

Abstract
SALM1 (SALM (synaptic adhesion-like molecule), also known as LRFN2 (leucine rich repeat and fibronectin type III domain containing), is a postsynaptic density (PSD)-95-interacting synaptic adhesion molecule implicated in the regulation of NMDA receptor (NMDAR) clustering largely based on in vitro data, although its in vivo functions remain unclear. Here, we found that mice lacking SALM1/LRFN2 (Lrfn2−/− mice) show a normal density of excitatory synapses but altered excitatory synaptic function, including enhanced NMDAR-dependent synaptic transmission but suppressed NMDAR-dependent synaptic plasticity in the hippocampal CA1 region. Unexpectedly, SALM1 expression was detected in both glutamatergic and GABAergic neurons and Lrfn2−/− CA1 pyramidal neurons showed decreases in the density of inhibitory synapses and the frequency of spontaneous inhibitory synaptic transmission. Behaviorally, ultrasonic vocalization was suppressed in Lrfn2−/− pups separated from their mothers and acoustic startle was enhanced, but locomotion, anxiety-like behavior, social interaction, repetitive behaviors, and learning and memory were largely normal in adult male Lrfn2−/− mice. These results suggest that SALM1/LRFN2 regulates excitatory synapse function, inhibitory synapse development, and social communication and startle behaviors in mice.
SIGNIFICANCE STATEMENT Synaptic adhesion molecules regulate synapse development and function, which govern neural circuit and brain functions. The SALM/LRFN (synaptic adhesion-like molecule/leucine rich repeat and fibronectin type III domain containing) family of synaptic adhesion proteins consists of five known members for which the in vivo functions are largely unknown. Here, we characterized mice lacking SALM1/LRFN2 (SALM1 KO) known to associate with NMDA receptors (NMDARs) and found that these mice showed altered NMDAR-dependent synaptic transmission and plasticity, as expected, but unexpectedly also exhibited suppressed inhibitory synapse development and synaptic transmission. Behaviorally, SALM1 KO pups showed suppressed ultrasonic vocalization upon separation from their mothers and SALM1 KO adults showed enhanced responses to loud acoustic stimuli. These results suggest that SALM1/LRFN2 regulates excitatory synapse function, inhibitory synapse development, social communication, and acoustic startle behavior.
Keywords: excitatory synaptic function, GABAergic neurons, inhibitory synapses, Lrfn2, NMDA receptor, social communication
Introduction
Synaptic adhesion molecules, with the prototypical molecules being neuroligins and neurexins, have been shown to regulate the development, function, and plasticity of neuronal synapses (Dalva et al., 2007; Biederer and Stagi, 2008; Shen and Scheiffele, 2010; Siddiqui and Craig, 2011; Krueger et al., 2012; Missler et al., 2012; Valnegri et al., 2012; Takahashi and Craig, 2013; Um and Ko, 2013; Bemben et al., 2015; Ko et al., 2015; de Wit and Ghosh, 2016; Jang et al., 2017; Südhof, 2017; Um and Ko, 2017; Yuzaki, 2017). Among such synaptic adhesion molecules is the leucine-rich repeat (LRR)-containing SALM/LRFN (synaptic adhesion-like molecule/leucine rich repeat and fibronectin type III domain containing) family comprising five known members: SALM1/LRFN2, SALM2/LRFN1, SALM3/LRFN4, SALM4/LRFN3, and SALM5/LRFN5 (Ko et al., 2006; Morimura et al., 2006; Wang et al., 2006; Nam et al., 2011; Lie et al., 2018).
SALM family proteins share a similar domain structure consisting of six LRRs, an Ig domain, and a fibronectin type III domain in the extracellular region, followed by a single transmembrane domain and a cytoplasmic region. The extreme C-terminal tails of SALM1-3, but not SALM4/5, contain a PDZ domain-binding motif that interacts with the PDZ domains of PSD-95, an abundant postsynaptic scaffolding protein (Sheng and Kim, 2011; Won et al., 2017). The cytoplasmic regions of individual SALMs share minimal amino acid sequence identities, except for the C-terminal PDZ-binding motif, suggestive of functional diversity. However, SALMs associate with each other to form various homomeric and heteromeric complexes in vivo (Seabold et al., 2008; Lie et al., 2016), perhaps for concerted actions.
Functionally, SALMs regulate synapse development. SALM3 and SALM5, but not other SALMs, promote synapse development by interacting with presynaptic LAR family receptor protein tyrosine phosphatases (Mah et al., 2010; Li et al., 2015; Choi et al., 2016). Mice lacking SALM3/LRFN4 display suppressed excitatory synapse development (Li et al., 2015). SALM4/LRFN3, despite lacking synaptogenic activity, interacts in cis with SALM3 and inhibits SALM3-dependent synapse development (Lie et al., 2016).
SALMs also regulate excitatory synaptic transmission and plasticity. SALM1 associates with NMDA receptors (NMDARs) and induces their dendritic clustering through a mechanism that requires its C-terminal PDZ-binding motif in vitro (Wang et al., 2006), but it does not interact with AMPA receptors (AMPARs). SALM2 promotes excitatory synapse maturation by associating with both NMDARs and AMPARs (Ko et al., 2006). Mice lacking SALM1/LRFN2 have recently been reported to display impairments in excitatory synapse maturation and enhancements in long-term potentiation (LTP) that are associated with autistic-like social deficits and stereotypies as well as enhanced learning and memory (Morimura et al., 2017).
SALMs have also been implicated in human brain disorders. SALM1/LRFN2 has been associated with autism spectrum disorders (ASD; Voineagu et al., 2011; Morimura et al., 2017), schizophrenia (Morimura et al., 2017), working memory deficits (Thevenon et al., 2016), and antisocial personality disorders (Rautiainen et al., 2016). However, how a SALM1/LRFN2 deficiency in humans leads to these abnormalities remains unclear.
In the present study, we further explored the in vivo functions of SALM1 using an independent Lrfn2-KO mouse line (Lrfn2−/− mice) and found that SALM1 is important for excitatory synaptic plasticity, inhibitory synapse development, and ultrasonic vocalization (USV) and acoustic startle.
Materials and Methods
cDNA constructs.
Full-length human SALM1 (aa 1–788) in pcDNA3.1 Myc HisA vector has been described previously (Mah et al., 2010).
Antibodies.
SALM1 (2022) guinea pig polyclonal antibodies were generated using the last 30 aa of mouse SALM1 as immunogen (NGMLLPFEESDLVGARGTFGSSEWVMESTV). The following antibodies have been described previously: GluA1 (#1193; Kim et al., 2009), GluA2 (#1195; Kim et al., 2009), Homer1 (#1133; Sala et al., 2001), PSD-93 (#1634; Kim et al., 2009), PSD-95 (#1688; Yang et al., 2011), SALM2 (#2058; Li et al., 2015), SALM3 (#2024; Lie et al., 2016), SALM4 (#2026; Li et al., 2015), and SALM5 (#1911; Mah et al., 2010). The following antibodies were purchased: GAD65 (Abcam ab85866, RRID:AB_1860505), GluN1 (Transduction Laboratories 556308, RRID:AB_2314954), GluN2A (Alomone Labs AGC-002, RRID:AB_2040025), GluN2B (Alomone Labs AGC-003, RRID:AB_2040028), mGluR5 (Millipore AB5675, RRID:AB_2295173), synapsin I (Millipore AB1543, RRID:AB_2200400), synaptophysin (Santa Cruz Biotechnology sc-9116, RRID:AB_2199007), VGluT1 (Synaptic Systems 135 303, RRID:AB_887875), VGAT (Synaptic Systems 131 003, RRID:AB_887869), gephyrin (Synaptic Systems 147 111, RRID:AB_887719), α-tubulin (Sigma-Aldrich T5168, RRID:AB_477579), and NeuN (Millipore MAB377, RRID:AB_2298772).
Crude synaptosomes.
Crude synaptosomes from the WT and Lrfn2−/− hippocampus [postnatal day 20 (P20)–P23; males and females] were prepared as described previously (Huttner et al., 1983). Briefly, hippocampal homogenates were centrifuged at 1000 × g to remove nuclear and other debris, followed by resuspension and centrifugation of the pellet at 10,000 × g to obtain the P2 (crude synaptosomal) fraction.
Generation and characterization of Lrfn2−/− mice.
Lrfn2-deficient mice were generated by Biocytogen by targeting the exon 2 of the Lrfn2 gene under the genetic background of C57BL/6J. To remove the EGFP + Neo cassette, mice were crossed with Protamine-Flp mice. For global Lrfn2 KO in the whole body, mice removed of the EGFP + Neo cassette were crossed with Protamine-Cre mice. The resulting mice were crossed with WT mice to produce heterozygous mice (Lrfn2+/−). Male and female Lrfn2+/− mice were crossed to produce WT and Lrfn2−/− mice for all the experiments performed except for X-gal experiments. Mice were PCR-genotyped using the following primers: for WT allele: 5′-ATGGAGACTCTGCTTGGTGGGC-3′ (forward) and 5′-GTTAGCAAGGAAGCCTGGGAGC-3′ (reverse); for KO allele: 5′-CCAAGTAACTAGGTTGTTCTGGGC-3′ and 5′-TGAGGAATCTGGAACCGACCAG-3′. The primers for RT-PCR were 5′-AATAAGCTGCTCAGGGCTCTC-3′ and 5′-CAGACAGATTCTGGCAGACG-3′. For X-gal staining, mouse sperm carrying the LacZ cassette in the Lrfn2 gene in the genetic background of C57BL/6N Tac was purchased from KOMP (VG15208) and used to produce progenies for X-gal staining using in vitro fertilization with oocytes in the C57BL/6J Tac background. Heterozygous male mice were used for X-gal staining.
X-gal staining.
Mice (P49) were perfused transcardially with heparinized 1× PBS and 4% paraformaldehyde. Vibratome brain sections (250 μm thickness) were incubated in staining solution containing 5 mm K3Fe(CN)6, 5 mm K4Fe(CN)6•3H2O, 2 mm MgCl2, 0.01% deoxycholate,1 mg/ml X-gal, and 0.02% NP-40 in PBS for 3 h at room temperature.
Electron microscopy.
WT and Lrfn2−/− mice were deeply anesthetized with sodium pentobarbital (80 mg/kg, intraperitoneal) and intracardially perfused with 10 ml of heparinized normal saline, followed by 50 ml of a freshly prepared fixative of 2.5% glutaraldehyde and 1% paraformaldehyde in 0.1 m phosphate buffer (PB, pH 7.4). Hippocampus was removed from the whole brain, postfixed in the same fixative for 2 h, and stored in PB overnight at 4°C. Sections were cut transversely on a Vibratome at 70 μm. The sections were osmicated with 0.5% osmium tetroxide (in PB) for 1 h, dehydrated in graded alcohols, flat embedded in Durcupan ACM (Fluka), and cured for 48 h at 60°C. Small pieces containing stratum radiatum of hippocampal CA1 region were cut out of the wafers and glued onto the plastic block by cyanoacrylate. Ultrathin sections were cut and mounted on Formvar-coated single slot grids. For quantification of excitatory synapses, sections were stained with uranyl acetate and lead citrate and examined with an electron microscope (Montclair Microscopy and Microanalysis Research Laboratory, RRID:SCR_012453) at 80 kV accelerating voltage. For quantification of inhibitory synapse, sections were further immunogold stained for GABA.
Postembedding immunogold staining for GABA.
Sections were immunostained for GABA by postembedding immunogold method, as described previously (Paik et al., 2007) with some modifications. In brief, the grids were treated for 5 min in 1% periodic acid to etch the resin and for 8 min in 9% sodium periodate to remove the osmium tetroxide, and then washed in distilled water, transferred to Tris-buffered saline containing 0.1% Triton X-100 (TBST; pH 7.4) for 10 min, and incubated in 2% human serum albumin (HSA) in TBST for 10 min. The grids were then incubated with rabbit antiserum against GABA (GABA 990, 1:10,000) in TBST containing 2% HSA for 2 h at room temperature. The antiserum (a kind gift from professor O.P. Ottersen at the Center for Molecular Biology and Neuroscience, University of Oslo) was raised against GABA conjugated to bovine serum albumin with glutaraldehyde and formaldehyde (Kolston et al., 1992) and characterized by spot testing (Ottersen and Storm-Mathisen, 1984). To eliminate cross-reactivity, the diluted antiserum was preadsorbed overnight with glutaraldehyde (G)-conjugated glutamate (500 μm, prepared according as described previously; Ottersen et al., 1986). After extensive rinsing in TBST, grids were incubated for 3 h in goat anti-rabbit IgG coupled to 15 nm gold particles (1:25 in TBST containing 0.05% polyethylene glycol; BioCell). After a rinse in distilled water, the grids were counterstained with uranyl acetate and lead citrate and examined with an electron microscope (Montclair Microscopy and Microanalysis Research Laboratory, RRID:SCR_012453) at 80 kV accelerating voltage. To assess the immunoreactivity for GABA, gold particle density (number of gold particles per square millimeter) of each GABA-immunopositive (+) terminal was compared with gold particle density of terminals that containg round vesicles and make asymmetric synaptic contact with dendritic spines (background density). Terminals were considered GABA+ if the gold particle density over the vesicle-containing areas was at least five times higher than background density.
Quantitative analysis of excitatory and inhibitory synapses.
For quantification of excitatory synapses, 24 electron micrographs representing 368.9 μm2 neuropil regions in each mouse were taken at a 40,000×. The number of spines (PSD density), proportion of perforated spines, PSD length, and PSD thickness from each of three WT and Lrfn2−/− mice were quantified with ImageJ (RRID:SCR_003070). For quantification of inhibitory synapses, 24 electron micrographs representing 655.5 μm2 neuropil regions in each mouse were taken at 30,000×. Number of GABA+ terminals showing clear PSD (inhibitory synapse density), length and thickness of PSD contacting GABA+ terminals from each of three WT and Lrfn2−/− mice were quantified with ImageJ (RRID:SCR_003070). The measurements were all performed by an experimenter blinded to the genotype. Digital images were captured with Digital Micrograph software driving a CCD camera (SC1000 Orius; Gatan) and saved as TIFF files. Brightness and contrast of the images were adjusted using Adobe Photoshop 7.0 (RRID:SCR_014199).
In situ hybridization.
Mouse brains at various developmental stages [embryonic day 18 (E18) and P0, P7, P14, P21, and P42] were extracted and rapidly frozen in isopentane prechilled with dry ice. Brain sections were prepared with a cryostat and thaw-mounted onto gelatin-coated slides and fixed in 4% paraformaldehyde. Two independent hybridization probes targeted approximately the N- and C-terminal regions of the Lrfn2 gene (NM_027452.3). For probe 1, the forward sequence was TACGCCGGATCCGTGGGCTGCTGGCTTTT and reverse sequence was TACGCCGAATTCTGGCTGATGGTGTTCCTG. For probe 2, the forward sequence was TACGCCGGATCCTGCTCTTGCCCTTTGAGG and the reverse sequence was TACGCCGAATTCATGGGGAAGGGGGTGTAG.
FISH.
In brief, frozen sections (14 μm thick) were cut coronally through the hippocampal formation. Sections were thaw-mounted onto Superfrost Plus Microscope Slides (Fisher Scientific, 12-550-15). The sections were fixed in 4% formaldehyde for 10 min, dehydrated in increasing concentrations of ethanol for 5 min, and air-dried. Tissues were then pretreated for protease digestion for 10 min at room temperature. For RNA detection, incubations with different amplifier solutions were performed in a HybEZ hybridization oven (Advanced Cell Diagnostics) at 40°C. In this study, we used fluorescent probes to label Lrfn2, Gad1/2, and Vglut1/2; mixtures of Gad1 + Gad2 probes and Vglut1 + Vglut2 probes were used to collectively label GABAergic and glutamatergic neurons, respectively. Synthetic oligonucleotides in the probes were complementary to the following nucleotide regions in the target genes; Lrfn2, nucleotide sequence 542–2041 of Mm-Lrfn2-C1; Vglut1, 464-415 of Mm-Slc17a7-C2, Vglut2, 1986–2998 of Mm-Slc17a6-C2, Gad1, 62-3113 of Mm-Gad1-C3, Gad2, 552–1506 of Mm-Gad2-C3 (Advanced Cell Diagnostics), respectively. The labeled probes were conjugated to Atto 550 (C1), Atto 647 (C2), and Alexa Fluor 488 (C3). The sections were hybridized at 40°C with labeled probe mixtures (C1 + C2 + C3) per slide for 2 h. Then the nonspecifically hybridized probes were removed by washing the sections 3 times each in 1× wash buffer at room temperature for 2 min. Amplification steps involved sequential incubations with Amplifier 1-FL for 30 min, Amplifier 2-FL for 15 min, Amplifier 3-FL for 30 min, and Amplifier 4 Alt B-FL at 40°C for 15 min. Each amplifier solutions were removed by washing 3 times with 1× wash buffer for 2 min at RT. Fluorescent images were acquired using an LSM 700 microscope (Zeiss) and analyzed using ImageJ (RRID:SCR_003070).
Electrophysiology.
For whole-cell patch-clamp analysis, sagittal hippocampal and coronal mPFC slices (300 μm thick) from Lrfn2−/− mice and their WT littermates at P21–P24 were prepared using a vibratome in ice-cold section buffer containing the following (in mm): 212 sucrose, 25 NaHCO3, 5 KCl, 1.25 NaH2PO4, 0.5 CaCl2, 3.5 MgSO4, 10 d-glucose, 1.2 l-ascorbic acid, and 2 Na-pyruvate bubbled with 95% O2/5% CO2. The slices were recovered at 32°C for 30 min in normal artificial CSF (ACSF) containing the following (in mm): 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 10 glucose, 2.5 CaCl2, and 1.3 MgCl2 oxygenated with 95% O2/5% CO2. Stimulation and recording pipettes were pulled from borosilicate glass capillaries (Harvard Apparatus) using a micropipette electrode puller (Narishege). Whole-cell patch-clamp recordings were made using a MultiClamp 700B amplifier (Molecular Devices) and Digidata 1440A (Molecular Devices) under visual control with differential interference contrast illumination in an upright microscope. Signals were filtered at 2 kHz and digitized at 10 kHz. Miniature excitatory postsynaptic currents (mEPSCs) were obtained at a holding potential of −70 mV using patch electrode (3–4 MΩ) filled with an internal solution containing the following (in mm): 100 CsMeSO4, 10 TEA-Cl, 8 NaCl, 10 HEPES, 5 QX-314-Cl, 2 Mg-ATP, 0.3 Na-GTP, and 10 EGTA, pH 7.25, 295 mOsm. TTX (0.5 μm) and picrotoxin (100 μm) were added to ACSF to inhibit spontaneous action potential-mediated synaptic currents and IPSCs, respectively. For spontaneous EPSCs (sEPSCs), the same experimental conditions used for mEPSC measurements were used, except for omitting TTX. To measure miniature inhibitory postsynaptic currents (mIPSCs), cells were also held at −70 mV and the pipette internal solution contained the following (in mm): 115 CsCl, 10 TEA-Cl, 8 NaCl, 10 HEPES, 5 QX-314-Cl, 4 Mg-ATP, 0.3 Na-GTP, and 10 EGTA, pH 7.35, 295 mOsm. To inhibit excitatory synaptic currents, TTX (0.5 μm), D-AP5 (25 μm), and NBQX (10 μm) were added to the ACSF. sIPSC measurements were made in the absence of TTX.
For measurements of the NMDA/AMPA ratio, sagittal hippocampal slices (300 μm thick) were prepared. The recording pipettes (3–4 MΩ) were filled with the same internal solution used for mEPSC measurements. Picrotoxin (100 μm) was added to the ACSF. CA1 pyramidal neurons were voltage clamped at −70 mV and EPSCs were evoked at every 15 s. AMPAR-mediated EPSCs were recorded at −70 mV and 20 consecutive responses were recorded after stable baseline. After recording AMPAR-mediated EPSCs, holding potential was changed to +40 mV to record NMDAR-mediated EPSCs. The NMDA component was measured at 60 ms after the stimulation. The NMDA/AMPA ratio was calculated by dividing the mean value of 20 NMDA-EPSC peak amplitudes by the mean value of 20 AMPA-EPSC peak amplitudes. Data were acquired using Clampex 10.4 (Molecular Devices) and analyzed using Clampfit 10.4 (NMDA/AMPA ratio; Molecular Devices).
To measure input/output responses of NMDA-EPSCs, synaptic currents were recorded at +40 mV with a pipette solution containing the following (in mm): 100 Cs-gluconate, 10 TEA-Cl, 10 CsCl, 8 NaCl, 10 NaCl, 10 EGTA, 4 Mg-ATP, 0.3 Na-GTP, 0.5 and QX-314-Cl, pH 7.36. The GABAAR antagonist picrotoxin (100 μm) and the AMPAR antagonist NBQX (10 μm) were added to the ACSF. The stimulating electrode was placed in the stratum radiatum ∼100–120 μm from the neuron under recording and synaptic responses were evoked by a series (20 s interval) of pulses with different stimulation intensities (0.5–4 V). To avoid potential confounding effects of propagated spike activity, the initial slopes rather than peak amplitudes of NMDA-EPSCs were analyzed offline (Kim et al., 2009). The data were discarded when the series resistance changed by >10% during the recordings.
For field recordings, sagittal hippocampal slices (400 μm) were prepared. The pipettes were filled with ACSF. For input/output and paired-pulse ratio experiments, CA1 field EPSP (fEPSP) was evoked every 20 s and the stable baseline was recorded for 10 min. For input/output recording, gradually increasing stimuli were delivered to induce fiber volley amplitudes of 0.05–0.3 mV/ms. For paired-pulse ratio recording, interstimulus intervals were 25, 50, 100, 200, and 300 ms. For long term potentiation (LTP) measurements, the Schaffer collateral pathway was stimulated every 20 s and a stable baseline was maintained for 20 min. LTP stimuli were high-frequency stimulation (HFS; 100 Hz, 1 s) or theta-burst stimulation (TBS; 10 trains of 4 pulses at 100 Hz, delivered at 5 Hz, repeated 4 times at 10 s interval). For long term depression (LTD) experiments, picrotoxin (100 μm) were added to ACS, and low-frequency stimulation (LFS; 1 Hz, 900 pulses) was given. After LTP or LTD stimulus, the responses were recorded for 1 h.
Animal behavioral tests.
Male mice at ∼2–6 months of age were used for all behavioral tests except the pup retrieval test. All mice were fed ad libitum, housed under a 12/12 h light/dark cycle, and all mouse behaviors were performed using mice at their light-off/dark periods. All procedures were approved by the Committee of Animal Research at KAIST (KA2012-19). Animal used in behavioral tests were generated from crossing male and female heterozygous mice. All tests used littermates or age-matched mice.
Open-field test.
Mice were placed into a white 40 × 40 × 40 cm open-field box and allowed to explore freely for 1 h under complete darkness (∼0 lux) or 110 lux. Mice movements were recorded using a video camera (infrared camera in the case of complete darkness) and analyzed using EthoVision XT 10 software (Noldus, RRID:SCR_000441).
LABORAS monitoring of movements.
Home cage locomotion behaviors of mice were recorded and analyzed using Laboratory Animal Behavior Observation Registration and Analysis System (LABORAS; Metris) for 72 consecutive hours.
Elevated plus-maze.
The maze consisted of two open arms, two closed arms, and a center zone elevated to a height 50 cm above the floor. Mice were initially placed on the center zone faced to the open arm and allowed to explore the space freely for 8 min. Light condition was ∼80 lux.
Light–dark test.
The apparatus had a dimension of 12 × 30 × 20 cm for the light chamber (∼600 lux) and 14 × 13 × 20 cm for the dark chamber (∼5 lux). Mice were placed in the center of the light chamber and allowed to explore the whole apparatus freely for 10 min. Time spent in the light chamber was analyzed using EthoVision XT 10 software.
Three-chamber test.
Subject mice were isolated for 3 d in their home cages before the experiment. The apparatus consisted of three-chamber and both side chambers had a steel wire cage in the corner to place inanimate objects or mice. First, mice were put in the center zone and allowed to explore the whole apparatus freely for 10 min. Next, a stranger mouse (S1) was placed in a wire cage in a side chamber and an inanimate object (O) was placed in another wire cage. Mice were then allowed to explore freely for 10 min. The stranger mouse was randomly positioned in the left or right chamber. Then, the object was replaced with a novel stranger mouse (S2) and mice were allowed to explore the apparatus freely for 10 min. Time spent in each chamber and time spent sniffing the wire cage containing O, S1, or S2 were analyzed using EthoVision XT 10 software.
Direct interaction test.
Subject mice were isolated for 4 d in their home cages. On day 1 for habituation, mice were individually placed in a gray box (30 × 30 × 40 cm) for 10 min. Twenty-four hours later, two age-matched mice in the same genotype that had never encountered each other before were put into the habituated box and allowed to interact with each other freely for 10 min. Physical interaction, nose-to-nose sniffing, and following behaviors were measured manually in a blinded manner.
USV test.
For pup USVs, pups were placed in a glass bowl in a recording chamber and a recording microphone was placed 20 cm above the pup. USVs from pups induced by separation from their mothers were recorded using Ultrasoundgate (Avisoft, 116Hb) system for 3 min. For adult USVs, male mice isolated for 3 d in their home cages and these cages with mice were placed in a chamber with a microphone 20 cm above the home cage. Age-matched female mice were introduced to the home cage for 5 min. Recorded sound files were analyzed using the SASLab Pro software (Avisoft, RRID:SCR_014438).
Pup retrieval assay.
Virgin female mice at 4 months of age were isolated in their home cages with nesting block for 4 d before the test. Three WT P1 pups were placed at three different corners away from the nesting block of the home cage of the test female mouse and the female mouse was allowed to retrieve the pups for 10 min. Latency to each pup retrieval to the nest was measured.
Repetitive behavior.
Self-grooming and digging tests were performed in mouse home cages with fresh bedding. Mice were individually placed into a home cage for 20 min and repetitive behaviors during the last 10 min were used to analyze self-grooming and digging behavior manually.
Marble-burying test.
Subjected mice were placed in a home cage with 5-cm-thick beddings and 21 metal marbles and allowed to explore freely for 30 min. The analysis counted the number of buried marbles in a blinded manner with marble burying counted when more than the two-thirds of marbles were buried.
Rotarod test.
Rotating speed of rod was gradually increased from 4 to 40 rpm over 5 min. Mice were placed gently on the rotating rod for 20 s, followed by the start of rod rotation. The experiment was performed for 5 consecutive days while measuring the latencies of mice to fall from the rod.
Object recognition test.
This test was performed in an open-field test apparatus. Mice were habituated in the apparatus for 1 h/d before training. For the displaced object recognition test (DORT), after exploring same 2 objects for 10 min, mice were put back into their home cages for 5 min. One of the two objects was translocated to a position opposite in the box. Then the mice were placed back in the apparatus and allowed to explore the objects for 10 min. Exploration time for the translocated object was measured. For the novel object recognition test (NORT), on the training day, mice were allowed to explore two same objects for 10 min. Twenty-four hours after training, one of the two objects was replaced with a new one and mice were allowed to explore both objects freely for 10 min. Object exploration was defined by the amount of time spent for each object, with the nose of the mouse touching or facing toward the objects within 2 cm from them.
Morris water maze.
This assay was performed in a stainless steel round tank (12 cm diameter) with a hidden platform. The tank was filled with tap water at temperature of ∼20–22°C made opaque with white watercolor paint. For memory acquisition, mice were trained to find the platform with 3 trials per day with an intertrial interval of 30 min for 5 d. When mice reached the platform, they were allowed to rest on the platform for 15 s before being put back into their home cages. If mice did not find the platform within 60 s, they were guided to the platform and allowed to rest there for 15 s. On day 6, for probe test, the platform was removed and mice were put in the center of the tank and allowed to explore for 1 min. Mice were retrained to find the hidden platform in the tank after the probe test to avoid memory extinction. On the next day (day 7), the platform was replaced to a site opposite to the original position and mice were trained for 4 d for reversal learning and memory. On day 11, another probe test was performed to test reversal learning and memory. The number of exact crossing over the platform region, quadrant occupancy, and swimming speed were analyzed using EthoVision XT 10 software.
Fear conditioning.
On training day, mice were placed in the fear chamber and allowed to explore the chamber freely for 2 min. Then, the mice went through 5 rounds of a 20 s tone with a 0.5 mA foot shock during the last 2 s, followed by 40 s of rest. The final shock was followed by a 2 min posttraining habituation. Twenty-four hours later, mice were returned to the same shock chamber for 5 min to test contextual fear conditioning. Four hours later, the mice were returned to the chamber with a different context to test cued fear conditioning. To change the context, mice were placed in a round-shaped tube added to the chamber and allowed to explore freely for 2 min. Then, a 3 min tone was given to test levels of cued fear conditioning.
Acoustic startle and prepulse inhibition.
Different mouse cohorts were used for acoustic startle responses and prepulse inhibition (Wells et al., 2016). To test acoustic startle responses, the session was preceded by a 5 min exposure to 65 dB background noise. Then each mouse received 92 trials with intertrial intervals ranging from 7 to 23 s in pseudorandom order. The trials included a presentation of eight pulse-alone trials (120 dB, 40 ms pulse, four were given at the beginning and four at the end of the test), 77 pulse trials (seven each of 70, 75, 80, 85, 85, 90, 95,100, 105, 110, 115, and 120 dB, 40 ms pulse), and seven trials each without pulse or prepulse inhibition. To test prepulse inhibition, each mouse received 57 trials with intertrial intervals ranging from 7 to 23 s presented in pseudorandom order. The trials included a presentation of eight pulse-alone trials (120 dB, 40 ms pulse, four were given at the beginning and four at the end of the test), 35 prepulse trials (seven each of 70, 75, 80, 85 and 90 dB, 20 ms prepulse given 100 ms before a 120 dB, 40 ms pulse), and seven trials each without pulse or prepulse presentation. The prepulse inhibition percentage was calculated as follows: 100 − (mean prepulse response/mean pulse response) × 100). Startle at each pulse level was averaged across trials.
PTZ-induced seizure.
Mice were injected with pentylenetetrazole (PTZ; 50 μg/weight in grams) into the intraperitoneal cavity and seizures recorded for 10 min. Seizures were scored blindly according to Racine scale designed for PTZ-induced seizures in mice (Ferraro et al., 1999).
Experimental design and statistical analysis.
The order of behavioral tests was designed in a way to minimize stress in animals. The behavioral experiments were performed in the following order using three independent cohorts: (1) cohort 1 underwent elevated plus-maze open-field test (110 lux), digging and grooming, light/dark test, three-chamber test, direct interaction test, and PPI and fear conditioning test; (2) cohort 2 underwent open-field test (0 lux), adult USV, acoustic startle response, and PTZ-induced seizure; and (3) cohort 3 underwent LABORAS test, displaced object recognition test, novel object recognition test, and Morris water maze. All statistical tests were performed using GraphPad Prism 7.0 software (RRID:SCR_002798). Normality of data distribution was assessed using the D'Agostino–Pearson omnibus test. Comparison of WT and KO data were performed using unpaired two-tailed Student's t test when the data showed Gaussian distribution; Mann–Whitney test or Wilcoxon test was used when the data followed a non-Gaussian distribution. Repeated-measures two-way ANOVA was used to determine between-subject variability (genotype) and within-subject variability (repeated-measures) for the analysis of the measures of input/output ratio, paired-pulse facilitation, open field, LABORAS, arotarod, Morris water maze, fear conditioning, acoustic startle, and prepulse inhibition tests. Bonferroni's test followed by ANOVA was used as a post hoc test for multiple comparisons. All data were displayed as mean ± SEM. The age, sex, and numbers of animals and all details of statistical results are shown in Fig. 1-1.
Results
Generation and characterization of Lrfn2−/− mice
To generate Lrfn2−/− mice, we used a mouse embryonic stem cell line that lacks exon 2 of the Lrfn2 gene encoding most of the extracellular region of SALM1 (Fig. 1A). This deletion was confirmed by PCR genotyping and qRT-PCR (Fig. 1B,C). Immunoblot analyses using an anti-SALM1 antibody raised against the last 30 aa residues of the protein confirmed the lack of SALM1 protein in the Lrfn2−/− hippocampus (Fig. 1D). These mice were born in normal Mendelian ratios and did not exhibit any gross anatomical abnormalities in the brain (Fig. 1E). The Lrfn2−/− brain displayed normal levels of other SALM family proteins (SALM2-5), glutamate receptor subunits (GluA1/2, GluN1/2A/2B, and mGluR5), and presynaptic and postsynaptic proteins (PSD-95, PSD-93,synapsin I, synaptophysin, Homer1, VGluT1, VGAT, GAD65, and gephyrin), compared with those in WT mice, as determined by immunoblot analysis of hippocampal crude synaptosomes (Fig. 1F; t(4) = 0.7226, p = 0.5099 for SALM2; t(4) = 0.2952, p = 0.7826 for SALM3; t(4) = 0.2437, p = 0.8194 for SALM4; t(4) = 1.054, p = 0.3514 for SALM5; t(6) = 1.747, p = 0.1312 for GluA1; t(6) = 2.281, p = 0.5471 for GluA2; t(6) = 0.638, p = 0.5471 for GluN1; t(6) = 0.3824, p = 0.7154 for GluN2A; t(6) = 0.2232, p = 0.8308 for GluN2B; t(6) = 0.6676, p = 0.5292 for mGluR5; t(4) = 0.1, p = 0.9251 for PSD-95; t(6) = 0.385, p = 0.7135 for PSD-93; t(6) = 0.4847, p = 0.6451 for synapsin I; t(6) = 0.5682, p = 0.5905 for synaptophysin; t(4) = 1.334, p = 0.2532 for Homer1; t(6) = 0.2666, p = 0.7987 for VGluT1; t(6) = 0.9887, p = 0.361 for VGAT; t(6) = 0.2988, p = 0.7752 for GAD65; t(4) = 0.1183, p = 0.9115 for gephyrin, unpaired t test for all).
Figure 1.

Generation and characterization of Lrfn2−/− mice. A, Strategy for the generation of Lrfn2−/− mice. B, PCR genotyping of Lrfn2−/− mice. HT, Heterozygous; C, Confirmation of Lrfn2 exon 2 deletion by qRT-PCR analysis of whole-brain mRNAs (P31). D, Lack of LRFN2 protein expression in the Lrfn2−/− brain (P21), as determined by immunoblot analysis of hippocampal lysates: total (Hp) and crude synaptosomal (Hp-P2). Untrans, Untransfected HEK293 cell lysates; SALM1-Myc, lysates of HEK293 cells transfected with SALM1-Myc. E, Normal gross morphology of the Lrfn2−/− brain (P28), as shown by staining for NeuN (a neuronal marker). Scale bar, 1 mm. F, Normal levels of other SALMs (SALM2-5), glutamate receptor subunits [AMPA (GluA1/A2), NMDA (GluN1/2A/2B), and metabotropic (mGluR5), and synaptic proteins (excitatory and inhibitory presynaptic and postsynaptic), compared with WT levels, as determined by immunoblot analyses of crude synaptosomal (P2) fractions from the WT and Lrfn2−/− hippocampus (P20–P23). Levels of the indicated proteins were normalized to those of α-tubulin for genotype comparisons. The dotted line indicates WT levels. n = 3 mice for WT and 4 mice for KO. NS, Student's t test (for additional details, see Figure 1-1).
Details of animals used and statistical results. Download Figure 1-1, DOCX file (567.1KB, docx)
Distribution patterns of SALM1 mRNA and protein
We next determined the distribution pattern of SALM1 mRNA at various developmental stages (E18, P0, P7, P14, P21, and P42) by in situ hybridization in mouse brain slices. SALM1 mRNA signals in sagittal and horizontal sections, as revealed by two independent probes, were relatively strong in cortical areas of the brain until P0 and gradually increased in other brain regions across postnatal developmental stages (Fig. 2A).
Figure 2.
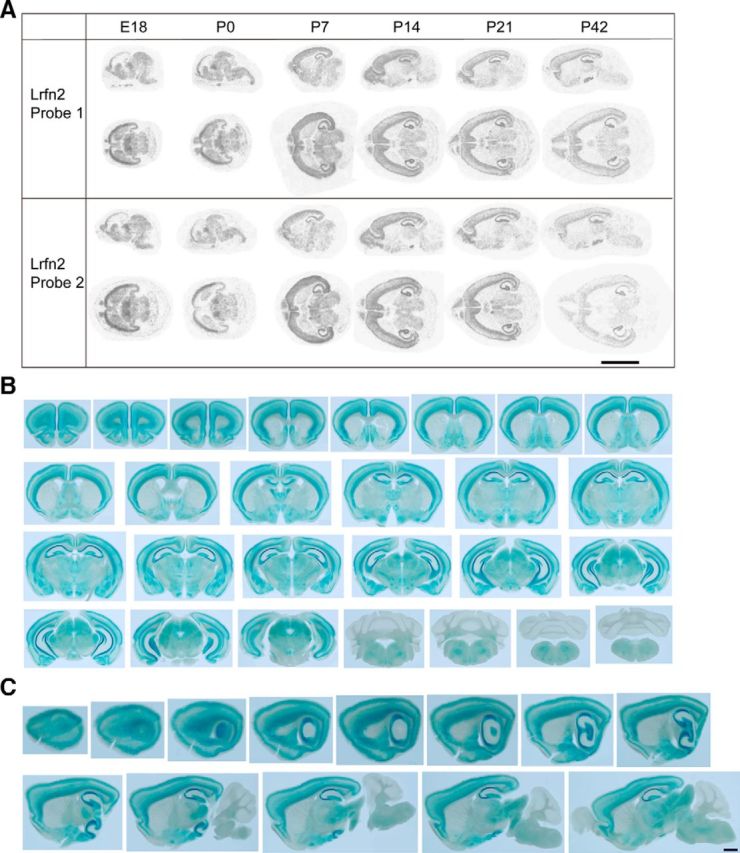
Distribution patterns of SALM1 mRNA and protein. A, In situ hybridization analysis of SALM1/Lrfn2 mRNA expression in mouse brain slices at different developmental stages using two independent probes against two different regions of SALM1/Lrfn2 mRNA. Scale bar, 10 mm. B, C, Distribution pattern of SALM1 protein, as determined by X-gal staining of coronal and sagittal sections of brain from male heterozygous (Lrfn2+/−) mice (P46). Scale bar, 1 mm.
To determine the distribution pattern of SALM1 protein in the brain, we used another line of transgenic mice in which the entire open reading frame of the Lrfn2 gene was replaced with a LacZ cassette (KOMP VG15208; termed Lrfn2-LacZ mice; Valenzuela et al., 2003). X-gal staining of coronal and sagittal sections from Lrfn2-LacZ mice (P46; male heterozygous) revealed that SALM1 protein is highly expressed in various brain regions, including the cortex, hippocampus, amygdala, thalamus, and hypothalamus (Fig. 2B,C). SALM1 protein was more abundant in layers II/III and VI of the cortex relative to the middle layers and in CA1 and CA3 regions of the hippocampus relative to the dentate gyrus. In contrast, SALM1 protein was minimally detected in the striatum and cerebellum.
Suppressed excitatory and inhibitory synaptic transmission in Lrfn2−/− mice
To determine whether Lrfn2 deletion has any effects on synapse development and function, we first determined spontaneous excitatory and inhibitory synaptic transmission in the CA1 region of the hippocampus, where SALM1 is highly expressed. We found that both the frequency and amplitude of mEPSCs in Lrfn2−/− CA1 pyramidal neurons (P21–P23) were comparable to those from WT mice (Fig. 3A; t(30) = 0.286, p = 0.7769 for frequency; t(30) = 0.799, p = 0.4306 for amplitude, unpaired t test).
Figure 3.

Suppressed excitatory and inhibitory synaptic transmission in Lrfn2−/− mice. A, mEPSCs measured in WT and Lrfn2−/− CA1 pyramidal neurons (P20–P23). n = 17 cells from 4 WT mice and 15 cells from 6 KO mice. NS, Student's t test. B, mIPSCs in WT and Lrfn2−/− CA1 pyramidal neurons (P20–P23). n = 15, 3 for WT and 15, 3 for KO. *p < 0.05, NS, Mann–Whitney U test and Student's t test. C, sEPSCs in WT and Lrfn2−/− CA1 pyramidal neurons (P20–P23). n = 17, 5 for WT and 16, 5 for KO. NS, Student's t test. D, sIPSCs in WT and Lrfn2−/− CA1 pyramidal neurons (P20–P23). n = 18, 3 for WT and 16, 3 for KO. **p < 0.01, NS, Student's t test. E, mEPSCs in WT and Lrfn2−/− mPFC prelimbic layer II/III pyramidal neurons (P20–P23). n = 15, 3 for WT and 16, 3 for KO. **p < 0.01, NS, Mann–Whitney U test and Student's t test. F, mIPSCs in WT and Lrfn2−/− mPFC prelimbic layer II/III pyramidal neurons (P20–P23). n = 17, 3 for WT and 19, 3 for KO. NS, Mann–Whitney U test and Student's t test.
In contrast, the frequency, but not amplitude, of mIPSCs was significantly reduced in Lrfn2−/− CA1 pyramidal neurons (P20–P23; Fig. 3B; U = 54.5, p = 0.015, Mann–Whitney U test for frequency; t(28) = 0.7699, p = 0.4478, unpaired t test for amplitude). Similar results were obtained for sEPSCs and sIPSCs, revealing a specific decrease in sIPSC frequency (Fig. 3C,D; t(31) = 0.4689, p = 0.6418, unpaired t test for sEPSC frequency; t(31) = 1.354, p = 0.1857, unpaired t test for sEPSC amplitude; t(32) = 2.824, p = 0.0081, unpaired t test for sIPSC frequency; t(32) = 0.3834, p = 0.7039, unpaired t test for sIPSC amplitude); these latter recordings were obtained in the absence of TTX to allow action potential firing and network activities. These results suggest that SALM1 deficiency leads to a decrease in the frequency of inhibitory, but not excitatory, synaptic transmission in the hippocampal CA1 region and that this decrease is not compensated by network activities.
To further test whether Lrfn2 deletion leads to similar changes in other brain regions, we measured mEPSCs and mIPSCs from layer II/III pyramidal neurons in the prelimbic region of the medial prefrontal cortex (mPFC). We found that the frequency, but not amplitude, of mEPSCs, was significantly reduced, whereas mIPSCs were unaltered (Fig. 3E,F; t(29) = 3.082, p = 0.0045, unpaired t test for mEPSC frequency; U = 99, p = 0.4232, Mann–Whitney U test for mEPSC amplitude; U = 168, p = 0.0864, Mann–Whitney U test for mIPSC frequency; t(42) = 1.596, p = 0.118, unpaired t test for mIPSC amplitude), a finding that contrasts with the results obtained in the hippocampus. These results collectively suggest that Lrfn2 deletion suppresses the frequency of excitatory synaptic transmission in the mPFC, but the frequency of inhibitory transmission in the hippocampus.
Decreased inhibitory synapse density in the Lrfn2−/− hippocampus
To further understand the mechanism underlying the suppressed synaptic transmission in Lrfn2−/− mice, we analyzed the density and morphology of excitatory and inhibitory synapses using electron microscopy (EM). Morphologically, excitatory and inhibitory synapses were defined by PSDs apposed to presynaptic axon terminals and PSDs apposed to GABA immunopositive inhibitory axon terminals, respectively. We found no significant changes in the density of excitatory synapses in the CA1 stratum radiatum region of the Lrfn2−/− hippocampus compared with WT controls (Fig. 4A,B; t(4) = 0.09004, p = 0.9326 for PSD density; t(4) = 0.5, p = 0.6433 for PSD length; t(4) = 0, p > 0.9999 for PSD thickness; t(4) = 0.03165, p = 0.9763 for perforated synapse frequency, unpaired t test for all). In addition, there were no changes in the length, thickness, or perforation of Lrfn2−/− PSDs.
Figure 4.

Decreased inhibitory synapse density in the Lrfn2−/− hippocampus. A, B, Decreased density of inhibitory, but not excitatory, synapses in the stratum radiatum region of the CA1 region in Lrfn2−/− mice (P21), as determined by EM analysis. Excitatory synapses were defined by PSD structures apposed to axon terminals (arrows and double arrowheads for nonperforated and perforated PSD, respectively) and inhibitory synapses were defined by PSDs apposed to presynaptic axon terminals with immunopositive GABA signals (arrows). Asterisks, GABA immunopositive axon terminals; d, postsynaptic dendrites; single arrowhead, GABA-immunopositive axon. Scale bar, 500 nm. n = 3 mice for WT and KO. **p < 0.01, NS, Student's t test.
In contrast, an analysis of inhibitory synapses indicated a significant decrease in the density, but not the length or thickness, of PSDs (Fig. 4C,D; t(4) = 6.115, p = 0.0036 for inhibitory PSD density; t(4) = 1.512, p = 0.2051 for inhibitory PSD length; t(4) = 2, p = 0.1161 for inhibitory thickness, unpaired t test for all). These results suggest that SALM1 deficiency leads to a decrease in the density of inhibitory, but not excitatory, synapses in the hippocampus. Together with the synaptic transmission results, these findings suggest that the reduced inhibitory synapse number contributes to the reduced frequency of inhibitory synaptic transmission. This conclusion apparently contrasts with the normal levels of inhibitory synaptic proteins such as VGAT and gephyrin in the Lrfn2−/− hippocampus, determined by immunoblot analyses (Fig. 1F). However, this could be attributable to the fact that immunoblot methods may be less sensitive than electrophysiological or EM methods or that synaptic levels of these proteins might have been increased to compensate for the loss of inhibitory synapses.
SALM1 expression in both glutamatergic and GABAergic neurons
The decrease in inhibitory synapse density associated with SALM1 deficiency might be attributable to cell-autonomous changes in CA1 pyramidal neurons and/or alterations in presynaptic GABAergic neurons that express SALM1 protein. To investigate this, we performed double-FISH experiments for SALM1/Lrfn2 and the glutamatergic neuron marker Vglut1/2 (vesicular glutamate transporter-1/2) and the GABAergic neuron marker Gad1/2 (glutamate decarboxylase-1/2). Signals for SALM1/Lrfn2 mRNA were detected strongly in both hippocampal and cortical areas, where they were detected in cell bodies positive for Vglut1/2 as well as Gad1/2 (Fig. 5). These results suggest that SALM1/Lrfn2 is expressed in both glutamatergic and GABAergic neurons in the cortex and hippocampus.
Figure 5.

Lrfn2 mRNA is detected in both glutamatergic and GABAergic neurons. Expression of SALM1/Lrfn2 mRNA in glutamatergic and GABAergic neurons was determined by FISH. Coronal sections from mouse brains (male, 8 weeks) were triply stained for SALM1/Lrfn2 and Vglut1/2 (glutamatergic neuron markers) and Gad1/2 (GABAergic neuron markers) and counterstained with the nuclear dye DAPI. A mixture of two probes (Vglut1 + Vglut2 or Gad1 + Gad2) was used to label all glutamatergic or GABAergic neurons. The indicated cortical and hippocampal regions in the image at left were enlarged on the right to highlight the expression of Lrfn2 mRNA in both glutamatergic and GABAergic neurons; Lrfn2 expression in GABAergic neurons is further highlighted by arrows. Scale bar, 50 μm.
Increased NMDAR-mediated synaptic transmission and suppressed NMDAR-dependent synaptic plasticity in the Lrfn2−/− hippocampus
Although we found that SALM1 deficiency has no effect on excitatory synapse density or spontaneous synaptic transmission in the hippocampus, given that SALM1 forms a complex with PSD-95 and NMDARs in vitro and in vivo (Ko et al., 2006; Morimura et al., 2006; Wang et al., 2006), it is possible that SALM1 deficiency might alter other aspects of excitatory synapse development and function. To test this possibility, we first measured the ratio of evoked NMDAR- and AMPAR-mediated EPSCs (NMDA/AMPA ratio). Interestingly, these experiments revealed an increase in the NMDA/AMPA ratio at Lrfn2−/− Schaffer collateral-CA1 pyramidal (SC-CA1) synapses (Fig. 6A; t(17) = 2.69, p = 0.0155, unpaired t test). In contrast, there were no genotype differences in basal excitatory synaptic transmission (input/output relationship) or paired-pulse facilitation, as shown by field recordings (Fig. 6B,C; for input/output ratio, interaction, F(5,70) = 0.04395, p = 0.9988, fiber volley, F(5,70) = 450.3, p < 0.0001, genotype, F(1,14) = 0.2457, p = 0.6353, repeated-measures two-way ANOVA; for paired-pulse ratio, interaction, F(5,35) = 0.07325, p = 0.9959, interval, F(5,35) = 39.53, p < 0.0001, genotype, F(1,7) = 0.1602, p = 0.7009, repeated-measures two-way ANOVA), suggesting that AMPAR-mediated excitatory synaptic transmission and the probability of presynaptic glutamate release are normal at Lrfn2−/− SC-CA1 synapses. However, there was a significant increase in the NMDAR component of excitatory synaptic transmission, as shown by NMDAR-mediated EPSCs (Fig. 6D; interaction, F(7,224) = 1.341, p = 0.2320, stimulus, F(7,224) = 189.9, p < 0.0001, genotype, F(1,32) = 5.106, p = 0.0308, repeated-measures two-way ANOVA; for each stimulus point, 0.5, p = 0.0635; 1.0, p = 0.0331; 1.5, p = 0.0210; 2.0, p = 0.0251; 2.5, p = 0.0163; 3.0, p = 0.0591; 3.5, p = 0.0766; 4.0, p = 0.0644, one-way ANOVA). These results suggest that the deletion of Lrfn2 in mice leads to an increase in NMDAR-mediated, but not AMPAR-mediated, excitatory synaptic transmission.
Figure 6.

Increased NMDAR-mediated synaptic transmission and suppressed NMDAR-dependent synaptic plasticity in the Lrfn2−/− hippocampus. A, Increased ratio of NMDAR/AMPAR-mediated synaptic transmission at Lrfn2−/− SC-CA1 synapses (P21–P24), measured as AMPA and NMDA EPSCs evoked at holding potentials of −70 and +40 mV, respectively. n = 9 slices from 8 mice for WT and 10 slices from 8 mice for KO. *p < 0.05, Student's t test. B, Normal basal transmission at Lrfn2−/− SC-CA1 synapses (P30–P35), as shown by the input/output relationship between fiber volley and fEPSP slopes. n = 8, 3 for WT and KO, repeated-measures two-way ANOVA. C, Normal paired-pulse facilitation at Lrfn2−/− SC-CA1 synapses (P30–P35), as shown by the relationship between interpulse intervals and fEPSP slopes. n = 8, 3 for WT and KO, repeated-measures two-way ANOVA. D, Increased NMDAR EPSCs at Lrfn2−/− SC-CA1 synapses (P22–P27), as shown by the relationship between stimulus intensities and initial slopes of NMDAR EPSCs. N = 18, 5 for WT and 16, 4 for KO. *p < 0.05, repeated-measures two-way ANOVA and one-way ANOVA. E, Normal TBS-LTP at Lrfn2−/− SC-CA1 synapses (P28–P36). Bar graphs represent average values during the last 10 min. n = 13, 5 for WT and 10, 6 for KO. NS, Student's t test. F, Suppressed HFS-LTP at Lrfn2−/− SC-CA1 synapses (P28–P36). Bar graphs represent average values during the last 10 min. n = 15, 7 for WT and 16, 7 for KO. *p < 0.05, Student's t test. G, Suppressed LFS-LTD at Lrfn2−/− SC-CA1 synapses (P16–P20). Bar graphs represent average values during the last 10 min. n = 13, 5 for WT and 11, 6 for KO. *p < 0.05, Student's t test.
Given that NMDAR function regulates synaptic plasticity, affecting both LTP and LTD, we measured these parameters at Lrfn2−/− SC-CA1 synapses. Contrary to our initial expectation that NMDAR-dependent synaptic plasticity would be increased, we found that LTP induced by TBS (TBS-LTP) at Lrfn2−/− synapses was normal (Fig. 6E; t(21) = 0.866, p = 0.3963, unpaired t test). In contrast, LTP induced by HFS (HFS-LTP) and LTD induced by LFS (LFS-LTD) were significantly decreased at Lrfn2−/− synapses (Fig. 6F,G; for HFS-LTP, t(29) = 2.128, p = 0.042, unpaired t test; for LFS-LTD, t(22) = 2.239, p = 0.0356, unpaired t test). These results collectively suggest that SALM1 deficiency suppresses specific forms of NMDAR-dependent LTP and LTD without affection basal excitatory transmission or presynaptic neurotransmitter release.
Normal locomotion and anxiety-like behavior in Lrfn2−/− mice
To determine the impacts of SALM1 deletion on behaviors, we subjected Lrfn2−/− mice to a battery of behavioral tests. Lrfn2−/− mice showed normal locomotor activity in the open-field test under both bright-light (110 lux) and light-off (0 lux) conditions (Fig. 7A,B; 110 lux, interaction, F(5,125) = 1.007, p = 0.4162, time, F(5,125) = 72.97, p < 0.0001, genotype, F(1,25) = 0.2067, p = 0.6533, repeated-measures two-way ANOVA for distance moved; 0 lux, interaction, F(5,135) = 0.3176, p = 0.9017, time, F(5,135) = 67.05, p < 0.0001, genotype, F(1,27) = 0.02887, p = 0.8663, repeated-measures two-way ANOVA for distance moved). Continuous monitoring of mouse movements for 3 d in a home cage-like environment (LABORAS cage) revealed no abnormalities in the locomotor activity of Lrfn2−/− mice (Fig. 7C; interaction, F(35,945) = 1.02, p = 0.4376, time, F(35,945) = 61.15, p < 0.0001, genotype, F(1,27) = 0.3567, p = 0.5553, repeated-measures two-way ANOVA).
Figure 7.

Normal locomotion and anxiety-like behavior in Lrfn2−/− mice. A, B, Normal locomotor activity and anxiety-like behavior of Lrfn2−/− mice (3 months) in open-field tests at two different light intensities (110 and 0 lux), as shown by distance moved and time spent in the center region of the open-field arena. n = 13 mice for WT and 14 mice for KO (110 lux), and 13 for WT and 16 for KO (0 lux). NS, repeated-measures two-way ANOVA and Student's t test. C, Normal locomotor activity of Lrfn2−/− mice (2 months) in LABORAS cages, where mouse movements were monitored for 3 consecutive days. n = 13 for WT and 16 for KO. NS, repeated-measures two-way ANOVA. D, Normal anxiety-like behavior of Lrfn2−/− mice (4 months) in the elevated plus-maze test. n = 14 for WT and 17 for KO. NS, Student's t test. E, Normal anxiety-like behavior of Lrfn2−/− mice (4 months) in the light/dark test. n = 14 for WT and 16 for KO. NS, Student's t test.
Lrfn2−/− mice showed normal anxiety-like behaviors in the elevated plus-maze (Fig. 7D; t(29) = 0.3131, p = 0.7564 for time spent in closed arms; t(29) = 1.304, p = 0.2026 for time spent in open arms, unpaired t test for all), the light/dark test (Fig. 7E; t(29) = 0.7436, p = 0.4631, unpaired t test), and the open-field test (center time; Fig. 7A,B; 100 lux, t(25) = 0.8096, p = 0.4258; 0 lux, t(27) = 0.5809, p = 0.5661, unpaired t test for all). These results collectively suggest that SALM1 deficiency has minimal effects on locomotor activity and anxiety-like behavior.
Suppressed USVs in Lrfn2−/− pups, but normal social interaction and repetitive behaviors, in adult Lrfn2−/− mice
We next examined social behaviors. Lrfn2−/− mice displayed normal social interaction and social novelty recognition in the three-chamber test (Fig. 8A; O1 vs S1, chamber time, t(13) = 8.066, p < 0.0001, paired t test for WT; W = 143, p = 0.0002, Wilcoxon matched-pairs signed-rank test for KO; sniffing time, t(13) = 11.36, p < 0.0001, paired t test for WT, t(16) = 9.846, p < 0.0001, paired t test for KO; S1 vs S2, chamber time, t(13) = 2.287, p = 0.0396, paired t test for WT, t(15) = 2.314, p = 0.0325, paired t test for KO; sniffing time, t(13) = 3.898, p = 0.0018, paired t test for WT, W = 143, p = 0.0002, Wilcoxon matched-pairs signed-rank test for KO), and the direct social interaction test (Fig. 8B; nose-to-nose, t(13) = 0.2337, p = 0.8188; following, t(13) = 0.1277, p = 0.9004; total, t(13) = 1.633, p = 0.12165, unpaired t test for all). Measurements of USVs revealed that female encounters elicited normal levels of USVs in adult male Lrfn2−/− mice (Fig. 8C; call number, t(27) = 0.8255, p = 0.4163; call duration, t(27) = 0.4588, p = 0.6501, unpaired t test for all). In addition, adult female Lrfn2−/− mice retrieved pups to an extent similar to that of WT females (Fig. 8D; pup1, U = 240, p = 0.9768; pup2, U = 226.5, p = 0.7315; pup3, U = 213, p = 0.5106, Mann–Whitney U test for all). In contrast, USVs emitted by Lrfn2−/− pups separated from their mothers were diminished, as shown by the number of calls and individual call duration (Fig. 8E; call number, P4, t(36) = 0.4562, p = 0.651, unpaired t test; P6, U = 90.5, p = 0.0088, Mann–Whitney U test; P8, t(30) = 1.123, p = 0.2702, unpaired t test; P10, U = 79, p = 0.0216, Mann–Whitney U test; call duration, P4, U = 175, p = 0.9884, Mann–Whitney U test; P6, t(36) = 1.448, p = 0.1564, unpaired t test; P8, t(30) = 1.878, p = 0.0702, unpaired t test; P10, t(33) = 1.492, p = 0.1452, unpaired t test).
Figure 8.

Suppressed USVs in pups, but normal social interaction, communication, and repetitive behaviors in adult Lrfn2−/− mice. A, Normal social approach and social novelty recognition in Lrfn2−/− mice (3–4 months) in the 3-chamber test, as shown by time spent in the chamber and sniffing. n = 14 mice for WT and 17 mice for KO. *p < 0.05, **p < 0.01, ***p < 0.001, paired t test and Wilcoxon test. B, Normal social interaction in Lrfn2−/− mice (3–4 months) in the direct social interaction test. n = 7 WT mouse pairs and 8 KO mouse pairs. NS, Student's t test. C, Normal USVs of male Lrfn2−/− mice (4–5 months) induced by encounter with a female. n = 13 mice for WT and 16 mice for KO. NS, Student's t test. D, Normal pup retrieval in female Lrfn2−/− mice (3–5 months) induced by WT pups (P1) separated from their mothers, as shown by the time taken to retrieve first, second, and third pups. n = 23 mice for WT and 21 mice for KO. NS, Mann–Whitney U test. E, Suppressed USVs in Lrfn2−/− pups (P4, P6, P8, and P10) separated from their mothers, as shown by number of calls and individual call duration. For P4, n = 16 mice for WT and 22 mice for KO; for P6, n = 17 for WT and 21 for KO; for P8, n = 11 for WT and 21 for KO; for P10, n = 14 for WT and 21 for KO. *p < 0.05, **p < 0.01, NS, Student's t test and Mann–Whitney U test. F, G, Normal repetitive behaviors of Lrfn2−/− mice (3–4 months), as shown by self-grooming, digging, and marble burying. For digging and grooming, n = 14 mice for WT and 17 mice for KO; for marble burying, n = 13 for WT and 16 for KO. NS, Student's t test and Mann–Whitney U test. H, Normal motor coordination of Lrfn2−/− mice (4–5 months) in the rotarod test. n = 14 mice for WT and 17 mice for KO. NS, repeated-measures two-way ANOVA.
In tests measuring repetitive behaviors, Lrfn2−/− mice showed normal levels of self-grooming, digging, and marble burying in home cages (Fig. 8F,G; digging, U = 95, p = 0.3561, Mann–Whitney U test; self-grooming, t(29) = 0.5169, p = 0.6092, unpaired t test; marble burying, t(27) = 0.5795, p = 0.5671, unpaired t test). In addition, Lrfn2−/− mice showed normal motor coordination and learning in the rotarod test (Fig. 8H; interaction, F(4,116) = 0.1178, p = 0.9760; time, F(4,116) = 23.56, p < 0.0001; genotype, F(1,29) = 0.1653, p = 0.6873, repeated-measures two-way ANOVA). Collectively, these results suggest that SALM1 deficiency does not affect social interaction or repetitive behaviors, but does affect USVs, a form of social communication, in pups but not in adult mice.
Normal learning and memory in Lrfn2−/− mice
Turning to learning and memory behaviors, we found that Lrfn2−/− mice performed normally in the novel object recognition test and displaced object recognition tests (Fig. 9A,B; for novel object test, t(33) = 1.436, p = 0.1603, unpaired t test; for displaced object recognition test, t(30) = 0.7224, p = 0.4756, unpaired t test). In the Morris water maze test, Lrfn2−/− mice showed normal levels of learning and memory in learning, probe, and reversal phases (Fig. 9C–H; for escape latency, interaction, F(8,136) = 0.6172, p = 0.7623, time, F(8,136) = 45.03, p < 0.0001, genotype, F(1,17) = 0.02015, p = 0.8888, repeated-measures two-way ANOVA; for quadrant occupancy, t(17) = 1.287, p = 0.2155, unpaired t test for target; t(17) = 1.732, p = 0.1014, unpaired t test for left; t(17) = 0.248, p = 0.8071, unpaired t test for right; t(17) = 0.148, p = 0.8841, unpaired t test for opposite; for crossing number, U = 34, p = 0.3796, Mann–Whitney U test; for velocity, t(17) = 0.1917, p = 0.8503, unpaired t test; for reversal quadrant occupancy, t(17) = 2.665, p = 0.0163, unpaired t test for target; t(17) = 1.393, p = 0.1815, unpaired t test for pre_target; t(17) = 1.432, p = 0.1702, unpaired t test for left; t(17) = 0.8738, p = 0.3944, unpaired t test for right; for reversal crossing number, t(17) = 0.3929, p = 0.6993, unpaired t test). In fear conditioning tests, in which mice were exposed to foot shocks in a spatial context combined with a tone, Lrfn2−/− mice showed normal levels of freezing in the same spatial context 24 h after acquisition of fear memory (Fig. 9I,J; interaction, F(4,116) = 0.2241, p = 0.9245, time, F(4,116) = 37.34, p < 0.0001, genotype, F(1,39) = 0.00663, p = 0.9357, repeated-measures two-way ANOVA; for pre-training, U = 98.5, p = 0.3437, Mann–Whitney U test; t(29) = 1.163, p = 0.2545, unpaired t test). Freezing induced by the same sound cues (Cs) 28 h after fear memory acquisition was also normal in Lrfn2−/− mice (Fig. 9K; t(29) = 1.595, p = 0.1215, unpaired t test for pre-Cs 24 h; t(29) = 1.596, p = 0.1212, unpaired t test for Cs 24 h). These results collectively suggest that SALM1 deficiency has minimal effects on learning and memory behaviors.
Figure 9.

Normal learning and memory in Lrfn2−/− mice. A, Normal novel object recognition memory in Lrfn2−/− mice (2–3 months). n = 17 mice for WT and 18 mice for KO. NS, Student's t test. B, Normal displaced object recognition memory in Lrfn2−/− mice (2–3 months). n = 16 mice for WT and KO. NS, Student's t test. C–H, Normal learning and memory in Lrfn2−/− mice (3–4 months) in the learning, probe, and reversal phases of the Morris water maze test. Pre-T, Previous target. n = 9 mice for WT and 10 mice for KO. *p < 0.05, NS, repeated-measures two-way ANOVA, Student's t test, and Mann–Whitney U test. I–K, Normal fear learning and memory in Lrfn2−/− mice (4–5 months). Mice with acquired fear memory in a spatial context combined with a sound cue (I) were tested for contextual fear memory 24 h after training (J) and for cued fear memory 28 h after training (K). n = 14 mice for WT and 17 mice for KO. NS, repeated-measures two-way ANOVA, Student's t test, and Mann–Whitney U test.
Enhanced acoustic startle, but normal prepulse inhibition and susceptibility to induced seizure, in Lrfn2−/− mice
Finally, to assess behaviors in sensory and motor domains, we first measured acoustic startle responses. Lrfn2−/− mice showed enhanced startle responses to stimuli in a high loudness range (>110 dB; Fig. 10A; interaction, F(12,504) = 2.926, p = 0.0006, time, F(12,504) = 133, p < 0.0001, genotype, F(1,42) = 3.192, p = 0.0812; 0 dB, p > 0.9999; 65, p > 0.9999; 70, p > 0.9999; 75, p > 0.9999; 80, p > 0.9999; 85, p > 0.9999; 90, p > 0.9999; 95, p > 0.9999; 100, p > 0.9999; 105, p > 0.9999; 110, p > 0.9999; 115, p > 0.9999; 120, p > 0.9999, repeated-measures two-way ANOVA, Bonferroni's multiple comparison test). In contrast, prepulse inhibition was normal in Lrfn2−/− mice, despite a tendency toward a decrease (Fig. 10B; interaction, F(4,116) = 0.4163, p = 0.7966, time, F(4,116) = 130, p < 0.0001, genotype, F(1,29) = 2.908, p = 0.0988, repeated-measures two-way ANOVA), suggesting that sensory motor integration is normal.
Figure 10.

Enhanced acoustic startle, but normal prepulse inhibition and susceptibility to induced seizure, in Lrfn2−/− mice. A, Enhanced acoustic startle responses of Lrfn2−/− mice (4–5 months) in a high loudness range (>110 dB). n = 20 mice for WT and 24 mice for KO. **p < 0.01, ***p < 0.001, repeated-measures two-way ANOVA with Bonferroni test. B, Normal prepulse inhibition in Lrfn2−/− mice (4–5 months). n = 14 mice for WT and 17 mice for KO. Repeated-measures two-way ANOVA. C, Normal susceptibility to a seizure induced by PTZ (40 mg/kg, i.p.) in Lrfn2−/− mice (5–6 months). The numbers in pie charts indicate the proportions of mice that reached the indicated stages of seizure at the end of the test (10 min). n = 13 mice for WT and 16 mice for KO. NS, χ2 analysis.
In a test measuring seizure susceptibility, Lrfn2−/− mice showed normal susceptibility to seizures induced by PTZ compared with WT mice (Fig. 10C; p = 0.7513, χ2 analysis). These results collectively suggest that SALM1 deficiency leads to sensory hypersensitivity, but has a minimal impact on sensory motor integration or seizure susceptibility.
Discussion
SALM1 deficiency alters NMDAR-dependent synaptic plasticity
SALM1 has been shown to form a complex with NMDARs in vitro and in vivo and to regulate dendritic surface clustering of NMDARs in cultured hippocampal neurons (Wang et al., 2006). We thus expected that SALM1 deficiency in mice would lead to a reduction in NMDAR function. However, our results indicated that SALM1 deficiency causes an increase in NMDAR function in the hippocampus, as evidenced by an increase in NMDA/AMPA ratio and NMDA-EPSCs with normal AMPAR-mediated basal synaptic transmission.
This unexpected increase in NMDAR function led us to predict enhanced NMDAR-dependent synaptic plasticity in Lrfn2−/− mice. However, we found that, although TBS-LTP was normal, NMDAR-dependent HFS-LTP and LFS-LTD were significantly reduced. What might explain these results? One possibility is that abnormally enhanced NMDAR-mediated synaptic transmission at Lrfn2−/− excitatory synapses caused secondary changes in the molecular pathways downstream of NMDAR activation, suppressing NMDAR activation-dependent recruitment or removal of AMPARs during synaptic plasticity. Alternatively, the increase in NMDAR function may merely represent a compensatory change induced by defective synaptic plasticity. These possibilities, which are not mutually exclusive, would create a situation in which NMDAR-mediated synaptic transmission is enhanced and NMDAR-dependent synaptic plasticity is suppressed. Given that SALM1 associates with NMDARs in vivo and promotes dendritic clustering of NMDARs in cultured neurons (Wang et al., 2006), it is unlikely that SALM1 deletion would markedly increase NMDAR-mediated synaptic transmission, making the latter possibility more likely. Although further details remain to be determined, our in vivo results clearly point to the possibility that SALM1 is required for bidirectional activity-dependent changes in AMPAR-mediated synaptic transmission during NMDAR-dependent synaptic plasticity.
In contrast to these significant changes in NMDAR-dependent synaptic plasticity, multiple lines of evidence suggest that SALM1 deficiency has little effect on the development of excitatory synapses, at least in the hippocampus. First, the density of excitatory synapses was normal in the Lrfn2−/− hippocampus, as supported by the normal density of excitatory synapses in EM analyses and the normal frequencies of mEPSCs and sEPSCs. In addition, the size and shape of excitatory synapses were minimally affected, as supported by the normal length, thickness, and perforation of PSDs in EM analyses and the normal amplitudes of mEPSCs and sEPSCs. Therefore, an Lrfn2 deficiency appears to have a greater effect on excitatory synaptic plasticity than on excitatory synapse development.
The findings reported here regarding excitatory synapses differ somewhat from those observed in the hippocampus of another Lrfn2−/− mouse line (Morimura et al., 2017). The investigators in that study reported major abnormalities in the morphology of excitatory synapses and dendritic spines in the hippocampus, with longer and thinner spines and more frequent oddly shaped spinule-like structures (Morimura et al., 2017). Our data, however, indicate no changes in the morphology of excitatory synapses, again, as supported by the normal length, thickness, and perforation of the PSD, results that are consistent with the normal amplitude of mEPSCs.
In terms of NMDAR function, this latter study found increases in the NMDA/AMPA ratio similar to those found here, but reported that LTP, an NMDAR-dependent function, was changed in the opposite direction: whereas our study found suppressed HFS-LTP, this previous study reported enhanced HFS-LTP. This reported increase in LTP could be attributable to the increased number of silent synapses observed in their mice because silent synapses would have more room to accommodate incoming AMPARs during LTP. Our Lrfn2−/− mice, however, appeared to have normal silent synapses, as supported by the normal frequency of mEPSCs. Therefore, silent synapses could not explain our LTP phenotype; moreover, LTP was suppressed rather than enhanced in our mice. However, it should be noted that NMDAR and LTP phenotypes in the two mouse lines arguably involve a similar defect, limited NMDAR-dependent synaptic delivery of AMPARs, although these defects appear to occur at different stages of synapse development: an early unsilencing stage of excitatory synapses and a relatively late post-unsilencing stage of excitatory synapse maturation.
Both studies used mice that lacked the same exon 2 of the Lrfn2 gene and had the same genetic background (C57BL/6J). Possible explanations for discrepancies in synapse phenotypes could include differences in conditions in which mice were bred and handled, the ages of mice used for slice recordings (P21–P35 in our study and 3–6 months in the prior study), the method for preparing brain slices, or electrophysiology conditions (e.g., buffer solutions).
SALM1 deficiency suppresses inhibitory synapse development
An unexpected finding of our study was that an Lrfn2 deficiency leads to suppression of inhibitory synapse development in the hippocampus, as supported by the decreased density of inhibitory synapses in EM analyses and the decreased frequency of mIPSCs and sIPSCs (Figs. 3, 4). It is possible that SALM1 could be targeted to inhibitory postsynaptic sites in CA1 pyramidal neurons, in addition to excitatory synapses, where it may regulate inhibitory synapse development, perhaps by participating in trans-synaptic adhesion with as yet unknown presynaptic ligands. Although SALM1 is known to associate with excitatory postsynaptic proteins such as PSD-95 and NMDARs (Ko et al., 2006; Morimura et al., 2006; Wang et al., 2006), SALM1 might also be targeted to inhibitory postsynaptic sites through cis interactions with other SALMs such as SALM4 and SALM5 (Wang et al., 2008; Lie et al., 2016, 2018), which do not have PSD-95-binding C-terminal tails and might be localized to inhibitory synapses. In addition, these cis interactions might be mediated by the dimerization of SALMs recently identified in SALM5 and SALM2 by x-ray crystallographic studies (Goto-Ito et al., 2018; Lin et al., 2018). However, proteomic studies using proximity biotinylation have identified SALM1 at excitatory but not inhibitory synapses (Loh et al., 2016; Uezu et al., 2016), although these results do not preclude the possibility that SALM1 localizes at inhibitory synaptic sites.
Alternatively, SALM1 may be expressed in presynaptic GABAergic neurons. Indeed, our FISH experiments revealed the presence of SALM1/Lrfn2 mRNA signals in cortical and hippocampal GABAergic neurons in addition to glutamatergic neurons. In GABAergic neurons, SALM1 may be targeted to presynaptic axon terminals, where it could participate in trans-synaptic adhesion or regulate presynaptic differentiation. Consistent with this possibility, SALM1 immunogold EM signals have been detected in presynaptic nerve terminals (Thevenon et al., 2016). Another possibility is that SALM1 in GABAergic neurons may be targeted to dendritic excitatory synapses. A lack of SALM1 at these sites might suppress excitatory synapse development or function, consequently suppressing the output functions of GABAergic neurons.
SALM1 deficiency alters social communication and startle behavior
In our study, we found that Lrfn2 KO led to suppression of USVs in pups and enhancement of acoustic startle responses in adults. Our Lrfn2−/− mice, however, were largely normal in other behavioral domains, including locomotion, anxiety-like behavior, social interaction, repetitive behavior, learning and memory, and seizure propensity. Behavioral test results reported for the previous Lrfn2−/− mouse line (Morimura et al., 2017) show some overlap with ours, but are largely dissimilar. The most similar reported behavior is enhanced acoustic startle; the concurrence of the two studies on this point strongly suggests that SALM1 is required for normal development of auditory startle responses. The prior study did not observe any changes in pup USVs, another strong phenotype observed in our mice. Behaviors that were uniquely observed in the previously reported Lrfn2−/− mouse line include suppressed social interaction, enhanced repetitive behavior, and enhanced learning and memory (Morimura et al., 2017). Again, these discrepancies could be attributable to differences in experimental conditions, including mouse breeding and handling. Importantly, the time at which behavioral experiments were performed differed between the two studies: during a lights-off period for our study and during a lights-on period for the prior study.
The acoustic startle response has been shown to involve oligosynaptic circuits and a small group of giant neurons in the caudal nucleus of the pontine reticular formation (PnC) (Yeomans and Frankland, 1995; Koch and Schnitzler, 1997), a brain region where SALM1/Lrfn2 mRNA signals are detectable, albeit at relatively low levels; moreover, current images lack sufficient cellular resolution to provide reliable information on this point (Fig. 3 and Allen Brain Atlas). In addition, inhibition of NMDARs in the PnC by local infusion of AP5 has been shown to suppress the acoustic startle response markedly in rats (Miserendino and Davis, 1993). Given the abnormally enhanced NMDAR function in the Lrfn2−/− hippocampus, it is tempting to speculate that SALM1 deletion in the PnC might lead to abnormal increases in NMDAR function and acoustic startle response.
USVs are an important mode of social communication in rodents and deficits in USVs have been observed in many mouse models of ASD (Scattoni et al., 2009; Wöhr, 2014). Intriguingly, our Lrfn2−/− mice showed suppressed isolation-induced USVs in pups, but normal female-induced USVs in adults. This difference may be attributable to the distinct nature of the two USV types: pup USVs are more anxiety- and development-related behaviors, whereas adult USVs are more reproduction related (Scattoni et al., 2009). Indeed, distinct pup and adult USV phenotypes have been reported in neuroligin-2-KO mice, which also show suppressed pup USVs but normal adult USVs (Wöhr et al., 2013).
Pup USVs are known to involve many brain regions, including the periaqueductal gray (PAG) and amygdala (Hofer, 1996), where SALM1/Lrfn2 mRNA is expressed (Fig. 2 and Allen Brain Atlas). In addition, NMDAR inhibition has been shown to suppress isolation-induced pup USVs in rats and mice (Winslow et al., 1990). Mouse pups also respond similarly, although certain NMDAR antagonists seem to induce biphasic responses; enhanced and suppressed USVs at low and high concentrations, respectively (Takahashi et al., 2009). Therefore, SALM1 deletion in mice might abnormally enhance pup USVs through NMDAR hyperactivity in brain regions that include the PAG and amygdala. In addition, because SALM1/LRFN2 has been implicated in ASD (Morimura et al., 2017), the reduction in pup USVs in Lrfn2−/− mice may represent a novel autistic-like behavior. This is a potentially valuable tool because early symptoms are relatively common in human ASDs, but early behavioral measures are rare in animal models of ASD (Silverman et al., 2010; Wöhr and Scattoni, 2013).
In conclusion, our results suggest that SALM1 is important for NMDAR function at excitatory synapses and is required for normal synapse development at inhibitory synapses. Behaviorally, SALM1 is required for normal pup USVs and acoustic startle, but not for the other behaviors tested.
Footnotes
This work was supported by the National Research Foundation of Korea (Ministry of Science, ICT and Future Planning Grant NRF-2017M3C7A1048566 to H.K., NRF Global PhD Fellowship Program Grant NRF-2015H1A2A1033937 to R.K., Grant NRF-2017R1A5A2015391 to Y.C.B., Grant 2016R1A2B200682 to J.K., and Grant NRF-2017R1D1A1B03032935 to M.H.K) and the Institute for Basic Science (Grant IBS-R002-D1 to E.K.).
The authors declare no competing financial interests.
References
- Bemben MA, Shipman SL, Nicoll RA, Roche KW (2015) The cellular and molecular landscape of neuroligins. Trends Neurosci 38:496–505. 10.1016/j.tins.2015.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biederer T, Stagi M (2008) Signaling by synaptogenic molecules. Curr Opin Neurobiol 18:261–269. 10.1016/j.conb.2008.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y, Nam J, Whitcomb DJ, Song YS, Kim D, Jeon S, Um JW, Lee SG, Woo J, Kwon SK, Li Y, Mah W, Kim HM, Ko J, Cho K, Kim E (2016) SALM5 trans-synaptically interacts with LAR-RPTPs in a splicing-dependent manner to regulate synapse development. Sci Rep 6:26676. 10.1038/srep26676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalva MB, McClelland AC, Kayser MS (2007) Cell adhesion molecules: signalling functions at the synapse. Nat Rev Neurosci 8:206–220. 10.1038/nrn2075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit J, Ghosh A (2016) Specification of synaptic connectivity by cell surface interactions. Nat Rev Neurosci 17:22–35. 10.1038/nrn.2015.1 [DOI] [PubMed] [Google Scholar]
- Ferraro TN, Golden GT, Smith GG, St Jean P, Schork NJ, Mulholland N, Ballas C, Schill J, Buono RJ, Berrettini WH (1999) Mapping loci for pentylenetetrazol-induced seizure susceptibility in mice. J Neurosci 19:6733–6739. 10.1523/JNEUROSCI.19-16-06733.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Ito S, Yamagata A, Sato Y, Uemura T, Shiroshima T, Maeda A, Imai A, Mori H, Yoshida T, Fukai S (2018) Structural basis of trans-synaptic interactions between PTPdelta and SALMs for inducing synapse formation. Nat Commun 9:269. 10.1038/s41467-017-02417-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer MA. (1996) Multiple regulators of ultrasonic vocalization in the infant rat. Psychoneuroendocrinology 21:203–217. 10.1016/0306-4530(95)00042-9 [DOI] [PubMed] [Google Scholar]
- Huttner WB, Schiebler W, Greengard P, De Camilli P (1983) Synapsin I (protein I), a nerve terminal-specific phosphoprotein. III. Its association with synaptic vesicles studied in a highly purified synaptic vesicle preparation. J Cell Biol 96:1374–1388. 10.1083/jcb.96.5.1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S, Lee H, Kim E (2017) Synaptic adhesion molecules and excitatory synaptic transmission. Curr Opin Neurobiol 45:45–50. 10.1016/j.conb.2017.03.005 [DOI] [PubMed] [Google Scholar]
- Kim MH, Choi J, Yang J, Chung W, Kim JH, Paik SK, Kim K, Han S, Won H, Bae YS, Cho SH, Seo J, Bae YC, Choi SY, Kim E (2009) Enhanced NMDA receptor-mediated synaptic transmission, enhanced long-term potentiation, and impaired learning and memory in mice lacking IRSp53. J Neurosci 29:1586–1595. 10.1523/JNEUROSCI.4306-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko J, Kim S, Chung HS, Kim K, Han K, Kim H, Jun H, Kaang BK, Kim E (2006) SALM synaptic cell adhesion-like molecules regulate the differentiation of excitatory synapses. Neuron 50:233–245. 10.1016/j.neuron.2006.04.005 [DOI] [PubMed] [Google Scholar]
- Ko J, Choii G, Um JW (2015) The balancing act of GABAergic synapse organizers. Trends Mol Med 21:256–268. 10.1016/j.molmed.2015.01.004 [DOI] [PubMed] [Google Scholar]
- Koch M, Schnitzler HU (1997) The acoustic startle response in rats-circuits mediating evocation, inhibition and potentiation. Behav Brain Res 89:35–49. 10.1016/S0166-4328(97)02296-1 [DOI] [PubMed] [Google Scholar]
- Kolston J, Osen KK, Hackney CM, Ottersen OP, Storm-Mathisen J (1992) An atlas of glycine- and GABA-like immunoreactivity and colocalization in the cochlear nuclear complex of the guinea pig. Anat Embryol (Berl) 186:443–465. [DOI] [PubMed] [Google Scholar]
- Krueger DD, Tuffy LP, Papadopoulos T, Brose N (2012) The role of neurexins and neuroligins in the formation, maturation, and function of vertebrate synapses. Curr Opin Neurobiol 22:412–422. 10.1016/j.conb.2012.02.012 [DOI] [PubMed] [Google Scholar]
- Li Y, Zhang P, Choi TY, Park SK, Park H, Lee EJ, Lee D, Roh JD, Mah W, Kim R, Kim Y, Kwon H, Bae YC, Choi SY, Craig AM, Kim E (2015) Splicing-dependent trans-synaptic SALM3-LAR-RPTP interactions regulate excitatory synapse development and locomotion. Cell Rep 12:1618–1630. 10.1016/j.celrep.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie E, Ko JS, Choi SY, Roh JD, Cho YS, Noh R, Kim D, Li Y, Kang H, Choi TY, Nam J, Mah W, Lee D, Lee SG, Kim HM, Kim H, Choi SY, Um JW, Kang MG, Bae YC, et al. (2016) SALM4 suppresses excitatory synapse development by cis-inhibiting trans-synaptic SALM3-LAR adhesion. Nat Commun 7:12328. 10.1038/ncomms12328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie E, Li Y, Kim R, Kim E (2018) SALM/Lrfn family synaptic adhesion molecules. Front Mol Neurosci 11:105. 10.3389/fnmol.2018.00105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Z, Liu J, Ding H, Xu F, Liu H (2018) Structural basis of SALM5-induced PTPdelta dimerization for synaptic differentiation. Nat Commun 9:268. 10.1038/s41467-017-02414-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh KH, Stawski PS, Draycott AS, Udeshi ND, Lehrman EK, Wilton DK, Svinkina T, Deerinck TJ, Ellisman MH, Stevens B, Carr SA, Ting AY (2016) Proteomic analysis of unbounded cellular compartments: synaptic clefts. Cell 166:1295–1307.e21. 10.1016/j.cell.2016.07.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mah W, Ko J, Nam J, Han K, Chung WS, Kim E (2010) Selected SALM (synaptic adhesion-like molecule) family proteins regulate synapse formation. J Neurosci 30:5559–5568. 10.1523/JNEUROSCI.4839-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miserendino MJ, Davis M (1993) NMDA and non-NMDA antagonists infused into the nucleus reticularis pontis caudalis depress the acoustic startle reflex. Brain Res 623:215–222. 10.1016/0006-8993(93)91430-Z [DOI] [PubMed] [Google Scholar]
- Missler M, Südhof TC, Biederer T (2012) Synaptic cell adhesion. Cold Spring Harb Perspect Biol 4:a005694. 10.1101/cshperspect.a005694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimura N, Inoue T, Katayama K, Aruga J (2006) Comparative analysis of structure, expression and PSD95-binding capacity of lrfn, a novel family of neuronal transmembrane proteins. Gene 380:72–83. 10.1016/j.gene.2006.05.014 [DOI] [PubMed] [Google Scholar]
- Morimura N, Yasuda H, Yamaguchi K, Katayama KI, Hatayama M, Tomioka NH, Odagawa M, Kamiya A, Iwayama Y, Maekawa M, Nakamura K, Matsuzaki H, Tsujii M, Yamada K, Yoshikawa T, Aruga J (2017) Autism-like behaviours and enhanced memory formation and synaptic plasticity in Lrfn2/SALM1-deficient mice. Nat Commun 8:15800. 10.1038/ncomms15800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam J, Mah W, Kim E (2011) The SALM/Lrfn family of leucine-rich repeat-containing cell adhesion molecules. Semin Cell Dev Biol 22:492–498. 10.1016/j.semcdb.2011.06.005 [DOI] [PubMed] [Google Scholar]
- Ottersen OP, Storm-Mathisen J (1984) Neurons containing or accumulating transmitter amino acids. Handb Chem Neuroanat 3:141–246. [Google Scholar]
- Ottersen OP, Storm-Mathisen J, Madsen S, Skumlien S, Strømhaug J (1986) Evaluation of the immunocytochemical method for amino acids. Med Biol 64:147–158. [PubMed] [Google Scholar]
- Paik SK, Bae JY, Park SE, Moritani M, Yoshida A, Yeo EJ, Choi KS, Ahn DK, Moon C, Shigenaga Y, Bae YC (2007) Developmental changes in distribution of gamma-aminobutyric acid- and glycine-immunoreactive boutons on rat trigeminal motoneurons. I. Jaw-closing motoneurons. J Comp Neurol 503:779–789. 10.1002/cne.21423 [DOI] [PubMed] [Google Scholar]
- Rautiainen MR, Paunio T, Repo-Tiihonen E, Virkkunen M, Ollila HM, Sulkava S, Jolanki O, Palotie A, Tiihonen J (2016) Genome-wide association study of antisocial personality disorder. Transl Psychiatry 6:e883. 10.1038/tp.2016.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M (2001) Regulation of dendritic spine morphology and synaptic function by shank and homer. Neuron 31:115–130. 10.1016/S0896-6273(01)00339-7 [DOI] [PubMed] [Google Scholar]
- Scattoni ML, Crawley J, Ricceri L (2009) Ultrasonic vocalizations: a tool for behavioural phenotyping of mouse models of neurodevelopmental disorders. Neurosci Biobehav Rev 33:508–515. 10.1016/j.neubiorev.2008.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seabold GK, Wang PY, Chang K, Wang CY, Wang YX, Petralia RS, Wenthold RJ (2008) The SALM family of adhesion-like molecules forms heteromeric and homomeric complexes. J Biol Chem 283:8395–8405. 10.1074/jbc.M709456200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Scheiffele P (2010) Genetics and cell biology of building specific synapse connectivity. Annu Rev Neurosci 33:473–507. 10.1146/annurev.neuro.051508.135302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Kim E (2011) The postsynaptic organization of synapses. Cold Spring Harb Perspect Biol 3: pii: a005678. 10.1101/cshperspect.a005678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiqui TJ, Craig AM (2011) Synaptic organizing complexes. Curr Opin Neurobiol 21:132–143. 10.1016/j.conb.2010.08.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silverman JL, Yang M, Lord C, Crawley JN (2010) Behavioural phenotyping assays for mouse models of autism. Nat Rev Neurosci 11:490–502. 10.1038/nrn2851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. (2017) Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171:745–769. 10.1016/j.cell.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, Yap JJ, Bohager DZ, Faccidomo S, Clayton T, Cook JM, Miczek KA (2009) Glutamatergic and GABAergic modulations of ultrasonic vocalizations during maternal separation distress in mouse pups. Psychopharmacology 204:61–71. 10.1007/s00213-008-1437-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Craig AM (2013) Protein tyrosine phosphatases PTPdelta, PTPsigma, and LAR: presynaptic hubs for synapse organization. Trends Neurosci 36:522–534. 10.1016/j.tins.2013.06.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thevenon J, Souchay C, Seabold GK, Dygai-Cochet I, Callier P, Gay S, Corbin L, Duplomb L, Thauvin-Robinet C, Masurel-Paulet A, El Chehadeh S, Avila M, Minot D, Guedj E, Chancenotte S, Bonnet M, Lehalle D, Wang YX, Kuentz P, Huet F, et al. (2016) Heterozygous deletion of the LRFN2 gene is associated with working memory deficits. Eur J Hum Genet 24:911–918. 10.1038/ejhg.2015.221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uezu A, Kanak DJ, Bradshaw TW, Soderblom EJ, Catavero CM, Burette AC, Weinberg RJ, Soderling SH (2016) Identification of an elaborate complex mediating postsynaptic inhibition. Science 353:1123–1129. 10.1126/science.aag0821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, Ko J (2013) LAR-RPTPs: synaptic adhesion molecules that shape synapse development. Trends Cell Biol 23:465–475. 10.1016/j.tcb.2013.07.004 [DOI] [PubMed] [Google Scholar]
- Um JW, Ko J (2017) Neural glycosylphosphatidylinositol-anchored proteins in synaptic specification. Trends Cell Biol 27:931–945. 10.1016/j.tcb.2017.06.007 [DOI] [PubMed] [Google Scholar]
- Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas J, Yasenchak J, Chernomorsky R, Boucher M, Elsasser AL, Esau L, Zheng J, Griffiths JA, Wang X, Su H, Xue Y, Dominguez MG, et al. (2003) High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol 21:652–659. 10.1038/nbt822 [DOI] [PubMed] [Google Scholar]
- Valnegri P, Sala C, Passafaro M (2012) Synaptic dysfunction and intellectual disability. Adv Exp Med Biol 970:433–449. 10.1007/978-3-7091-0932-8_19 [DOI] [PubMed] [Google Scholar]
- Voineagu I, Wang X, Johnston P, Lowe JK, Tian Y, Horvath S, Mill J, Cantor RM, Blencowe BJ, Geschwind DH (2011) Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature 474:380–384. 10.1038/nature10110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Chang K, Petralia RS, Wang YX, Seabold GK, Wenthold RJ (2006) A novel family of adhesion-like molecules that interacts with the NMDA receptor. J Neurosci 26:2174–2183. 10.1523/JNEUROSCI.3799-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PY, Seabold GK, Wenthold RJ (2008) Synaptic adhesion-like molecules (SALMs) promote neurite outgrowth. Mol Cell Neurosci 39:83–94. 10.1016/j.mcn.2008.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wells MF, Wimmer RD, Schmitt LI, Feng G, Halassa MM (2016) Thalamic reticular impairment underlies attention deficit in Ptchd1(Y/-) mice. Nature 532:58–63. 10.1038/nature17427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winslow JT, Insel TR, Trullas R, Skolnick P (1990) Rat pup isolation calls are reduced by functional antagonists of the NMDA receptor complex. Eur J Pharmacol 190:11–21. 10.1016/0014-2999(90)94107-9 [DOI] [PubMed] [Google Scholar]
- Wöhr M. (2014) Ultrasonic vocalizations in shank mouse models for autism spectrum disorders: detailed spectrographic analyses and developmental profiles. Neurosci Biobehav Rev 43:199–212. 10.1016/j.neubiorev.2014.03.021 [DOI] [PubMed] [Google Scholar]
- Wöhr M, Scattoni ML (2013) Behavioural methods used in rodent models of autism spectrum disorders: current standards and new developments. Behav Brain Res 251:5–17. 10.1016/j.bbr.2013.05.047 [DOI] [PubMed] [Google Scholar]
- Wöhr M, Silverman JL, Scattoni ML, Turner SM, Harris MJ, Saxena R, Crawley JN (2013) Developmental delays and reduced pup ultrasonic vocalizations but normal sociability in mice lacking the postsynaptic cell adhesion protein neuroligin2. Behav Brain Res 251:50–64. 10.1016/j.bbr.2012.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won S, Levy JM, Nicoll RA, Roche KW (2017) MAGUKs: multifaceted synaptic organizers. Curr Opin Neurobiol 43:94–101. 10.1016/j.conb.2017.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Seo J, Nair R, Han S, Jang S, Kim K, Han K, Paik SK, Choi J, Lee S, Bae YC, Topham MK, Prescott SM, Rhee JS, Choi SY, Kim E (2011) DGKiota regulates presynaptic release during mGluR-dependent LTD. EMBO J 30:165–180. 10.1038/emboj.2010.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeomans JS, Frankland PW (1995) The acoustic startle reflex: neurons and connections. Brain Res Brain Res Rev 21:301–314. 10.1016/0165-0173(96)00004-5 [DOI] [PubMed] [Google Scholar]
- Yuzaki M. (2017) The C1q complement family of synaptic organizers: not just complementary. Curr Opin Neurobiol 45:9–15. 10.1016/j.conb.2017.02.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Details of animals used and statistical results. Download Figure 1-1, DOCX file (567.1KB, docx)