAR-mediated currents relative to AMPAR-mediated currents. These results are in line with a 20% reduction in protein levels of NR1 NMDAR subunits in the hippocampus of Ts65Dn mice (Souchet et al., 2014, 2015). A similar reduction of NR1 subunits relative to GluA1/A2 receptors was also found in Dyrk1a-overexpressing mice (Souchet et al., 2014), suggesting that the triplication of Dyrk1a contributes to the reduced NMDAR expression in DS. By contrast, levels of GluA1 AMPAR subunits were not significantly different from control. Another potential pathway could involve APP, as triplication of APP, also located on human chromosome 21 and mouse chromosome 16, has been suggested to promote increased endocytosis of NMDARs in Ts65Dn mice (Snyder et al., 2005; Netzer et al., 2010). As we observed a reduced NMDA-AMPA ratio only of SC inputs stimulated in SR, but not of inputs onto basal dendrites stimulated in SO, there may be other factors in addition to APP and Dyrk1a triplication contributing to the observed differentially localized deficits. Overall, a relatively large network of proteins related to NMDAR signaling is potentially modified in DS (Block et al., 2018).
Taken together, these data suggest that, in addition to enhanced dendritic inhibition, NMDAR-mediated EPSCs in CA1 pyramidal cells are also reduced in DS. Although triplication of Dyrk1a probably plays a major role in downregulation of synaptic NMDARs, several converging pathways may be involved. This may include dynamic regulation by activity-dependent associative or homeostatic synaptic plasticity during development.
Treatment options to restore NMDAR-dependent plasticity and learning
Chronic treatment with negative modulators of α5-GABAARs not only restored NMDAR-dependent LTP but also ameliorated disturbed hippocampus-dependent learning in Ts65Dn mice (Braudeau et al., 2011; Martínez-Cué et al., 2013). In addition, decreased inhibitory markers were observed in the hippocampus of Ts65Dn mice treated with the α5-NAM RO4938581 (Martínez-Cué et al., 2013). Similarly, in earlier studies, low concentrations of the nonselective GABAA antagonist PTZ rescued disturbed LTP in the dentate gyrus and deficits in hippocampus-dependent learning (Fernandez et al., 2007). These encouraging preclinical data supported the initiation of clinical trials in DS with basmisanil, a selective GABAA α5-NAM (www.clinicaltrials.gov). However, the Phase 2 trials were terminated due to lack of efficacy (Roche Press Release, June 28, 2016; http://www.roche.com/media/store/statements.htm), indicating that our understanding of the cognitive deficits in DS is incomplete.
The reasons for the inconclusive clinical results may be manifold. First, the reduction in synaptic NMDAR currents, which we have found in Ts65Dn mice, might be larger in patients relative to the mouse model. Therefore, in addition to counterbalancing dendritic inhibition via an α5-NAM, further treatment to upregulate NMDAR responses might be useful to restore the E/I balance and neuronal plasticity in the DS brain. Alternatively, intervention might be necessary at much earlier time points in brain development. Furthermore, it is also possible that the Ts65Dn mouse model does not fully recapitulate the brain deficits in DS and more triplicated genes contribute to the human condition. Proof for increased inhibitory transmission in individuals with DS is still missing and therefore additional studies are needed. In case GABAergic inhibition is indeed increased in DS patients, additional medication on top of an α5-NAM might be necessary to successfully improve cognitive deficits in patients.
In conclusion, the present study indicates that deficits in synaptic plasticity in Ts65Dn mice is the combined effect of a smaller NMDAR complement and increased nonlinear dendritic inhibition. Hence, pharmacological intervention specifically targeting dendritic inhibition via α5-GABAARs may be well suited to restore the fundamentally important mechanism of synaptic plasticity in the hippocampus without adverse side effects.
Footnotes
This work was supported by a Roche Postdoctoral Fellowship and Swiss National Science Foundation Project 31003A_176321. We thank Tom Otis for helpful comments on the manuscript; Selma Becherer and Martine Schwager for mouse genotyping, histochemical stainings, and technical assistance; Marie-Claire Pflimlin for some electrophysiological recordings; and Andrew Thomas for RO4938581 supply.
F.K. and M.-C. H. are employed by F. Hoffmann-La Roche.
References
- Ballard TM, Knoflach F, Prinssen E, Borroni E, Vivian JA, Basile J, Gasser R, Moreau JL, Wettstein JG, Buettelmann B, Knust H, Thomas AW, Trube G, Hernandez MC (2009) RO4938581, a novel cognitive enhancer acting at GABAA alpha5 subunit-containing receptors. Psychopharmacology 202:207–223. 10.1007/s00213-008-1357-7 [DOI] [PubMed] [Google Scholar]
- Barry PH. (1994) JPCalc, a software package for calculating liquid junction potential corrections in patch-clamp, intracellular, epithelial and bilayer measurements and for correcting junction potential measurements. J Neurosci Methods 51:107–116. 10.1016/0165-0270(94)90031-0 [DOI] [PubMed] [Google Scholar]
- Bartesaghi R, Haydar TF, Delabar JM, Dierssen M, Martínez-Cué C, Bianchi DW (2015) New perspectives for the rescue of cognitive disability in Down syndrome. J Neurosci 35:13843–13852. 10.1523/JNEUROSCI.2775-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko NP, Belichenko PV, Kleschevnikov AM, Salehi A, Reeves RH, Mobley WC (2009) The “Down syndrome critical region” is sufficient in the mouse model to confer behavioral, neurophysiological, and synaptic phenotypes characteristic of Down syndrome. J Neurosci 29:5938–5948. 10.1523/JNEUROSCI.1547-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best TK, Cramer NP, Chakrabarti L, Haydar TF, Galdzicki Z (2012) Dysfunctional hippocampal inhibition in the Ts65Dn mouse model of Down syndrome. Exp Neurol 233:749–757. 10.1016/j.expneurol.2011.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischofberger J, Engel D, Li L, Geiger JR, Jonas P (2006) Patch-clamp recording from mossy fiber terminals in hippocampal slices. Nat Protoc 1:2075–2081. 10.1038/nprot.2006.312 [DOI] [PubMed] [Google Scholar]
- Bittner KC, Grienberger C, Vaidya SP, Milstein AD, Macklin JJ, Suh J, Tonegawa S, Magee JC (2015) Conjunctive input processing drives feature selectivity in hippocampal CA1 neurons. Nat Neurosci 18:1133–1142. 10.1038/nn.4062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block A, Ahmed MM, Rueda N, Hernandez MC, Martinez-Cué C, Gardiner KJ (2018) The GABA(A)α5-selective modulator, RO4938581, rescues protein anomalies in the Ts65Dn mouse model of Down syndrome. Neuroscience 372:192–212. 10.1016/j.neuroscience.2017.12.038 [DOI] [PubMed] [Google Scholar]
- Bloss EB, Cembrowski MS, Karsh B, Colonell J, Fetter RD, Spruston N (2016) Structured dendritic inhibition supports branch-selective integration in CA1 pyramidal cells. Neuron 89:1016–1030. 10.1016/j.neuron.2016.01.029 [DOI] [PubMed] [Google Scholar]
- Braat S, Kooy RF (2015) The GABAA receptor as a therapeutic target for neurodevelopmental disorders. Neuron 86:1119–1130. 10.1016/j.neuron.2015.03.042 [DOI] [PubMed] [Google Scholar]
- Braudeau J, Delatour B, Duchon A, Pereira PL, Dauphinot L, de Chaumont F, Olivo-Marin JC, Dodd RH, Hérault Y, Potier MC (2011) Specific targeting of the GABA-A receptor α5 subtype by a selective inverse agonist restores cognitive deficits in Down syndrome mice. J Psychopharmacol 25:1030–1042. 10.1177/0269881111405366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brickley SG, Mody I (2012) Extrasynaptic GABA(A) receptors: their function in the CNS and implications for disease. Neuron 73:23–34. 10.1016/j.neuron.2011.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, Pearce RA (2011) GABA A,slow: causes and consequences. Trends Neurosci 34:101–112. 10.1016/j.tins.2010.10.005 [DOI] [PubMed] [Google Scholar]
- Caraiscos VB, Elliott EM, You-Ten KE, Cheng VY, Belelli D, Newell JG, Jackson MF, Lambert JJ, Rosahl TW, Wafford KA, MacDonald JF, Orser BA (2004) Tonic inhibition in mouse hippocampal CA1 pyramidal neurons is mediated by alpha5 subunit-containing gamma-aminobutyric acid type A receptors. Proc Natl Acad Sci U S A 101:3662–3667. 10.1073/pnas.0307231101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Galdzicki Z, Haydar TF (2007) Defects in embryonic neurogenesis and initial synapse formation in the forebrain of the Ts65Dn mouse model of Down syndrome. J Neurosci 27:11483–11495. 10.1523/JNEUROSCI.3406-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti L, Best TK, Cramer NP, Carney RS, Isaac JT, Galdzicki Z, Haydar TF (2010) Olig1 and Olig2 triplication causes developmental brain defects in Down syndrome. Nat Neurosci 13:927–934. 10.1038/nn.2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa AC, Grybko MJ (2005) Deficits in hippocampal CA1 LTP induced by TBS but not HFS in the Ts65Dn mouse: a model of Down syndrome. Neurosci Lett 382:317–322. 10.1016/j.neulet.2005.03.031 [DOI] [PubMed] [Google Scholar]
- Costa AC, Scott-McKean JJ, Stasko MR (2008) Acute injections of the NMDA receptor antagonist memantine rescue performance deficits of the Ts65Dn mouse model of Down syndrome on a fear conditioning test. Neuropsychopharmacology 33:1624–1632. 10.1038/sj.npp.1301535 [DOI] [PubMed] [Google Scholar]
- Dierssen M. (2012) Down syndrome: the brain in trisomic mode. Nat Rev Neurosci 13:844–858. 10.1038/nrn3314 [DOI] [PubMed] [Google Scholar]
- Engel J, Schultens HA, Schild D (1999) Small conductance potassium channels cause an activity-dependent spike frequency adaptation and make the transfer function of neurons logarithmic. Biophys J 76:1310–1319. 10.1016/S0006-3495(99)77293-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez F, Morishita W, Zuniga E, Nguyen J, Blank M, Malenka RC, Garner CC (2007) Pharmacotherapy for cognitive impairment in a mouse model of Down syndrome. Nat Neurosci 10:411–413. 10.1038/nn1860 [DOI] [PubMed] [Google Scholar]
- Gardiner KJ. (2014) Pharmacological approaches to improving cognitive function in Down syndrome: current status and considerations. Drug Des Devel Ther 9:103–125. 10.2147/DDDT.S51476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geiger JR, Bischofberger J, Vida I, Fröbe U, Pfitzinger S, Weber HJ, Haverkampf K, Jonas P (2002) Patch-clamp recording in brain slices with improved slicer technology. Pflugers Arch 443:491–501. 10.1007/s00424-001-0735-3 [DOI] [PubMed] [Google Scholar]
- Guzman SJ, Schlögl A, Schmidt-Hieber C (2014) Stimfit: quantifying electrophysiological data with python. Front Neuroinform 8:16. 10.1515/bmt-2013-4181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karayannis T, Elfant D, Huerta-Ocampo I, Teki S, Scott RS, Rusakov DA, Jones MV, Capogna M (2010) Slow GABA transient and receptor desensitization shape synaptic responses evoked by hippocampal neurogliaform cells. J Neurosci 30:9898–9909. 10.1523/JNEUROSCI.5883-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Villar AJ, Epstein CJ, Malenka RC, Mobley WC (2004) Hippocampal long-term potentiation suppressed by increased inhibition in the Ts65Dn mouse, a genetic model of Down syndrome. J Neurosci 24:8153–8160. 10.1523/JNEUROSCI.1766-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleschevnikov AM, Belichenko PV, Faizi M, Jacobs LF, Htun K, Shamloo M, Mobley WC (2012) Deficits in cognition and synaptic plasticity in a mouse model of Down syndrome ameliorated by GABAB receptor antagonists. J Neurosci 32:9217–9227. 10.1523/JNEUROSCI.1673-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis DA. (2014) Inhibitory neurons in human cortical circuits: substrate for cognitive dysfunction in schizophrenia. Curr Opin Neurobiol 26:22–26. 10.1016/j.conb.2013.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lien CC, Martina M, Schultz JH, Ehmke H, Jonas P (2002) Gating, modulation and subunit composition of voltage-gated K+ channels in dendritic inhibitory interneurons of rat hippocampus. J Physiol 538:405–419. 10.1113/jphysiol.2001.013066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lott IT, Dierssen M (2010) Cognitive deficits and associated neurological complications in individuals with Down's syndrome. Lancet Neurol 9:623–633. 10.1016/S1474-4422(10)70112-5 [DOI] [PubMed] [Google Scholar]
- Lovett-Barron M, Turi GF, Kaifosh P, Lee PH, Bolze F, Sun XH, Nicoud JF, Zemelman BV, Sternson SM, Losonczy A (2012) Regulation of neuronal input transformations by tunable dendritic inhibition. Nat Neurosci 15:423–430, S1–S3. 10.1038/nn.3024 [DOI] [PubMed] [Google Scholar]
- Maccaferri G, Roberts JD, Szucs P, Cottingham CA, Somogyi P (2000) Cell surface domain specific postsynaptic currents evoked by identified GABAergic neurones in rat hippocampus in vitro. J Physiol 524:91–116. 10.1111/j.1469-7793.2000.t01-3-00091.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Cué C, Martínez P, Rueda N, Vidal R, García S, Vidal V, Corrales A, Montero JA, Pazos Á, Flórez J, Gasser R, Thomas AW, Honer M, Knoflach F, Trejo JL, Wettstein JG, Hernández MC (2013) Reducing GABAA α5 receptor-mediated inhibition rescues functional and neuromorphological deficits in a mouse model of Down syndrome. J Neurosci 33:3953–3966. 10.1523/JNEUROSCI.1203-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez-Cué C, Delatour B, Potier MC (2014) Treating enhanced GABAergic inhibition in Down syndrome: use of GABA α5-selective inverse agonists. Neurosci Biobehav Rev. 46:218–227. 10.1016/j.neubiorev.2013.12.008 [DOI] [PubMed] [Google Scholar]
- Milstein AD, Bloss EB, Apostolides PF, Vaidya SP, Dilly GA, Zemelman BV, Magee JC (2015) Inhibitory gating of input comparison in the CA1 microcircuit. Neuron 87:1274–1289. 10.1016/j.neuron.2015.08.025 [DOI] [PubMed] [Google Scholar]
- Netzer WJ, Powell C, Nong Y, Blundell J, Wong L, Duff K, Flajolet M, Greengard P (2010) Lowering beta-amyloid levels rescues learning and memory in a Down syndrome mouse model. PLoS One 5:e10943. 10.1371/journal.pone.0010943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pocklington AJ, Rees E, Walters JT, Han J, Kavanagh DH, Chambert KD, Holmans P, Moran JL, McCarroll SA, Kirov G, O'Donovan MC, Owen MJ (2015) Novel findings from CNVs implicate inhibitory and excitatory signaling complexes in schizophrenia. Neuron 86:1203–1214. 10.1016/j.neuron.2015.04.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prenosil GA, Schneider Gasser EM, Rudolph U, Keist R, Fritschy JM, Vogt KE (2006) Specific subtypes of GABAA receptors mediate phasic and tonic forms of inhibition in hippocampal pyramidal neurons. J Neurophysiol 96:846–857. 10.1152/jn.01199.2005 [DOI] [PubMed] [Google Scholar]
- Raveau M, Polygalov D, Boehringer R, Amano K, Yamakawa K, McHugh TJ (2018) Alterations of in vivo CA1 network activity in Dp(16)1Yey Down syndrome model mice. Elife 7:e31543. 10.7554/eLife.31543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond CR, Redman SJ (2006) Spatial segregation of neuronal calcium signals encodes different forms of LTP in rat hippocampus. J Physiol 570:97–111. 10.1113/jphysiol.2005.098947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves RH, Irving NG, Moran TH, Wohn A, Kitt C, Sisodia SS, Schmidt C, Bronson RT, Davisson MT (1995) A mouse model for Down syndrome exhibits learning and behaviour deficits. Nat Genet 11:177–184. 10.1038/ng1095-177 [DOI] [PubMed] [Google Scholar]
- Reinholdt LG, Ding Y, Gilbert GT, Czechanski A, Solzak JP, Roper RJ, Johnson MT, Donahue LR, Lutz C, Davisson MT (2011) Molecular characterization of the translocation breakpoints in the Down syndrome mouse model Ts65Dn. Mamm Genome 22:685–691. 10.1007/s00335-011-9357-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Royer S, Zemelman BV, Losonczy A, Kim J, Chance F, Magee JC, Buzsáki G (2012) Control of timing, rate and bursts of hippocampal place cells by dendritic and somatic inhibition. Nat Neurosci 15:769–775. 10.1038/nn.3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Knoflach F (2011) Beyond classical benzodiazepines: novel therapeutic potential of GABAA receptor subtypes. Nat Rev Drug Discov 10:685–697. 10.1038/nrd3502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolph U, Möhler H (2014) GABAA receptor subtypes: therapeutic potential in Down syndrome, affective disorders, schizophrenia, and autism. Annu Rev Pharmacol Toxicol 54:483–507. 10.1146/annurev-pharmtox-011613-135947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz JM, Knoflach F, Hernandez MC, Bischofberger J (2018) Dendrite-targeting interneurons control synaptic NMDA-receptor activation via nonlinear α5-GABAA receptors. Nat Commun 9:3576. 10.1038/s41467-018-06004-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serwanski DR, Miralles CP, Christie SB, Mehta AK, Li X, De Blas AL (2006) Synaptic and nonsynaptic localization of GABAA receptors containing the alpha5 subunit in the rat brain. J Comp Neurol 499:458–470. 10.1002/cne.21115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P (2005) Regulation of NMDA receptor trafficking by amyloid-beta. Nat Neurosci 8:1051–1058. 10.1038/nn1503 [DOI] [PubMed] [Google Scholar]
- Souchet B, Guedj F, Sahún I, Duchon A, Daubigney F, Badel A, Yanagawa Y, Barallobre MJ, Dierssen M, Yu E, Herault Y, Arbones M, Janel N, Créau N, Delabar JM (2014) Excitation/inhibition balance and learning are modified by Dyrk1a gene dosage. Neurobiol Dis 69:65–75. 10.1016/j.nbd.2014.04.016 [DOI] [PubMed] [Google Scholar]
- Souchet B, Guedj F, Penke-Verdier Z, Daubigney F, Duchon A, Herault Y, Bizot JC, Janel N, Créau N, Delatour B, Delabar JM (2015) Pharmacological correction of excitation/inhibition imbalance in Down syndrome mouse models. Front Behav Neurosci 9:267. 10.3389/fnbeh.2015.00267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Magee JC (2009) Pathway interactions and synaptic plasticity in the dendritic tuft regions of CA1 pyramidal neurons. Neuron 62:102–111. 10.1016/j.neuron.2009.03.007 [DOI] [PubMed] [Google Scholar]
- Tricoire L, Pelkey KA, Daw MI, Sousa VH, Miyoshi G, Jeffries B, Cauli B, Fishell G, McBain CJ (2010) Common origins of hippocampal ivy and nitric oxide synthase expressing neurogliaform cells. J Neurosci 30:2165–2176. 10.1523/JNEUROSCI.5123-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal V, García-Cerro S, Martínez P, Corrales A, Lantigua S, Vidal R, Rueda N Ozmen L, Hernández MC, Martínez-Cué C (2018) Decreasing the expression of GABA(A) α5 subunit-containing receptors partially improves cognitive, electrophysiological, and morphological hippocampal defects in the Ts65Dn model of Down syndrome. Mol Neurobiol 55:4745–4762. 10.1007/s12035-017-0675-3 [DOI] [PubMed] [Google Scholar]
- Zarnowska ED, Keist R, Rudolph U, Pearce RA (2009) GABAA receptor alpha5 subunits contribute to GABAA,slow synaptic inhibition in mouse hippocampus. J Neurophysiol 101:1179–1191. 10.1152/jn.91203.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]