

 Mechanisms of antifolate resistance in bacterial and mammalian cells.
Mechanisms of antifolate resistance in bacterial and mammalian cells.
Abstract
In prokaryotes and eukaryotes, folate (vitamin B9) is an essential metabolic cofactor required for all actively growing cells. Specifically, folate serves as a one-carbon carrier in the synthesis of amino acids (such as methionine, serine, and glycine), N-formylmethionyl-tRNA, coenzyme A, purines and thymidine. Many microbes are unable to acquire folates from their environment and rely on de novo folate biosynthesis. In contrast, mammals lack the de novo folate biosynthesis pathway and must obtain folate from commensal microbiota or the environment using proton-coupled folate transporters. The essentiality and dichotomy between mammalian and bacterial folate biosynthesis and utilization pathways make it an ideal drug target for the development of antimicrobial agents and cancer chemotherapeutics. In this minireview, we discuss general aspects of folate biosynthesis and the underlying mechanisms that govern susceptibility and resistance of organisms to antifolate drugs.
1. Introduction
De novo folate biosynthesis begins with synthesis of para-aminobenzoic acid (PABA, Fig. 1) and 6-hydroxymethyl-7,8-dihydropterin pyrophosphate (DHPPP, Fig. 2). Both pathways are absent in mammals and essential in bacteria, protozoa and fungi. There are no antimicrobial agents in clinical use that target PABA or DHPPP biosynthesis. As described below, recent efforts have revealed many challenges that must be overcome to target these pathways.
Fig. 1. para-Aminobenzoic acid biosynthesis pathway in bacteria with known inhibitors.

Fig. 2. Pterin biosynthesis and inhibitors in bacteria and mammals.

Later steps in folate synthesis involve the conversion of 7,8-dihydropteroate (DHP) to 5,6,7,8-tetrahydrofolate (THF) in three ordered steps by DHP synthase (FolP), dihydrofolate synthase (FolC), and dihydrofolate reductase (DHFR in mammals and FolA in bacteria) (Fig. 3). The only approved therapeutic agents in clinical use that target this portion of the pathway inhibit FolP and DHFR. In this review, we discuss the therapeutic agents that are currently used to target folate biosynthesis in bacteria, protozoa, fungi and mammals.
Fig. 3. Folate biosynthesis and inhibitors in bacteria and eukaryotes.

Antifolates, agents that impair folate biosynthesis, were the first class of antimetabolites to enter clinical use. They were used to treat a variety of cancers and inflammatory disorders in humans and were among the first antimicrobial agents used to treat bacterial infections. Historically, antifolates have targeted the enzymes FolP (sulfonamides and sulfones) and DHFR (methotrexate, proguanil and trimethoprim). Resistance to antifolates was reported shortly after their introduction into clinical use. Since some antifolates can be associated with adverse reactions, some have been replaced with safer and more effective drugs. With the rise in resistance to many chemotherapeutics and antimicrobial agents and with the advent of better-tolerated antifolates, there has been a renewed interest in using this drug class to treat many diseases.
Sulfonamides were introduced into the clinic in 1935 to treat a variety of bacterial infections. However, some sulfonamides, such as sulfamethoxazole (SMX), were associated with rare adverse reactions such as blood dyscrasias, hypoglycemia, hyperkalemia, and inhibition of cytochrome P450.1–3 In 1962, TMP was introduced into the clinic as a bacterial FolA inhibitor. It was noted that SMX and TMP showed potent synergistic activity, permitting the use of lower dosages of SMX that resulted in fewer adverse reactions.4 Consequently, co-administration of SMX with TMP (co-trimoxazole) became standard of care for treating many infections. Since co-trimoxazole is generally well-tolerated, relatively inexpensive and can target a broad spectrum of microbes, it is commonly used for prophylactic treatment against opportunistic infections in immunocompromised individuals.
Herein, we primarily focus on antifolates that target microbes and will briefly cover inhibition of mammalian folate metabolism. Further, we examine PABA, DHPPP, and later steps in folate biosynthesis as drug targets and the most recent strategies used to target enzymes in the respective pathways. As all of the antifolates in clinical use are associated with multiple mechanisms of resistance, there is a need to understand the differences in susceptibility to each antifolate.
2. Targeting para-aminobenzoic acid biosynthesis in microbes
In most bacteria, fungi, protozoans and plants, folates are synthesized from the convergence of the PABA and pterin biosynthesis pathways. The PABA biosynthesis pathway is absent in the metazoan lineage. The first step to synthesize PABA involves the conversion of chorismate to aminodeoxychorismate (ADC) (Fig. 1). Chorismate is the product of the shikimate pathway, from which aromatic amino acids, quinones and salicylic acid are also derived. In the synthesis of PABA, chorismate and glutamine are converted to ADC and glutamate by the action of ADC synthase. ADC synthase is comprised of two enzymatic subunits PabA and PabB encoded by pabA and pabB, respectively. Both enzymes require physical interaction for optimal catalysis. PabA catalyzes glutamine lyase activity and is typically only active in the presence of stoichiometric amounts of PabB.5,6 In the absence of PabA and glutamine, PabB is able to catalyze the amination of chorismate in the presence of vast molar excess ammonia.6 Both pabA and pabB are predicted to be essential in many bacterial species.
Interestingly, some bacteria do not encode a defined PabA homolog and instead there is functional overlap with an analogous enzyme TrpG from the tryptophan biosynthetic pathway. In tryptophan biosynthesis TrpG functions in conjunction with TrpE to catalyze synthesis of anthranilate from chorismate and glutamine in a reaction similar to that for synthesis of ADC.7–11 As TrpG and PabA show a high degree of sequence conservation and have mechanistically identical enzymatic function,7,11 organisms such as Bacillus subtilis, Acinetobacter calcoaceticus and Salmonella enterica express a bi-functional (amphibolic) glutamine ammonium lyase that operates as a subunit of both anthranilate synthase and ADC synthase.
The last step in PABA biosynthesis is the conversion of ADC to PABA by ADC lyase encoded by pabC (Fig. 1).12,13 This reaction proceeds with the elimination of the pyruvyl moiety of ADC enabling subsequent aromatization to PABA. Since elimination of pyruvate from ADC can occur spontaneously, pabC is not predicted to be essential in many bacteria.
In Mycobacterium tuberculosis disruption of PABA biosynthesis, specifically through mutagenesis of pabB or pabC, results in enhanced susceptibility to antifolates, including sulfa-drugs and the diaminophenylsulfone dapsone.14 There are multiple natural products that inhibit PabB including abyssomicin C and anthelminthicin C and their derivatives (Fig. 4).15–19 These compounds show inhibitory activity against M. tuberculosis and methicillin resistant Staphylococcus aureus (MRSA) in whole cell assays.17–21 Evaluation of their activity in animal models of infection has not been reported. Abyssomicin C and some of its derivatives display a high level of cytotoxicity against mammalian cells.18 Although, atrop-O-benzyl-desmethylabyssomicin C was found to have significantly reduced cytotoxicity whilst retaining activity against MRSA and may represent a promising lead (Fig. 4).19 In addition to these derivatized natural products, the synthetic dichloronitrophenyl propanone, MAC173979, was found to inhibit PABA biosynthesis in E. coli both enzymatically and in whole cell assays.22 MAC173979 exhibited time-dependent inhibition in a one-pot assay using purified recombinant PabA, PabB, and PabC, yet, the exact target of MAC173979 is not known (Fig. 4).22 MAC173979 also showed potent activity against M. tuberculosis that could be antagonized by exogenously supplied PABA.14 Consistent with genetic studies described above, synergistic inhibition of growth was observed when M. tuberculosis was treated with MAC173979 in combination with various sulfa-drugs or dapsone.14 Taken together, the PABA biosynthesis pathway remains a promising drug target especially in combination with sulfa-drugs.
Fig. 4. Chemical structures of inhibitors of PABA biosynthesis.

3. Targeting pterin biosynthesis in prokaryotes and eukaryotes
Prokaryotes and eukaryotes synthesize pterins, albeit, the metabolic functions they serve can be quite distinct. For instance, in humans, guanine cyclohydrolase (encoded by GCH1) catalyzes GTP hydrolysis, isomerization and cyclization to form 7,8-dihydroneopterin triphosphate (Fig. 3), a precursor in tetrahydrobiopterin synthesis. Tetrahydrobiopterin is a critical co-factor for the synthesis of aromatic amino acids and multiple neurotransmitters.23 Deficiencies in the production of tetrahydrobiopterin results in dystonia, or sustained muscle contractions. Bacteria, fungi, protozoans and plants also encode a guanine hydrolase (FolE, encoded by folE) that performs the same biochemical function as GCH1. However, in this context, 7,8-dihydroneopterin triphosphate is essential for folate biosynthesis (Fig. 2). FolE requires a zinc ion to be bound that allows for subsequent binding of the purine ring of GTP. Following binding, GTP is cleaved and isomerized to allow for the closure of the dihydropyrazine ring.24–26 Interestingly, 8-aminoguanosine triphosphate and other structural analogs of GTP, can competitively inhibit bacterial FolE and GCH1 by mimicking the transition state between GTP and 7,8-dihydroneopterin triphosphate (Fig. 5).24,27
Fig. 5. Chemical structures of pterin biosynthesis inhibitors.

In addition to FolE, bacteria encode other distinct classes of GTP glycohydrolases, including GCH1b and RibA.26 GCH1b is found in about 20% of eubacteria and lacks sequence similarity with FolE. Unlike FolE, GCH1b is not zinc-dependent and can utilize a variety of divalent cations for catalytic activity.26 Interestingly, in eubacteria encoding both FolE and GCH1b, FolE is constitutively expressed while GCH1b is only expressed during zinc starvation.26,28 Since GCH1b is not present in humans, it represents a novel antibacterial target, specifically for Staphylococcus aureus and Neisseria gonorrhoeae, as both organisms lack GCH1.28
RibA was first characterized in E. coli as a novel enzyme that catalyzed the release of 2,5-diamino-6-β-ribosyl-4(3H)-pyrimidinone 5′-phosphate (DARP) and formate from GTP.29 DARP is required for the synthesis of riboflavin, an essential cellular cofactor required for growth of bacteria. RibA contains no amino acid sequence homology to FolE. Although, like FolE, RibA is also a zinc dependent enzyme that is responsible for the opening of the guanine ring.30 In most bacteria, RibA catalyzes the first step in riboflavin biosynthesis, producing DARP.31 Many pathogenic bacteria lack the ability to transport riboflavin from the extracellular environment making RibA essential. Since RibA is absent in higher eukaryotes, it represents an attractive target for antimicrobial drug discovery.
In the next step of DHPPP synthesis, 7,8-dihydroneopterin triphosphate is dephosphorylated by a cytoplasmic Nudix hydrolase to produce 7,8-dihydroneopterin (Fig. 2). Nudix hydrolases form a superfamily of pyrophosphatases that are often involved in detoxifying toxic metabolites. The first 7,8-dihydroneopterin triphosphate pyrophosphatase Nudix hydrolase enzyme to be described was YlgG, encoded by ylgG, in Lactococcus lactis.32 Since YlgG did not show sufficient sequence similarity to Nudix enzymes of bacteria with known genome sequences, identifying other Nudix hydrolases involved in pterin synthesis has been quite challenging. The product of the gene orf17, NudB (or NtpA), was the first dihydroneopterin triphosphate pyrophosphatase described in E. coli.33,34 Nudix enzymes hydrolyze substrates through the use of a nucleophilic substitution and require a metal ion for coordination of GTP, though the specific metal varies between Nudix enzymes.32–35 Since dihydroneopterin triphosphate pyrophosphatases have only been recently discovered, inhibitors of this enzyme have yet to be identified. Nevertheless, deletion of nudB in E. coli has been shown to dramatically enhance susceptibility to both SMX and TMP.36,37 Thus, these recent discoveries open avenues for novel therapeutic discovery.
Once formed, 7,8-dihydroneopterin is converted to 6-hydroxymethyl-7,8-dihydropterin by dihydroneopterin aldolase (FolB) yielding glyceraldehyde as a byproduct (Fig. 2). FolB, encoded by folB, is predicted to be essential in bacteria. Most aldolases require a Schiff base for the cleavage of the carbon–carbon double bond and a zinc ion for catalysis, both of these features are targets for selective aldolase inhibitors.38 FolB is a unique aldolase as it requires neither a Schiff base for cleavage nor a zinc ion for catalysis.39,40 Due to its small active site binding pocket and unique catalytic mechanism, designing FolB inhibitors has been challenging. In some organisms, dihydroneopterin aldolase is a bi-, or in some parasites, a tri-functional enzyme. For instance, in Streptococcus pneumonia, dihydroneopterin aldolase (SulD, encoded by sulD) is a bi-functional enzyme that encodes 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase that can convert 7,8-dihydroneopterin to 6-hydroxymethyl-7,8-dihydropterin pyrophosphate.41,42 The opportunistic pathogen Pneumocystis jiroveci encodes a tri-functional enzyme that encodes a dihydroneopterin aldolase and 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase, as well as a dihydropteroate synthase.43 These multi-functional enzymes have been extensively targeted for development of antimicrobial inhibitors.44
Most FolB inhibitors mimic the pterin or pyrimidine moiety of 7,8-dihydroneopterin.45 A high-throughput X-ray crystallography screen and structure-directed lead optimization discovered multiple potent inhibitors of dihydroneopterin aldolase from S. aureus including 9-methylguanine, 2-amino-5-carboxyethylpyrimidine-4-one, 8-amino-1,3-dimethyl-3,7-dihydropurine-2,6-dione, and 2-amino-5-bromo-6-phenylpyrimidine-4-one46 (Fig. 5). Unfortunately these compounds do not possess whole cell inhibitory activity against S. aureus, Haemophilus influenzae, E. coli, Enterococcus faecium and Streptococcus pneumoniae.
The last step in pterin biosynthesis for folates requires the conversion of 6-hydroxymethy-7,8-dihydropterin to DHPPP via the enzyme 6-hydroxymethyl-7,8-dihydropterin pyrophosphokinase (FolK, encoded by folK in bacteria) (Fig. 2). In bacteria, disruption of FolK results in a pronounced growth defect.47 FolK is a unique pyrophosphokinase because it transfers a pyrophosphate from the beta phosphate of ATP rather than a more conical gamma phosphate yielding AMP.48 FolK has been studied for the development of novel bisubstrate analog inhibitors allowing for selective inhibition of this target.49–51 Most of the bisubstrate inhibitors developed mimic the ATP phosphate donor and acceptor (serine, threonine, or tyrosine). Some such compounds include P1-(6-hydroxymethylpterin)-P4-(50-adenosyl)tetraphosphate (HP4A) and 50-S-[1-(2-{[(2-amino-7,7-dimethyl-4-oxo-3,4,7,8-tetrahydropteridin-6-yl)carbonyl]amino}ethyl)piperidin-4-yl]-50-thioadenosine (HP-18) (Fig. 5).52 Despite both compounds potently inhibiting purified FolK, they showed limited whole cell activity. 8-Mercaptoguanine (Fig. 5) has also been shown to inhibit S. aureus FolK by acting as a non-competitive inhibitor.53,54 Although 8-mercaptoguanine is a potential scaffold for discovery, its activity against S. aureus has not been reported and it is likely to show polypharmacology through interaction with other proteins with purine binding domains. In bacteria, the pterin biosynthetic pathway is highly conserved, however, the enzymes are structurally distinct and may ultimately allow for selective targeting.55
4. Targeting folate biosynthesis in prokaryotes and eukaryotes
DHP is produced from the condensation of PABA and DHPPP with elimination of pyrophosphate via DHP synthase (DHPS, encoded by folP) (Fig. 3). FolP is a homodimer and is the a target of many antimicrobial agents including sulfonamides and diaminodiphenyl sulfones (Fig. 6). Both drug classes are analogs of PABA. Sulfonamides, such as sulfamethoxazole (SMX), are broad spectrum antimicrobial agents that inhibit FolP by creating the dead-end product sulfamethoxazole-dihydropterin.56–58 Another inhibitor of FolP is 8-mercaptoguanine that has been shown to have promiscuous activity by inhibiting many enzymes that have a substrate bearing a purine moiety.59 Since FolP is an important drug target, there have been many structural and biochemical studies focused on designing more potent FolP inhibitors60,61 during the catalytic cycle DHPPP binds first, and the pyrophosphate is removed in a magnesium dependent reaction.57,58,61,62 Once the pyrophosphate is eliminated, the PABA binding pocket opens and allows for binding of PABA, sulfonamides or diaminodiphenyl sulfones.57,61,63,64 Sulfonamides and sulfones bind the PABA binding pocket that is separate from the pterin binding pocket. To develop novel classes of inhibitors, there is an increased interest in targeting the pterin binding pocket. Recently, a series of novel 4,5-dioxo-1,4,5,6-tetrahydropyrimido[4,5-c]pyridazine compounds were designed to inhibit the pterin binding pocket of purified recombinant Bacillus anthracis FolP (Fig. 6).65 These compounds potently inhibited through binding within the pterin binding pocket and provided structure activity relationship information for designing new inhibitors. Furthermore, pterin–sulfonamide conjugates have been found to competitively inhibit FolP and inhibition was enhanced in the presence of pyrophosphate.66 The pterin–sulfonamide conjugates displayed antimicrobial activity against E. coli that could be antagonized by PABA, and to a lesser extent by methionine.66
Fig. 6. Chemical structures of folate biosynthesis inhibitors.

One challenge in designing small molecules that inhibit the pterin binding pocket results in pterin-like molecules that have poor solubility.65 Therefore, there is an interest in inhibitors that bind outside of the active site. Hammoudeh and colleagues performed a fragment based lead discovery and found fragments bound at the dimer interface.67 Specifically, N-(4-(trifluoromethyl)-benzylidene)-1-(4-(trifluoromethyl)benzylamine) inhibited multiple FolP enzymes from B. anthracis, Yersina pestis, and S. aureus; however, the compound lacked antimicrobial activity.67
Many sulfonamides and sulfonylurea-based drugs, such as sulfasalazine, sulfathiazole, sulfapyridine, sulfamethoxazole, and chloropropamide, are associated with adverse reactions, most commonly allergies, that can limit the usage in HIV patients. Furthermore, sulfa-drugs can cause toxicity through off target effects in humans. These compounds have been found to inhibit the NADPH dependent enzyme sepiapterin reductase.68,69 Sepiapterin reductase is an essential enzyme that is required for synthesis of tetrahydropterin, a critical co-factor for neurotransmitter synthesis in mammals.
DHP is converted to 7,8 dihydrofolate (DHF) by the ligation of l-glutamate using the ATP-dependent enzyme DHF-synthase (FolC, encoded by folC) (Fig. 3). In many organisms, such as E. coli, Corynebacterium sp., and P. falciparum, this enzyme is bi-functional and can catalyze the gamma linkage of additional l-glutamate residues to fully reduced folate species.70–72 Folylpolyglutamates are important in bacteria for methionine synthesis.73 In humans, folates are polyglutamylated by the enzyme downstream of dihydrofolate reductase, folylpolyglutamate synthase (FPGS). In eukaryotes, polyglutamylation is important for retaining reduced folates in subcellular compartments. The role of polyglutamylation has not been extensively studied in bacteria, but, it is conceivable that the negatively charged polyglutamate tail may prevent folate species from diffusing across the cytoplasmic membrane.
Both FolC and FPGS are members of the Mur superfamily of enzymes and both contain an ATPase domain.55,74 ATP is positioned in a narrow channel and is stabilized by two magnesium ions.75 In the crystal structure of many apo-FolC enzymes the pterin binding site is not well resolved, however, the active site becomes more ordered upon the binding of DHP.74–76 Interestingly, in bacteria, FolC has an induced fit pterin binding pocket to allow mono- and poly-glutamylated folates to bind.77–79 The conversion of DHP to DHF generates a tetrahedral intermediate through the transfer of a phosphate from ATP to DHP.80,81 Glutamate will react with the intermediate to produce DHF. Attempts to target the tetrahedral transition state have yielded initial hits against purified FolC but none have shown whole cell activity.82,83
The last step in folate biosynthesis is the reduction of DHF to THF by the NADPH dependent enzyme dihydrofolate reductase (FolA in bacteria, DHFR in eukaryotes) (Fig. 3). Although most enzymes in the folate biosynthesis pathway are essential, FolA in bacteria can be conditionally essential. In E. coli, folA can be deleted following disruption of the gene for thymidylate synthase,84 a folate-dependent enzyme required for thymidine synthesis. Unlike other folate-dependent enzymes thymidylate synthase yields DHF rather than THF. Thus, while elimination of this activity confers thymidine auxotrophy, it decreases the demand for folate reduction which can be inefficiently catalyzed by FolM.85 In E. coli, FolM functions as a dihydromonapterin reductase. FolA reduces DHF using NADPH as an electron donor to THF and with the release of NADP+. Under steady state reaction conditions, the catalytic mechanism of DHFR is stepwise.86,87 The reaction begins with NADPH bound to the enzyme (E : NADPH), next DHF binds which creates the Michaelis complex (E : NADPH : DHF). Once DHF binds, the hydride transfer occurs (E : NADP+ : THF). The exact biochemical mechanism of the hydride transfer is not well understood. To date there are two proposed mechanisms, the first states the hydride transfer is pH dependent and occurs by an ionizable side chain in the active site.88 The second mechanism states the pH dependent hydride transfer is solvent dependent.89 NADP+ is released (E : THF), another NADPH binds (E : NADPH : THF), and finally THF is released in the rate determining step to prime the enzyme for another catalytic cycle.
There are many drugs that selectively inhibit DHFR in humans, bacteria, fungi, and protozoa. DHFR inhibitors in humans and protozoans generally mimic the substrate folate. One class of inhibitors, the diaminopteridines, include methotrexate (MTX) and aminopterin (Fig. 6), both are used to treat cancers and autoimmune disorders.90 Diaminopteridines are transported into human cells by folate receptors. Since, folate receptors are highly upregulated by as much as 100-fold in cancer cells, MTX and other diaminopteridines potently target these cells compared to non-cancerous cells.91 Not only do folate receptors recognize folate and folate analogs but they also recognize polyglutamylated folate species. As described above, polyglutamylated species are retained within cellular compartments. Thus, this mechanism for retention of folates has been co-opted to promote accumulation DHFR inhibitors, such as pralatrexate (Fig. 6), within target cells.92
Aminopterin (Fig. 6) was the first human DHFR inhibitor to enter clinical use for the treatment of childhood lymphocytic leukemia. Aminopterin was associated with high toxicity and was quickly replaced with MTX (Fig. 6) which was found to be as effective, but less toxic. MTX is known to inhibit all purified DHFRs, including those from bacteria, fungi and protozoans, and is characterized as a slow, time-dependent, tight-binding competitive inhibitor.93,94 Binding of MTX results in a slow conformational change in the enzyme active site and is known to bind almost identically to folate by forming similar hydrogen bonding within the active site. One notable difference occurs when the pterin ring of MTX is flipped 180° with respect to DHF. This flip allows hydrogen bonding with the ionizable group in the active site.95 From a mechanistic perspective, this flip within the active site provided key insight that a keto–enol equilibrium pertaining to the transition state of the enzyme.96 Furthermore, this flip suggests that THF release is assisted by NADPH binding versus NADP+ binding in NADP+ rich environments of bacteria.97
Since most pathogenic bacteria lack folate receptors and cannot actively take up this cofactor, antibacterial FolA inhibitors are not close structural analogs of folate. In general, bacterial FolA inhibitors must diffuse across the cell envelope or require folate independent transport.98,99 The structure of many of these inhibitors leaves them vulnerable to numerous efflux pumps.98,99 One notable bacterial FolA inhibitor, trimethoprim (TMP, Fig. 6), contains a 2,4-diaminopyrimidine ring that allows for critical hydrogen bonding in the active site. TMP binds much more tightly (about 105) to bacterial FolA than mammalian DHFR.100 The benzyl ring of TMP undergoes a thermodynamically unfavorable flip in the apo-enzyme. However, in the presence of NADPH the flipping of the benzyl ring is decreased. Therefore, in bacteria, TMP is a competitive inhibitor with respect to DHF and a non-competitive inhibitor with respect to NADPH. Conversely, mammalian DHFR can bind TMP but cannot form key hydrogen bond contacts within the active site.101
TMP was first introduced into the clinic in 1962. Resistance was described as early as 1968, therefore, TMP derivatives were synthesized and explored for antimicrobial activity. Two notable TMP derivatives are tetroxoprim used for the treatment of Pneumocystis pneumonia and iclaprim for treatment of methicillin and vancomycin resistant strains of Staphylococcus aureus as well as drug resistant strains of Streptococcus species (Fig. 6).102,103 Trimetrexate is a quinazoline derivative and is a chimera of pterin moiety of methotrexate and contains the benzyl ring of TMP. Iclaprim, like TMP, is a diaminopyrimidine that contains a chromene ring in place of the benzyl ring of TMP that favors more hydrophobic interactions in the active site.102,104
The first anti-malarial antifolate drug discovered was the biguanide proguanil which is one of the components of the widely used malaria prophylactic drug malarone. Shortly after its discovery, proguanil was found to be a pro-drug that is metabolized by the host CYP2C19 to cycloguanil (Fig. 6).105,106 This activated form was found to inhibit Plasmodium falciparum, the most prevalent causative agent of malaria, through targeting of DHFR. Many proguanil derivatives, including the cycloguanil structural analog pyrimethamine, have since been developed to counter drug resistant strains of P. falciparum.107 Pyrimethamine (Fig. 6) showed excellent efficacy in humans because of the low dosage required for treatment and paucity of side effects.108 However, pyrimethamine resistance quickly emerged which diminished its use in to treatment of malaria. Pyrimethamine has been repurposed to treat toxoplasmosis and is used in combination with dapsone to treat Pneumocystis pneumonia in HIV infected individuals.
The dihydrotriazine, WR99210, is an anti-malarial DHFR inhibitor that was developed after emergence of resistance to pyrimethamine and cycloguanil (Fig. 6). WR99210 potently inhibits DHFR at very low concentrations, in the nano- to picomolar range. Unfortunately, in human clinical trials, daily administration of 200 mg of WR99210 for three days resulted in severe gastrointestinal distress. As a result, further clinical investigation into WR99210 stalled. To counter WR99210 toxicity, the biguanide pro-form PS-15 (N-(3-(2,4,5-trichlorophenoxy)propyloxy)-N′-(1-methylethyl)-imidocarbonimidic diamide hydrochloride) was synthesized based on proguanil that could be activated in vivo (Fig. 6).109 PS-15 showed improved antimalarial activity and it is anticipated that it will be better tolerated than WR99210. Importantly, PS-15 was found to selectively inhibit P. falciparum DHFR and had little activity against mammalian DHFR. Unfortunately, large-scale synthesis of PS-15 is challenging for drug production and has caused its development to lag. However, with this scaffold, other WR99210 analogs have been explored as novel DHFR inhibitors not only in Plasmodia species, but also in M. tuberculosis, Mycobacterium avium and P. jiroveci.110,111
The last FolA inhibitor that we will discuss is para-aminosalicylic acid (PAS, Fig. 6). PAS is an antitubercular agent used to treat drug resistant strains of M. tuberculosis.112 PAS is a structural analog of PABA, possessing a hydroxyl-group in the ortho-position to the carboxylic acid. Soon after its discovery, PAS was thought to disrupt folate biosynthesis based on the observation that exogenous PABA could antagonize its inhibition of M. tuberculosis growth.113 Initially, PAS was thought to act as a FolP inhibitor similar to sulfonamides. However, no cross-resistance between PAS and sulfonamides in M. tuberculosis was observed.114 Further, in contrast to sulfonamides, PAS was found to only weakly inhibit purified FolP enzymatic activity and was later demonstrated to be converted to a hydroxylated version of DHP, hydroxy-DHP.115–117 FolC was found to catalyze the glutamylation of hydroxy-DHP to hydroxy-DHF (Fig. 3 and 6), which suggested that FolC is not the target of bioactivated PAS and the target was likely downstream within the folate pathway.115–117 It was predicted and later confirmed that hydroxy-DHF could potently inhibit FolA.115,117,118 Interestingly, PAS has been shown to only have activity against M. tuberculosis, although the mechanistic basis for this high level of selectivity is not known.119–123
5. Antifolate synergy
Currently, TMP and SMX are rarely given in monotherapy. When used in combination, TMP and SMX (co-trimoxazole) exhibit highly synergistic antimicrobial activity. Due to differences in drug metabolism rates, they are given in a 1 : 5 (TMP : SMX) ratio to achieve serum concentrations that are typically observed to be synergistic. Co-trimoxazole is used to treat a variety of microbial infections including respiratory tract infections, otitis media, urinary tract infections, MRSA infections, and cholera. With the emergence of HIV, co-trimoxazole is often given as lifetime prophylactic therapy to treat and prevent opportunistic infections, such as Pneumocystis pneumonia and toxoplasmosis, in individuals with acquired immunodeficiency syndrome. When SMX is not well-tolerated, dapsone is used as a surrogate. Given the success and global availability of highly active antiretroviral therapy for HIV, there is ongoing debate regarding the continued need for lifetime co-trimoxazole prophylactic therapy.
For several decades it was presumed that the basis for synergy between SMX and TMP was exclusively due to the ability of SMX to prevent DHF synthesis and enhance the ability of TMP to inhibit FolA.37,124,125 While this model explains how SMX is able to enhance microbial susceptibility to TMP, it is not sufficient to explain the ability of TMP to enhance susceptibility of microbes to SMX.126 It was recently demonstrated that TMP treatment results in depletion of DHPPP through impairment of GTP synthesis and is the basis for potentiation of SMX susceptibility.37 Thus, it is the cyclic nature of the folate biosynthetic pathway that enables the potent synergy between SMX and TMP.37
6. Antifolate resistance
While folate metabolism remains a powerful target for drug discovery and development, there has been a surge in resistance to many anti-folates. This section will focus on the broad resistance mechanisms to sulfonamides, TMP, PAS and MTX.
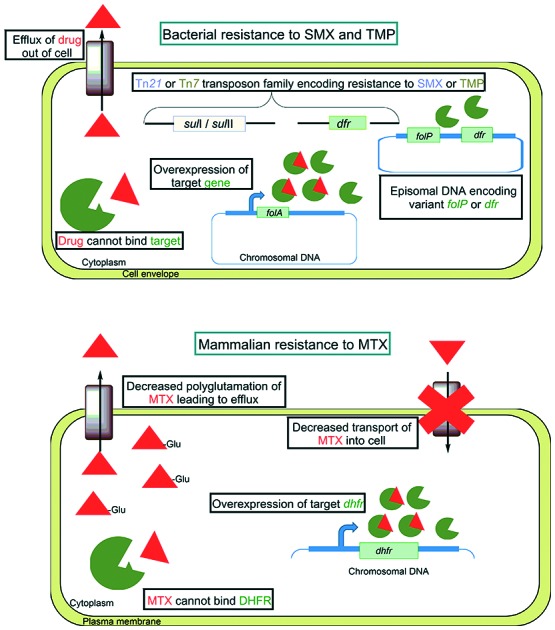
There are multiple mechanisms that can confer broad resistance to sulfonamides and were found to emerge shortly after entrance of this drug class into clinical use. One such resistance mechanism involves single amino acid point mutations in the chromosomal folP (Fig. 7). Among clinically resistant isolates, many have been characterized that have mutations that result in alteration of the ternary structure and prevent productive interaction with sulfonamides.127 In the case of the pathogen S. pyogenes, clinical resistance to sulfonamides emerged much faster than in other most organisms and was the result of horizontal acquisition of a plasmid encoding a sulfonamide resistant FolP variant.128 Similarly, in many Gram-negative bacteria, resistance to sulfonamides has been traced to mobile genetic elements bearing sul genes that encode fully functional FolP variants that fail to interact with sulfonamides (Fig. 7).129 In early reports of this resistance mechanism, E. coli was found to transfer sulfonamide resistance to Shigella species via a Tn21 family transposon encoding sul1 or sul2.130 Sul1 and Sul2 both have dihydropteroate synthase activity.131 While Sul1 and Sul2 were found to bind PABA with similar affinity as FolP, their affinity for sulfonamides was 10 000 times lower.131 Tn21 family transposons have since been found to be broadly distributed amongst enteric bacteria that are resistant to sulfonamides.132 Recent evidence suggests that the sul genes originated in the Gram-negative bacterial orders Rhodobiaceae and Leptospiraceae prior to the development of sulfonamides and were subsequently distributed amongst human associated microbes with broad clinical use of sulfonamides.133
Fig. 7. Mechanisms of antifolate resistance in bacterial and mammalian cells.

Another factor that limits the use of sulfonamides is intrinsic resistance through methionine-associated metabolism. Shortly after discovery of the sulfonamides, it was found that exogenously supplied methionine could potently antagonize the antimicrobial action of these drugs in numerous bacterial species.134 While the mechanism governing this antagonism has not been fully elucidated, it has been presumed to occur through methylation-dependent inactivation. In the context of M. tuberculosis, sulfonamides are known to inhibit purified recombinant FolP, yet, they show poor activity against intact bacilli.135 The related drug dapsone was initially developed as an antitubercular agent and also inhibits purified recombinant M. tuberculosis FolP, but, like sulfonamides shows limited activity against whole cells.135 In support of a model for methyl inactivation, it has been demonstrated that M. tuberculosis rapidly metabolizes sulfonamides and dapsone to N-methyl and N,N-dimethyl species.117 In addition to methyl inactivation, it has recently been demonstrated in E. coli and M. tuberculosis that methionine also mediates antagonism of antifolates through affecting synthesis of folate precursors.37,136 Beyond the problem of intrinsic resistance, sulfonamides show limited activity against intracellular bacilli, suggesting that it may be difficult to achieve therapeutically relevant concentrations needed to target organisms like M. tuberculosis in their in vivo niche.137
Like SMX, TMP resistance can occur by a variety of mechanisms including intrinsic resistance, target modification or overexpression, and acquisition of mobile genetic elements (Fig. 7). Many bacterial species are naturally resistant to therapeutically relevant concentrations of TMP including M. tuberculosis, Bacteroides species, Clostridium species, and Neisseria species.138,139 In the case of M. tuberculosis, it was found that the native FolA Tyr100 residue was responsible for the weak binding of TMP.140 Modification of Tyr100 to phenylalanine resulted in increased affinity for TMP.140 It has been reported that some strains of E. coli, S. aureus, and H. influenzae harbor spontaneous mutations in the promoter region of folA that confer resistance through increased expression of FolA.138,141,142 In addition, it has been shown that TMP resistance in S. aureus, S. pneumonia and H. influenzae can arise from mutations that result in a single amino acid substitutions in FolA.141,143,144 The mechanism that is primarily associated with TMP resistance in both Gram-positive and Gram-negative bacteria involves acquisition of mobile genetic elements encoding variants of FolA (encoded by dhfr or more recently dfr).145,146 To date there are over 30 different mobile genetic elements encoding unique dfr genes and are classified as either type I or type II variants.147 The type I variants share sequence similarity with bacterial folA and are typically located on the site-specific integron Tn7.148 In contrast, type II variants are often found on conjugal R plasmids and that lack discernable sequence similarity to folA.149
Resistance to PAS in M. tuberculosis arises exclusively through occurrence of spontaneous mutations in genes related to folate synthesis and metabolism. Most frequently, PAS resistance mutations are identified in folC, ribD and thyA.115,116,150,151 Mutations in folC are the most prevalent mutations identified in PAS resistant clinical isolates.115,116,150 folC mutations typically map to positions corresponding to substrate binding and nucleoside binding pockets of the ATP domain in FolC.116,150 Both of these positions are essential for proper enzymatic function. Purified recombinant FolC variants from resistant strains were found to have reduced enzymatic activity (10 to 20% of wild type activity).116 When these variants were analyzed for the ability to glutamylate hydroxy-DHP, no detectable hydroxy-DHF was observed.116 Similarly, PAS resistant M. tuberculosis folC mutant strains were found to produce substantially less hydroxy-DHF than the wild type control.115 Thus, folC-linked PAS resistance occurs via impaired synthesis of hydroxy-DHF.
Another common PAS resistance mechanism involves loss-of-function mutations in thyA, encoding a non-essential thymidylate synthase (ThyA) in M. tuberculosis that is responsible for the 5,10-methylene-tetrahydrofolate dependent conversion of dUMP to dTMP.116,150,151 When functional, ThyA releases DHF that must be re-reduced to be utilized in folate metabolism. M. tuberculosis encodes an alternate thymidylate synthase (ThyX, encoded by thyX) that regenerates THF from 5,10-methylene-THF following catalysis.152 Thus, in contrast to ThyA, ThyX places limited demand of FolA for maintaining reduced folate pools. In contrast to other pathogens that do not encode a ThyX ortholog and are attenuated upon mutational inactivation of thyA,153,154 M. tuberculosis thyA loss-of-function mutants are not compromised for fitness growth and survival in vivo.
To date, few folA missense mutations associated with PAS resistance have been reported.152 Numerous studies have reported that alterations in the FolA active site can be highly deleterious to the overall enzymatic function. Thus, it could be rare for folA point mutations to confer resistance to PAS without compromising FolA enzymatic activity. One paper recently described five independent clinical PAS resistant isolates containing a deletion of the folA–thyA coding sequence.155 These strains were 26 times more resistant to PAS than susceptible control strains. Unlike many bacteria, M. tuberculosis does not encode a FolM ortholog that would be expected to compensate for loss of FolA activity. Instead, this compensation likely comes from weak DHF reductase activity that can be catalyzed by the 5-amino-6-(5-phosphoribosylamino)uracil reductase RibD that normally functions in riboflavin synthesis.72,116 Consistent with this model, ribD promoter mutations have been described that confer PAS resistance and were shown to render folA as non-essential in M. tuberculosis.116,150
Like TMP, PAS is prone to exclusion from the cytoplasm by the action of efflux pumps (Fig. 7). Overexpression of the major facilitator superfamily protein Tap (Rv1258c) in Mycobacterium bovis BCG was found to confer resistance to PAS.156 Furthermore, overexpression of Tap in M. tuberculosis, M. fortuitum and M. bovis BCG was found to confer resistance to a variety of other antimicrobial agents that do not target folate biosynthesis including streptomycin, vancomycin, and tetracycline.157–159
In humans resistance to MTX can arise and can lead to treatment failure. Several MTX resistance mechanisms have been described to date. One common mechanism for acquired MTX resistance stems from a defect in transport of the drug into target cells and is frequently observed in the context of acute lymphocytic leukemia.160 One of the most frequently observed MTX resistance mechanism associated with acute non-lymphoblastic leukemia involves impaired polyglutamylation, a process that is normally associated with cytosolic retention of folates.92,161–163 To overcome this resistance mechanism, analogs of MTX that show better intracellular retention independent of polyglutamylation, such as pralatrexate, have been employed164 other mammalian antifolate resistance mechanisms involve overexpression and missense mutations in DHFR.165,166 Interestingly, increased DHFR expression has been linked to mutations in p53 (a tumor suppressor gene), E2F (a transcription factor in cell cycle regulation), and genes that are involved in regulation of thymidylate synthase.166–168 Similar to that which is observed for PAS resistance in M. tuberculosis, DHFR active site mutations that confer MTX resistance are rare but have been observed in the laboratory.169,170
7. Conclusions and prospective
Antimicrobial agents and cancer chemotherapeutics have transformed treatment of disease in humans. With the critical role of folate as a cofactor in synthesis of DNA, RNA, protein and other essential cellular metabolites, folate biosynthesis is a highly attractive target for drug discovery. There has been a renewed interest in targeting the folate biosynthesis pathway for treating infectious diseases and cancer due to resistance to other chemotherapeutics. Selective inhibition of mammalian or bacterial enzymes is an achievable goal due to fundamental differences in microbial and mammalian pathways for synthesis and utilization of this cofactor. Even structural differences in bacterial enzymes within the folate biosynthesis pathway allow for selective bacterial targeting and facilitate development of inhibitors with fewer off target effects on the microbiome and the human host.
Some outstanding questions exist regarding the selectivity of antifolates. Why are some bacteria naturally resistant to certain FolA inhibitors? For instance, shortly after introduction of TMP into clinic use it was noted that several bacteria, especially Gram-negative enteric bacteria, showed intrinsic resistance. Further, the antitubercular FolA inhibitor PAS, is exquisitely selective for M. tuberculosis. Since TMP and PAS are not naturally occurring molecules what was the basis for emergence of distinct FolA structures and can these distinct features be exploited for selective targeting of microbes? Understanding the mechanisms that drive susceptibility and resistance could allow for design of highly selective next generation inhibitors.
With the increasing interest in the association between commensal microbiota and human health, it would be interesting to understand the consequences of antifolate treatment on the gut microbiota. For instance, in humans, bacteria of the large intestine are an important source of folate. During antifolate treatment, what are the consequences of decreased folate production on colonic enterocytes? Recent studies have found that both an increase and decrease in folate production in the colon are linked to colorectal cancer.171,172 Conversely, since there is interplay between bacteria found in the gastrointestinal tract, mucosal immunity and intestinal enterocytes, what are the consequences of antifolate treatment on this dynamic relationship?
Rapid evolution of antimicrobial drug resistance continues to threaten global healthcare. Resistance to antifolates has been broadly observed during cancer chemotherapy and in treating microbial infections. Our review highlights the recent understanding of the mechanisms that govern susceptibility and resistance. Despite the increasing scourge of resistance the folate biosynthesis pathway provides novel selective avenues of chemotherapeutic drug discovery.
Conflicts of interest
The authors declare no competing interest.
Acknowledgments
This work was supported by startup funds from the University of Minnesota, an Academic Health Center Faculty Research Development Program award and NIH grant AI123146 to ADB, and a University of Minnesota Doctoral Dissertation Fellowship awarded to SLK.
References
- Wen X., Wang J.-S., Backman J. T., Laitila J., Neuvonen P. J. Drug Metab. Dispos. 2002;30:631–635. doi: 10.1124/dmd.30.6.631. [DOI] [PubMed] [Google Scholar]
- Strevel E. L., Kuper A., Gold W. L. Lancet Infect. Dis. 2006;6:178–182. doi: 10.1016/S1473-3099(06)70414-5. [DOI] [PubMed] [Google Scholar]
- Keisu M., Wiholm B.-E., Palmblad J. J. Intern. Med. 1990;228:353–360. doi: 10.1111/j.1365-2796.1990.tb00245.x. [DOI] [PubMed] [Google Scholar]
- Smilack J. Mayo Clin. Proc. 1999;74:730–734. doi: 10.4065/74.7.730. [DOI] [PubMed] [Google Scholar]
- Viswanathan V. K., Green J. M., Nichols B. P. J. Bacteriol. 1995;177:5918–5923. doi: 10.1128/jb.177.20.5918-5923.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux B., Walsh C. T. Biochemistry. 1993;32:3763–3768. doi: 10.1021/bi00065a031. [DOI] [PubMed] [Google Scholar]
- Slock J., Stahly D. P., Han C. Y., Six E. W., Crawford I. P. J. Bacteriol. 1990;172:7211–7226. doi: 10.1128/jb.172.12.7211-7226.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan J. B., Goncharoff P., Seibold A. M., Nichols B. P. Mol. Biol. Evol. 1984;1:456–472. doi: 10.1093/oxfordjournals.molbev.a040331. [DOI] [PubMed] [Google Scholar]
- Knöchel T., Ivens A., Hester G., Gonzalez A., Bauerle R., Wilmanns M., Kirschner K., Jansonius J. N. Proc. Natl. Acad. Sci. U. S. A. 1999;96:9479–9484. doi: 10.1073/pnas.96.17.9479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugumbate G., Abrahams K. A., Cox J. A. G., Papadatos G., Van Westen G., Leli J., Calus S. T., Loman N. J., Ballell L. PLoS One. 2015;10:1–17. doi: 10.1371/journal.pone.0121492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morollo A. A., Eck M. J. Nat. Struct. Biol. 2001;8:243–247. doi: 10.1038/84988. [DOI] [PubMed] [Google Scholar]
- Green J. M., Nichols B. P. J. Biol. Chem. 1991;266:12971–12975. [PubMed] [Google Scholar]
- Green J. M., Merkel W. K., Nichols B. P. J. Bacteriol. 1992;174:5317–5323. doi: 10.1128/jb.174.16.5317-5323.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiede J. M., Kordus S. L., Turman B. J., Buonomo J. A., Aldrich C. C., Minato Y., Baughn A. D. Sci. Rep. 2016;6:38083. doi: 10.1038/srep38083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller S., Nicholson G., Drahl C., Sorensen E., Fiedler H.-P., Süssmuth R. D. J. Antibiot. 2007;60:391. doi: 10.1038/ja.2007.54. [DOI] [PubMed] [Google Scholar]
- Keller S., Schadt H. S., Ortel I., Süssmuth R. D. Angew. Chem., Int. Ed. 2007;46:8284–8286. doi: 10.1002/anie.200701836. [DOI] [PubMed] [Google Scholar]
- Freundlich J. S., Lalgondar M., Wei J.-R., Swanson S., Sorensen E. J., Rubin E. J., Sacchettini J. C. Tuberculosis. 2010;90:298–300. doi: 10.1016/j.tube.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bihelovic F., Karadzic I., Matovic R., Saicic R. N. Org. Biomol. Chem. 2013;11:5413–5424. doi: 10.1039/c3ob40692j. [DOI] [PubMed] [Google Scholar]
- Matovic R., Bihelovic F., Gruden-Pavlovic M., Saicic R. N. Org. Biomol. Chem. 2014;12:7682–7685. doi: 10.1039/c4ob01436g. [DOI] [PubMed] [Google Scholar]
- Wang J. F., Dai H. Q., Wei Y. L., Zhu H. J., Yan Y. M., Wang Y. H., Long C. L., Zhong H. M., Zhang L. X., Cheng Y. X. Chem. Biodiversity. 2010;7:2046–2053. doi: 10.1002/cbdv.201000072. [DOI] [PubMed] [Google Scholar]
- Riedlinger J., Reicke A., Zahner H., Krismer B., Bull A. T., Maldonado L. A., Ward A. C., Goodfellow M., Bister B., Bischoff D., Sussmuth R. D., Fiedler H.-P. J. Antibiot. 2004;57:271–279. doi: 10.7164/antibiotics.57.271. [DOI] [PubMed] [Google Scholar]
- Zlitni S., Ferruccio L. F., Brown E. D. Nat. Chem. Biol. 2013;9:796–804. doi: 10.1038/nchembio.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappock T. J., Caradonna J. P. Chem. Rev. 1996;96:2659–2756. doi: 10.1021/cr9402034. [DOI] [PubMed] [Google Scholar]
- Tanaka Y., Nakagawa N., Kuramitsu S., Yokoyama S., Masui R. J. Biochem. 2005;138:263–275. doi: 10.1093/jb/mvi120. [DOI] [PubMed] [Google Scholar]
- Auerbach G., Herrmann A., Bracher A., Bader G., Gutlich M., Fischer M., Neukamm M., Garrido-Franco M., Richardson J., Nar H., Huber R., Bacher A. Proc. Natl. Acad. Sci. U. S. A. 2000;97:13567–13572. doi: 10.1073/pnas.240463497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gräwert T., Fischer M., Bacher A. IUBMB Life. 2013;65:310–322. doi: 10.1002/iub.1153. [DOI] [PubMed] [Google Scholar]
- Blau N., Niederwieser A. Biochim. Biophys. Acta. 1986;880:26–31. doi: 10.1016/0304-4165(86)90115-7. [DOI] [PubMed] [Google Scholar]
- Sankaran B., Bonnett S. A., Shah K., Gabriel S., Reddy R., Schimmel P., Rodionov D. A., de Crecy-Lagard V., Helmann J. D., Iwata-Reuyl D., Swairjo M. A. J. Bacteriol. 2009;191:6936–6949. doi: 10.1128/JB.00287-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foor F., Brown G. M. J. Biol. Chem. 1975;250:3545–3551. [PubMed] [Google Scholar]
- Kaiser J., Schramek N., Eberhardt S., Püttmer S., Schuster M., Bacher A. Eur. J. Biochem. 2002;269:5264–5270. doi: 10.1046/j.1432-1033.2002.03239.x. [DOI] [PubMed] [Google Scholar]
- Bacher A., Eberhardt S., Kis K., Richter G. Annu. Rev. Nutr. 2000;20:153–167. doi: 10.1146/annurev.nutr.20.1.153. [DOI] [PubMed] [Google Scholar]
- Klaus S. M. J., Wegkamp A., Sybesma W., Hugenholtz J., Gregory J. F., Hanson A. D. J. Biol. Chem. 2005;280:5274–5280. doi: 10.1074/jbc.M413759200. [DOI] [PubMed] [Google Scholar]
- Gabelli S. B., Bianchet M. A., Xu W., Dunn C. A., Niu Z. D., Amzel L. M., Bessman M. J. Structure. 2007;15:1014–1022. doi: 10.1016/j.str.2007.06.018. [DOI] [PubMed] [Google Scholar]
- O'Handley S. F., Frick D. N., Dunn C. A., Bessman M. J. J. Biol. Chem. 1998;273:3192–3197. doi: 10.1074/jbc.273.6.3192. [DOI] [PubMed] [Google Scholar]
- Hill S. E., Nguyen E., Ukachukwu C. U., Freeman D. M., Quirk S., Lieberman R. L. PLoS One. 2017;7:1–16. doi: 10.1371/journal.pone.0180241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li K., Li T., Yang S.-S., Wang X.-D., Gao L., Wang R.-Q., Gu J., Zhang X. E., Deng J. Y. Antimicrob. Agents Chemother. 2017;61:1–11. doi: 10.1128/AAC.02378-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato Y., Dawadi S., Kordus S. L., Sivanandam A., Aldrich C. C., Baughn A. D. Nat. Commun. 2018;9:1–6. doi: 10.1038/s41467-018-03447-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher R., Therisod M. ACS Med. Chem. Lett. 2010;1:101–104. doi: 10.1021/ml100017c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk J., Li Y., Gan J., Yan H., Ji X. J. Mol. Biol. 2007;368:161–169. doi: 10.1016/j.jmb.2007.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., Li Y., Yan H. Biochemistry. 2006;45:15232–15239. doi: 10.1021/bi060949j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garçon A., Levy C., Derrick J. P. J. Mol. Biol. 2006;360:644–653. doi: 10.1016/j.jmb.2006.05.038. [DOI] [PubMed] [Google Scholar]
- Lopez P., Lacks S. A. J. Bacteriol. 1993;175:2214–2220. doi: 10.1128/jb.175.8.2214-2220.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe F., Ballantine S. P., Delves C. J. Eur. J. Biochem. 1993;458:449–458. doi: 10.1111/j.1432-1033.1993.tb18163.x. [DOI] [PubMed] [Google Scholar]
- Pemble C. W., Mehta P. K., Mehra S., Li Z., Nourse A., Lee R. E., White S. W. PLoS One. 2010;5:e14165. doi: 10.1371/journal.pone.0014165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerman M., Tolman R., Morman H., Graham D., Rogers E. J. Med. Chem. 1977;20:1213–1215. doi: 10.1021/jm00219a021. [DOI] [PubMed] [Google Scholar]
- Sanders W. J., Nienaber V. L., Lerner C. G., McCall J. O., Merrick S. M., Swanson S. J., Harlan J. E., Stoll V. S., Stamper G. F., Betz S. F., Condroski K. R., Meadows R. P., Severin J. M., Walter K. A., Magdalinos P., Jakob C. G., Wagner R., Beutel B. A. J. Med. Chem. 2004;47:1709–1718. doi: 10.1021/jm030497y. [DOI] [PubMed] [Google Scholar]
- Griffin J. E., Gawronski J. D., DeJesus M. A., Ioerger T. R., Akerley B. J., Sassetti C. M. PLoS Pathog. 2011;7:e1002251. doi: 10.1371/journal.ppat.1002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaszczyk J., Shi G., Yan H., Ji X. Structure. 2000;8:1049–1058. doi: 10.1016/s0969-2126(00)00502-5. [DOI] [PubMed] [Google Scholar]
- Shi G., Shaw G., Liang Y. H., Subburaman P., Li Y., Wu Y., Yan H., Ji X. Bioorg. Med. Chem. 2012;20:47–57. doi: 10.1016/j.bmc.2011.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parang K., Cole P. A. Pharmacol. Ther. 2002;93:145–157. doi: 10.1016/s0163-7258(02)00184-5. [DOI] [PubMed] [Google Scholar]
- Shi G., Shaw G., Li Y., Wu Y., Yan H., Ji X. Bioorg. Med. Chem. 2012;20:4303–4309. doi: 10.1016/j.bmc.2012.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G., Shaw G., Li Y., Wu Y., Yan H., Ji X. Bioorg. Med. Chem. 2012;20:4303–4309. doi: 10.1016/j.bmc.2012.05.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra S., Barlow N., Dolezal O., Hattarki M. K., Newman J., Peat T. S., Graham B., Swarbrick J. D. PLoS One. 2013;8:e59535. doi: 10.1371/journal.pone.0059535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra S., Dolezal O., Collins B. M., Newman J., Simpson J. S., Macreadie I. G., Fernley R., Peat T. S., Swarbrick J. D. PLoS One. 2012;7:e29444. doi: 10.1371/journal.pone.0029444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne C. Antibiotics. 2014;3:1–28. doi: 10.3390/antibiotics3010001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer A. C., Kishony R. Nat. Commun. 2014;5:1–8. doi: 10.1038/ncomms5296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevener K. E., Yun M., Qi J., Kerr I. D., Babaoglu K., Julian G., Balakrishna K., White S. W., Lee R. E. J. Med. Chem. 2011;53:166–177. doi: 10.1021/jm900861d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Shadrick W. R., Wallace M. J., Wu Y., Griffith E. C., Qi J., Yun M.-K., White S. W., Lee R. E. Bioorg. Med. Chem. Lett. 2016;26:3950–3954. doi: 10.1016/j.bmcl.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis M. L., Lee M. D., Harjani J. R., Ahmed M., DeBono A. J., Pitcher N. P., Wang Z.-C., Chhabra S., Barlow N., Rahmani R., Cleary B., Dolezal O., Hattarki M., Aurelio L., Shonberg J., Graham B., Peat T. S., Baell J. B., Swarbrick J. D. Chem. – Eur. J. 2018;24:1922–1930. doi: 10.1002/chem.201704730. [DOI] [PubMed] [Google Scholar]
- Hammoudeh D. I., Zhao Y., White S. W., Lee R. E. Future Med. Chem. 2013;5:1331–1340. doi: 10.4155/fmc.13.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun M.-K., Wu Y., Li Z., Zhoa Y., Waddell M. B., Ferreira A. M., Lee R. E., Bashford D., White S. W. Science. 2012;335:1110–1115. doi: 10.1126/science.1214641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baca A. M., Sirawaraporn R., Turley S., Sirawaraporn W., Hol W. G. J. J. Mol. Biol. 2000;302:1193–1212. doi: 10.1006/jmbi.2000.4094. [DOI] [PubMed] [Google Scholar]
- Levy C., Minnis D., Derrick J. P. Biochem. J. 2008;412:379–388. doi: 10.1042/BJ20071598. [DOI] [PubMed] [Google Scholar]
- Babaoglu K., Qi J., Lee R. E., White S. W. Structure. 2004;12:1705–1717. doi: 10.1016/j.str.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Zhao Y., Hammoudeh D., Yun M. K., Qi J., White S. W., Lee R. E. ChemMedChem. 2012;7:861–870. doi: 10.1002/cmdc.201200049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y., Shadrick W. R., Wallace M. J., Wu Y., Griffith E. C., Qi J., Yun M. K., White S. W., Lee R. E. Bioorg. Med. Chem. Lett. 2016;26:3950–3954. doi: 10.1016/j.bmcl.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammoudeh D. I., Daté M., Yun M. K., Zhang W., Boyd V. A., Viacava Follis A., Griffith E., Lee R. E., Bashford D., White S. W. ACS Chem. Biol. 2014;9:1294–1302. doi: 10.1021/cb500038g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S., Jan Y.-H., Mishin V., Richardson J. R., Hossain M. M., Heindel N. D., Heck D. E., Laskin D. L., Laskin J. D. J. Pharmacol. Exp. Ther. 2015;352:529–540. doi: 10.1124/jpet.114.221572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haruki H., Pedersen M. G., Gorska K. I., Pojer F., Johnsson K. Science. 2013;340:987–991. doi: 10.1126/science.1232972. [DOI] [PubMed] [Google Scholar]
- Bognar A. L., Osborne C., Shane B., Singer S. C., Ferone R. J. Biol. Chem. 1985;260:5625–5630. [PubMed] [Google Scholar]
- Shane B. J. Biol. Chem. 1979;255:5655–5662. [PubMed] [Google Scholar]
- Salcedo E., Cortese J. F., Plowe C. V., Sims P. F. G., Hyde J. E. Mol. Biochem. Parasitol. 2001;112:239–252. doi: 10.1016/s0166-6851(00)00370-4. [DOI] [PubMed] [Google Scholar]
- Watson M., Liu J.-W., Ollis D. Rev. Geophys. 2007;274:2661–2671. doi: 10.1111/j.1742-4658.2007.05801.x. [DOI] [PubMed] [Google Scholar]
- Mathieu M., Debousker G., Vincent S., Viviani F., Bamas-Jacques N., Mikol V. J. Biol. Chem. 2005;280:18916–18922. doi: 10.1074/jbc.M413799200. [DOI] [PubMed] [Google Scholar]
- Young P., Smith C., Metcalf P., Baker E. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2008;64:745–753. doi: 10.1107/S0907444908012262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Y., Khanam N., Tsaksis Y., Shi X., Lu Q., Bognar A. L. Biochemistry. 2008;47:2388–2396. doi: 10.1021/bi701670y. [DOI] [PubMed] [Google Scholar]
- Sun X., Cross J. A., Bognar A. L., Baker E. N., Smith C. A. J. Mol. Biol. 2001;310:1067–1078. doi: 10.1006/jmbi.2001.4815. [DOI] [PubMed] [Google Scholar]
- Smith C. A., Cross J. A., Bognar A. L., Sun X. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006;62:548–558. doi: 10.1107/S0907444906009796. [DOI] [PubMed] [Google Scholar]
- Sheng Y., Khanam N., Tsaksis Y., Shi X., Lu Q., Bognar A. L. Biochemistry. 2008;47:2388–2396. doi: 10.1021/bi701670y. [DOI] [PubMed] [Google Scholar]
- Sun X., Cross J. A., Bognar A. L., Baker E. N., Smith C. A. J. Mol. Biol. 2001;310:1067–1078. doi: 10.1006/jmbi.2001.4815. [DOI] [PubMed] [Google Scholar]
- Smith C. A., Cross J. A., Bognar A. L., Sun X. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2006;62:548–558. doi: 10.1107/S0907444906009796. [DOI] [PubMed] [Google Scholar]
- Banerjee R. V., Shane B., McGuire J. J., Coward J. K. Biochemistry. 1988;27:9062–9070. doi: 10.1021/bi00425a027. [DOI] [PubMed] [Google Scholar]
- Wang P., Wang Q., Yang Y., Coward J. K., Nzila A., Sims P. F. G., Hyde J. E. Mol. Biochem. Parasitol. 2010;172:41–51. doi: 10.1016/j.molbiopara.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell E. E., Foster P. G., Foster L. M. J. Bacteriol. 1988;170:3040–3045. doi: 10.1128/jb.170.7.3040-3045.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giladi M., Altman-Price N., Levin I., Levy L., Mevarech M. J. Bacteriol. 2003;185:7015–7018. doi: 10.1128/JB.185.23.7015-7018.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnell J. R., Dyson H. J., Wright P. E. Annu. Rev. Biophys. Biomol. Struct. 2004;33:119–140. doi: 10.1146/annurev.biophys.33.110502.133613. [DOI] [PubMed] [Google Scholar]
- Fierke C. A., Johnson K. A., Benkovic S. J. Biochemistry. 1987;26:4085–4092. doi: 10.1021/bi00387a052. [DOI] [PubMed] [Google Scholar]
- Cannon W. R., Garrison B. J., Benkovic S. J. J. Am. Chem. Soc. 1997;119:2386–2395. [Google Scholar]
- Chen Y.-Q., Kraut J., Blakley R. L., Callender R. Biochemistry. 1994;33:7021–7026. doi: 10.1021/bi00189a001. [DOI] [PubMed] [Google Scholar]
- Cronstein B. N. Pharmacol. Rev. 2005;57:163–172. doi: 10.1124/pr.57.2.3. [DOI] [PubMed] [Google Scholar]
- Sudimack J., Lee R. J. Adv. Drug Delivery Rev. 2000;41:147–162. doi: 10.1016/s0169-409x(99)00062-9. [DOI] [PubMed] [Google Scholar]
- Visentin M., Unal E. S., Zhao R., Goldman I. D. Cancer Chemother. Pharmacol. 2013;72:597–606. doi: 10.1007/s00280-013-2231-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams J. W., Morrison J. F., Duggleby R. G. Biochemistry. 1979;18:2567–2573. doi: 10.1021/bi00579a021. [DOI] [PubMed] [Google Scholar]
- Morrison J. F., Walsh C. T. Adv. Enzymol. Relat. Areas Mol. Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- Bennett B., Langan P., Coates L., Mustyakimov M., Schoenborn B., Howell E. E., Dealwis C. Proc. Natl. Acad. Sci. U. S. A. 2006;103:18493–18498. doi: 10.1073/pnas.0604977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Reyes V. M., Kraut J. Biochemistry. 1996;35:7012–7020. doi: 10.1021/bi960028g. [DOI] [PubMed] [Google Scholar]
- Sawaya M. R., Kraut J. Biochemistry. 1997;36:586–603. doi: 10.1021/bi962337c. [DOI] [PubMed] [Google Scholar]
- Kok M., Michea-Hamzehpour M., Plesiat P., Gotoh N., Nishino T., Curty L. K., Pechere J. C., Köhler T., Kok M., Michea-Hamzehpour M., Plesiat P., Gotoh N., Nishino T., Curty L. K., Pechere J. C. Antimicrob. Agents Chemother. 1996;40:2288–2290. doi: 10.1128/aac.40.10.2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd J. L., Smith K. P., Kumar S. H., Floyd J. T., Varela M. F. Antimicrob. Agents Chemother. 2010;54:5406–5412. doi: 10.1128/AAC.00580-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchall J. J., Hitchings G. H. Mol. Pharmacol. 1965;1:126–136. [PubMed] [Google Scholar]
- Matthews D. A., Bolin J. T., Burridge J. M., Filman D. J., Volz K. W., Kraut J. J. Biol. Chem. 1985;260:392–399. [PubMed] [Google Scholar]
- Sincak C. A., Schmidt J. M. Ann. Pharmacother. 2009;43:1107–1114. doi: 10.1345/aph.1L167. [DOI] [PubMed] [Google Scholar]
- Aschhoff H. S., Vergin H. J. Antimicrob. Chemother. 1979;5:19–25. doi: 10.1093/jac/5.supplement_b.19. [DOI] [PubMed] [Google Scholar]
- Oefner C., Bandera M., Haldimann A., Laue H., Schulz H., Mukhija S., Parisi S., Weiss L., Lociuro S., Dale G. E. J. Antimicrob. Chemother. 2009;63:687–698. doi: 10.1093/jac/dkp024. [DOI] [PubMed] [Google Scholar]
- Carrington H. C., Crowther A. F., Davey D. G., Levi A. A., Rose F. Nature. 1951;168:1080. doi: 10.1038/1681080a0. [DOI] [PubMed] [Google Scholar]
- Desta Z., Zhao X., Shin J.-G., Flockhart D. A. Clin. Pharmacokinet. 2002;41:913–958. doi: 10.2165/00003088-200241120-00002. [DOI] [PubMed] [Google Scholar]
- Hitchings G. H., Elion G. B., Falco E. A., Russell P. B., VanderWerff H. Ann. N. Y. Acad. Sci. 1950;52:1318–1335. doi: 10.1111/j.1749-6632.1950.tb54032.x. [DOI] [PubMed] [Google Scholar]
- Hoekenga M. T. Am. J. Trop. Med. Hyg. 1954;3:833–836. doi: 10.4269/ajtmh.1954.3.833. [DOI] [PubMed] [Google Scholar]
- Canfield C. J., Milhous W. K., Ager A. L., Rossan R. N., Sweeney T. R. Am. J. Trop. Med. Hyg. 1993;49:121–126. doi: 10.4269/ajtmh.1993.49.121. [DOI] [PubMed] [Google Scholar]
- Gerum A., Ulmer J., Jacobus D., Sherman D., Sibley C. Antimicrob. Agents Chemother. 2002;46:3362–3369. doi: 10.1128/AAC.46.11.3362-3369.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar A., Guardia A., Colmenarejo G., Pérez E., Gonzalez R. R., Torres P., Calvo D., Gómez R. M., Ortega F., Jiménez E., Gabarro R. C., Rullás J., Ballell L., Sherman D. R. ACS Infect. Dis. 2015;1:604–614. doi: 10.1021/acsinfecdis.5b00063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHO, Who, Global report 2017, 2017.
- Youmans G. P., Raleigh G. W., Youmans A. S. J. Bacteriol. 1947;54:409–416. doi: 10.1128/jb.54.4.409-416.1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yegian D., Long R. T. J. Bacteriol. 1951;61:747–749. doi: 10.1128/jb.61.6.747-749.1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J., Rubin E. J., Bifani P., Mathys V., Lim V., Au M., Jang J., Nam J., Dick T., Walker J. R., Pethe K., Camacho L. R. J. Biol. Chem. 2013;288:23447–23456. doi: 10.1074/jbc.M113.475798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F., De Wang X., Erber L. N., Luo M., Guo A. Z., Yang S. S., Gu J., Turman B. J., Gao Y. R., Li D. F., Cui Z. Q., Zhang Z. P., Bi L. J., Baughn A. D., Zhang X. E., Deng J. Y. Antimicrob. Agents Chemother. 2014;58:1479–1487. doi: 10.1128/AAC.01775-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S., Gruber T., Barry C., Boshoff H., Rhee K. Science. 2013;339:88–91. doi: 10.1126/science.1228980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawadi S., Kordus S. L., Baughn A. D., Aldrich C. C. Org. Lett. 2017;19:5220–5223. doi: 10.1021/acs.orglett.7b02487. [DOI] [PubMed] [Google Scholar]
- Sievers O. Sven. Lakartidn. 1946;204:2041. [Google Scholar]
- Ivanovics G., Csabi I., Diczfalusy E. Hung. Acta Physiol. 1948;1:171–179. [PubMed] [Google Scholar]
- Wyss O. J. Bacteriol. 1943;52:346. doi: 10.1128/jb.46.5.483-484.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragaz L. Schweiz. Med. Wochenschr. 1948;78:332–334. [PubMed] [Google Scholar]
- Tobie W., Jones M. J. Bacteriol. 1949;57:573. doi: 10.1128/jb.57.5.573-573.1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wormser G., Keusch G., Heel R. Drugs. 1982;24:459–518. doi: 10.2165/00003495-198224060-00002. [DOI] [PubMed] [Google Scholar]
- Bushby M., Hitchings G. H. Br. J. Pharmacol. Chemother. 1968;90:72–90. doi: 10.1111/j.1476-5381.1968.tb00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey R. J. J. Theor. Biol. 1978;74:411–437. doi: 10.1016/0022-5193(78)90223-0. [DOI] [PubMed] [Google Scholar]
- Padayachee T., Klugman K. P. Antimicrob. Agents Chemother. 1999;43:2225–2230. doi: 10.1128/aac.43.9.2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swedberg G., Ringertz S., Sköld O. Antimicrob. Agents Chemother. 1998;42:1062–1067. doi: 10.1128/aac.42.5.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sköld O. Drug Resist. Updates. 2000;3:155–160. doi: 10.1054/drup.2000.0146. [DOI] [PubMed] [Google Scholar]
- Akiba T., Koyama K., Ishiki Y., Kimura S., Fukushima T. Jpn. J. Microbiol. 1960;4:219–227. doi: 10.1111/j.1348-0421.1960.tb00170.x. [DOI] [PubMed] [Google Scholar]
- Swedberg G., Skold O. J. Bacteriol. 1980;142:1–7. doi: 10.1128/jb.142.1.1-7.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H. W., Lim J., Kim S., Kim J., Kwon G. C., Koo S. H. J. Microbiol. Biotechnol. 2014;25:137–142. doi: 10.4014/jmb.1409.09041. [DOI] [PubMed] [Google Scholar]
- Sánchez-Osuna M., Cortés P., Barbé J., Erill I. Front. Microbiol. 2019;9:1–15. doi: 10.3389/fmicb.2018.03332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry R. J. Am. Chem. Soc. 1944;66:459–464. [Google Scholar]
- Nopponpunth V., Sirawaraporn W., Greene P. J., Santi D. V. J. Bacteriol. 1999;181:6814–6821. doi: 10.1128/jb.181.21.6814-6821.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe M. D., Kordus S. L., Cole M. S., Bauman A. A., Aldrich C. C., Baughn A. D., Minato Y. Front. Cell. Infect. Microbiol. 2018;8:399. doi: 10.3389/fcimb.2018.00399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsman L. D., Schön T., Simonsson U. S. H., Bruchfeld J., Larsson M., Juréen P., Sturegård E., Giske C. G., Ängebyh K. Antimicrob. Agents Chemother. 2014;58:7557–7559. doi: 10.1128/AAC.02995-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huovinen P., Sundstrom L., Swedberg G., Skold O. Antimicrob. Agents Chemother. 1995;39:279–289. doi: 10.1128/aac.39.2.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato Y., Thiede J. M., Kordus S. L., Mcklveen E. J., Turman B. J., Baughn D. Antimicrob. Agents Chemother. 2015;59:5097–5106. doi: 10.1128/AAC.00647-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias M. V. B., Tyrakis P., Domingues R. R., Leme A. F. P., Blundell T. L. Structure. 2014;22:94–103. doi: 10.1016/j.str.2013.09.022. [DOI] [PubMed] [Google Scholar]
- de Groot R., Sluijter M., Bruyn A., Smith A. L., Hermans P. W. M. Antimicrob. Agents Chemother. 1996;40:2131–2136. doi: 10.1128/aac.40.9.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurjadi D., Olalekan A. O., Layer F., Shittu A. O., Alabi A., Ghebremedhin B., Schaumburg F., Hofmann-eifler J., Van Genderen P. J. J. J. Antimicrob. Chemother. 2014;27:2361–2368. doi: 10.1093/jac/dku174. [DOI] [PubMed] [Google Scholar]
- Dale G. E., Broger C., D'Arcy A., Hartman P. G., DeHoogt R., Jolidon S., Kompis I., Labhardt A. M., Langen H., Locher H., Page M. G. P., Stüber D., Then R. L., Wipf B., Oefner C. J. Mol. Biol. 1997;266:23–30. doi: 10.1006/jmbi.1996.0770. [DOI] [PubMed] [Google Scholar]
- Pikis A., Donkersloot J., Rodriguez W., Keith J. J. Infect. Dis. 1998;178:700–706. doi: 10.1086/515371. [DOI] [PubMed] [Google Scholar]
- Reeve S. M., Scocchera E. W., G-Dayanadan N., Keshipeddy S., Krucinska J., Hajian B., Ferreira J., Aeschlimann J., Wright D. L., Nailor M., Anderson A. C. Cell Chem. Biol. 2016;23:1458–1467. doi: 10.1016/j.chembiol.2016.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann R., van der Linden M., Chhatwal G. S., Nitsche-Schmitz D. P. Antimicrob. Agents Chemother. 2014;58:2281–2288. doi: 10.1128/AAC.02282-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seputiene V., Povilonis J., Ruz M., Pavilonis A., Suziedeliene E. J. Med. Microbiol. 2010;59:315–322. doi: 10.1099/jmm.0.015008-0. [DOI] [PubMed] [Google Scholar]
- Heikkila E., Sundstrom L., Skurnik M., Huovinen P. Antimicrob. Agents Chemother. 1991;35:1562–1569. doi: 10.1128/aac.35.8.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone D., Smith S. L. J. Biol. Chem. 1979;254:10857–10861. [PubMed] [Google Scholar]
- Zhang X., Liu L., Zhang Y., Dai G., Huang H., Jina Q. Antimicrob. Agents Chemother. 2015;59:1320–1324. doi: 10.1128/AAC.03695-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathys V., Wintjens R., Lefevre P., Bertout J., Singhal A., Kiass M., Kurepina N., Wang X. M., Mathema B., Baulard A., Kreiswirth B. N., Bifani P. Antimicrob. Agents Chemother. 2009;53:2100–2109. doi: 10.1128/AAC.01197-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllykallio H., Lipowski G., Leduc D., Filee J., Forterre P., Liebl U. Science. 2002;297:105–107. doi: 10.1126/science.1072113. [DOI] [PubMed] [Google Scholar]
- Kok M., Bühlmann E., Pechère J. C. Microbiology. 2001;147:727–733. doi: 10.1099/00221287-147-3-727. [DOI] [PubMed] [Google Scholar]
- Cersini A., Salvia A. M., Bernardini M. L. Infect. Immun. 1998;66:549–557. doi: 10.1128/iai.66.2.549-557.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradigaravand D., Grandjean L., Martinez E., Li H., Zheng J., Coronel J., Moore D. Antimicrob. Agents Chemother. 2016;60:3864–3867. doi: 10.1128/AAC.00253-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramón-García S., Mick V., Dainese E., Martín C., Thompson C. J., De Rossi E., Manganelli R., Aínsa J. A. Antimicrob. Agents Chemother. 2012;56:2074–2083. doi: 10.1128/AAC.05946-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aínsa J. A., Blokpoel M. C. J., Otal I., Young D. B., De Smet K. A. L., Martín C. J. Bacteriol. 1998;180:5836–5843. doi: 10.1128/jb.180.22.5836-5843.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramón-García S., Mick V., Dainese E., Martín C., Thompson C. J., De Rossi E., Manganelli R., Aínsa J. A. Antimicrob. Agents Chemother. 2012;56:2074–2083. doi: 10.1128/AAC.05946-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqi N., Das R., Pathak N., Banerjee S., Ahmed N., Katoch V. M., Hasnain S. E. Infection. 2004;32:109–111. doi: 10.1007/s15010-004-3097-x. [DOI] [PubMed] [Google Scholar]
- Trippett T., Schlemmer S., Elisseyeff Y., Goker E., Wachter M., Steinherz P., Tan C., Berman E., Wright J., Rosowsky A. Blood. 1992;80:1158–1163. [PubMed] [Google Scholar]
- Rhee M. S., Wang Y., Nair M. G., Galivan J. Cancer Res. 1993;53:2227–2230. [PubMed] [Google Scholar]
- Li W. W., Lin J. T., Tong W. P., Trippett T. M., Brennan M. F., Beitino J. R. Cancer Res. 1992;52:1434–1438. [PubMed] [Google Scholar]
- Lin J. T., Tong W. P., Trippett T. M., Niedzwiecki D., Tao Y., Tan C., Steinherz P., Schweitzer B. I., Bertino J. R. Leuk. Res. 1991;15:1191–1196. doi: 10.1016/0145-2126(91)90189-z. [DOI] [PubMed] [Google Scholar]
- Gonen N., Assaraf Y. G. Drug Resist. Updates. 2012;15:183–210. doi: 10.1016/j.drup.2012.07.002. [DOI] [PubMed] [Google Scholar]
- Schimke R. T. J. Biol. Chem. 1988;263:5989–5992. [PubMed] [Google Scholar]
- Göker E., Waltham M., Kheradpour A., Trippett T., Mazumdar M., Elisseyeff Y., Schnieders B., Steinherz P., Tan C., Berman E. Blood. 1995;86:677–684. [PubMed] [Google Scholar]
- Banerjee D., Mayer-Kuckuk P., Capiaux G., Budak-Alpdogan T., Gorlick R., Bertino J. R. Biochim. Biophys. Acta, Mol. Basis Dis. 2002;1587:164–173. doi: 10.1016/s0925-4439(02)00079-0. [DOI] [PubMed] [Google Scholar]
- Li W., Fan J., Hochhauser D., Banerjee D., Zielinski Z., Almasan A., Yin Y., Kelly R., Wahl G. M., Bertino J. R. Proc. Natl. Acad. Sci. U. S. A. 1995;92:10436–10440. doi: 10.1073/pnas.92.22.10436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srimatkandada S., Schweitzer B. I., Moroson B. A., Dube S., Bertino J. R. J. Biol. Chem. 1989;264:3524–3528. [PubMed] [Google Scholar]
- Ercikan-Abali E. A., Mineishi S., Tong Y., Nakahara S., Waltham M. C., Banerjee D., Chen W., Sadelain M., Bertino J. R. Cancer Res. 1996;56:4142–4145. [PubMed] [Google Scholar]
- Kok D. E., Steegenga W. T., Smid E. J., Zoetendal E. G., Ulrich C. M., Kampman E. Crit. Rev. Food Sci. Nutr. 2018;0:1–13. doi: 10.1080/10408398.2018.1522499. [DOI] [PubMed] [Google Scholar]
- Mason J. B., Tang S. Y. Mol. Aspects Med. 2017;53:73–79. doi: 10.1016/j.mam.2016.11.010. [DOI] [PubMed] [Google Scholar]