

Abstract
Prominent 7–12 Hz oscillations in frontal cortical networks in rats have been reported. However, the mechanism of generation and the physiological function of this brain rhythm have not yet been clarified. Multichannel extracellular field potentials of the ACC were recorded and analyzed using the current source density method in halothane-anesthetized rats. Spontaneous high-current spikes (HCSs) were localized in the deep part of layer II/III and upper part of layer V of the ACC. The frequency of HCSs in the ACC was 7–12 Hz, with an amplitude of 6.5 ± 0.76 mV/mm2 and duration of 55.24 ± 2.43 ms. The power density significantly decreased (84.56 ± 6.93%, p < 0.05, t test) after pinching the hindpaw and significantly increased (149.28 ± 15.96%) after treatment with morphine. The suppressive effect of pinching was reversed by naloxone (0.7 mg/kg, i.p.). HCSs coincided with initiation of the depolarization of cingulate neurons and remained in a depolarized upstate. The occurrence of cingulate HCSs was persistently preceded by a hyperpolarization phase and a burst of multiunit spike activity in the medial dorsal thalamic nucleus. Spontaneous field-potential oscillations changed from 10 Hz to a lower band (i.e., ∼7.5 Hz) when a central poststroke pain condition was induced. The central poststroke pain group had a higher average coherence coefficient compared with the control group. Our results indicate that spontaneous cingulate cortical HCSs could be initiated by thalamocortical synaptic inputs from the medial dorsal thalamic nucleus and maintained by intracortical neuronal upstate mechanisms in physiological and pathological pain states.
SIGNIFICANCE STATEMENT This study elucidated the mechanism of generation and physiological function of prominent 7–12 Hz frequency oscillations in frontal cortical networks in rats. Spontaneous cingulate cortical high-current spikes in anesthetized rats could be initiated by thalamocortical synaptic inputs from the medial dorsal thalamic nucleus and maintained by intracortical neuronal upstate mechanisms. Suppression of the anterior cingulate cortex-filtered EEG during noxious stimulation may have resulted from the desynchronization of high-current spikes in the ACC. The enhancement of fast Fourier transform power after a systemic morphine injection suggested that the opioid system may play an important role in synchronizing cingulate cortical neuronal networks. Spontaneous cingulate high-current spikes may also play an important role in thalamocortical dysrhythmia in central poststroke pain.
Keywords: anterior cingulate cortex, brain oscillation, central poststroke pain, current source density, medial thalamus, morphine
Introduction
Nociceptive pain is evoked in response to a noxious stimulus. In contrast, inflammatory and neuropathic pain may arise from innocuous stimuli and result in pain hypersensitivity. Our current understanding of the precise cortical dynamics of chronic pain is limited because previous studies were mostly based on the analysis of cortical responses to experimental phasic noxious stimuli that induce a time-locked cortical response. EEG studies of cortical responses to tonic pain stimuli have shown that EEG oscillations are influenced by different patterns of pain stimulation, such as intramuscular painful stimulation or ice-cold pain treatment (Chen et al., 1989; Chang et al., 2002; Egsgaard et al., 2009). The main α rhythm in humans decreases over occipital regions and the medial cortex (Chen and Rappelsberger, 1994; Cantero et al., 2002). The cause of this decrease in the α band appears to be related to the characteristics of painful stimuli (Chang et al., 2002). A decrease in α-band EEG oscillations after exposure to noxious stimuli was related to an increase in cellular excitability in the thalamocortical system (Steriade and Llinás, 1988; Pfurtscheller and Lopes da Silva, 1999).
Sensory information that is gated by thalamocortical loops is based on the source of the signal (i.e., sensory perception and information about the current state). Sensory information that enters into specific thalamic nuclei is signaled as perceptual inputs. Concurrent inputs that enter into nonspecific thalamic nuclei provide contextual information from internal brain states (Llinás et al., 1998). Thalamocortical neurons that carry both types of information to cortical pyramidal neurons are thought to integrate outside signals into the current mental state context. Signals from nociceptive pain can forward discriminatory aspects of sensory information through the specific thalamocortical system. However, the possibility that chronic pain can be represented by the internal state of the brain and signaled by nonspecific thalamic nuclei has not yet been explored.
The medial dorsal thalamic nucleus (MD) is a nonspecific thalamic nucleus that relays affective/emotional components of nociceptive information to the ACC. The function of the ACC is related to pain and pain-related emotions. In humans and rats, PET and MRI studies have indicated that the ACC is activated by neuropathic and inflammatory pain (Vogt et al., 1996; Tseng et al., 2013). The EEG power density of the α2 band (10–12 Hz) in the frontal cortex decreased after tonic pain stimulation (Ferracuti et al., 1994). However, the mechanism by which ACC α-band oscillations decrease after noxious stimuli is unclear. Our hypothesis is that the thalamocingulate pathway may represent information flow that signals the internal state of the pain condition. The relationship between medial thalamic nuclei and the ACC may be manifested by spontaneously continuous and ever-present oscillations that can be used to gauge chronically occurring pain. In the present study, we found that spontaneously occurring high-amplitude current oscillations in the ACC were triggered and mediated by synchronous discharges of MD unit activity. Noxious stimulation and morphine treatment influenced the synchronicity of the MD and ACC and changed the oscillatory state of the ACC. High-amplitude current oscillations in the ACC were initiated by transient membrane depolarization, preceded by membrane hyperpolarization, and the membrane potential was shifted by noxious stimulation and morphine treatment. Spontaneous high-current spikes (HCSs) in the ACC and cortical coherence were altered in pathological pain sates. This dynamic aspect of oscillations is consistent with Llinás' view of chronic pain as a thalamocortical dysrhythmia (Llinás et al., 1999), in which a slow rhythm is produced by the disruption of thalamocortical feedback.
Materials and Methods
Preparation of animals.
Male Sprague Dawley rats (300–400 g) were housed in an air-conditioned room (21°C–23°C, 50% humidity, 12 h/12 h light/dark cycle, lights on at 8:00 A.M.) with free access to food and water. All of the experiments were performed in accordance with the guidelines of the Academia Sinica Institutional Animal Care and Utilization Committee.
The rats were initially anesthetized with 4% halothane (in pure O2) in an acrylic box. A PE-240 tube was inserted by tracheotomy, and EMLA cream (2.5% lidocaine and 2.5% prilocaine; AstraZeneca) was smeared over the wound. The animals were then placed in a stereotaxic apparatus and maintained under anesthesia with 2% halothane in 30%/70% nitrous oxide/oxygen during surgery. Body temperature was maintained at 37.5°C–36.5°C with a homeothermic blanket system (model 50–7079, Harvard Apparatus). Craniotomies were performed over two or three target areas. To expose the ACC, craniotomy that was centered 2.5 mm anterior to bregma and 1 mm lateral to the midline was performed. To expose the primary somatosensory cortex (S1), craniotomy that was centered 1 mm posterior to bregma and 3 mm lateral to the midline was performed. To expose the motor cortex, craniotomy that was centered 2.5 mm anterior to bregma and 2.5 mm lateral to the midline was performed. To expose the medial thalamus (MT), craniotomy that was centered 2.2–3.5 mm posterior to bregma and 0.5–1.5 mm lateral to the midline was performed. A small piece of the dura over each target area was carefully removed, and warm paraffin was applied to keep the cortical surface moist. Heart rate was monitored by electrocardiography. The animals were subsequently kept anesthetized with 0.75%–1.5% halothane in a nitrous oxide/oxygen mixture (10%/90%) during the recording session. The depth of anesthesia was checked and verified periodically by pinching the tail and confirming that no overt reflexive movements or heart rate accelerations were observed.
Electrical stimulation of the sciatic nerve.
The sciatic nerve was exposed, and a custom-made stainless-steel cuff electrode was attached to the nerve. A biphasic electrical current (0.03–10 mA, 0.5 ms duration, 0.1 Hz) was delivered by an isolated pulse stimulator (model 2100, A-M Systems). The anode electrode was placed ∼1 cm distal to the cathode electrode. The minimal intensity for inducing hindpaw digit responses was regarded as the threshold value. Stimulation was delivered at intensities that were multiples of the threshold current value. We previously showed that innocuous A-β fibers are excited by twice the intensity at which muscle twitches are elicited and that nociceptive A-δ and C fibers are recruited and excited by 10× and 20× the threshold, respectively (Chang and Shyu, 2001).
Mechanical stimulation.
For brush stimulation, a brush was repetitively applied to the skin of the hindpaw. For pinching stimulation, a toothed forceps was applied to the skin of the hindpaw. The pinching pressure on the hindpaw was a force that could induce minimal contraction of the hindlimb. When brushing was applied to the experimenter's hand, it did not induce any uncomfortable feeling, but the pinching stimulus produced a painful pinch sensation.
Recording of evoked multichannel field potentials and unit activity.
A Michigan probe with 16 contact points (150 μm interval spacing, NeuroNexus) was used to record extracellular field potentials in the left ACC (∼2.5 mm anterior and 1 mm lateral to bregma; probe inserted 50° from the vertical line). Another Michigan probe was used to record the left M1(∼2.5 mm anterior and 2.5 mm lateral to bregma; probe inserted perpendicular to the cortical surface) or MT (∼2.2–3.5 mm posterior and 0.5–1.0 mm lateral to bregma; probe inserted perpendicular to the cortical surface). An Ag-AgCl reference electrode was placed in the nasal cavity. The sampling rate of recorded analog signals was 6 kHz, and the data were processed using a multichannel data acquisition system (Tucker-Davis Technologies). The multiunit activity of spontaneous responses was obtained by filtering the multichannel field potentials (200–3000 Hz). The square root of the mean basal activity over a 100 ms period of multiunit activity was calculated. This value was multiplied by 4, and the product was set as the spike threshold. Spikes that exceeded the threshold were marked as the digital signal “1.” Subthreshold signals were marked as the digital signal “0.” The multiunit activity of spontaneous responses was also obtained using a custom sorting program. The sampling rate of the custom sorting program was 24 kHz, and the threshold of spike sorting was >2-fold greater than the baseline noise background.
Intracellular recording.
Intracellular electrodes were filled with 3 m potassium acetate and 5% biocytin (80–120 mΩ impedance). Recordings were made using an Axoclamp 2A (Molecular Devices) microelectrode amplifier with 10× amplification. The amplified signals were digitized by an A/D converter card in a Tucker-Davis Technologies system and analyzed using a custom-made MATLAB program (The MathWorks). The input resistance was calculated by measuring Vm deflections that were produced in response to constant hyperpolarizing current pulses (1–1.5 nA, 500 ms). Only cells with a stable resting membrane potential >−60 mV were used.
Biocytin labeling of recorded neurons.
Intracellularly recorded cells were injected with biocytin using 1–2 nA depolarizing pulses that were delivered with 300 ms on/off cycles for at least 10 min. The recording electrodes were carefully withdrawn, and biocytin was allowed to diffuse for an additional 30 min. The rats were perfused with 0.1 m PBS and 4% PFA in 0.1 m PBS. The isolated brains were kept in 4% PFA in 0.1 m PBS at 4°C overnight and then cryoprotected by immersion in 30% sucrose in 0.1 m PBS for at least 3 d. The brains were cut into 60-μm-thick coronal sections using a cryostat (model CM 3050, Leica Microsystems). The sections were washed several times in PBS and then incubated in 1% H2O2 in PBS for 15 min to block endogenous peroxidase activity and reacted with 0.3% Triton X-100 in 0.1 m PBS for 1 h. Immunohistochemistry was performed using avidin-biotin elite solution at 4°C overnight (ABC kit, Vector Laboratories). Cell staining was visualized using 0.3% ammonium nickel sulfate, 0.03% DAB, and 0.07% H2O2 in Tris buffer, pH 8.1.
Current-source density (CSD) analysis.
We used CSD analysis to determine the underlying current generators of spontaneous oscillations or evoked field potentials (Nicholson and Freeman, 1975; Mitzdorf, 1985). The ACC is dominated by parallel-aligned pyramidal cells whose apical dendrites extend along the axis across cortical layers. Spontaneous oscillations or electrical stimulation produces the synchronous excitation of ensembles of pyramidal cells, which results in a major current flow perpendicular to the cortical layers. Under the assumption of homogeneous cortical activity and constant extracellular electrical conductivity, the CSD can be estimated from the second spatial derivative of the recorded field potentials in the axis parallel to the cortical layers. Thus, the procedure that was used to obtain the CSD data consisted of recording field potentials at equidistant, linearly positioned electrode contacts using Michigan probe electrodes that vertically penetrated the cortical layers. A long period of continuous field potential was recorded. With regard to the time span and sampling variations in each recording session, we adopted a 5 point formula (Nicholson and Freeman, 1975) to smooth the spatial sampling variability. The membrane current, Im, was derived from the second spatial derivations of the extracellular field potentials and was calculated by the finite-difference formula as follows:
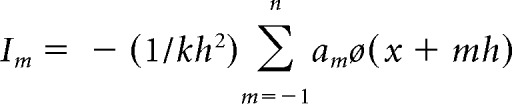 |
The formula was simplified as follows:
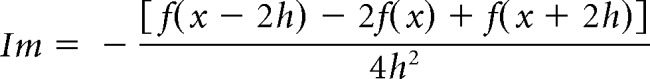 |
where h is the distance between successive measuring points (150 μm in the present study) and x is the coordinate perpendicular to the cortical layer. The remaining constants were as follows: n = 2, k = 4, a0 = −2, a ± 1 = 0, and a ± 2 = 1; (x − 2 h), (x − h), x, (x + h), and (x + 2 h) each correspond to the recording electrode positions (a − 2, a − 2, a, a + 1, and a + 2, respectively).
Drug application.
The μ-opioid receptor agonist morphine (5–8 mg/kg) and μ-opioid receptor antagonist naloxone (0.7 mg/kg) were administered intraperitoneally.
Electrolytic lesions of the medial thalamic nucleus.
The location of the MT was functionally identified by recording responses to electrical stimulation of the contralateral sciatic nerve. A Michigan probe was inserted vertically into the MT ipsilateral to the ACC that was recorded. Thalamic activity was monitored and examined as the electrode advanced, and electrical stimulation was applied to the sciatic nerve. Once the locations of stimulus-responding thalamic unit responses were found, the Michigan probe was withdrawn. A tungsten electrode was reintroduced along the same track and lowered to the depth that was previously reached by the Michigan probe. To deactivate thalamic responses, a 100 μA, 100 s direct current was delivered using a constant-current pulse generator (model 2100, A-M Systems).
Verification of recording and lesion sites.
At the end of the experiment, a small lesion was made by passing an anodal current (30 μA for 5 s) to the 15th contact lead. Another lesion was made at the same lead after the Michigan probe was withdrawn for 900 μm. The rats were fixed by perfusion with normal saline followed by 10% formalin or 10% PFA. The brains were cut into 60-μm-thick coronal sections using a cryostat, and the sections were stained with cresyl violet. The rat brain atlas (Paxinos and Watson, 1998) was used as a reference to estimate the cortical layer structures during histological examination. The positions of the remaining recording sites in the cortex were estimated by determining their distances from the two lesions.
Preparation of animal model of central poststroke pain (CPSP).
CPSP was induced by a thalamic hemorrhagic lesion as described in our recent studies (Kuan et al., 2015; Shih et al., 2017). The rats were prepared under the same anesthesia procedures as in the electrophysiological experiment. The CPSP group of rats was injected with 0.125 U Type 4 collagenase in 0.5 μl saline in the ventral posteromedial thalamic nucleus and ventral posterolateral thalamic nucleus: 3.0–3.5 mm posterior and 3.0–3.5 mm lateral to bregma, 5.5–5.8 mm depth. The sham control group of rats was injected with 0.5 μl sterile saline at the same coordinates. The animals were injected with antibiotics (6 mg/kg gentamycin, i.p.) to prevent infection, and they were weighed before and after the lesion procedure. Only rats whose body weights recovered after surgery were included in the experiment.
Data analysis.
The peak latencies and amplitudes of sink currents that were evoked by electrical, mechanical, and laser stimuli were determined from the CSD data. To calculate the number of HCSs, the spontaneous CSD trace of 180 s in deep layer II/III and upper layer V was selected according to the maximum current sink that was evoked by electrical stimulation of the sciatic nerve. The selected CSD trace was filtered (5–50 Hz) and then squared. We calculated the SD of the filtered and squared CSD traces and set sevenfold of the SD as the threshold to detect the number of HCSc. The changes in sink currents were measured and compared before and after drug application. The sink amplitudes between groups were analyzed using Student's t test. The coherence coefficient method is described in detail in our previous study (Shih et al., 2017). Spontaneous local field potentials that were recorded in deep layer II/III or upper layer V underwent FFT. The coherence coefficient of two different frequency power spectra, XA(t) and XB(t), was calculated using the Pearson correlation coefficient method. By looping calculation of the 1–50 Hz coherence coefficient of 10–15 min of recorded data, a 2D image of the coherence coefficient between 1 and 50 Hz spontaneous local field potential activity was generated. For intracellular spike counts, the z test was performed. The z score borderline value was ±2.56. One- or two-way ANOVA was used to analyze the effects of the drugs, and noxious stimuli on the evoked sink currents. Tukey's post hoc test was used to detect the sources of group differences in the ANOVAs.
Results
Identification of spontaneous HCS activity in the ACC
Local field potentials across cingulate cortical layers were simultaneously recorded by multichannel electrodes that were inserted into the ACC. The electrode track was penetrated obliquely from the medial cortical surface. It was approximately perpendicular to the cingulate cortical layers as shown in the reconstructed track overlaid with the histological coronal plane (Fig. 1Ai). Various frequency components could be observed in the spontaneous field potential sweeps. Some high-frequency (large-amplitude) spike activities were riding over the lower-frequency waves. These components were extended over most of the recorded channels (Fig. 1Bi). The CSD was analyzed from evenly spaced multichannel field potentials across the cortical layers. These spontaneous high-amplitude sink/source currents were more localized between deep layer II/III and upper layer V in the ACC (Fig. 1Bii). A typical CSD trace was selected from layer II/III (Fig. 1Bii, red dot) and filtered (5 Hz low cutoff and 50 Hz high cutoff; Fig. 1Ci) and then squared (Fig. 1Cii). HCSs were defined as an amplitude of current sink that was >7-fold of the SD of the total recording period after it was filtered and squared (Fig. 1Cii). HCSs were depicted clearly in the time series frequency color plot, and the main frequency of HCSs was in the α band (Fig. 1Ciii, pseudocolor map). To illustrate the distinctive feature of HCSs, the low-frequency bands were filtered. The amplitude of HCSs was 6.51 ± 0.76 mV/mm2, with a duration of 55.24 ± 22.43 ms. The main frequency band of spontaneous HCSs was 9.66 ± 0.27 Hz (n = 10). Active current signals were selected from layers II/III and analyzed by FFT, and a peak in the α band was noted (Fig. 1D). The power density of the α band was modulated by the level of anesthesia. The intensity of the α band was vigorous at halothane concentrations of 0.6%–1.0% but decreased after the halothane concentration increased to 2.5% (n = 4; Fig. 1D). Different frequency bands (δ, θ, α, β, and γ) of spontaneous CSD activities were tested under different halothane concentrations. The power density of each respective band was gradually decreased as the halothane concentration increased from 1% to 4% (Fig. 1E).
Figure 1.

HCSs in the ACC were analyzed based on multichannel field potentials under different anesthesia conditions. A, Field potentials in different layers of the ACC were recorded simultaneously with a 16 channel electrode probe. B, Multichannel field potential (Bi) analyzed using the CSD method. Diagram represents a high-amplitude sink between deep layer II/III and upper layer V (Bii). Ci, Top, Blue trace represents the CSD trace with a high-amplitude sink trace selected from Bii. Middle, Blue trace represents the CSD trace filtered with a 5 Hz low cutoff and 50 Hz high cutoff. Cii, Bottom, Blue trace represents the squared value of the filtered CSD trace. Ciii, Color map of the FFT power density of the selected CSD trace. D, Power density of FFT of selected CSD traces in different anesthesia states (1.0%, 1.5%, and 2.5%). E, Power density of CSD in different anesthesia conditions (1%–4%) was differentiated into δ, θ, α, β, and γ bands.
Medial dorsal thalamic unit activity preceded HCSs in the ACC
We previously showed that the MD provides afferent inputs to the ACC (Wang and Shyu, 2004). To investigate the thalamic origin of HCSs in the ACC, spontaneous extracellular field potentials in the ACC and multiunit activity in the MT were recorded simultaneously. Michigan probes were inserted into the ACC and MT separately, as shown in two histological coronal slices (Fig. 2Ai). A segment of the spontaneous CSD trace (2 s) that contained HCSs in the ACC was aligned with multiunit activity that was recorded from multiple traces of the MT (Fig. 2Aii). Time points of the occurrence of peaks of HCSs were identified from the CSD sweeps (Fig. 2Aii, blue dots). The detected times of occurrence of each targeted HCS were used to set the time point for averaging multiunit activity that was recorded from the MT. HCS-correlated multiunit activity in the MT was analyzed from 23 tracks (Fig. 2Bi). The apex of summed multiunits in the MD that were related to HCSs was earlier than the apex of averaged HCSs (−3.17 ± 1.47 ms in the medial dorsal lateral thalamic nucleus, −2.46 ± 3.44 ms in the medial dorsal medial thalamic nucleus, and −2.62 ± 0.65 ms in the medial dorsal central thalamic nucleus), whereas it was delayed in the paracentral thalamic nucleus (13.27 ± 10.32 ms). Multiunit activity that was recorded in the MT along 23 electrode tracks was pooled and is illustrated in serial coronal sections (Fig. 2C). The percentages of multiunit activity (<15%, 15%–25%, 25%–35%, 35%–45% and >45%) in the MT that was triggered by HCSs at each location are presented as different levels of shades of gray dots. HCSs that triggered MT multiunit activity were distributed in layer II/III of area 24b.
Figure 2.

Medial dorsal thalamic unit activity preceded HCSs in the ACC. Ai, Coronal sections of the ACC and MT with the electrode tracks marked. Aii, CSD trace with high-amplitude sink in the ACC (top) and simultaneously recorded multiunit activity in the MT (bottom). Bi, HCSs in the ACC (yellow) could trigger spikes in the MD. Red vertical line indicates the peak of HCSs. Bii, Comparison of the latency between HCSs in the ACC and HCS-triggered spikes in the MT. C, Serial recording tracks in the ACC and MT. Colored dots represent the correlation between HCSs in the ACC and multiunit activity in the MT. MDL, Medial dorsal lateral thalamic nucleus; MDC, medial dorsal central thalamic nucleus; MDM, medial dorsal medial thalamic nucleus; PC, paracentral thalamic nucleus; VL, ventral lateral thalamic nucleus; M1, primary motor cortex; M2, secondary motor cortex; Cg1, cingulate cortex, area 1; PrL, prelimbic cortex; IL, infralimbic cortex.
Medial dorsal thalamic unit activity was followed by HCSs in the ACC
Spike-triggered average data showed that HCSs in the ACC were preceded by multiunit activity in the MD. We further investigated whether HCSs in the ACC could be driven by unit activity in the MD. The averaged CSD sweeps across ACC layers were selected, aligned, and determined by times of occurrence of the peak of unit activity that was identified from the multiunit activity that was recorded in the MD (Fig. 3Bi). The result of 272 traces was superimposed in gray lines, and the black line indicated the averaged result (Fig. 3Bi, bottom). The percentage of HCSs in the ACC that were triggered by multiunit activity in the MD is presented by grayscale dots (Fig. 3A, Cg1). The depth profile of triggered HCSs had a similar feature as the spontaneously recorded HCS distribution pattern (i.e., high amplitude of sink current distributed in deep part of layer II/III or upper part of layer V and corresponding source current in deep part of layer V; Fig. 3Bii). The grand average of the HCS amplitude in the ACC that was triggered by multiunit activity in the medial dorsal lateral thalamic nucleus was 1.5 ± 0.2 mV/mm2. The distribution of multiunit activity in the MD that could trigger HCSs in the ACC is illustrated in serial coronal sections of the thalamic region (Fig. 3C). The percentages of multiunit activity (<15%, 15%–25%, 25%–35%, 35%–45% and >45%) in the MT that triggered HCSs at each location are presented as different levels of shades of gray dots.
Figure 3.

Medial dorsal thalamic unit activity was followed by HCSs in the ACC. A, All recording tracks in the ACC. Colored dots represent the correlation between multiunit activity in the MD and HCSs in the ACC. Bi, Multiunit activity in the MD (top) could trigger a high-amplitude sink in layers II/III in the ACC. Arrow indicates one example of an averaged HCS overlaid with original multiple HCS sweeps. Bii, Profile of MD spikes that triggered HCSs in different layers of the ACC. C, All recorded tracks in the MT. Colored dots represent the correlation between multiunit activity in the MT and the amplitude of HCSs in the ACC. M1, Primary motor cortex; M2, secondary motor cortex; Cg1, cingulate cortex, area 1; PrL, prelimbic cortex; IL, infralimbic cortex.
HCSs in the ACC were significantly reduced after MT lesions
To test the specificity of the occurrence of HCSs in the thalamocingulate pathway, we compared EEGs that were recorded in the ACC and adjacent primary motor cortex (M1). Two Michigan probes were used to record local field potentials in the ACC and M1 simultaneously. After 30 min of control recording and functional verification of the site of the MD, nociceptive responses to noxious electrical stimulation of the contralateral sciatic nerve were first determined, and electrolytic lesions were then made (Fig. 4A, posterior to bregma, P −2.5 to 3.2 mm, lateral to bregma, L 0.5 to 1 mm and 1 nA, 100 s). The pattern of HCS oscillations in the ACC and M1 was transformed by FFT. After the MD lesion, example traces showed that the α band in the ACC rapidly decreased but did not influence the oscillation pattern in the M1 (Fig. 4B). The number of HCSs (15.88 ± 1.93 spikes/min) in the ACC decreased after a large lesion area was made in the MT (3.38 ± 1.56 spikes/min; Fig. 4Ci, paired t test, t = 8.31, p = 0.001, n = 6). The FFT power density of the CSD in the α band in the ACC also decreased after the MT lesion (39.83 ± 9.82%; Fig. 4Cii, one-way ANOVA, F(5,29) = 4.00, p = 0.02, n = 5). However, the lesion did not influence the number of HCSs in the M1 (17.43 ± 1.12 spikes/min before lesion; 31.89 ± 10.71 spikes/min after lesion; Fig. 4Ci, paired t test, t = 2.16, p = 0.08, n = 6) or the FFT power density (118.60 ± 20.36%; Fig. 4Cii, one-way ANOVA, F(5,29) = 2.12, p = 0.98, n = 5). These results indicated the specific connection between the MD and ACC, and the oscillation of the ACC was critically mediated by the MD input.
Figure 4.

HCSs in the ACC were significantly reduced after lesion of the MT. A, Gray areas represent the lesion margins in the MT in serial coronal sections. B, Effect of lesion of the MT on FFT power density of HCSs in the ACC and M1. Ci, Effect of lesions of the MT on the number of HCSs in the ACC and M1. #p < 0.05, compared with control group (paired t test). Cii, Effect of lesions of the MT on the power density ratio of different frequency bands (control as 100%) in the ACC and M1. #p < 0.05, compared with control group (one-way ANOVA followed by post hoc test).
Spontaneous HCSs in the ACC were regulated by noxious inputs and morphine treatment
Our previous studies showed that distinct sink currents in the ACC could be evoked by noxious stimuli, and these evoked responses could be enhanced by morphine treatment (Yang et al., 2006; Chen et al., 2008). In the present study, we further examined the effect of noxious stimuli and morphine on spontaneous HCSs. The main frequency band of spontaneous HCSs was in the frequency range of 7–12 Hz (Fig. 5A). Innocuous brushing of the hindlimb did not influence the frequency band or the number of spontaneous spikes (Fig. 5A, Bru, paired t test, t = −1.52, p = 0.17, n = 8). The frequency band decreased during pinching of the hindpaw. The number of large spontaneous current spikes was also significantly decreased by noxious pinching of the hindpaw (Fig. 5A, Pin, paired t test, t = 4.73, p = 0.001, n = 8). The frequency band significantly increased ∼20 min after morphine treatment (5 mg/kg, i.p.). The number of spontaneous HCSs also significantly increased (Fig. 5B, mor, paired t test, t = −3.65, p = 0.01, n = 8). The effect of noxious stimuli on the EEG band and number of HCS was abolished by morphine treatment (Fig. 5C, Pin, paired t test, t = 1.86, p = 0.11, n = 8). The effect of morphine was reversed by naloxone treatment (0.7 mg/kg; Fig. 5B, nal, paired t test, t = 2.40, p = 0.05, n = 8). These results indicate that oscillations of the ACC were influenced by peripheral noxious inputs and the opioid system.
Figure 5.

HCSs in the ACC were mediated by noxious inputs and morphine treatment. A, FFT power density of HCSs and HCS traces in the ACC with control, brush, and pinch stimulation. B, FFT power density of HCSs and HCS traces in the ACC with morphine and naloxone treatments. C, Effect of peripheral stimulation under morphine treatment. #p < 0.05, compared with control group (paired t test). $p < 0.05, compared with morphine group (paired t test).
Synchronicity of medial dorsal thalamic unit activity and cingulate HCSs was altered by noxious inputs and morphine treatment
The coinciding unit activity in the MT and HCSs in the ACC and large variety of amplitudes of spontaneously occurring HCSs led us to examine the relationship between the synchronicity of MT unit activity and amplitude of HCSs. The activity of spontaneous HCSs in the ACC and multiunit activity in the MD were simultaneously recorded with two multichannel electrodes (Fig. 6A, top traces). The HCS oscillation in the ACC (Fig. 6A, bottom) was correlated with multiunit activity in the MD (Fig. 6A, top multiple traces). The synchronized bursting of multiunit activity in the MD that was recorded in the same probe track may have resulted in a higher amplitude of integrated activity. Less-synchronized multiunit activity in the MD that was recorded along the recording track would have a lower amplitude of integrated activity because of the spreading of unit activity over time. Multiunit activity in the MD along the recording track was summed and integrated (Fig. 6A, blue trace, arrow) and transferred to a smooth waveform (Fig. 6A, black trace, arrowhead). The correlation between peak amplitudes of integrated MD units and the coinciding peak amplitude of HCSs was calculated. A positive correlation was found between different folds of high-amplitude sink currents and the corresponding amplitude of integrated multiunit activity (R2 = 0.26; Fig. 6Ci, black). The morphine injection increased HCSs in the ACC and unit activity in the MD (Fig. 6Bi). The enhancement of MD unit activity was also reflected by the level of synchronicity. The correlation between ACC and MD activity was also increased by morphine treatment (R2 = 0.42; Fig. 6Ci, blue). HCSs in the ACC were decreased after noxious stimulation (Fig. 6Bii), and the correlation was also significantly decreased by noxious stimulation (R2 = 0.06; Fig. 6Ci, red). After noxious stimulation, multiunit activity in the MD correlated with a lower amplitude of HCSs in the ACC (Fig. 6Cii, Nox, paired t test, t = 3.78, p = 0.001, n = 200), and this was reversed by morphine treatment, in which multiunit activity in the MD correlated with a larger amplitude of HCSs in the ACC (Fig. 6Cii, Mor, paired t test, t = −8.28, p = 0.001, n = 200). The increase or decrease in HCSs or MD unit activity could be translated to the synchronization of thalamic and cingulate activities. These results indicate that noxious stimulation and the opioid system modulated oscillations of the thalamocingulate pathway, and the activity of HCSs was mediated by the synchronous activity of thalamic unit activity.
Figure 6.

Synchronicity of spontaneous medial dorsal thalamic unit activity and HCSs in the ACC. A, Spontaneous MD multiunit activity (tops) and ACC CSD oscillations (bottom, yellow) were recorded with multichannel probes simultaneously. Signals of multiunit activity in the MD were selected (channels 9–16), and the total multiunit number of selected channels was averaged (blue line trace, arrow) over time. The average was transferred to a waveform trace (arrowhead). B, Simultaneous MD multiunit activity and ACC CSD oscillations with morphine treatment (Bi) or noxious stimulation (Bii). Ci, Relationship between ACC CSD amplitude and number of MD multiunit activity: control (black), morphine treatment (blue), and noxious stimulation (red). Cii, Comparison of the ratio of the CSD and multiunit activity in the control, morphine treatment, and noxious stimulation groups. #p < 0.05, compared with control group (paired t test).
HCSs in the ACC were correlated with the upstate of cingulate cortex neurons
The cellular mechanism of HCSs was further investigated in the cingulate cortex. Intracellular recordings of ACC neurons were obtained simultaneously with spontaneous ACC field potentials that were recorded from 10 neurons in 8 rats. All of the recorded neurons were located in layer II/III and layer V. Figure 7Ai shows an example of a recorded neuron and the multichannel electrode mapping site. The location and reconstruction of the neuron are shown in Figure 7Aii. The membrane potential of spontaneous neuronal activity showed upstate and downstate oscillations (Fig. 7B,Ci). Spontaneous high-amplitude sinks in the ACC (Fig. 7B, top) were correlated with the upstate of recorded neurons (Fig. 7B, bottom). After calculating the distribution of HCSs, we found that HCS oscillations were almost all distributed in the upstate of neuronal activity (91.6 ± 2.7%, n = 10; Fig. 7Cii). The high correlation between HCSs and the upstate of neuronal activity means that HCS oscillations were attributable to the co-oscillation of neurons. However, unclear is whether the HCS oscillation was attributable to intrinsic coworking of the neurons or induced by output stimulation. Current-voltage (I–V) curves in response to −0.6 to 0.6 nA stimulation of the MD in different rats (n = 15 cells) are shown in Figure 7D. The hyperpolarized currents of −0.2, −0.4, and −0.8 nA (200 ms duration) were applied while MD stimuli were applied during the induction of hyperpolarization (Fig. 7E). Synaptic responses were evoked by MD stimulation with −0.2 nA (green), −0.4 nA (red), and −0.8 nA (blue). Rebound action potentials were observed upon the cessation of hyperpolarized currents.
Figure 7.

HCSs in the ACC were correlated with upstate of cingulate neurons. Ai, Anatomical location of recording sites of 16 channel probe and position of recorded cell in the ACC. Aii, Morphology of recorded cell. B, Simultaneous profile of CSD and single-cell membrane activities. Ci, Two-state plot for intracellular recording. Cii, Distribution of upstate and downstate of spontaneous neuronal activity. D, I–V curves in response to −0.6 to 0.6 nA MD stimulation in different rats (n = 15 cells). E, Three examples of I–V curve after hyperpolarization induced by negative current. Synaptic responses that were evoked by MD stimulation with −0.2 nA (green), −0.4 nA (red), and −0.8 nA (blue) are shown.
Cross-correlated cellular events: unit activity in the MD, HCSs in the ACC, and cingulate neuronal activity
To investigate the effects of possible inputs of the thalamocortical pathway on the initiation of HCSs, intracellular recordings of ACC neurons were obtained simultaneously with spontaneous ACC field potentials and MD unit activity (Fig. 8A). Simultaneous event data on HCSs, intracellular recordings, and multiunit activity in the ACC and MD were collected, and 10 neurons were recorded successfully. The timing was realigned to the peak of the maximal amplitude of HCSs (Fig. 8B). Spontaneous HCSs of the averaged trace coincided with depolarization and were preceded by the lasting hyperpolarization of intracellular membrane potential of the averaged trace. The summation of multiunit activity in the ACC or MD according to the occurrence of HCSs significantly increased, followed by HCSs (Fig. 8B). The peak unit activity in the MD was clearly preceded by the peak of HCSs and high-passed ACC unit activity. It also preceded depolarization of the cellular response.
Figure 8.

Correlations between medial dorsal thalamic unit activity, ACC HCSs, and cingulate neuronal activity. Ai, Anatomical location of recording sites of 16 channel probe and intracellular cell position in the ACC. Aii, Reconstructed morphology of recorded cell. Aiii, Anatomical location of recording site of 16 channel probe in the MD. Bi, Multiple traces of HCSs in the ACC. HCSs were selected and realigned by the lowest negative amplitude as the zero time point. Bii, Average of simultaneously recorded intracellular membrane potentials. Biii, Multiunit summation of all selected HCS traces. Biv, Multiunit summation of all selected MD multiunit activities.
Membrane potentials of cingulate neurons influenced by noxious inputs and morphine treatment
The influence of noxious stimulation and the opioid system on individual cingulate neurons was analyzed in simultaneous extracellular and intracellular recordings (Fig. 9A,B). Examples of changes in neuronal activity after innocuous and noxious stimulation are shown in Figure 9A. The average resting membrane potential of neurons was −60.30 ± 3.06 mV (n = 6, Fig. 9Ai,D, Con). After noxious stimulation, the average resting membrane potential shifted to −59.13 ± 2.54 mV (Fig. 9Aii,D, Pin, paired t test, t = −1.50, p = 0.19, n = 6). Figure 9B shows the influence of morphine and naloxone on cingulate neurons (Fig. 9Bii,Biii). The average resting membrane potential was also decreased by morphine (−68.67 ± 4.11 mV; Fig. 9Bii,D, Mor, paired t test, t = 3.30, p = 0.02, n = 6) and noxious stimulation (−68.16 ± 4.41 mV; Fig. 9D, Mor+Pin, paired t test, t = 2.73, p = 0.04, n = 6). The average duration of neuronal hyperpolarization was −244.0 ± 72.8 ms (Fig. 9E, Con, n = 6), which was decreased by noxious stimulation (150.1. ± 49.74 ms; Fig. 9E, Pin, paired t test, t = 2.88, p = 0.04, n = 6). The duration of hyperpolarization of HCS was not influenced by morphine (259.8 ± 102.77 ms; Fig. 9E, Mor, paired t test, t = −0.40, p = 0.71, n = 6) or morphine plus noxious stimulation (227.7 ± 73.4 ms; Fig. 9E, Mor+Pin, paired t test, t = 0.54, p = 0.62, n = 6). The rebound of hyperpolarization to hypopolarization in control cells was 5.7 ± 0.6 mV. No difference was found between groups (Fig. 9F). In Figure 9C, the sites of 17 recorded neurons are indicated by black dots, most of them in the Cg1.
Figure 9.

Correlations between HCSs and single-neuron responses in the ACC. Ai, Superimposition of HCSs in the ACC (top) and averaged single-neuron spontaneous activity realigned by HCS (bottom). Aii, Relationship between HCS and averaged single-neuron activity with pinch stimulation of the hindpaw. B, Example of superimposition of HCS in the ACC (top) and averaged single-neuron spontaneous activity realigned by HCS (bottom): spontaneous (Bi), morphine treatment (Bii), naloxone treatment (Biii). C, The distribution of recorded neurons in the ACC. D, Comparison of resting membrane potentials in control, pinch, morphine, and morphine + pinch groups. E, Comparison of duration of hyperpolarization in control, pinch, morphine, and morphine + pinch groups. F, Comparison of rebound potential in control, pinch, morphine, and morphine + pinch groups. #p < 0.05, compared with control group (paired t test).
Spontaneous cingulate HCSs and thalamocortical dysrhythmia in CPSP
The animal model of CPSP was established by injecting collagenase in the lateral thalamus (Fig. 10A). The mixed two-way (group vs day) ANOVA indicated significant effects of group (F(1,22) = 6.29, p = 0.02 0.05, n = 12) and day (F(4,88) = 5.29, p = 0.000.05, n = 12) on mechanical pain sensitivity and a significant group × day interaction (F(4,88) = 6.52, p = 0.00, n = 12). Tukey's Honestly Significant Difference (HSD) post hoc test indicated that CPSP rats had significantly lower mechanical pain thresholds only on day 21 and day 28 (both p < 0.05). Therefore, the mechanical pain threshold of the left hindlimb in the CPSP group significantly decreased in the von Frey test compared with the control group after the thalamic lesion, indicating allodynic effects (Fig. 10B). Furthermore, the characteristics of spontaneous field potential oscillations were different between the control and CPSP groups. In contrast to the control group, the peak of spontaneous field potential oscillations shifted from 10 Hz to a lower band (i.e., ∼7.5 Hz) when CPSP occurred (Fig. 10C). In the coherence coefficient tests for 10–50 Hz, the CPSP group had a higher coherence coefficient compared with the control group, indicating a so-called “edge effect” (Fig. 10D). Furthermore, the coherence coefficient between 10 and 50 Hz was significantly higher in the CPSP group than in the control group (Fig. 10E; independent t test, t = −2.89, p = 0.02, n = 6). These results indicate that crucial features of CPSP include a “peak shift” and an “edge effect” for EEG oscillations.
Figure 10.

Dysrhythmia in CPSP. A, Thalamic lesion induced by collagenase injection (0.125 U/0.5 μl). B, von Frey test of the left hindpaw in the control and CPSP groups. The threshold for the left hindpaw decreased 3 weeks after the lesion. C, Power density of spontaneous field potential oscillation. The peak of 10 Hz was shown in the control group (blue arrow) but slightly shifted left in the CPSP group. D, Coherence coefficient map of field potentials in the control and CPSP groups. Compared with the control group, the correlation of spontaneous field potential oscillations was higher in the CPSP group. E, Total average correlation coefficient between 1 and 50 Hz in the control and CPSP groups. #p < 0.05 (t test). F, Number of HCSs in the ACC in the control and CPSP groups. The animals were subjected to pinch stimuli and a morphine injection (8 mg/kg, i.p.). G, Multiunit activity in the MD in the control and CPSP groups. The animals were subjected to pinch stimuli and a morphine injection (8 mg/kg, i.p.). #p < 0.05, significant difference between groups (mixed two-way ANOVA followed by post hoc test).
To test spontaneous ACC HCSs in the control and CPSP groups, we performed a mixed two-way (group vs treatment) ANOVA. We found no effect of group (F(1,15) = 2.29, p = 0.15, n = 6), a significant effect of treatment (F(3,45) = 24.76, p = 0.00, n = 6), and no group × treatment interaction (F(3,45) = 2.54, p = 0.07, n = 6). Thus, the CPSP group exhibited a decrease in the number of spontaneous HCSs in the ACC, and morphine and morphine + pinching increased the number of spontaneous HCSs in the ACC in both the control and CPSP groups. Moreover, Tukey's HSD post hoc test indicated that the number of spontaneous HCSs in the CPSP group significantly decreased compared with the control group (p < 0.05), and morphine and morphine + pinching reversed this effect in the CPSP group (both p < 0.05; Fig. 10F).
Multiunit activity in the MD and HCSs in the ACC were simultaneously recorded (Fig. 10G). The mixed two-way (group vs treatment) ANOVA indicated significant effects of group (F(1,11) = 5.55, p = 0.04, n = 6) and treatment (F(3,33) = 7.18, p = 0.001, n = 6) but no group × treatment interaction (F(3,33) = 0.95, p = 0.43, n = 6). Tukey's HSD post hoc test indicated that unit number of spontaneous (p = 0.01), morphine (p = 0.03), and morphine + pinching (n = 0.02) in the CPSP group significantly increased compared with controls (all p values <0.05, n = 6). However, unit number of pinching was not a significant difference between the CPSP and control groups (p = 0.15, n = 6).
Discussion
In the present study, we found that spontaneous HCS oscillations in the ACC were regulated by a specific thalamocortical circuit from the MD. Noxious stimuli and the opioid system influenced HCS oscillations by increasing or decreasing neuronal activity. HCSs are initiated by transient membrane depolarization, which coincides with upstate membrane depolarization and is driven by medial thalamic inputs.
The CSD method is more meaningful than traditional voltage recordings because it provides cellular-level information, high-resolution spatial location, and the time course of extracellular net current flow (Agmon and Connors, 1991). The relationship between thalamocortical neuronal and cortex oscillations, including sleep spindles and high-voltage spike-and-wave patterns (HVSs), has been analyzed using the CSD method (Kandel and Buzsáki, 1997). In the present study, we used the CSD method along the one-dimensional electrode array to analyze the spatial distribution of spontaneous EEG oscillations. HCSs were always located in the deep part of layer II/III and upper part of layer V. HCSs correlated with multiunit activity in the MD. Although the CSD method cannot identify the neuronal morphology of related current flow (McCormick et al., 1985), we combined single intracellular recordings and found that HCS current flow was mediated by pyramidal neurons.
Spontaneous HVS oscillations were described by Klingberg and Pickenhain (1968), and HVSs have been observed by other investigators (Vergnes et al., 1987). Multichannel superficial EEG recording data indicated that HVSs are distributed in the whole brain, especially in the somatosensory cortex. Basic features of HVSs include a single or series of sharp and high-amplitude voltage spikes on EEG. The amplitude is 3–5 times greater than the background desynchronized EEG, with a rhythm of 6–12 Hz, incidence of 1–50/h, and duration of 0.8–60 s (Aporti et al., 1986; Vergnes et al., 1986; Buzsáki et al., 1988a,b). The HVS oscillation was shown to be influenced by different states. HVS oscillations decreased after large voice stimuli or airpuffs to the eye (Wiest and Nicolelis, 2003). The regulation of HVSs in the cortex has also been considered that HVSs are regulated by thalamocortical circuits, including relay neurons and GABAergic interneurons in the nucleus reticularis thalami (nRT). It has been suggested that, if HVSs are triggered by a local cortical network, then a longer interspike interval would follow a larger amplitude spike compared with a smaller spike (Buzsáki, 1991). The results of analyses of HVS interspike intervals indicated that the main interspike interval is 160 ms, and 300 and 400 ms intervals mean that the HVS oscillation is regulated by an output trigger. The correlation between HVSs in the cortex and unit activity in the thalamus was also suggested to be regulated by the thalamocortical circuit (Buzsáki, 1991; Inoue et al., 1993; Seidenbecher and Pape, 2001). Cortical HVSs correlated with thalamic multiunit burst firing but not with single-unit activity in the thalamus. The relationship between HVSs and pain function was reported by Walker and Yaksh (1986). A local microinjection of D-Ala2-Leu5-encephalin (an analog of the endogenous δ-opioid peptide encephalin) into the thalamic nucleus caused sudden burst HVS activity in the cortex, which transferred to an active HVS state within a few minutes. Decreases in mechanical and thermal noxious responses were also observed in the active HVS state.
The HCSs we observed in the ACC in halothane-anesthetized rats were not all the same as HVSs, with a duration of <0.5 s and incidence of ∼20/min in the α band. The oscillation of HCSs was also regulated by sensory inputs. The number of HCSs was decreased by noxious stimuli, and this decrease was reversed by morphine. The results of this experiment, including spike-trigger averaging and the cross-correlation of firing events of single neurons and multiunit activity in the ACC and MD, also indicate that HCSs are regulated by thalamocortical input. Only ACC currents, and not primary motor cortex spikes, were eliminated by the medial thalamic lesion experiments. Therefore, other cortical regions that receive medial thalamic projections may also be involved in HCSs. Our findings suggest that the thalamocortical circuit from the MD to the ACC, noxious stimuli, and the opioid system modulate such oscillations.
The projection terminal of the MD has been well characterized by light microscopy and electron microscopy. Most afferents from the MD to area 24 were in layer IIIb, with fewer in layer I (Vogt et al., 1981; Wang and Shyu, 2004). Primary projections to layers V and VI in area 24 come from the parafascicular nucleus and callosal (Vogt et al., 1981). Previous studies showed that electrostimulation of the MD immediately triggered paired-pulse facilitation responses in the ACC (Yang et al., 2006). The oscillation pattern in area 24 was changed after lesions of the MD, indicating that neuronal activity in the MD plays an important role in spontaneous oscillations in area 24.
Electroencephalographic α oscillations are considered to be attributable to thalamocortical and intrinsic properties of thalamic relay neurons. The intrinsic properties of thalamic neurons can spontaneously generate 6–10 Hz oscillations (Jahnsen and Llinás, 1984a,b). After disconnection of nRT synaptic transmission, the α spindle oscillations decreased in the cortex (Steriade et al., 1985). The existence of an intact thalamocortical feedback circuit is an important factor in generating cortical α EEG oscillations. Simple single-spike oscillations at 6–14 Hz (relay mode) of thalamic relay neurons can be transformed to bursting high-frequency oscillations (oscillatory mode) after ascending information increases (Pfurtscheller and Lopes da Silva, 1999). Cortical states can be changed by inputs of the thalamocortical circuit, in which α spindles appear during the unconscious state and are correlated with the blockade of thalamocortical synaptic transmission and anesthesia treatment (Steriade and Llinás, 1988). nRT inhibitory neurons generate sequences of long-lasting IPSCs on thalamocortical relay neurons, and then thalamocortical relay neurons present rhythmic hyperpolarization oscillations. Such hyperpolarization induces the deinactivation of low-threshold calcium currents. Consequently, action potentials are generated in thalamocortical relay neurons that lead to the feedback activation of nRT neurons. Thus, the oscillatory state of thalamocortical relay neurons is established and influences the cortical oscillation state. Many previous studies indicated that acute morphine treatment increases the spectral power of EEG in the total spectrum or α or β band (Khazan and Colasanti, 1971; Sala et al., 1995; Zuo et al., 2007), but the mechanism is unclear. Although we did not measure nRT neuron activity in the present study, we found that multiunit activity in the MD was decreased by morphine treatment. The enhanced synchronicity of MD multiunit activity may be attributable to the IPSC regulation of nRT neurons, which then triggers high-amplitude HCSs in the ACC that the α oscillation was enhanced. A similar result also suggested that the inhibition of thalamic neuron activity increases α oscillations in the cortex (Destexhe et al., 1996).
With pinch stimuli, multiunit activity in the MD and ACC both increased. The increase in neuronal activity in the ACC may occur through a noxious information pathway from the MD. The increase in neuronal activity appears to desynchronize MD neuron activity. The appearance of cortical HCSs correlated with thalamic multiunit burst firing but not single-unit activity. This may explain why desynchronizing the MD did not trigger HCSs in the ACC and decreased α oscillations. Cortical multiunit activity in the ACC also increased after noxious pinch stimuli. Higher frequency but not synchronized bursting may also influence the power spectrum of α oscillations. However, we did not observe an increase in burst firing in the β or γ band after pinch stimuli. This may be attributable to the recording settings or characteristics of the recording electrode. A previous study indicated that after a fentanyl injection, the spectral power EEG was increased, and the increase in the α to γ band and decrease in the δ band that were induced by noxious stimuli were inhibited by the opioid system (Peng et al., 2010).
Shifts in the low-frequency band and increase in EEG coherence coefficients in neurological patients with chronic pain have been shown to be caused by the dysrhythmia of thalamocortical oscillations (Stern et al., 2006; Sarnthein and Jeanmonod, 2008). Coherent low-frequency θ activity results from a resonant interaction between the thalamus and cortex. Alterations of synaptic inputs that are caused by deafferentation after neuronal injury hyperpolarize cells sufficiently to deinactivate T-type calcium channels in thalamic cells, resulting in thalamic oscillations in the θ range. Such low-threshold spike bursts are produced and lock the related thalamocortical circuits in low-frequency resonance. In various pathological neurological disorders, the mechanism of dysrhythmia is triggered from the thalamus toward the cortex or triggered by a reduction of the corticothalamic input, resulting in excess inhibition or dis-facilitation and generating thalamic cell membrane hyperpolarization and low-frequency oscillation (Llinás et al., 1999). Abnormal cortical oscillation patterns were thought to be attributable to overenhancement of the GABAergic system in the thalamus (Gwak and Hulsebosch, 2011). Consistent with this possibility, we previously found that the GABAergic system's role changes in CPSP (Shih et al., 2017). The characteristics of the EEG oscillation pattern and coherence coefficient in the CPSP group were highly similar to human patients (Llinás et al., 1999, 2005; Sarnthein et al., 2006). TrkB-Fc treatment confirmed that changes in the GABAergic system are a causal factor for thalamocortical dysrhythmia (Kuan et al., 2015; Shih et al., 2017). The role of the glutamatergic system in the “edge effect” has not been previously investigated. The high coherence coefficient (10–50 Hz) in CPSP animals was completely abolished by BBG treatment (Kuan et al., 2015). We found that P2X7 receptor antagonist treatment alters glutamatergic transmission. BBG targets the P2X7 receptor but also inhibits the P2X4 receptor, which is related to the overexpression of BDNF after chronic pain (Trang et al., 2012; Kuan et al., 2015; Shih et al., 2017). Thus, the glutamatergic system may also play an important role in the dysrhythmia of thalamocortical oscillations after CPSP. The abolishment of the coherence coefficient between 20 and 50 Hz by P2X7 receptor and BDNF inhibition was not well differentiated. Therefore, the differential effects of BBG and TrkB-FC on the “edge effect” suggest that the involvement of P2X7 receptors and BDNF in CPSP is different.
Footnotes
This work was supported by Ministry of Science and Technology Grants 105-2325-B-001-010, 105-2320-B-001-025-MY2, and 106-2321-B-001-043 to B.-C.S. This work was conducted at the Institute of Biomedical Sciences, which received funding from Academia Sinica. We thank the Taiwan Mouse Clinics for suggestions on the behavioral tests; and Neural Circuit Electrophysiology Core at Academia Sinica for technical support.
The authors declare no competing financial interests.
References
- Agmon A, Connors BW (1991) Thalamocortical responses of mouse somatosensory (barrel) cortex in vitro. Neuroscience 41:365–379. 10.1016/0306-4522(91)90333-J [DOI] [PubMed] [Google Scholar]
- Aporti F, Borsato R, Calderini G, Rubini R, Toffano G, Zanotti A, Valzelli L, Goldstein L (1986) Age-dependent spontaneous EEG bursts in rats: effects of brain phosphatidylserine. Neurobiol Aging 7:115–120. 10.1016/0197-4580(86)90149-1 [DOI] [PubMed] [Google Scholar]
- Buzsáki G. (1991) The thalamic clock: emergent network properties. Neuroscience 41:351–364. 10.1016/0306-4522(91)90332-I [DOI] [PubMed] [Google Scholar]
- Buzsáki G, Bickford RG, Ponomareff G, Thal LJ, Mandel R, Gage FH (1988a) Nucleus basalis and thalamic control of neocortical activity in the freely moving rat. J Neurosci 8:4007–4026. 10.1523/JNEUROSCI.08-11-04007.1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzsáki G, Bickford RG, Armstrong DM, Ponomareff G, Chen KS, Ruiz R, Thal LJ, Gage FH (1988b) Electric activity in the neocortex of freely moving young and aged rats. Neuroscience 26:735–744. 10.1016/0306-4522(88)90095-4 [DOI] [PubMed] [Google Scholar]
- Cantero JL, Atienza M, Salas RM (2002) Human α oscillations in wakefulness, drowsiness period, and REM sleep: different electroencephalographic phenomena within the α band. Neurophysiol Clin 32:54–71. 10.1016/S0987-7053(01)00289-1 [DOI] [PubMed] [Google Scholar]
- Chang C, Shyu BC (2001) A fMRI study of brain activations during non-noxious and noxious electrical stimulation of the sciatic nerve of rats. Brain Res 897:71–81. 10.1016/S0006-8993(01)02094-7 [DOI] [PubMed] [Google Scholar]
- Chang PF, Arendt-Nielsen L, Chen AC (2002) Dynamic changes and spatial correlation of EEG activities during cold pressor test in man. Brain Res Bull 57:667–675. 10.1016/S0361-9230(01)00763-8 [DOI] [PubMed] [Google Scholar]
- Chen AC, Rappelsberger P (1994) Brain and human pain: topographic EEG amplitude and coherence mapping. Brain Topogr 7:129–140. 10.1007/BF01186771 [DOI] [PubMed] [Google Scholar]
- Chen AC, Dworkin SF, Haug J, Gehrig J (1989) Human pain responsivity in a tonic pain model: psychological determinants. Pain 37:143–160. 10.1016/0304-3959(89)90126-7 [DOI] [PubMed] [Google Scholar]
- Chen TC, Cheng YY, Sun WZ, Shyu BC (2008) Differential regulation of morphine antinociceptive effects by endogenous enkephalinergic system in the forebrain of mice. Mol Pain 4:41. 10.1186/1744-8069-4-41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Destexhe A, Bal T, McCormick DA, Sejnowski TJ (1996) Ionic mechanisms underlying synchronized oscillations and propagating waves in a model of ferret thalamic slices. J Neurophysiol 76:2049–2070. 10.1152/jn.1996.76.3.2049 [DOI] [PubMed] [Google Scholar]
- Egsgaard LL, Wang L, Arendt-Nielsen L (2009) Volunteers with high versus low α EEG have different pain-EEG relationship: a human experimental study. Exp Brain Res 193:361–369. 10.1007/s00221-008-1632-1 [DOI] [PubMed] [Google Scholar]
- Ferracuti S, Seri S, Mattia D, Cruccu G (1994) Quantitative EEG modifications during the cold water pressor test: hemispheric and hand differences. Int J Psychophysiol 17:261–268. 10.1016/0167-8760(94)90068-X [DOI] [PubMed] [Google Scholar]
- Gwak YS, Hulsebosch CE (2011) GABA and central neuropathic pain following spinal cord injury. Neuropharmacology 60:799–808. 10.1016/j.neuropharm.2010.12.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Duysens J, Vossen JM, Coenen AM (1993) Thalamic multiple-unit activity underlying spike-wave discharges in anesthetized rats. Brain Res 612:35–40. 10.1016/0006-8993(93)91641-5 [DOI] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R (1984a) Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol 349:205–226. 10.1113/jphysiol.1984.sp015153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinás R (1984b) Ionic basis for the electro-responsiveness and oscillatory properties of guinea-pig thalamic neurones in vitro. J Physiol 349:227–247. 10.1113/jphysiol.1984.sp015154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel A, Buzsáki G (1997) Cellular-synaptic generation of sleep spindles, spike-and-wave discharges, and evoked thalamocortical responses in the neocortex of the rat. J Neurosci 17:6783–6797. 10.1523/JNEUROSCI.17-17-06783.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khazan N, Colasanti B (1971) Decline in the mean integrated electroencephalogram voltage during morphine abstinence in the rat. J Pharmacol Exp Ther 177:491–499. [PubMed] [Google Scholar]
- Klingberg F, Pickenhain L (1968) Occurrence of “spindle discharges” in the rat in relation to behavior. Acta Biol Med Ger 20:45–54. [PubMed] [Google Scholar]
- Kuan YH, Shih HC, Tang SC, Jeng JS, Shyu BC (2015) Targeting P(2)X(7) receptor for the treatment of central post-stroke pain in a rodent model. Neurobiol Dis 78:134–145. 10.1016/j.nbd.2015.02.028 [DOI] [PubMed] [Google Scholar]
- Llinás RR, Ribary U, Jeanmonod D, Kronberg E, Mitra PP (1999) Thalamocortical dysrhythmia: a neurological and neuropsychiatric syndrome characterized by magnetoencephalography. Proc Natl Acad Sci U S A 96:15222–15227. 10.1073/pnas.96.26.15222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Ribary U, Contreras D, Pedroarena C (1998) The neuronal basis for consciousness. Philos Trans R Soc Lond B Biol Sci 353:1841–1849. 10.1098/rstb.1998.0336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Urbano FJ, Leznik E, Ramirez RR, van Marle HJ (2005) Rhythmic and dysrhythmic thalamocortical dynamics: GABA systems and the edge effect. Trends Neurosci 28:325–333. 10.1016/j.tins.2005.04.006 [DOI] [PubMed] [Google Scholar]
- McCormick DA, Connors BW, Lighthall JW, Prince DA (1985) Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. J Neurophysiol 54:782–806. 10.1152/jn.1985.54.4.782 [DOI] [PubMed] [Google Scholar]
- Mitzdorf U. (1985) Current source-density method and application in cat cerebral cortex: investigation of evoked potentials and EEG phenomena. Physiol Rev 65:37–100. 10.1152/physrev.1985.65.1.37 [DOI] [PubMed] [Google Scholar]
- Nicholson C, Freeman JA (1975) Theory of current source-density analysis and determination of conductivity tensor for anuran cerebellum. J Neurophysiol 38:356–368. 10.1152/jn.1975.38.2.356 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C (1998) A stereotaxic atlas of the rat brain. San Diego: Academic. [Google Scholar]
- Peng YZ, Li XX, Wang YW (2010) Effects of Parecoxib and Fentanyl on nociception-induced cortical activity. Mol Pain 6:3. 10.1186/1744-8069-6-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfurtscheller G, Lopes da Silva FH (1999) Event-related EEG/MEG synchronization and desynchronization: basic principles. Clin Neurophysiol 110:1842–1857. 10.1016/S1388-2457(99)00141-8 [DOI] [PubMed] [Google Scholar]
- Sala M, Leone MP, Lampugnani P, Braida D, Frattola D, Gori E (1995) EEG power spectra and behavioral correlates in rats given chronic morphine. lack of residual long-term EEG and neuronal changes. Pharmacol Res 32:95–103. 10.1016/S1043-6618(95)80015-8 [DOI] [PubMed] [Google Scholar]
- Sarnthein J, Jeanmonod D (2008) High thalamocortical theta coherence in patients with neurogenic pain. Neuroimage 39:1910–1917. 10.1016/j.neuroimage.2007.10.019 [DOI] [PubMed] [Google Scholar]
- Sarnthein J, Stern J, Aufenberg C, Rousson V, Jeanmonod D (2006) Increased EEG power and slowed dominant frequency in patients with neurogenic pain. Brain 129:55–64. 10.1093/brain/awh631 [DOI] [PubMed] [Google Scholar]
- Seidenbecher T, Pape HC (2001) Contribution of intralaminar thalamic nuclei to spike-and-wave-discharges during spontaneous seizures in a genetic rat model of absence epilepsy. Eur J Neurosci 13:1537–1546. 10.1046/j.0953-816x.2001.01537.x [DOI] [PubMed] [Google Scholar]
- Shih HC, Kuan YH, Shyu BC (2017) Targeting brain-derived neurotrophic factor in the medial thalamus for the treatment of central poststroke pain in a rodent model. Pain 158:1302–1313. 10.1097/j.pain.0000000000000915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Llinás RR (1988) The functional states of the thalamus and the associated neuronal interplay. Physiol Rev 68:649–742. 10.1152/physrev.1988.68.3.649 [DOI] [PubMed] [Google Scholar]
- Steriade M, Deschênes M, Domich L, Mulle C (1985) Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J Neurophysiol 54:1473–1497. 10.1152/jn.1985.54.6.1473 [DOI] [PubMed] [Google Scholar]
- Stern J, Jeanmonod D, Sarnthein J (2006) Persistent EEG overactivation in the cortical pain matrix of neurogenic pain patients. Neuroimage 31:721–731. 10.1016/j.neuroimage.2005.12.042 [DOI] [PubMed] [Google Scholar]
- Trang T, Beggs S, Salter MW (2012) ATP receptors gate microglia signaling in neuropathic pain. Exp Neurol 234:354–361. 10.1016/j.expneurol.2011.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng MT, Chiang MC, Chao CC, Tseng WY, Hsieh ST (2013) fMRI evidence of degeneration-induced neuropathic pain in diabetes: enhanced limbic and striatal activations. Hum Brain Mapp 34:2733–2746. 10.1002/hbm.22105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C, Depaulis A, Micheletti G, Warter JM (1986) Ontogeny of spontaneous petit mal-like seizures in Wistar rats. Brain Res 395:85–87. 10.1016/S0006-8993(86)80011-7 [DOI] [PubMed] [Google Scholar]
- Vergnes M, Marescaux C, Depaulis A, Micheletti G, Warter JM (1987) Spontaneous spike and wave discharges in thalamus and cortex in a rat model of genetic petit mal-like seizures. Exp Neurol 96:127–136. 10.1016/0014-4886(87)90174-9 [DOI] [PubMed] [Google Scholar]
- Vogt BA, Rosene DL, Peters A (1981) Synaptic termination of thalamic and callosal afferents in cingulate cortex of the rat. J Comp Neurol 201:265–283. 10.1002/cne.902010210 [DOI] [PubMed] [Google Scholar]
- Vogt BA, Derbyshire S, Jones AK (1996) Pain processing in four regions of human cingulate cortex localized with co-registered PET and MR imaging. Eur J Neurosci 8:1461–1473. 10.1111/j.1460-9568.1996.tb01608.x [DOI] [PubMed] [Google Scholar]
- Walker GE, Yaksh TL (1986) Studies on the effects of intrathalamically injected DADL and morphine on nociceptive thresholds and electroencephalographic activity: a thalamic delta-receptor syndrome. Brain Res 383:1–14. 10.1016/0006-8993(86)90002-8 [DOI] [PubMed] [Google Scholar]
- Wang CC, Shyu BC (2004) Differential projections from the mediodorsal and centrolateral thalamic nuclei to the frontal cortex in rats. Brain Res 995:226–235. 10.1016/j.brainres.2003.10.006 [DOI] [PubMed] [Google Scholar]
- Wiest MC, Nicolelis MA (2003) Behavioral detection of tactile stimuli during 7–12 Hz cortical oscillations in awake rats. Nat Neurosci 6:913–914. 10.1038/nn1107 [DOI] [PubMed] [Google Scholar]
- Yang JW, Shih HC, Shyu BC (2006) Intracortical circuits in rat anterior cingulate cortex are activated by nociceptive inputs mediated by medial thalamus. J Neurophysiol 96:3409–3422. 10.1152/jn.00623.2006 [DOI] [PubMed] [Google Scholar]
- Zuo YF, Wang JY, Chen JH, Qiao ZM, Han JS, Cui CL, Luo F (2007) A comparison between spontaneous electroencephalographic activities induced by morphine and morphine-related environment in rats. Brain Res 1136:88–101. 10.1016/j.brainres.2006.11.099 [DOI] [PubMed] [Google Scholar]