

Abstract
Synucleinopathies are characterized by the accumulation of insoluble α-synuclein (αSyn). To test whether αSyn aggregates modulate synaptic activity, we used a recently developed model in primary neurons for inducing αSyn pathology. We demonstrated that preformed fibrils (PFFs) generated with recombinant human αSyn compromised synaptic activity in a time- and dose-dependent manner and that the magnitude of these deficits correlated with the formation of αSyn pathology in cultured excitatory hippocampal neurons from both sexes of mice. Remarkably, acute passive infusion of αSyn PFFs from whole-cell patch-clamp pipette decreased mEPSC frequency within 10 min followed by induction of αSyn pathology within 1 d. Moreover, by direct addition of αSyn PFFs into culture medium, the formation of misfolded αSyn inclusions dramatically compromised the colocalization of synaptic markers and altered dynamic changes of dendritic spines, but the viability of neurons was not affected up to 7 d post-treatment with αSyn PFFs. Our data indicate that intraneuronal αSyn fibrils impaired the initiation of synaptogenesis and their physiological functions, thereby suggesting that targeting synaptic dysfunction in synucleinopathies may provide a promising therapeutic direction.
SIGNIFICANCE STATEMENT Under pathological conditions, the presynaptic protein α-synuclein (αSyn) aggregates to form intraneuronal inclusions. To understand how and to what extent αSyn aggregates modulate synaptic activity before neuron loss, we demonstrate that αSyn preformed fibrils (PFFs) reduced synaptic activity in a dose- and time-dependent manner. The magnitude of these deficits correlated with the deposition of αSyn pathology, which dramatically compromised the colocalization of synaptic markers and altered the dendritic spine dynamics. The present work further highlights the impact of αSyn PFFs on synaptogenesis and physiological function, which may be applicable to other types of synucleinopathies.
Keywords: α-synuclein, dendritic spine, preformed fibrils, synaptic function, synucleinopathy
Introduction
α-Synuclein (αSyn) is a soluble presynaptically-localized protein that together with the soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex and the cysteine string protein α (CSPα) chaperone (Chandra et al., 2005; Burré et al., 2010), facilitates vesicle pool refilling and vesicle release at the active zone (Murphy et al., 2000; Nemani et al., 2010; Scott and Roy, 2012; Burré et al., 2014). Under pathological conditions, this soluble protein undergoes several post-translational modifications (Burré et al., 2018) and forms higher-order oligomeric and fibrillar species, leading to conformationally and biologically distinct strains (Peng et al., 2018). These inclusions are referred to as Lewy bodies (LBs) and Lewy neurites (LNs) in Parkinson's disease (PD), PD with dementia (PDD), and dementia with Lewy bodies (DLB) or glial cytoplasmic inclusions (GCIs) in multiple system atrophy. Furthermore, neurodegenerative diseases with αSyn pathology are termed synucleinopathies, but LBs and LNs also occur commonly in Alzheimer's disease (AD; Irwin et al., 2013). In addition, αSyn monomers might also be involved in synucleinopathies by altering stress signaling as shown in yeast and human cells (Wang et al., 2012).
Mounting evidence from postmortem studies, animal models, and primary culture systems, as well as from biochemical and biophysical investigations, have established that the presence of αSyn oligomers and/or fibrillar structures are essential for the pathogenesis of synucleinopathies, including the spreading of pathological αSyn (Dickson et al., 1989; Tsigelny et al., 2008; Desplats et al., 2009; Volpicelli-Daley et al., 2011; Luk et al., 2012a,b; Masuda-Suzukake et al., 2013). These pathogenic oligomeric or fibrillized αSyn species cause functional deficits and neuron loss both in vitro and in vivo (Volpicelli-Daley et al., 2011; Winner et al., 2011; Luk et al., 2012b; Peelaerts et al., 2015). Moreover, structurally distinct αSyn strains can also promote tau inclusion formation in vitro (Guo et al., 2013). However, the molecular mechanism of how αSyn accumulates into insoluble inclusions and contributes to neurodegeneration in these disorders remain elusive.
Extracellular αSyn affects synaptic transmission by either altering plasma membrane permeability (Pacheco et al., 2015) or fragmenting lipid rafts (Emanuele et al., 2016), resulting in an increase in intracellular calcium that can lead to neuron death. Prior biophysical and size-exclusion chromatography studies identified sodium dodecyl sulfate (SDS)-resistant oligomers as toxic forms of αSyn, which induced neurodegeneration and abnormal calcium currents in primary neuron cultures (Danzer et al., 2007). Oligomeric forms of αSyn have also been reported to be toxic in vivo (Winner et al., 2011) and we found that αSyn preformed fibrils (PFFs) derived from the aggregation of recombinant αSyn monomers can initiate the formation of αSyn pathogenic inclusions in nontransgenic or wild-type (WT) neurons and mice, as well as propagate pathological αSyn from one neuron to another (Volpicelli-Daley et al., 2011; Luk et al., 2012a,b). The αSyn fibrils found in LBs, LNs, and GCIs adopt the β-sheet conformation characteristic of amyloids (Tuttle et al., 2016), which could elicit a gain of toxic function (Volpicelli-Daley et al., 2011; Luk et al., 2012b). However, by what means and to what extent αSyn aggregates modulate synaptic activity before neuron loss remains unknown.
In the present study, we investigated the effects of αSyn PFF treatment on synaptic activity and neuron viability. We found that αSyn PFFs compromised both spontaneous as well as miniature excitatory and inhibitory synaptic responses, and the magnitude of this effect correlates with the degree of αSyn inclusion formation inside neurons. Following direct administration of αSyn PFFs via whole-cell patch-clamp pipettes into single neurons, synaptic activity decreased within 10 min. The compromised synaptic activity was a consequence of dysfunction of synapses and unbalanced dynamics of dendritic spines. These findings implicate αSyn fibrils-mediated synaptic dysfunction in the initial pathogenesis of PD, PDD, DLB, and AD with concomitant LBs.
Materials and Methods
Animals.
The use and care of animals were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and they were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Pregnant CD1 or αSyn knock-out mice (E16–E18) were used for isolation of hippocampal neurons. Mice were housed under a 12 h light/dark cycle with ad libitum access to food and water. Both male and female mice were used in this study.
Recombinant αSyn purification and in vitro fibrillization.
Full-length human αSyn (1–140) proteins were expressed in BL21 (DE3) RIL cells and purified as previously described (Giasson et al., 2001). Fibrillization was conducted by diluting recombinant αSyn to 5 mg/ml in sterile Dulbecco's PBS (Cellgro, Mediatech; pH adjusted to 7.0, without Ca2+ or Mg2+) followed by incubating this recombinant αSyn at 37°C with constant agitation at 1000 rpm for 7 d. Successful αSyn fibrillization was verified by electron microscopy, sedimentation studies and a thioflavin T-binding assay as described previously (Volpicelli-Daley et al., 2014). Assembled αSyn PFFs were aliquoted and stored at −80°C.
Primary neuronal cultures and fibrils transduction.
Primary mouse neurons were prepared from E16–E18 CD1 mouse embryos (Charles River Laboratories) and αSyn knock-out mice (Abeliovich et al., 2000) as previously described (Volpicelli-Daley et al., 2014). All procedures were performed according to the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Pennsylvania Institutional Animal Care and Use Committee. Dissociated hippocampal neurons were plated onto poly-d-lysine-coated coverslips (Carolina Biological Supply) with cell densities at 100,000/well per 24-well plate. Most experiments were performed at 14–19 d in vitro (DIV). αSyn PFFs were diluted in Dulbecco's PBS (without Ca2+ or Mg2+) and sonicated (QSonica Microson XL-2000; 60 pulses; setting 2–3; 0.5 s per pulse). Neurons were then treated with PBS or sonicated αSyn PFFs at 7–11 DIV and fixed for Immunocytochemistry (ICC) at 7 d post-treatment or performed whole-cell patch-clamp recording at indicated time points.
Electrophysiology and data analysis.
Hippocampal pyramidal neurons were selected for whole-cell patch-clamp recordings based on their morphology on an inverted microscope (Leica, DM-IRB). Data were collected with an Axopatch-700B amplifier, a Digidata-1440A digitizer and pClamp 10.2 software (Molecular Devices), low-pass filtered at 2 kHz, and digitally sampled at 10 kHz. Patch pipettes (4.0–6.0 MΩ) were pulled from borosilicate glass (Sutter Instruments) and filled with one of two types of pipette solution. For spontaneous EPSCs (sEPSC) or miniature EPSCs (mEPSC) recordings, the pipette solution contained the following (in mm): 120.0 Cs-methanesulfonate, 0.6 EGTA, 2.8 NaCl, 5.0 MgCl2, 2.0 ATP, 0.3 GTP, 20.0 HEPES, and 5.0 QX-314, and adjusted to pH 7.2 with CsOH (305–310 mOsm). The standard extracellular solution contained the following (in mm): 119.0 NaCl, 2.5 KCl, 2.0 CaCl2, 2.0 MgCl2, 25.0 HEPES, and 30.0 d-glucose. The pH was adjusted to 7.4 with NaOH. sEPSCs were recorded in the presence of 10 μm bicuculline to block GABAergic synaptic currents in the extracellular solution, whereas mEPSCs were recorded in the presence of both 10 μm bicuculline and 1 μm tetrodotoxin (TTX; Abcam) in the extracellular solution. Recordings were made at a holding potential of −70 mV. QX-314 was added to the pipette solution to block the GABAB-mediated currents and to prevent the generation of Na+-dependent action potentials. For miniature IPSCs (mIPSC) recordings, the pipette solution contained the following (in mm): 140.0 CsCl, 0.1 EGTA, 1.0 MgCl2, 4.0 NaCl, 2.0 MgATP, 0.3 Na2GTP, 10.0 HEPES, and 5.0 QX-314 (adjusted to pH 7.2 with CsOH, 305–310 mOsm). The standard extracellular solution contained the following (in mm): 146.0 NaCl, 2.5 KCl, 2.0 CaCl2, 3.0 MgCl2, 10.0 HEPES, and 10.0 d-glucose (pH was adjusted to 7.4 with NaOH). Recordings were made at a holding potential of −60 mV. mIPSC was recorded in the presence of 20 μm DNQX, 50 μm DL-AP5, and 1 μm TTX in the extracellular solution. QX-314, DNQX, DL-AP5 and bicuculline were purchased from Tocris Bioscience. sEPSC, mEPSC, and mIPSC were analyzed offline using Clampfit 10.7 (Molecular Devices) and Igor 5.3 (Wavemetrics). Recordings with series resistance >20 MΩ were excluded from analysis. Synaptic responses were analyzed as described previously (Zhu et al., 2017). Synaptic events >5 pA were detected after creating a unique template for each neuron. All recordings were performed at room temperature (25°C). All data are presented as mean ± SEM. Investigators were blinded to the treatments or genotypes of the animals during experiments.
Immunocytochemistry.
Neurons were fixed with 4% paraformaldehyde (PFA) and 4% sucrose in PBS for 15 min at room temperature (RT), followed by permeabilization with 0.1% Triton X-100 for 15 min, then blocked with 3% fetal bovine serum and 3% bovine serum albumin in PBS (blocking buffer) for 1 h at RT. Neurons were incubated overnight at 4°C in primary antibodies diluted in blocking buffer followed by AlexaFluor-conjugated secondary antibodies (Invitrogen) diluted in blocking buffer for 1.5 h at RT. Images were captured on an LSM 710 inverted confocal microscope (Carl Zeiss), and then processed with MBF ImageJ software (National Institutes of Health). Colocalization analysis was performed using ZEN 2010 (Carl Zeiss) with default M1 and M2 coefficients. For the imaging of dendritic spines, GFP-expressing lentivirus was transfected at 10 DIV, and PBS or αSyn PFFs added at 11–12 DIV. Neurons were fixed 1 week later and z-stack confocal imaging was performed. Images were z-projected for the analysis of spine densities and the spine subtypes detection with NeuronStudio software (Rodriguez et al., 2008). For the passive infusion of αSyn PFFs from the patch pipettes, sonicated αSyn PFFs (5 μm) were diluted into the pipette solution to assess the effect of αSyn PFFs on synaptic responses. After infusion, neurons were cultured for another 20 h, then fixed, and followed with ICC procedures.
Biocytin labeling.
During whole-cell patch-clamp recording, 5% AlexaFluor 594 biocytin (Sigma-Aldrich) was added into the pipette solution to label recorded neurons. The dye was allowed to infuse for 15–20 min under whole-cell patching condition, then the recording pipette was gently removed and cells were fixed with PFA followed by ICC.
Immuno-EM.
Immuno-electron microscopy (immuno-EM) was performed as previously described (Volpicelli-Daley et al., 2011).
Live imaging of spine dynamics.
Hippocampal neurons were transfected with GFP-expressing lentivirus at 10 DIV, whole-cell patch-clamp was performed 1 week later with 5 μm αSyn PFFs, αSyn monomers or PBS in the pipette solution. Laser scanning confocal microscopy (Olympus, FV-1000) equipped with Ar-ion laser (488 nm) and HeNe laser (532 nm) was used to capture spine dynamics. Confocal z-stack images were taken every 30 s during recording. Images were z-stack projected and analyzed with ImageJ software (National Institutes of Health).
Data analysis.
All experiments were performed with controls side-by-side and in random order and they were replicated at least three times. Data are shown as mean ± SEM. Statistical comparisons were performed with the two-tailed Student's t test, one-way ANOVA or two-sample Kolmogorov–Smirnov test (K–S test) as indicated. The distribution of the variables in each experimental group was approximately normal. All tests were conducted using Statistical Package for the Social Sciences (SPSS) version 20.0. Significant differences were accepted at p < 0.05.
Results
αSyn PFFs decreased synaptic activities in a dose- and time-dependent manner
Oligomeric or fibrillized conformations of αSyn have been reported to induce neuronal functional deficits and neuron loss both in vitro and in vivo (Volpicelli-Daley et al., 2011; Winner et al., 2011; Luk et al., 2012b; Peelaerts et al., 2015; Peng et al., 2018). When and how misfolded αSyn causes functional deficits and compromise neuronal survival remains elusive. To investigate whether αSyn PFFs impair synaptic activity, we exposed neurons with different dosages of αSyn PFFs at 7 DIV and recorded synaptic activity 1 week later (Fig. 1A). Compared with PBS controls, the mEPSC frequency was decreased when neurons were treated with αSyn PFFs at 0.5 μg/well or at higher dosages, but the peak amplitude remained unchanged at any dosages we tested (Fig. 1B,C). What's more, the mIPSC frequency was also significantly decreased at one-week post-αSyn PFFs treatment at low and higher dosages (Frequency: PBS, 2.53 ± 0.42 Hz; 0.5 μg PFF, 1.47 ± 0.31 Hz, **p < 0.01; 2 μg PFF, 1.55 ± 0.35 Hz, *p < 0.05). Next, we studied the effect of αSyn PFFs on sEPSC that are driven by action potentials. Both the peak amplitude and the sEPSC frequency were compromised at 0.5 μg/well PFFs and higher dosages after one-week of continuous exposure of the neurons to αSyn PFFs (Fig. 2), indicating that αSyn PFFs reduced either neuronal survival, neuronal excitability or the number of synapses (such as in the next section). What's more, the frequency of action potential (AP) was reduced ∼50% after PFF treatment for 7 d (number of AP spikes in PBS: PFF = 9.8 ± 0.7: 4.7 ± 0.8, ***p < 0.001), where both input resistance and whole-cell membrane capacitance remained unchanged [input resistance, PBS: PFF is 397.5 ± 53 MΩ: 408.4 ± 30 MΩ (no significance); whole-cell membrane capacitance in PBS: PFF is 85.2 ± 5.2 pF: 82.1 ± 3.3 pF (no significance)]. Therefore, as for the reduction of peak amplitude and frequency of sEPSC, this could result from the reduced AP firing. In addition, we assessed the αSyn pathology development at different αSyn PFFs dosages. Consistent with previous in vitro and primary culture studies (Wood et al., 1999; Volpicelli-Daley et al., 2011), low concentrations (0.1 μg/well and 0.25 μg/well) of αSyn PFFs triggered lower levels of pathological phosphorylated αSyn (p-αSyn) detected by the 81a monoclonal antibody (mAb) in the form of small puncta (Fig. 3A, top, C), but at dosages >0.5 μg/well, more LNs and LBs were visible (Fig. 3A, bottom, C). Thus, small quantities of αSyn PFFs are sufficient to trigger αSyn aggregate formation, and the amount of aggregation depends on the initial amount of αSyn PFFs or “seeds” added to the cultured neurons (Wood et al., 1999; Volpicelli-Daley et al., 2011). Therefore, 0.5 μg/well was used in the studies described in the next section unless otherwise specified. Together, these data suggested that αSyn PFFs induced endogenous neuronal αSyn aggregates, which attenuated both excitatory glutamatergic and inhibitory GABAergic neuronal synaptic activity in a dosage-dependent manner.
Figure 1.

Treatment of cultured hippocampal neurons with αSyn PFFs decreased synaptic activity in a dose- and time-dependent manner. A, Typical mEPSC recordings after αSyn PFFs treatment at different dosages for 1 week. B, Cumulative probabilities of peak amplitude and inter-event intervals from A. C, Statistics of peak amplitude and frequency of mEPSC at different doses of αSyn PFF treatments. (n = 11–14 cells per group). D, Typical mEPSC recordings after αSyn PFF treatment at different post-transduction time points. As shown in C, we used αSyn PFFs at a concentration of 0.5 μg/well. To record mEPSC from mature hippocampal neurons, we started the treatment at 11–12 DIV in D. E, Cumulative probabilities of peak amplitudes and inter-event intervals from D. F, Statistics of peak amplitude and frequency of mEPSC at different time points from E (n = 15–19 cells per group). Results shown as mean ± SEM; n = 4 independent batches of neurons for dose-dependence studies and n = 6 independent batches of neurons for time dependence studies. *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA with Tukey post hoc test (C), two-sample Kolmogorov–Smirnov test (K–S) test (B, E), and Student's t test (F).
Figure 2.

sEPSC frequency was also compromised by αSyn PFFs. Same as Figure 1, but sEPSCs were recorded after αSyn PFFs treatment. αSyn PFFs transduced for 7 d at different concentrations. A, Representative sEPSC recording traces treated with PBS or different doses of PFF for 7 d. B, C, Quantifications of amplitude and frequency of sEPSCs. Results shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA with Tukey post hoc test.
Figure 3.

ICC staining of p-αSyn (81a) after αSyn PFFs treatment at different dosages and time points. A, ICC staining of p-αSyn (81a; green) after αSyn PFFs treatment double-labeled with dendritic marker MAP2 (red) at different dosages. αSyn PFFs transduced at 7–9 DIV, fixed with 4% PFA 7 d later. B, ICC staining of p-αSyn (81a) after αSyn PFFs treatment at different time points. αSyn PFFs transduced at 11–12 DIV, fixed with 4% PFA at different post-transduction time points. Representative images from three different repeats. Scale bar, 100 μm. C, D, Quantification of p-αSyn (81a) after αSyn PFFs treatment at different dosages and time points. Experiments were repeated from 3 to 4 different cultures. **p < 0.01, ***p < 0.001; ns, no significance; one-way ANOVA with Tukey post hoc test.
Based on these results, we investigated more precisely when αSyn PFFs treatment started to have an effect on synaptic responses. To do this, we added αSyn PFFs (0.5 μg/well) at 11 DIV when neurons are mature enough to develop synaptic connections with other neurons and then recorded mEPSCs 1, 2, 4 and 7 d later (Fig. 1D). Consistent with treatments using different αSyn PFFs concentrations (Fig. 1C, top), the peak mEPSC amplitudes remained unchanged at any time points (Fig. 1E,F, top), whereas the mEPSC frequency decreased ∼30% at 4 d post-αSyn PFF treatment, and further decreased to ∼50% of the PBS controls at 7 d (Fig. 1E,F, bottom). No obvious 81a-positive αSyn pathology was detectable within 2 d after αSyn PFFs treatment, and αSyn inclusions began to emerge at 4 and 7 d (Fig. 3B,D). This observation is consistent with in vitro studies demonstrating that the seeding of αSyn aggregation is directly correlated with increasing developmental expression of endogenous αSyn in neurons (Volpicelli-Daley et al., 2011).
To confirm the specificity of the phenotypes we observed in αSyn PFF-treated neurons, we repeated the electrophysiological recordings and ICC staining in αSyn knock-out neurons. Prior studies have shown that in the absence of endogenous αSyn, no αSyn pathologies developed in vitro and in vivo after αSyn PFFs treatment (Volpicelli-Daley et al., 2011; Luk et al., 2012b). In contrast to WT neurons, no deficits in synaptic responses and no p-αSyn pathology were detected at low and high dosages of αSyn PFFs treatment, even at longer incubation time points (Fig. 4). These data suggest that αSyn PFFs alone could not modulate mEPSCs in the absence of endogenous αSyn. Thus, we conclude that αSyn PFFs compromised synaptic activity in a dose- and time-dependent manner, and these functional deficits are dependent on the presence of endogenous αSyn protein and likely because of the formation of intracellular αSyn aggregates, as there is a temporal correlation between the development of αSyn inclusions and synaptic dysfunction.
Figure 4.

No synaptic activity defects and pathology developed in αSyn KO neurons. A, B, Synaptic activity remained unchanged after αSyn PFFs treatment for 4 and 7 d in αSyn KO neurons. C, No αSyn pathology detected by ICC staining in αSyn KO neurons. Syn211 antibody was used to label the human αSyn PFFs seeds added to the cultures. 81a: p-αSyn, Syn211: human αSyn PFFs seeds, MAP2: dendrites. Electrophysiology data collected from three independent batches of cultures, ICC staining data collected from two independent batches of cultures. Scale bar, 50 μm.
Reduction in synaptic activity correlates with the development of αSyn pathology
As noted, there appears to be a correlation between the progressive accumulation of p-αSyn pathology inside neurons and the decrease in mEPSC frequency. To confirm this finding and to more carefully investigate the potential of misfolded αSyn to modulate neuronal physiology, we used recording micropipettes to introduce AlexaFluor 594 biocytin (biocytin594) dye into single neurons through passive infusion such that neurons can be identified after whole-cell patch-clamp recording. After infusion for 15–20 min, we were able to visualize the neurons we recorded (Fig. 5A–C). To determine whether these labeled neurons have developed αSyn pathology, we conducted ICC after completion of patch-clamp recordings, using the p-αSyn mouse antibody (mAb) 81a to immunostain both biocytin594-labeled and -unlabeled neurons. Confocal 3D reconstruction showed that some of the biocytin594-labeled neurons were 81a positive. In contrast and as expected, the PBS-treated neurons were 81a negative (Fig. 5C). Here, we summarized the criteria we used to characterize 81a pathologies: if the 81a staining was inside the biocytin594-labeled cells, we define this cell as 81a positive (Fig. 5A); if all the 81a puncta staining were outside of biocytin594-labeled cells, we define this neuron as 81a negative (Fig. 5B); the neuron in Figure 5C was transduced with PBS as a control and no 81a staining was detected. White arrows in Figure 5A indicate other 81a-positive staining inside biocytin594 labeled neurons. DAPI was merged with biocytin594 in Figure 5C.
Figure 5.

Reductions in mEPSC frequency correlated with the deposition of αSyn pathology. A–C, Confocal 3D scanning images of p-αSyn (detected by mAb 81a) in AlexaFluor 594 Biocytin (biocytin594)-labeled hippocampal neurons at 7 d after αSyn PFF treatment. The fluorescent dye biocytin594 (5% in pipette intracellular solution) was delivered via whole-cell patch-clamp pipettes to label αSyn PFF-infused hippocampal neurons. If the 81a staining was inside the biocytin594-labeled cells, we define this cell as 81a positive (A); if all the 81a puncta staining were outside of biocytin594-labeled cells, we define this neuron as 81a negative (B); the neuron in C was transduced with PBS as control. White arrows in A indicate other 81a-positive staining inside biocytin594-labeled neurons. DAPI was merged with biocytin594 in C. D, Corresponding mEPSC recordings from A–C. E, Statistics of mEPSC peak amplitude and frequency from D. n = 8 cells for PBS, n = 12 cells for 81a negative, n = 11 cells for 81a positive. F, Schematic diagram illustrating that with increasing deposition of αSyn pathology, the mEPSC frequency decreased accordingly. Please note that A and B were captured by confocal z-stack imaging and then z-projection into one focal plane, whereas Figure 3, A and B, were taken by conventional fluorescent microscopy displaying only one focal plane. This provides an explanation for why more 81a signals were found in A and B than Figure 3, A and B. Results shown as mean ± SEM; n = 5 independent batches of neurons, *p < 0.05, **p < 0.01, ***p < 0.001; one-way ANOVA with Tukey post hoc test (E).
Contemporaneous with the passive infusion of the biocytin594 dye to help identify neurons with p-αSyn pathology, we recorded the corresponding synaptic activity from these labeled neurons (Fig. 5D). Importantly, quantitative analysis showed that the peak amplitude of mEPSCs remained unchanged in neurons with 81A-positive αSyn pathology (Fig. 5E, left), wherein the mEPSC frequency showed significant reductions in 81a-positive neurons (Fig. 5E, right). We also found that biocytin594-labeled 81a-negative neurons in Figure 5E showed some reduction in mEPSC frequency that could result from the neighboring 81a-positive inputs, as mEPSCs are mediated by both presynaptic and postsynaptic contributions (Queenan et al., 2012). Thus, together with Figure 3, we found a correlation of reduction in mEPSC frequency with 81a burden in these neurons, such that an increased burden of αSyn pathology inside the neurons was associated with a more pronounced reduction in synaptic responses (Fig. 5F).
Passive infusion of αSyn PFFs from patch-clamp recording pipettes modulates both synaptic activity and αSyn pathology
Although there is a correlation between the develop of αSyn pathology and reduced mEPSC frequencies in the neuronal cultures, we cannot exclude the possibility that the added exogenous αSyn PFFs themselves induce a time-dependent change in neuronal electrophysiology. For example, it has been suggested that αSyn PFFs induce cellular oxidation (Dryanovski et al., 2013). To mitigate this potential confound, we passively infused either sonicated αSyn PFFs, αSyn monomers, or PBS via whole-cell patch-clamp recording pipettes into cultured hippocampal WT neurons (Fig. 6A) and then recorded synaptic responses at different time points (Fig. 6B). Consistent with previous findings in neurons bearing αSyn aggregates 7 d after αSyn PFF treatment (Fig. 1), the mEPSC peak amplitude in these three different groups were nearly the same when measured within 10 min post-infusion with αSyn PFFs (Fig. 6B, top). Interestingly, the mEPSC frequency decreased markedly within 5 min in αSyn PFF-infused neurons, whereas these changes were not observed after infusion of αSyn monomer or PBS (Fig. 6B, bottom). As a control experiment, we added biocytin594 into the pipette solution and found that 5 min was enough for infusion to occur from soma into the dendrites or axons (data not shown), suggesting that the αSyn PFFs in the pipette may infuse rapidly enough to modulate dendritic EPSC activity within the short time intervals (5–10 min) studied here (Fig. 6B). These data suggest that only the pathology-inducible αSyn PFFs, but not the αSyn monomers, compromised synaptic responses.
Figure 6.

Direct infusion of αSyn PFFs from patch-clamp pipettes modulated both synaptic activity and pathology. A, Schematic diagram showing the delivery of αSyn PFFs via patch-clamp pipette. The sonicated αSyn PFFs (5 μm in pipette) were passively infused into cultured hippocampal neurons by whole-cell patch-clamp and mEPSCs were recorded simultaneously, after which the formation of pathologies was examined in the infused neurons. B, mEPSC frequency was compromised after acute αSyn PFFs infusion, but not αSyn monomers or PBS. C, ICC staining of αSyn pathology in the infused neuron after culture for 20 h. DAPI: nucleic staining; 81a: p-αSyn; Syn211: human αSyn PFFs seeds; MAP2: dendrites staining. Scale bar, 20 μm. D, As control, neurons transduce with αSyn PFFs in the culture medium for 1 d where there is insufficient time to trigger pathology development. Scale bar, 50 μm. Results shown as mean ± SEM; n = 4 independent batches of neurons. *p < 0.05, **p < 0.01; one-way ANOVA with Tukey post hoc test (B).
We next asked whether the αSyn PFF-infused neurons would develop αSyn pathologies after additional incubation in culture. Unexpectedly, these neurons developed 81a-positive puncta near the somatodendritic compartments within 20 h after αSyn PFFs infusion (Fig. 6C), with smaller puncta detected in nascent dendrites as well. In a parallel control experiments where αSyn PFFs were added into the culture dish for 20 h, we only detected the added human αSyn PFFs in the extracellular space after immunostaining with the human αSyn-specific mAb Syn211, and we did not observe staining with the Ser129 phosphorylation-dependent mAb 81a antibody that only detects intracellular p-αSyn pathology (Fig. 6D).
Due to the fact that the internal solution is a homogeneously suspension of PFFs, it is not possible to directly measure the amount of PFFs that actually reached the distal compartment (dendrites and axons) of the patched cells. Despite that under these conditions, this small amount of PFFs was able to modulate synaptic response within minutes and induce pathology within 1 d. Thus, we have developed a neuron-based system that enabled us to demonstrate modulation of synaptic activity in response to intraneuronal αSyn PFFs at a single-cell level, and that neuronally-infused αSyn PFFs decreased mEPSC frequency within minutes (Fig. 6).
αSyn PFF treatment decreased synapse formation
To explore the mechanisms by which αSyn fibrils inhibited synaptic responses, we first asked whether addition of PFFs to the cultures could kill neurons directly. We added αSyn PFFs at 0.5 and 2 μg/well and incubated neurons for 4, 7, and 11 d in vitro, after which the cultures were immunostained with the neuronal marker NeuN to assess the total number of living neurons remaining after this treatment. No significant neuron loss was found within 1 week at either of the αSyn PFFs dosages, but there was a ∼10% loss of neurons at the higher dose of PFFs at 11 d post-treatment (Fig. 7D). As most of the experiments were completed within 1 week post-treatment with the low dose of αSyn PFFs (0.5 μg/well), we infer that the decreased mEPSC frequency is a direct consequence of αSyn PFFs induced intraneuronal αSyn aggregates rather than neuron loss. This conclusion is further supported by the rapid change in mEPSC frequency that we observed upon direct infusion of αSyn PFFs into the neurons (within 5 min), where we saw no evidence of acute neurotoxicity.
Figure 7.
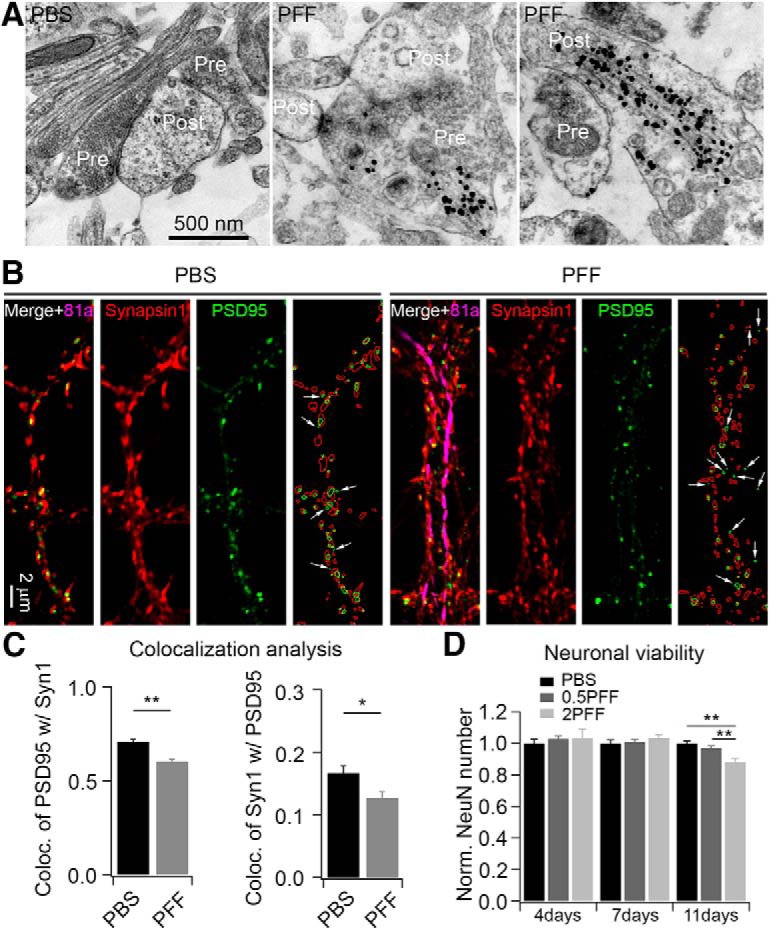
αSyn PFFs treatment decreased synapse formation. A, Ultrastructure of αSyn aggregates in the synapse revealed by immuno-EM. The 81a gold nanoparticles were found in both presynaptic and postsynaptic compartments (representative images from 3 independent batches of neurons). B, ICC staining of synapses in PBS and αSyn PFF-treated neurons. αSyn PFFs were transduced at 0.5 μg/well for 1 week. Synapsin 1 (red) and PSD-95 (green) were used to label presynaptic and postsynaptic compartments, respectively; 81a (magenta) was used to label αSyn pathology. White arrows indicate the PSD-95 puncta that are not colocalized with synapsin 1. ImageJ software was used to draw the outlines of synapsin 1 (red circles) and PSD-95 (green circles), and more green circles that are not colocalized with red ones in 81a-positive dendrites (rightmost panels in PBS- and PFF-treated cells). C, Quantification of colocalization of synaptic markers in PBS and 81a-positive dendritic segments. Left, Colocalization of PSD-95 with synapsin 1 (Syn1); right, colocalization of Syn1 with PSD-95. D, αSyn PFFs treatment decreased neuronal viability at a later stage. No significant neuronal loss was found within 1 week post-transduction. 0.5PFF, 0.5 μg/well αSyn PFFs; 2PFF, 2 μg/well αSyn PFFs. Results shown as mean ± SEM; n = 4 and 3 independent batches of neurons for colocalization and viability analysis, respectively. *p < 0.05, **p < 0.01; Student's t test (C) and one-way ANOVA with Tukey post hoc test (D).
Next, we performed immuno-EM to visualize the ultrastructure of synapses and 81a pathologies with gold nanoparticles in neurons treated for 7 d with αSyn PFFs (Fig. 7A). These images revealed gold nanoparticles in both presynaptic and postsynaptic compartments, which is consistent with previous reports that the αSyn is enriched at presynapses (Hsu et al., 1998; Murphy et al., 2000; Lashuel et al., 2013) and that the αSyn PFF-induced pathologies are transferred from axons to the dendrites over time (Volpicelli-Daley et al., 2011). Together, this suggests that αSyn PFFs might compromise synaptic function first before compromising viability of the neurons over time (Volpicelli-Daley et al., 2011; Peelaerts et al., 2015; Rey et al., 2018). To test this hypothesis, we used the presynaptic marker synapsin 1 and the postsynaptic marker PSD-95 to label synapses and analyzed the colocalization of these two markers. Figure 7B shows a reduction of colocalized synapsin 1 and PSD-95 in 81a-positive staining segments relative to those without αSyn pathology. Quantitative analysis confirmed that the colocalization of PSD-95 with synapsin 1 was decreased in αSyn PFF-treated neurons compared with PBS-treated neurons (Fig. 7C), which is consistent with postmortem studies wherein there were reduced numbers of synapses in the brains of PD and DLB patients (Zaja-Milatovic et al., 2005, 2006; Kramer and Schulz-Schaeffer, 2007). Thus, the reduction in mEPSC frequency is likely due to synaptic dysfunction before neuron loss occurs.
αSyn PFF treatment compromised the function of dendritic spines
Dendritic spines are specialized structures where the majority of excitatory synapses are localized (Chih et al., 2005; Lai and Ip, 2013), and the formation of synapses affects the function of dendritic spines (Toni et al., 1999; Nimchinsky et al., 2002). To investigate the effect of αSyn PFFs on the formation and maintenance of dendritic spines, we costained spines with the actin cytoskeleton labeling dye, AlexaFluor 488 phalloidin (green), and the spine-binding protein, drebrin (red; Fig. 8A). Quantitative analysis showed that the number of phalloidin and drebrin puncta per μm of dendrite length of cultured hippocampal neurons were both decreased after αSyn PFF treatment for 1 week (Fig. 8B,C, right), whereas the size and intensity of single puncta exhibited no significant difference (Fig. 8B,C, left and middle). The unchanged size and intensity of single puncta are consistent with the unchanged peak amplitudes of mEPSCs (Fig. 1), as the peak amplitude reflects the density of postsynaptic AMPA receptors. Furthermore, the shape of dendritic spines could regulate changes in synaptic strength (Frotscher et al., 2014). To assess this possibility, we infected neurons with a GFP-expressing lentivirus to label spines (Fig. 8D), which revealed single proximal and distal spines in both PBS- and αSyn PFF-treated neurons (Fig. 8E). The number of total GFP-labeled spines/μm decreased ∼50% after αSyn PFF treatment (Fig. 8F), which is consistent with the notion that the number of spines, and hence the number of synapses, is reflected in the frequency of mEPSCs (Fig. 1, 5, 6).
Figure 8.

αSyn PFFs treatment compromised formation of dendritic spines. A, ICC staining of dendritic spines with phalloidin and drebrin in PBS and PFF-treated neurons. Phalloidin (green) was used to label actin; drebrin (red) is an actin binding protein; 81a (white) was used to label αSyn pathology. αSyn PFFs were transduced at 0.5 μg/well for 1 week. B, Quantification of puncta size, fluorescence intensity, and number of drebrin/μm in PBS-treated and 81a-positive dendritic segments. C, Quantification of puncta size, fluorescence intensity, and number of phalloidin/μm in PBS-treated and 81a-positive dendritic segments. D, Dendritic spines were labeled with GFP in PBS and αSyn PFF-treated neurons. Scale bar, 20 μm. E, Enlarged images of proximal and distal spines from PBS and αSyn PFF-treated neurons. Scale bar, 5 μm. F, Quantification of number of spines/μm. G, Representative images of spine subtypes: mushroom, stubby, and thin in PBS and αSyn PFF-treated neurons. H, I, Quantifications of number and size of proximal and distal spines treated with either PBS or PFFs. Neurons were transfected with lentivirus overexpressing GFP to label spines at 4 DIV, PFFs were added at 7 DIV, and neurons were fixed at 14 DIV. Confocal z-stack imaging was performed with 1 μm serial scanning. Images were z-projected and analyzed with NeuronStudio software. Experiments were repeated with 4 different cultures, cell number n = 14 for PBS and n = 15 for PFF treatment. mush, Mushroom; stub, stubby. Results shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001; ns, no significant. Student's t test (B, C, F, H, I).
Dendritic spines act as compartmental calcium sinks during synaptic activity-induced rises in intracellular calcium (Yuste et al., 2000). Thus, spine density increases with enhanced synaptic activity, and shrinks or is replaced by thin/filopodia spines when synaptic activity is low (Segal et al., 2010). To this end, we analyzed the different subtypes of dendritic spines. Based on the morphologies and previous criteria for dendritic spines (Peters and Kaiserman-Abramof, 1970; Rodriguez et al., 2008), we characterized three different subtypes of spines in our cultured hippocampal neurons: i.e., mushroom, stubby, and thin (Fig. 8G). Mushroom spines, which are the most mature spine subtype that contacts with presynaptic boutons, decreased ∼40% after αSyn PFF treatment in both proximal and distal dendrites (Fig. 8H). Stubby spines, which may transform into thin spines and then change into mushroom subtypes after LTP (Popov et al., 2004), did not show significant changes following αSyn PFF treatment (Fig. 8H). Finally, the thin spines, which are an interphase subtype and quite dynamic, decreased ∼20% in proximal dendrites after αSyn PFF treatment (Fig. 8H). In addition to the number of spines, we also analyzed the size of distinct proximal and distal spines. No significant changes were found in PBS and PFF-treated neurons (Fig. 8I), which is consistent with previous immunostaining data where the size and intensity of single phalloidin and drebrin puncta exhibited no significant difference (Fig. 8B,C, left and middle). To exclude the possibility that short-term αSyn PFFs exposure alone can induce these changes, we treated neurons with PBS or PFFs for only 2 h and 1 d, during which time we saw no significant changes in spine morphologies between PBS- and PFF-treated neurons (data not shown). Thus, the altered morphology of dendritic spines was a consequence of long-term treatment with αSyn PFFs and the formation of αSyn pathology that then compromised synaptic responses.
Passive infusion of αSyn PFF from patch-clamp recording pipettes into cultured hippocampal neurons disrupted dendritic spine dynamics
Live imaging is advantageous in monitoring the time course of changes in spine structure (Dailey and Smith, 1996; Chen et al., 2008). Thus, we transfected neurons with lentivirus encoding GFP at 10–12 DIV to label individual spines and passively infused PBS, sonicated αSyn monomers or αSyn PFFs using a whole-cell patch-clamp pipette 10 d later following the protocol described in the previous section (Fig. 6). Using this approach, we observed the acute effects of αSyn PFFs on dendritic spine dynamics. In neurons infused with PBS, we detected a balanced formation and retraction of spines within 5–10 min (Fig. 9A), but in neurons infused with αSyn PFFs, we found less newly formed spines when compared with the initial time point (Fig. 9B,C). Quantitative analyses further supported that the formation and retraction of spines were quite dynamic and exhibited balanced changes in PBS and αSyn monomer infused neurons (Fig. 9D,E), although there were less newly formed spines in neurons infused with αSyn PFFs, even though the spine retraction speeds were nearly the same in these different treatments (Fig. 9D,E). Thus, together with the previous findings, infused αSyn PFFs modulated single spine dynamics within minutes (Fig. 9), and this in turn decreased mEPSC frequency followed by pathological αSyn induced neurodegeneration in the long-term (Fig. 10).
Figure 9.

Passive infusion of αSyn PFFs from patch-clamp pipettes altered dendritic spine dynamics. A, Time-lapse imaging of dynamic changes of dendritic spines in PBS-infused neurons. Dendritic spines were labeled with GFP-expressing lentivirus at 10 DIV and confocal z-stack images were taken at 17–20 DIV at 30 s intervals. Yellow stars indicate the newly formed spines; red stars indicate the retracted spines during infusion and imaging. B, Time-lapse imaging of dynamic changes of dendritic spines in αSyn PFF-infused neurons. For a better comparison of dynamic changes of dendritic spines, the “onset” of imaging was pseudocolor-labeled with green; the other time points after the onset were labeled with red. C, Merged images of onset and other time points from B. The red arrows indicate the newly formed spines during αSyn PFFs infusion; the white arrows indicate the retracted spines. Scale bars: A–C, 5 μm. D, Quantification of spine dynamic changes under different treatment conditions. E, Quantifications of the rate of retraction and formation of spines under different treatment conditions. Experiments have been performed with three independent repeats for PBS and monomer, four independent repeats for αSyn PFF. Cell number n = 8 for PBS, n = 10 for monomers, n = 17 for αSyn PFF. Results shown as mean ± SEM. *p < 0.05, **p < 0.01; Student's t test (D) and one-way ANOVA with Tukey post hoc test (E).
Figure 10.

Schematic summary of αSyn inclusion formation, dendritic spine dysfunction, and neuronal loss. In the presence of exogenous αSyn PFFs, soluble endogenous αSyn monomers are recruited into aggregates (1). These aggregates are distributed in the dendritic spines, induced spine shrinkage, and synaptic deficits (2). Long-term incubations lead to neurodegeneration in inclusion-bearing neurons (3).
Discussion
Here, we demonstrated that the addition of αSyn PFFs assembled from recombinant human αSyn monomers to primary neurons led to a time- and dose-dependent formation of intraneuronal aggregates comprised of mouse αSyn that compromised synaptic activity, followed by accelerated neuron death. Importantly, the degree of reduction in synaptic activity correlated with the deposition of misfolded αSyn as insoluble aggregates, suggesting that αSyn inclusions act as a pathogenic trigger that ultimately compromised synaptic function. Our findings further extended recent studies demonstrating that exposure to exogenous aggregated αSyn induces PD-like LB/LN pathologies, neuron loss and reduces the viability of neurons in αSyn transgenic and WT mice, as well as their lifespan (Volpicelli-Daley et al., 2011; Luk et al., 2012a,b). Notably, the time-dependent formation of αSyn pathology and associated reduction in mEPSC frequency observed after the addition of αSyn PFFs to neuron cultures can be bypassed by acute infusion of αSyn PFFs, but not αSyn monomers, into neurons via whole-cell patch-clamp, where a decrease of mEPSC frequency can be observed within 5 min after PFFs infusion. Moreover, the infused PFFs induce the initiation of 81A-positive pathology within 1 d post-infusion, which is much faster than previously reported for external exposure of neurons to αSyn PFFs (i.e., 3–4 d). This suggests that direct infusion of αSyn PFFs bypasses the multiple cellular processes of PFFs uptake and trafficking within the lysosomes (Volpicelli-Daley et al., 2011; Karpowicz et al., 2017), thereby facilitating seeding of endogenous mouse αSyn. This orthogonal experimental technique provided convincing evidence that αSyn fibrils modulate synaptic responses at the single cell level. Mechanistically, αSyn fibrils dramatically compromised the colocalization of synaptic markers and reduced formation of dendritic spines, thereby contributing to the neurodegenerative processes (Fig. 10), and hence recapitulating the phenotypes seen in clinical synucleinopathies, such as PD, PDD, and DLB (Zaja-Milatovic et al., 2005, 2006; Kramer and Schulz-Schaeffer, 2007; Irwin et al., 2017).
The studies of the effects of αSyn monomers, oligomers and fibrils on synaptic activity have been reported by multiple groups (Larsen et al., 2006; Nemani et al., 2010; Volpicelli-Daley et al., 2011; Diógenes et al., 2012; Wang et al., 2014; Lautenschläger et al., 2017; Logan et al., 2017; Froula et al., 2018). However, most of these prior studies either lacked vital controls or they used high levels of αSyn which are associated with confounds such as mitochondrial oxidative stress (Dryanovski et al., 2013). Moreover, in the studies that used transgenic animal models to mimic PD pathogenesis (Polymeropoulos et al., 1997; Krüger et al., 1998; Giasson et al., 2002; Zarranz et al., 2004; Kiely et al., 2013; Proukakis et al., 2013; Pasanen et al., 2014), these mutant αSyn-overexpressing models are limited by the fact that they develop αSyn pathologies at older ages (12–14 months), whereas most PD patients lack SNCA mutations or duplications showed the same amount of αSyn expression levels as age-matched controls (Su et al., 2017).
In the present work, we systematically quantified the dosages of αSyn PFFs that evoked activity deficits in primary neuron cultures. The power of this system is that we can determine the formation of PD-like LBs and LNs under physiological conditions using pure WT human αSyn PFFs that seed endogenous mouse αSyn without overexpression. Using this system, we revealed a relationship between the induction of inclusion formation by αSyn PFFs and reductions in synaptic responses that precede neuron loss. Thus, these findings suggest that a disruption of synaptic function may be one of the initial consequences of the formation of fibrillary αSyn pathology, and these synaptic alterations could contribute to neuronal dysfunction and ultimately neuronal toxicity. In addition, there could be loss of function mechanisms underlying the electrophysiological observations. Previous in vitro studies showed that formation of αSyn aggregates reduced synaptic protein levels, including the synaptic vesicle-associated SNARE proteins, and also progressively disrupted neuronal network activity (Volpicelli-Daley et al., 2011). What's more, exogenous αSyn PFFs were also reported to induce cellular oxidation (Dryanovski et al., 2013). Thus, loss-of-function defects must be taken into consideration in future investigations.
Another unique aspect of our work is the ability to technically modulate synaptic responses with passively infused αSyn PFFs at the single-cell level, without disturbances from other environmental factors. An unexpected result of our findings is that the direct infusion of αSyn PFFs induced the initiation of 81A-positive αSyn pathology as soon as 20 h after treatment, which is quite different from introducing αSyn PFFs into the culture media which takes 3–4 d to develop detectable pathologies (Volpicelli-Daley et al., 2011). Although we cannot preclude the possibility that the 81a-positive puncta that are observed shortly after αSyn PFFs infusion result from direct phosphorylation of the infused αSyn PFFs, the more likely explanation is that these represent recruited phosphorylated mouse αSyn that has been seeded by the infused PFFs. This rapidity of seeding of αSyn pathology could arise from the fact that, unlike the αSyn PFFs transduction system, the infused αSyn PFFs do not need to undergo uptake through the plasma membrane, trafficking along the endo-lysosomal pathway, processing and release from the lysosome into the cytosol before recruiting endogenous αSyn monomer into insoluble inclusions (Irwin et al., 2013; Guo and Lee, 2014; Karpowicz et al., 2017). This system provides the intriguing possibility of monitoring the trafficking of fluorescent-labeled αSyn seeds inside neurons and observing the interaction/recruitment of endogenous αSyn monomers into insoluble inclusions.
Finally, our model allowed us to determine the consequences of potential αSyn dysfunction resulting from its recruitment into LB/LN PD-like inclusions in both presynaptic and postsynaptic sites, leading to reduced colocalization of synaptic markers and inhibition of functional synapses. Indeed, we obtained evidence to demonstrate that there could be a direct interaction between αSyn PFFs and single dendritic spines. This is consistent with previous postmortem studies, where nearly 90% of αSyn aggregates were located at synapses in the frontal cortex of DLB patients (Schulz-Schaeffer, 2010), and significant synaptic pathology with almost complete loss of dendritic spines were observed at these postsynaptic areas (Zaja-Milatovic et al., 2005, 2006; Kramer and Schulz-Schaeffer, 2007). This was also recapitulated in long-term live imaging of spine dynamics (Blumenstock et al., 2017). Additional studies are needed to test the hypothesis that the maintenance of functional synapses in the presence of αSyn inclusions could preserve “healthier” neurons, and thus provide benefit in PD and related synucleinopathies.
Footnotes
This work was supported by NIH/NINDS Udall Center Grant NS53488, the Jeff and Anne Keefer Fund, and the Neurodegenerative Disease Research Fund. We thank Drs. Kelvin Luk and Kurt Brunden for helpful comments and editing of the paper, Anna Stieber for assistance in immnuno-EM samples preparation and imaging, Dr. Sharon Xie for power analysis of the data, and staff from the Cell and Developmental Biology Microscopy Core and the Electron Microscopy Resource Laboratory at the University of Pennsylvania for technical assistance in confocal and EM imaging.
The authors declare no competing financial interests.
References
- Abeliovich A, Schmitz Y, Fariñas I, Choi-Lundberg D, Ho WH, Castillo PE, Shinsky N, Verdugo JM, Armanini M, Ryan A, Hynes M, Phillips H, Sulzer D, Rosenthal A (2000) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25:239–252. 10.1016/S0896-6273(00)80886-7 [DOI] [PubMed] [Google Scholar]
- Blumenstock S, Rodrigues EF, Peters F, Blazquez-Llorca L, Schmidt F, Giese A, Herms J (2017) Seeding and transgenic overexpression of alpha-synuclein triggers dendritic spine pathology in the neocortex. EMBO Mol Med 9:716–731. 10.15252/emmm.201607305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC (2010) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329:1663–1667. 10.1126/science.1195227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Südhof TC (2014) Alpha-synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 111:E4274–E4283. 10.1073/pnas.1416598111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burré J, Sharma M, Südhof TC (2018) Cell biology and pathophysiology of alpha-synuclein. Cold Spring Harb Perspect Med 8:a024091. 10.1101/cshperspect.a024091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra S, Gallardo G, Fernández-Chacón R, Schlüter OM, Südhof TC (2005) Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 123:383–396. 10.1016/j.cell.2005.09.028 [DOI] [PubMed] [Google Scholar]
- Chen Y, Dubé CM, Rice CJ, Baram TZ (2008) Rapid loss of dendritic spines after stress involves derangement of spine dynamics by corticotropin-releasing hormone. J Neurosci 28:2903–2911. 10.1523/JNEUROSCI.0225-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chih B, Engelman H, Scheiffele P (2005) Control of excitatory and inhibitory synapse formation by neuroligins. Science 307:1324–1328. 10.1126/science.1107470 [DOI] [PubMed] [Google Scholar]
- Dailey ME, Smith SJ (1996) The dynamics of dendritic structure in developing hippocampal slices. J Neurosci 16:2983–2994. 10.1523/JNEUROSCI.16-09-02983.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danzer KM, Haasen D, Karow AR, Moussaud S, Habeck M, Giese A, Kretzschmar H, Hengerer B, Kostka M (2007) Different species of alpha-synuclein oligomers induce calcium influx and seeding. J Neurosci 27:9220–9232. 10.1523/JNEUROSCI.2617-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desplats P, Lee HJ, Bae EJ, Patrick C, Rockenstein E, Crews L, Spencer B, Masliah E, Lee SJ (2009) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106:13010–13015. 10.1073/pnas.0903691106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickson DW, Crystal H, Mattiace LA, Kress Y, Schwagerl A, Ksiezak-Reding H, Davies P, Yen SH (1989) Diffuse lewy body disease: light and electron microscopic immunocytochemistry of senile plaques. Acta Neuropathologica 78:572–584. 10.1007/BF00691284 [DOI] [PubMed] [Google Scholar]
- Diógenes MJ, Dias RB, Rombo DM, Vicente Miranda H, Maiolino F, Guerreiro P, Näsström T, Franquelim HG, Oliveira LM, Castanho MA, Lannfelt L, Bergström J, Ingelsson M, Quintas A, Sebastião AM, Lopes LV, Outeiro TF (2012) Extracellular alpha-synuclein oligomers modulate synaptic transmission and impair LTP via NMDA-receptor activation. J Neurosci 32:11750–11762. 10.1523/JNEUROSCI.0234-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryanovski DI, Guzman JN, Xie Z, Galteri DJ, Volpicelli-Daley LA, Lee VM, Miller RJ, Schumacker PT, Surmeier DJ (2013) Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J Neurosci 33:10154–10164. 10.1523/JNEUROSCI.5311-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuele M, Esposito A, Camerini S, Antonucci F, Ferrara S, Seghezza S, Catelani T, Crescenzi M, Marotta R, Canale C, Matteoli M, Menna E, Chieregatti E (2016) Exogenous alpha-synuclein alters pre- and post-synaptic activity by fragmenting lipid rafts. EBioMedicine 7:191–204. 10.1016/j.ebiom.2016.03.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Studer D, Graber W, Chai X, Nestel S, Zhao S (2014) Fine structure of synapses on dendritic spines. Front Neuroanat 8:94. 10.3389/fnana.2014.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froula JM, Henderson BW, Gonzalez JC, Vaden JH, Mclean JW, Wu Y, Banumurthy G, Overstreet-Wadiche L, Herskowitz JH, Volpicelli-Daley LA (2018) α-Synuclein fibril-induced paradoxical structural and functional defects in hippocampal neurons. Acta Neuropathol Commun 6:35. 10.1186/s40478-018-0537-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giasson BI, Murray IV, Trojanowski JQ, Lee VM (2001) A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J Biol Chem 276:2380–2386. 10.1074/jbc.M008919200 [DOI] [PubMed] [Google Scholar]
- Giasson BI, Duda JE, Quinn SM, Zhang B, Trojanowski JQ, Lee VM (2002) Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34:521–533. 10.1016/S0896-6273(02)00682-7 [DOI] [PubMed] [Google Scholar]
- Guo JL, Lee VM (2014) Cell-to-cell transmission of pathogenic proteins in neurodegenerative diseases. Nat Med 20:130–138. 10.1038/nm.3457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo JL, Covell DJ, Daniels JP, Iba M, Stieber A, Zhang B, Riddle DM, Kwong LK, Xu Y, Trojanowski JQ, Lee VM (2013) Distinct alpha-synuclein strains differentially promote tau inclusions in neurons. Cell 154:103–117. 10.1016/j.cell.2013.05.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu LJ, Mallory M, Xia Y, Veinbergs I, Hashimoto M, Yoshimoto M, Thal LJ, Saitoh T, Masliah E (1998) Expression pattern of synucleins (non-Aβ component of Alzheimer's disease amyloid precursor protein/alpha-synuclein) during murine brain development. J Neurochem 71:338–344. 10.1046/j.1471-4159.1998.71010338.x [DOI] [PubMed] [Google Scholar]
- Irwin DJ, Grossman M, Weintraub D, Hurtig HI, Duda JE, Xie SX, Lee EB, Van Deerlin VM, Lopez OL, Kofler JK, Nelson PT, Jicha GA, Woltjer R, Quinn JF, Kaye J, Leverenz JB, Tsuang D, Longfellow K, Yearout D, Kukull W, et al. (2017) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: a retrospective analysis. Lancet Neurol 16:55–65. 10.1016/S1474-4422(16)30291-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin DJ, Lee VM, Trojanowski JQ (2013) Parkinson's disease dementia: convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14:626–636. 10.1038/nrn3549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpowicz RJ Jr, Haney CM, Mihaila TS, Sandler RM, Petersson EJ, Lee VM (2017) Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J Biol Chem 292:13482–13497. 10.1074/jbc.M117.780296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiely AP, Asi YT, Kara E, Limousin P, Ling H, Lewis P, Proukakis C, Quinn N, Lees AJ, Hardy J, Revesz T, Houlden H, Holton JL (2013) Alpha-synucleinopathy associated with G51D SNCA mutation: a link between Parkinson's disease and multiple system atrophy? Acta Neuropathologica 125:753–769. 10.1007/s00401-013-1096-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer ML, Schulz-Schaeffer WJ (2007) Presynaptic alpha-synuclein aggregates, not Lewy bodies, cause neurodegeneration in dementia with lewy bodies. J Neurosci 27:1405–1410. 10.1523/JNEUROSCI.4564-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krüger R, Kuhn W, Müller T, Woitalla D, Graeber M, Kosel S, Przuntek H, Epplen JT, Schöls L, Riess O (1998) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18:106–108. 10.1038/ng0298-106 [DOI] [PubMed] [Google Scholar]
- Lai KO, Ip NY (2013) Structural plasticity of dendritic spines: the underlying mechanisms and its dysregulation in brain disorders. Biochim Biophys Acta 1832:2257–2263. 10.1016/j.bbadis.2013.08.012 [DOI] [PubMed] [Google Scholar]
- Larsen KE, Schmitz Y, Troyer MD, Mosharov E, Dietrich P, Quazi AZ, Savalle M, Nemani V, Chaudhry FA, Edwards RH, Stefanis L, Sulzer D (2006) α-Synuclein overexpression in PC12 and chromaffin cells impairs catecholamine release by interfering with a late step in exocytosis. J Neurosci 26:11915–11922. 10.1523/JNEUROSCI.3821-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashuel HA, Overk CR, Oueslati A, Masliah E (2013) The many faces of alpha-synuclein: from structure and toxicity to therapeutic target. Nat Rev Neurosci 14:38–48. 10.1038/nrn3406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lautenschläger J, Kaminski CF, Kaminski Schierle GS (2017) Alpha-synuclein: regulator of exocytosis, endocytosis, or both? Trends Cell Biol 27:468–479. 10.1016/j.tcb.2017.02.002 [DOI] [PubMed] [Google Scholar]
- Logan T, Bendor J, Toupin C, Thorn K, Edwards RH (2017) α-Synuclein promotes dilation of the exocytotic fusion pore. Nat Neurosci 20:681–689. 10.1038/nn.4529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm VM, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012a) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209:975–986. 10.1084/jem.20112457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luk KC, Kehm V, Carroll J, Zhang B, O'Brien P, Trojanowski JQ, Lee VM (2012b) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338:949–953. 10.1126/science.1227157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda-Suzukake M, Nonaka T, Hosokawa M, Oikawa T, Arai T, Akiyama H, Mann DM, Hasegawa M (2013) Prion-like spreading of pathological alpha-synuclein in brain. Brain 136:1128–1138. 10.1093/brain/awt037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy DD, Rueter SM, Trojanowski JQ, Lee VM (2000) Synucleins are developmentally expressed, and alpha-synuclein regulates the size of the presynaptic vesicular pool in primary hippocampal neurons. J Neurosci 20:3214–3220. 10.1523/JNEUROSCI.20-09-03214.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH (2010) Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 65:66–79. 10.1016/j.neuron.2009.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimchinsky EA, Sabatini BL, Svoboda K (2002) Structure and function of dendritic spines. Annu Rev Physiol 64:313–353. 10.1146/annurev.physiol.64.081501.160008 [DOI] [PubMed] [Google Scholar]
- Pacheco CR, Morales CN, Ramírez AE, Muñoz FJ, Gallegos SS, Caviedes PA, Aguayo LG, Opazo CM (2015) Extracellular alpha-synuclein alters synaptic transmission in brain neurons by perforating the neuronal plasma membrane. J Neurochem 132:731–741. 10.1111/jnc.13060 [DOI] [PubMed] [Google Scholar]
- Pasanen P, Myllykangas L, Siitonen M, Raunio A, Kaakkola S, Lyytinen J, Tienari PJ, Pöyhönen M, Paetau A (2014) Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson's disease-type pathology. Neurobiol Aging 35:2180.e1–5. 10.1016/j.neurobiolaging.2014.03.024 [DOI] [PubMed] [Google Scholar]
- Peelaerts W, Bousset L, Van der Perren A, Moskalyuk A, Pulizzi R, Giugliano M, Van den Haute C, Melki R, Baekelandt V (2015) Alpha-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522:340–344. 10.1038/nature14547 [DOI] [PubMed] [Google Scholar]
- Peng C, Gathagan RJ, Covell DJ, Medellin C, Stieber A, Robinson JL, Zhang B, Pitkin RM, Olufemi MF, Luk KC, Trojanowski JQ, Lee VM (2018) Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557:558–563. 10.1038/s41586-018-0104-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Kaiserman-Abramof IR (1970) The small pyramidal neuron of the rat cerebral cortex: the perikaryon, dendrites and spines. Am J Anat 127:321–355. 10.1002/aja.1001270402 [DOI] [PubMed] [Google Scholar]
- Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL (1997) Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045–2047. 10.1126/science.276.5321.2045 [DOI] [PubMed] [Google Scholar]
- Popov VI, Deev AA, Klimenko OA, Kraev IV, Kuz'minykh S, Medvedev NI, Patrushev IV, Popov RV, Rogachevskiı̆ VV, Khutsian SS, Stewart MG, Fesenko EE (2004) Three-dimensional reconstruction of synapse and dendritic spines in the hippocampus of rats and ground squirrels: new paradigms of the structure and function of a synapse[in Russian]. Zhurnal vysshei nervnoi deiatelnosti imeni I P Pavlova 54:120–129. [PubMed] [Google Scholar]
- Proukakis C, Dudzik CG, Brier T, MacKay DS, Cooper JM, Millhauser GL, Houlden H, Schapira AH (2013) A novel alpha-synuclein missense mutation in parkinson disease. Neurology 80:1062–1064. 10.1212/WNL.0b013e31828727ba [DOI] [PMC free article] [PubMed] [Google Scholar]
- Queenan BN, Lee KJ, Pak DT (2012) Wherefore art thou, homeo(stasis)? Functional diversity in homeostatic synaptic plasticity. Neural Plast 2012:718203. 10.1155/2012/718203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey NL, George S, Steiner JA, Madaj Z, Luk KC, Trojanowski JQ, Lee VM, Brundin P (2018) Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathologica 135:65–83. 10.1007/s00401-017-1792-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL (2008) Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3:e1997. 10.1371/journal.pone.0001997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. (2010) The synaptic pathology of alpha-synuclein aggregation in dementia with lewy bodies, Parkinson's disease and Parkinson's disease dementia. Acta Neuropathologica 120:131–143. 10.1007/s00401-010-0711-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D, Roy S (2012) α-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J Neurosci 32:10129–10135. 10.1523/JNEUROSCI.0535-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M, Vlachos A, Korkotian E (2010) The spine apparatus, synaptopodin, and dendritic spine plasticity. Neuroscientist 16:125–131. 10.1177/1073858409355829 [DOI] [PubMed] [Google Scholar]
- Su X, Fischer DL, Li X, Bankiewicz K, Sortwell CE, Federoff HJ (2017) Alpha-synuclein mRNA is not increased in sporadic PD and alpha-synuclein accumulation does not block GDNF signaling in Parkinson's disease and disease models. Mol Ther 25:2231–2235. 10.1016/j.ymthe.2017.04.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toni N, Buchs PA, Nikonenko I, Bron CR, Muller D (1999) LTP promotes formation of multiple spine synapses between a single axon terminal and a dendrite. Nature 402:421–425. 10.1038/46574 [DOI] [PubMed] [Google Scholar]
- Tsigelny IF, Sharikov Y, Miller MA, Masliah E (2008) Mechanism of alpha-synuclein oligomerization and membrane interaction: theoretical approach to unstructured proteins studies. Nanomedicine 4:350–357. 10.1016/j.nano.2008.05.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle MD, Comellas G, Nieuwkoop AJ, Covell DJ, Berthold DA, Kloepper KD, Courtney JM, Kim JK, Barclay AM, Kendall A, Wan W, Stubbs G, Schwieters CD, Lee VM, George JM, Rienstra CM (2016) Solid-state NMR structure of a pathogenic fibril of full-length human alpha-synuclein. Nat Struct Mol Biol 23:409–415. 10.1038/nsmb.3194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Patel TP, Tanik SA, Riddle DM, Stieber A, Meaney DF, Trojanowski JQ, Lee VM (2011) Exogenous alpha-synuclein fibrils induce lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72:57–71. 10.1016/j.neuron.2011.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli-Daley LA, Luk KC, Lee VM (2014) Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 9:2135–2146. 10.1038/nprot.2014.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Das U, Scott DA, Tang Y, McLean PJ, Roy S (2014) Alpha-synuclein multimers cluster synaptic vesicles and attenuate recycling. Curr Biol 24:2319–2326. 10.1016/j.cub.2014.08.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Xu B, Liou LC, Ren Q, Huang S, Luo Y, Zhang Z, Witt SN (2012) α-Synuclein disrupts stress signaling by inhibiting polo-like kinase Cdc5/Plk2. Proc Natl Acad Sci U S A 109:16119–16124. 10.1073/pnas.1206286109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winner B, Jappelli R, Maji SK, Desplats PA, Boyer L, Aigner S, Hetzer C, Loher T, Vilar M, Campioni S, Tzitzilonis C, Soragni A, Jessberger S, Mira H, Consiglio A, Pham E, Masliah E, Gage FH, Riek R (2011) In vivo demonstration that alpha-synuclein oligomers are toxic. Proc Natl Acad Sci U S A 108:4194–4199. 10.1073/pnas.1100976108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SJ, Wypych J, Steavenson S, Louis JC, Citron M, Biere AL (1999) Alpha-synuclein fibrillogenesis is nucleation-dependent. implications for the pathogenesis of Parkinson's disease. J Biol Chem 274:19509–19512. 10.1074/jbc.274.28.19509 [DOI] [PubMed] [Google Scholar]
- Yuste R, Majewska A, Holthoff K (2000) From form to function: calcium compartmentalization in dendritic spines. Nat Neurosci 3:653–659. 10.1038/76609 [DOI] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Milatovic D, Schantz AM, Zhang J, Montine KS, Samii A, Deutch AY, Montine TJ (2005) Dendritic degeneration in neostriatal medium spiny neurons in Parkinson disease. Neurology 64:545–547. 10.1212/01.WNL.0000150591.33787.A4 [DOI] [PubMed] [Google Scholar]
- Zaja-Milatovic S, Keene CD, Montine KS, Leverenz JB, Tsuang D, Montine TJ (2006) Selective dendritic degeneration of medium spiny neurons in dementia with Lewy bodies. Neurology 66:1591–1593. 10.1212/01.wnl.0000216137.09685.c1 [DOI] [PubMed] [Google Scholar]
- Zarranz JJ, Alegre J, Gómez-Esteban JC, Lezcano E, Ros R, Ampuero I, Vidal L, Hoenicka J, Rodriguez O, Atarés B, Llorens V, Gomez Tortosa E, del Ser T, Muñoz DG, de Yebenes JG (2004) The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann Neurol 55:164–173. 10.1002/ana.10795 [DOI] [PubMed] [Google Scholar]
- Zhu F, Wu Q, Li J, Grycel K, Liu B, Sun X, Zhou L, Jiao R, Song R, Khan YM, Wang Q, Wang L, Xu Y, Li J, Zhang B, Zhou Z (2017) A single dose of cocaine potentiates glutamatergic synaptic transmission onto locus ceruleus neurons. Cell Calcium 67:11–20. 10.1016/j.ceca.2017.07.007 [DOI] [PubMed] [Google Scholar]