

Abstract
Mutations or altered protein levels of SHANK3 are implicated in neurodevelopmental disorders such as Phelan–McDermid syndrome, autism spectrum disorders, and schizophrenia (Guilmatre et al., 2014). Loss of SHANK3 in mouse models results in decreased synapse density and reduction in the levels of multiple synaptic proteins (Jiang and Ehlers, 2013). The family of SHANK scaffolding molecules are among the most heavily ubiquitinated proteins at the postsynaptic density. The ubiquitin-dependent proteasome degradation of SHANK is regulated by synaptic activity and may contribute to activity-dependent synaptic remodeling (Ehlers, 2003; Shin et al., 2012). However, the identity of the specific deubiquitinating enzymes and E3 ligases that regulate SHANK ubiquitination at synapses are unknown. Here we identify USP8/UBPY as a deubiquitinating enzyme that regulates SHANK3 and SHANK1 ubiquitination and protein levels. In primary rat neurons, USP8 enhances SHANK3 and SHANK1 protein levels via deubiquitination and increases dendritic spine density. Additionally, USP8 is essential for changes in SHANK3 protein levels following synaptic activity modulation. These data identify USP8 as a key modulator of SHANK3 downstream of synaptic activity.
SIGNIFICANCE STATEMENT Precise regulation of the protein levels of the postsynaptic scaffolding protein SHANK3 is essential for proper neurodevelopment. Mutations of SHANK3 have been identified in Phelan–McDermid syndrome, autism spectrum disorders, and schizophrenia (Guilmatre et al., 2014). In this research, we identify USP8 as a key enzyme that regulates SHANK3 protein levels in neurons. USP8 acts to deubiquitinate SHANK3, which prevents its proteasomal-mediated degradation and enhances overall dendritic spine stability. In the future, the modulation of USP8 deubiquitinating activity could potentially be used to titrate the protein levels of SHANK3 to ameliorate disease.
Keywords: dendritic spine, deubiquitinating enzyme, SHANK1, SHANK3, ubiquitination, USP8
Introduction
The ubiquitin–proteasome system (UPS) is a central mechanism for protein degradation and turnover. Proteins are tagged for degradation through a series of enzymatic reactions culminating with the covalent attachment of K48 polyubiquitin chains to specific lysine residues of target proteins via E3 ligases. This polyubiquitination acts as a molecular tag to direct proteins to the proteasome for proteolysis (Bhattacharyya et al., 2014). Ubiquitin is also used to regulate numerous nonproteolytic cellular functions via conjugation as mono-ubiquitin or non-K48 linkages (Swatek and Komander, 2016). Due to its important roles in fundamental cellular processes, the ubiquitination of proteins is counterbalanced by a set of deubiquitinating enzymes (DUBs) that remove ubiquitin from target proteins and thus inhibit their degradation (Nijman et al., 2005).
In neurons, the UPS is an important mechanism used to regulate activity-dependent protein turnover and structural changes at excitatory synapses (Ehlers, 2003; Bingol and Schuman, 2005; Shin et al., 2012). The morphology of dendritic spines, the postsynaptic actin-based protrusions that house excitatory synapses, changes bidirectionally in response to synaptic activity, and the size and shape of dendritic spines correlates with their synaptic strength (Matsuzaki et al., 2004; Nägerl et al., 2004; Sheng and Hoogenraad, 2007; Araki et al., 2015). Postsynaptic density (PSD) scaffolding molecules, particularly the SHANK and GKAP/SAPAP family of proteins, are some of the most highly ubiquitinated proteins at synapses, and their protein levels are controlled bidirectionally by synaptic activity (Ehlers, 2003; Hung et al., 2010; Shin et al., 2012). Blocking synaptic activity results in a reduction of SHANK ubiquitination and enhanced SHANK protein levels, while increasing excitatory synaptic activity leads to heightened ubiquitination and loss of SHANK protein via proteasomal degradation (Ehlers, 2003). SHANK proteins are abundant and highly concentrated in the PSD, where they interact with GKAP and PSD-95 family members. Together, these scaffolds undergird the structure of the PSD and coordinate intracellular responses to extracellular cues by binding to multiple membrane receptors including NMDA and metabotropic glutamate receptors, as well as intracellular cytoskeletal modulators and signaling enzymes including cortactin and CaMKII (Naisbitt et al., 1999; Tu et al., 1999; Sheng and Hoogenraad, 2007; MacGillavry et al., 2016). Based on its central position and functional effects, the SHANK family has been hypothesized to play a master regulatory role in PSD organization and protein turnover at synapses (Sala et al., 2001; Shin et al., 2012).
The SHANK family of proteins consists of three members, SHANK1, SHANK2, and SHANK3. Notably, multiple mutations in SHANK3 (heterozygous gene deletions, gene duplications, and missense mutations) have been identified in neurodevelopmental disorders, including Phelan–McDermid syndrome, autism spectrum disorders (ASDs), and schizophrenia (Guilmatre et al., 2014). Recently, SHANK1 and SHANK2 mutations have also been implicated in neurodevelopmental disorders (Berkel et al., 2010, 2012; Sato et al., 2012; Leblond et al., 2014). The human genetics findings, together with numerous SHANK3 knock-out and transgenic mouse studies (Peça et al., 2011; Han et al., 2013; Lee et al., 2013; Schmeisser, 2015; Mei et al., 2016; Reim et al., 2017), highlight the importance of the regulation of SHANK3 protein levels in vivo. However, the enzymes that regulate SHANK3 protein levels through ubiquitination are poorly understood (Hung et al., 2010).
Due to the strong link to human disease and the known regulation of SHANK3 turnover by UPS, we performed an unbiased screen of DUBs that might regulate the protein level of SHANK3. We identified USP8/UBPY as a regulator of SHANK3 ubiquitination and protein levels in heterologous cells and neurons. USP8 controls SHANK3 and SHANK1 protein levels (but not GKAP or PSD-95) via deubiquitination, which in turn controls dendritic spine density in neurons. Furthermore, the knockdown of USP8 in neurons prevents bidirectional changes in SHANK3 protein levels induced by altered synaptic activity. Together, these data identify USP8 as a critical enzyme that deubiquitinates PSD scaffold proteins SHANK3 and SHANK1, thereby regulating synapse number and size.
Materials and Methods
Animal use.
All rodent experiments were approved by the Genentech Animal Care and Use Committee and following the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
DNA constructs.
A FLAG-tagged library of 114 human DUB cDNAs was used for the DUB overexpression screen in pRK5 vectors, as described previously (Bingol et al., 2014). The USP8 dead vector was generated using QuikChange II XL (Agilent Technologies) to introduce the point mutation C786S. Human SHANK3 cDNA was synthesized at GENEWiz into the phosphorylated (p) CDNA/FRT/TO (Invitrogen) for stable cell line generation and into pCDNA vector with a TdTomato tag for overexpression and immunofluorescence in cultured neurons. SHANK1 HA-tagged pGW1 was provided by Dr. Eunjoon Kim's laboratory at the Korea Advanced Institute of Science and Technology (Daejeon, South Korea). Human SHANK2 Myc-tagged pCMV, human GKAP pCMV, and Myc-tagged rat USP8 cDNAs were purchased (OriGene Technologies). β-galactosidase (βGAL) and PSD-95 cDNA was described previously (Bingol et al., 2014). Rat USP8 shRNA constructs were purchased from OriGene Technologies and targeted the following sequences: USP8_A (ATCTTGACCTGTCACAGTATGTTATTGGC), USP8_B (TTCGGACATCTCCGTGTCTTCTGTGAGGT), USP8_C (ATGAGAACTGGCTCCTGTGCTATCCACAG), USP8_D (TCCGCCTGAGATGGCTCCTTCGTCTGTGC), and nontargeting shRNA (catalog #TR30008, OriGene Technologies). For knockdown in HEK293 cells, pools of four human siRNAs were purchased (Dharmacon), and targeted the following sequences: USP8 (TGAAATACGTGACTGTTTA, GGACAGGACAGTATAGATA, AAATAAAGCTCAACGAGAA, GGCAAGCCATTTAAGATTA), SHANK3 (GATACAAGCGGCGAGTTTA, GGGCTTCACCTGACTACAA, CGAGATTAGCTCATTGTTT, CGACAAACCAACTGTGATC), and nontargeting pool #1 (D-001206-13-05). For knockdown in primary cortical neuron cultures, the following Accell siRNAs were purchased (Dharmacon): nontargeting control pool #1 D-001206, rat SHANK3 SMARTpool E-080173, USP3 SMARTpool E-101133, UCHL1 SMARTpool E-089822, USP22 SMARTpool E-083656, HDAC6 SMARTpool E-095518, ATXN3 SMARTpool E-096577, JOSD1 SMARTpool E-081141, and USP8 SMARTpool E-092861.
Antibodies and reagents.
The following antibodies and reagents were used: SHANK3 (catalog #162304, Synaptic Systems), guinea pig polyclonal antiserum used for immunofluorescence of cultured neurons 1:1000; SHANK3 (Clone S72-16 SAB5200047, Sigma-Aldrich), a monoclonal mouse antibody used for immunoblotting 1:500; SHANK1 (catalog #NB300-167 162013, Novus Biologicals), a polyclonal rabbit antibody used for immunoblot and immunofluorescence at 1:1000; SHANK2 [Clone N23B/6 75-088, NeuroMab at University of California, Davis (UC Davis), Davis, CA; RRID: AB_2254586], a monoclonal mouse antibody used for immunoblotting and immunofluorescence at 1:1000; Pan SHANK (Clone N23B/49 75-089, NeuroMab UC Davis; RRID:AB_11213314), a monoclonal mouse antibody used for immunoblotting 1:1000; β-actin-HRP conjugate (Clone 13E5 5125, Cell Signaling Technology; RRID:AB_1903890), a monoclonal rabbit antibody used for immunoblotting 1:5000; FLAG (Clone M2 F1804, Sigma-Aldrich; RRID:AB_262044), a monoclonal mouse antibody used for immunoblotting 1:2000; Myc-tag HRP conjugate (Clone 9B11, Cell Signaling Technology; RRID:AB_10707162), mouse monocolonal antibody used for immunoblotting 1:2000; PSD-95 (Clone K28/43, NeuroMab UC Davis; RRID:AB_2292909), a monoclonal mouse antibody used for immunoblotting and immunofluorescence of cultured neurons 1:1000; GKAP (catalog #13602, Cell Signaling Technology), a polyclonal rabbit antibody used for immunoblotting 1:2000; GKAP (Clone S127-31 NBP1-47612, Novus Biologicals; RRID:AB_10010569), a monoclonal mouse antibody used for immunofluorescence of cultured neurons 1:1000; HOMER1A (catalog #8231, Cell Signaling Technology), a polyclonal rabbit antibody used for immunofluorescence of cultured neurons 1:1000; USP8 (Clone US872 SAB4200527, Sigma-Aldrich), a mouse monoclonal antibody used for immunoblotting 1:100; ubiquitinated proteins (Clone FK2 04263, Millipore; RRID:AB_612093), a monoclonal mouse antibody used for immunoblotting 1:1000; HA-Tag (Clone C29F4, Cell Signaling Technology; RRID:AB_1549585), a rabbit monoclonal antibody used for immunoblotting at 1:2000; MG132 (catalog #S2619, Selleckchem; DMSO (catalog #D8418, Sigma-Aldrich; tetrodotoxin (TTX; BML-NA120, Enzo Life Sciences); bicuculline (BICC; catalog #14343, Sigma-Aldrich); PR-619 (catalog #SI9619, LifeSensors); 1,10-phenanthroline (catalog #SI9649, LifeSensors); N-ethylmaleimide (NEM; catalog #E3876, Sigma-Aldrich); papain (catalog #LS003119, Worthington Biochemical Corporation); Neurobasal medium (catalog #21103-049, Invitrogen; poly-d-lysine (catalog #P7280, Sigma-Aldrich); mouse laminin (catalog #23017-015, Invitrogen); B27 medium (catalog #17504-044, Invitrogen); GlutaMAX (catalog #35050, Invitrogen); Pen/Strep (catalog #15140, Invitrogen); Lipofectamine 2000 (catalog #11668027, Invitrogen); FLAG magnetic beads (catalog #M8823, Sigma-Aldrich); Pierce Protein A/G Agarose Beads (catalog #20423, Thermo Fisher Scientific; Tandem Ubiquitin Binding Entity (TUBE) magnetic beads (catalog #UM402M, LifeSensors); recombinant USP8 (E-520, BostonBiochem); TRIzol Reagent (catalog #15596026, Thermo Fisher Scientific); iScript cDNA Synthesis Kit (catalog #1708890, Bio-Rad); and SHANK3 and HPRT1 TaqMan Probes (Applied Biosystems).
Generation of stable cell line.
Stable SHANK3 doxycycline-inducible cell lines were generated using Flp-In T-Rex Core Kit (catalog #K6500-01, Invitrogen). Briefly, pOG44 Flp-recombinase pCDNA and SHANK3 human pCDNA/FRT/TO were transfected into HEK293 Flp-In T-REx cells at a 9:1 ratio. Forty-eight hours post-transfection, cell lines were transferred to selection media containing 50 μg/ml hygromycin and 15 μg/ml blasticidin. Media were exchanged every 3–4 d for ∼2 weeks before the development of cell foci. These foci were placed into independent cultures, expanded, and tested for dox-inducible expression of SHANK3. Positive clones were picked and maintained in selection media. A single stable clone was used for all experiments presented in this article.
Deubiquitinating enzyme overexpression screen.
Stable SHANK3 HEK293 cells were plated in 24 wells with doxycycline, and each well was transfected with a separate DUB from the library. βGAL was transfected as a baseline control, and βGAL-transfected cells were treated with 1 μm MG132 for 24 h as a positive control. Cell lysates were collected 76 h post-transfection and resolved via SDS-PAGE, then were analyzed by immunoblotting for SHANK3, FLAG-DUB, and ACTIN. Band intensity was measured using ImageLab software (Bio-Rad), and SHANK3 intensity was normalized to ACTIN as a loading control. The screen was run twice with two technical replicates each time, and data were pooled, resulting in 11 SHANK3 DUB hits that significantly increased SHANK3 expression over that of the control. A subsequent, more stringent screen was run to further narrow down the list of candidate DUBs. The 11 significant DUBs were transfected into stable SHANK3 cells for 24 h and then assayed as described above for SHANK3 protein expression. The screen was run three times with two technical replicates each time, and data were pooled, resulting in seven statistically significant SHANK3 candidate DUBs. To determine whether MG132 and USP8 overexpression had an additive effect, we transfected USP8 or βGAL control in stable SHANK3 HEK293 cells for 24 h and then treated them with 1 μm MG132 or vehicle for an additional 24 h. SHANK3 protein expression levels were then assayed as described above.
RNA extraction, RT-PCR, and TaqMan real-time PCR assay.
Stable SHANK3 HEK293 cells were plated in six-well plates with doxycycline, and each well was transfected with either βGAL control or USP8 for 48 h. Total RNA was extracted with TRIzol reagent following the manufacturer instructions. Two micrograms of RNA was used for cDNA synthesis using the iScript kit. Then a TaqMan gene expression assay was run for each cDNA sample in triplicate with a standard curve and SHANK3 mRNA levels were normalized to the housekeeping gene HPRT1.
Primary neuronal culture.
Embryos from timed pregnant Sprague Dawley rats were removed at embryonic day 18 and decapitated into cold HBSS/HEPES buffer. The hippocampus or cortex and hippocampus (as indicated in figure legends) were dissected and placed into 5 ml of HBSS/HEPES buffer. Tissue was washed four times before digestion at 37°C for 20 min with papain (0.5 ml at 200 U/ml into 4.5 ml HBSS/HEPES buffer with tissue). Tissue was washed four times with warm HBSS/HEPES buffer and once with Neurobasal medium. Dissociated tissue was triturated and passed through a 70 μm filter before plating on poly-d-lysine/laminin-coated plates or coverglass with Neurobasal medium supplemented with B27, Glutamax, and Pen/Strep media. Cells were plated at a density of 120,000 cells/well in 24-well plates with coverglass for imaging or 250,000 cells/well in 24-well plates and 8 million cells/6 cm plate for biochemical experiments. Media were exchanged 100% 3–5 h after initial plating and then exchanged 50% once per week.
Knockdown and analysis of DUBs in primary neuronal cultures.
Accell siRNAs from Dharmacon were added to primary hippocampal neuronal cultures at day in vitro 10 (DIV10) at a final concentration of 1 μm and refreshed at DIV14. Cultures were lysed at DIV18 and resolved on 4–8% Tris-acetate gels. Gels were analyzed by quantitative immunoblotting using ImageLab software (Bio-Rad), and SHANK3 intensity was normalized to actin as a loading control. The quantifications are from four independent cultures with two to four replicates from each culture.
Transfection, immunofluorescence, and quantification of primary neuronal cultures.
Primary neuronal cultures were transfected between DIV12 and DIV14 with Lipofectamine 2000. Neurons were transfected by incubating cells with 2 μl of Lipofectamine, 400–1000 ng of DNA, and 500 μl of Neurobasal medium for 45 min. In some experiments, neurons were treated with the following drugs before fixation: DMSO (3 or 24 h, vehicle control used at same volume as drug treatment), MG132 (3 h, 1 μm), TTX (24 h, 1 μm), and BICC (24 h, 40 μm). Cells were fixed 1–3 d post-transfection, as indicated in figure legends, using 4% paraformaldehyde in PBS for 12 min at room temperature or 4% paraformaldehyde in PBS for 2 min at room temperature followed by 10 min at −20°C with 100% methanol. Cells were permeabilized and blocked using GDB buffer (0.2% gelatin, 0.5% Triton X-100, 0.8 m NaCl in PBS) and incubated with primary antibodies overnight at 4°C. Primary antibodies were visualized using Alexa Fluor dye-conjugated secondary antibodies. For experiments using fluorescent tags (GFP, TdTomato), antibodies were not used to visualize these tags. Images were acquired by a single experimenter blinded to transfection or treatment using a Zeiss LSM780 laser scanning confocal microscope with a 100× oil-objective (0.5 μm confocal z-step size). Images were quantified by a single experimenter blinded to transfection or treatment using ImageJ software. Image z-stacks were collapsed into maximum projections and thresholded before analysis. In Figure 1A, the fluorescence intensity of the entire field was quantified (81.92 × 81.92 μm). For all other figures, a segment of secondary dendrite (20 × 5 μm) was selected for analysis. For Figure 1, E and F, and Figure 2, A, B, E, G, and H, the entire fluorescence intensity was measured in the SHANK3 channel and normalized to the entire fluorescence intensity in the GFP channel. Spine density was calculated by counting the number of dendrite protrusions <2 μm in length using the GFP channel. For Figure 3, F and G, Figure 4A, and Figure 6, A and B, images were processed using the “Analyze Particles” macro in ImageJ to obtain a fluorescence intensity per puncta, size of individual puncta, and puncta number per 20 μm. The cumulative frequency of puncta intensity and size was calculated using Prism 6 software for Figure 3G.
Figure 1.
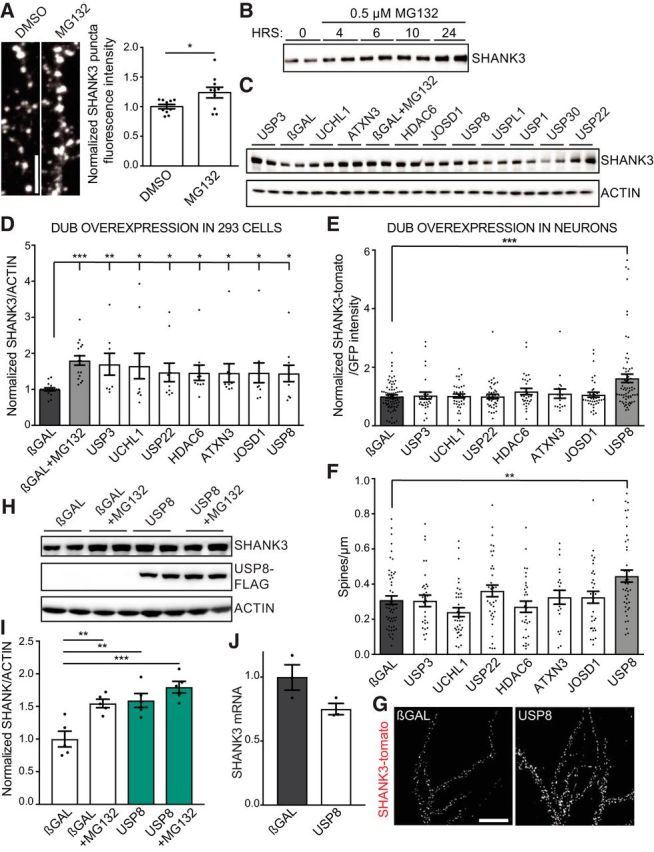
Screen for deubiquitinating enzymes that modulate SHANK3. A, DIV21 dissociated rat cortical neurons were treated for 3 h with DMSO (vehicle) or 1 μm MG132 (proteasome inhibitor) and were stained for endogenous SHANK3. SHANK3 immunofluorescence intensity increases with MG132 treatment. Scale bar, 5 μm. n = 10 images; 1 culture; Student's t test, p = 0.0248. B, Stable human SHANK3-expressing cell line shows increase in SHANK3 protein levels upon proteasome inhibition with 0.5 μm MG132 over time. C, Representative Western blots of SHANK3 and actin after overexpression of individual FLAG-tagged DUBs from a human cDNA library for 76 h in stable SHANK3-expressing cells. Blots show βGAL control, βGAL with MG132 treatment as a positive control, the seven DUBs that increase SHANK3 protein levels, and three control DUBs that do not show a difference with control, USPL1, USP1, USP30. D, Quantification of SHANK3/ACTIN intensity reveals seven DUBs with statistically significant increases compared with βGAL control. βGAL transfected cells treated with MG132 was used as a positive control. n = 5 biological replicates with n = 2–3 technical replicates per experiment. One-way ANOVA: F = 2.316, p < 0.0001; post hoc Student's t test, βGAL vs MG132, p = 0.0001; βGAL vs USP3, p = 0.0033; βGAL vs USP22, p = 0.0279; βGAL vs HDAC6, p = 0.0112; βGAL vs ATXN3, p = 0.0332; βGAL vs UCHL1, p = 0.0249; βGAL vs JOSD1, p = 0.0427; βGAL vs USP8, p = 0.021. E–G, DIV15 rat hippocampal neurons were transfected with GFP, SHANK3-TdTomato, and individual FLAG-tagged DUBs (as indicated) for 24 h. Confocal microscope images were obtained by observers blinded to transfection conditions, and collapsed z-stack images were quantified by a single blinded experimenter. E, F, Overexpression of USP8 results in a significant increase in SHANK3-TdTomato/GFP intensity (E) and dendritic spine density (F). n = 3–5 independent cultures, ∼10 neurons quantified for each condition per culture. Intensity: one-way ANOVA: F = 3.093, p = 0.0146; post hoc Student's t test: βGAL vs USP8, p = 0.0002; spine density: one-way ANOVA: F = 4.595, p = 0.0011; post hoc Student's t test: βGAL vs USP8, p = 0.0068. G, Representative images. Scale bar, 20 μm. H, I, Overexpression of USP8 in combination with 1 μm MG132 treatment (USP8+MG132) for 24 h does not have an additive effect compared with USP8 or MG132 alone. One-way ANOVA: F = 12.18, p = 0.0002; post hoc Student's t test: βGAL vs βGAL + MG132, p = 0.004; βGAL vs USP8, p = 0.0061; βGAL vs USP8 + MG132, p = 0.0007. J, Quantification by TaqMan qPCR of total SHANK3 mRNA relative to HPRT1 mRNA in stable human-SHANK3-expressing HEK293 cells following the overexpression of either βGAL or USP8. Student's t test, p = 0.077. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 2.
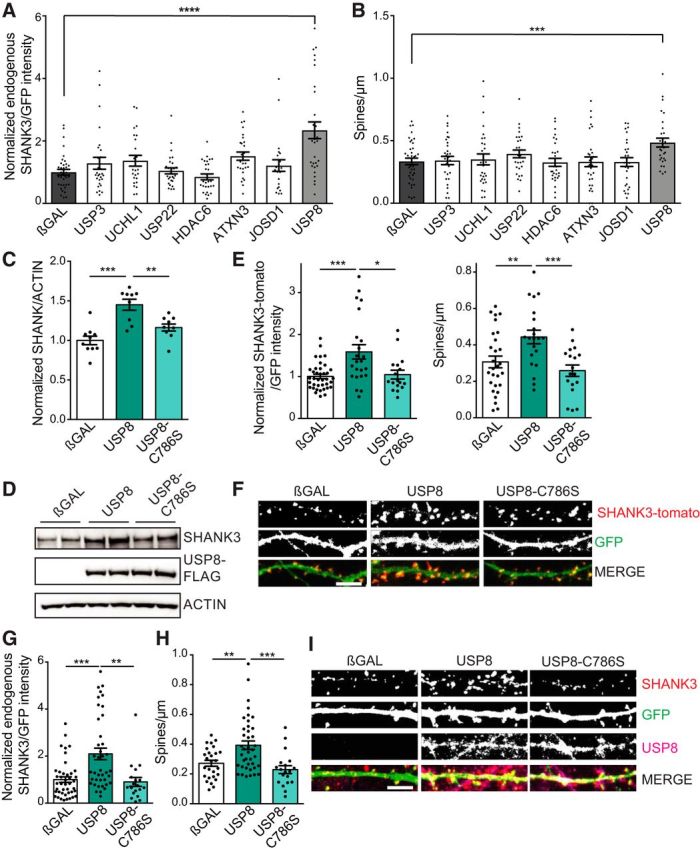
Overexpression of deubiquitinating enzymes in dissociated neurons. A, B, DIV15 rat hippocampal neurons were transfected with GFP and individual FLAG-tagged DUBs (as indicated) for 24 h. Confocal microscope images were obtained by an observer blinded to transfection conditions, and collapsed z-stack images were quantified by a single blinded experimenter. A, B, Overexpression of USP8 results in a significant increase in endogenous SHANK3/GFP intensity (A) and dendritic spine density (B). n = 3–5 independent cultures; ∼10 neurons quantified for each condition per culture. Intensity: one-way ANOVA: F = 8.605, p < 0.0001; post hoc Student's t test: βGAL vs USP8, p < 0.0001; spine density: one-way ANOVA: F = 2.478, p = 0.018; post hoc Student's t test: βGAL vs USP8, p = 0.0008. C, D, Overexpression of catalytically dead mutant USP8–C786S in the human SHANK3-expressing stable cells does not increase SHANK3 protein levels like WT USP8. n = 9 independent transfections per construct. One-way ANOVA: F = 16.28, p < 0.0001; post hoc Student's t test: βGAL vs USP8, p < 0.0001; βGAL vs C786S, p = 0.1062; USP8 vs C786S, p = 0.0022. E, F, Overexpression of WT USP8 in DIV15 rat hippocampal cultures results in an increase in SHANK3-TdTomato intensity and dendritic spine density compared with βGAL control or USP8–C786S (enzyme dead). n = 3 independent cultures, ∼10 neurons/culture. Intensity: one-way ANOVA: F = 9.338, p = 0.0002; post hoc Student's t test: βGAL vs USP8, p = 0.0002; βGAL vs C786S: p = 0.6652; USP8 vs C786S, p = 0.0176; spine density: one-way ANOVA: F = 7.175, p = 0.0015; post hoc Student's t test: βGAL vs USP8, p = 0.0079; βGAL vs C786S, p = 0.3168; USP8 vs C786S, p = 0.0007. F, Representative images. Scale bar, 5 μm. G–I, DIV15 rat hippocampal neurons were transfected with GFP and USP8 or USP8–C786S (enzyme dead) for 24 h and stained for endogenous SHANK3. Confocal microscope images were obtained by observers blinded to transfection conditions and collapsed z-stack images were quantified by a single blinded experimenter. G, H, Overexpression of USP8, but not USP8–C786S, results in an increase in immunofluorescence intensity of endogenous SHANK3 clusters (G) and in dendritic spine density (H) compared with control. n = 3 independent cultures, ∼10 neurons quantified for each condition per culture. Intensity: one-way ANOVA: F = 11.46, p < 0.0001; post hoc Student's t test: βGAL vs USP8, p = 0.0001; βGAL vs C786S, p = 0.6596; USP8 vs C786S, p = 0.0027; spine density: one-way ANOVA: F = 9.888, p = 0.0001; post hoc Student's t test: βGAL vs USP8, p = 0.0026; βGAL vs C786S, p = 0.2037; USP8 vs C786S, p = 0.0007. I, Representative images. Scale bar, 5 μm. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Figure 3.
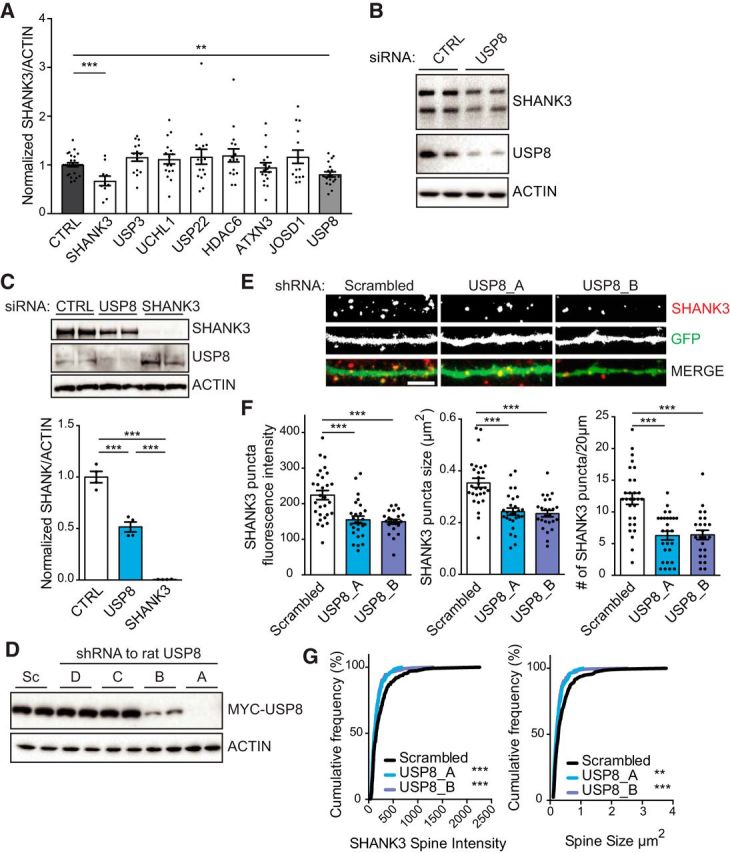
USP8 regulates SHANK3 protein levels and dendritic spine density in neurons. A, Primary hippocampal neurons were treated from DIV10 to DIV18 with Accell siRNA to knock down individual DUBs (as indicated) or SHANK3. SHANK3 protein levels were measured by immunoblot and normalized to actin. Knockdown of SHANK3 or USP8 resulted in a significant loss of SHANK3 protein levels relative to control. One-way ANOVA: F = 2.899, p = 0.0051; post hoc Student's t test: Control vs SHANK3, p = 0.0007; Control vs USP8, p = 0.0046. B, Representative Western blots from knockdown with control (CTRL) or USP8 siRNA. C, Knockdown of USP8 or SHANK3 with Dharmacon ON-TARGET siRNA pools in stable SHANK3-expressing cells for 72 h significantly reduces SHANK3 protein levels. n = 4 independent cultures. One-way ANOVA: F = 137.1, p < 0.0001; post hoc Student's t test: CTRL vs USP8, p = 0.0006; CTRL vs SHANK3, p < 0.0001; USP8 vs SHANK3, p < 0.0001. D, HEK293 cells were transfected with MYC-tagged rat USP8 and four shRNA constructs targeting rat USP8 or a scrambled shRNA control for 72 h. Cell lysates were blotted for& MYC and actin loading control. shRNA constructs A and B efficiently knocked down USP8 65–90% and were used in all of the following experiments. E, Representative images. Scale bar, 5 μm. F, Rat hippocampal dissociated neurons were transfected at DIV14 with a scrambled shRNA or one of two shRNAs that target rat USP8 (USP8_A, USP8_B) for 72 h. At DIV17, neurons were fixed and stained for endogenous SHANK3. Collapsed z-stack images were thresholded, and a mask was developed to identify SHANK3 puncta by a single blinded experimenter in a 20 × 5 μm region of interest. Knockdown of USP8 results in decreased SHANK3 puncta intensity, reduced SHANK3 puncta size, and fewer SHANK3 puncta/20 μm of dendrite. n = 3 independent cultures, ∼10 neurons quantified for each condition per culture. Puncta intensity: one-way ANOVA: F = 15.89, p < 0.0001; post hoc Student's t test: scrambled vs USP8_A, p < 0.0001; scrambled vs USP8_B, p < 0.0001; USP8_A vs USP8_B, p = 0.9341; puncta size: one-way ANOVA: F = 19.68, p < 0.0001; post hoc Student's t test: scrambled vs USP8_A, p < 0.0001; scrambled vs USP8_B, p < 0.0001; USP8_A vs USP8_B, p = 0.9352; puncta count: one-way ANOVA: F = 18.50, p < 0.0001; post hoc Student's t test: scrambled vs USP8_A, p < 0.0001; scrambled vs USP8_B, p < 0.0001; USP8_A vs USP8_B, p = 0.9943. G, Cumulative frequency of SHANK3 puncta intensity and puncta size was significantly reduced in USP8 knock-down neurons relative to control. Cumulative frequency puncta intensity: scrambled, n = 374; USP8_A, n = 184; USP8_B, n = 19; Kolmogorov–Smirnov test: scrambled vs USP8_A, p = 0.0007; scrambled vs USP8_B, p = 0.0002; USP8_A vs USP8_B, p = 0.9473; cumulative frequency puncta size: scrambled, n = 375; USP8_A, n = 184; USP8_B, n = 196; scrambled vs USP8_A, p = 0.0012; scrambled vs USP8_B, p < 0.0001; USP8_A vs USP8_B, p = 0.9410. **p < 0.01, ***p < 0.001.
Figure 4.

Knockdown of USP8 leads to a loss of synaptic puncta and regulates SHANK1 puncta size and intensity. A, Rat hippocampal dissociated neurons were transfected at DIV14 with a scrambled shRNA or one of two shRNAs that target rat USP8 (USP8_A, USP8_B) for 72 h. At DIV17, neurons were fixed and stained for endogenous synaptic proteins with antibodies to SHANK1, SHANK2, PSD-95, GKAP, and HOMER1A. B, Collapsed z-stack images were thresholded and a mask was developed to identify synaptic protein puncta by a single blinded experimenter in a 20 × 5 μm region of interest. Knockdown of USP8 causes a reduction in SHANK1 synaptic puncta intensity and size, while SHANK2, PSD-95, GKAP, and HOMER1A puncta are unaffected. The number of synaptic protein puncta per 20 μm are reduced for all, with the exception of HOMER1A not reaching significance. n = 3–5 independent cultures, ∼10 neurons quantified for each condition per culture. Puncta intensity SHANK1: one-way ANOVA: F = 12.37, p < 0.0001; post hoc Student's t test: scrambled vs USP8_A, p < 0.0001; scrambled vs USP8_B p = 0.0014; USP8_A vs USP8_B, p = 0.5416; puncta intensity SHANK2: one-way ANOVA: F = 0.5540, p = 0.5772; puncta intensity PSD-95: one-way ANOVA: F = 2.583, p = 0.0837; puncta intensity GKAP: one-way ANOVA: F = 0.4559, p = 0.6356; puncta intensity HOMER1A: one-way ANOVA: F = 1.899, p = 0.1566; puncta size SHANK1: one-way ANOVA: F = 4.553, p = 0.0147; post hoc Student's t test: scrambled vs USP8_A, p = 0.0371; scrambled vs USP8_B, p = 0.0304; USP8_A vs USP8_B, p = 0.9995; puncta size SHANK2: one-way ANOVA: F = 0.5615, p = 0.5728; puncta size PSD-95: one-way ANOVA: F = 1.804, p = 0.1732; puncta size GKAP: one-way ANOVA: F = 0.7936, p = 0.4559; puncta size HOMER1A: one-way ANOVA: F = 1.222, p = 0.3002; puncta number SHANK1: one-way ANOVA: F = 9.892, p = 0.0002; post hoc Student's t test: scrambled vs USP8_A, p = 0.0002; scrambled vs USP8_B, p = 0.0054; USP8_A vs USP8_B, p = 0.4991; puncta number SHANK2: one-way ANOVA: F = 6.297, p = 0.0030; post hoc Student's t test: scrambled vs USP8_A, p = 0.0031; scrambled vs USP8_B, p = 0.0245; USP8_A vs USP8_B, p = 0.7621; puncta number PSD-95: one-way ANOVA: F = 13.88, p < 0.0001; post hoc Student's t test: scrambled vs USP8_A, p = 0.0006; scrambled vs USP8_B, p < 0.0001; USP8_A vs USP8_B, p = 0.5508; puncta number GKAP: one-way ANOVA: F = 5.567, p = 0.0054; post hoc Student's t test: scrambled vs USP8_A, p = 0.0066; scrambled vs USP8_B, p = 0.0302; USP8_A vs USP8_B, p = 0.8321; puncta number HOMER1A: one-way ANOVA: F = 1.621, p = 0.2040. B, Representative images. Scale bar, 5 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
Figure 6.
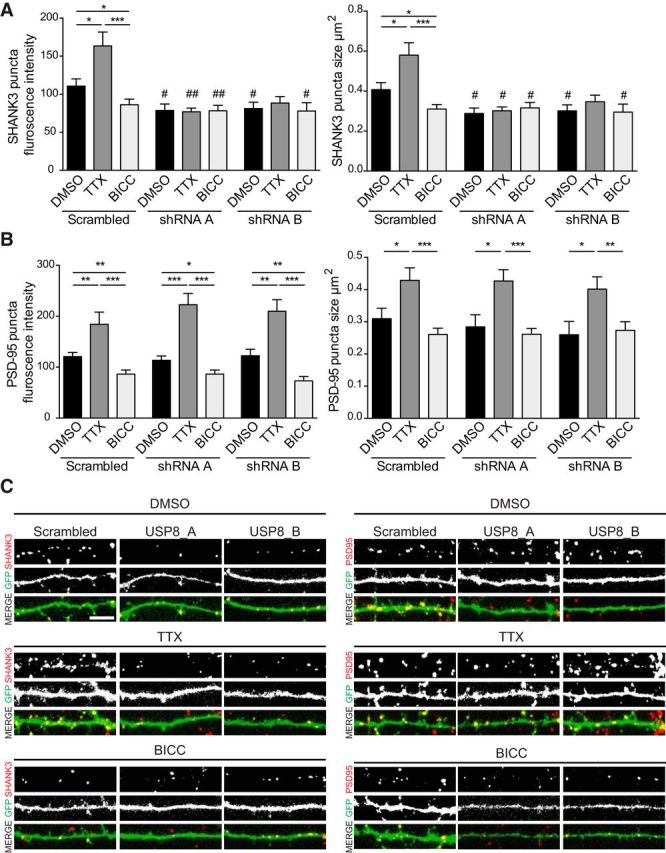
Loss of USP8 occludes the bidirectional changes in SHANK3 puncta intensity and size in response to chronic synaptic activity modulation. A, B, Dissociated rat cortical neurons were transfected with GFP, and a scrambled shRNA or one of two shRNAs that target rat USP8 (USP8_A, USP8_B) for 72 h. Cultures were treated with DMSO, TTX (1 μm), or BICC (40 μm) for the final 24 h before fixation. Neurons were stained for endogenous synaptic proteins with antibodies to SHANK3 or PSD-95. Collapsed z-stack images were thresholded and a mask was created to identify synaptic puncta. All quantifications were done by a single blinded experimenter. A, Knockdown of USP8 causes a significant reduction (#) in SHANK3 puncta fluorescence intensity and size, as shown previously (Fig. 3F). Additionally, knockdown of USP8 blocks the significant bidirectional changes (*) observed upon treatment with TTX or BICC in control neurons. n = 3 independent cultures, ∼8 neurons quantified for each condition per culture. Puncta intensity within drug treatment, p = *: one-way ANOVA: F = 8.273, p < 0.0001; post hoc Student's t test: scrambled DMSO vs scrambled TTX, p = 0.0174; scrambled DMSO vs scrambled BICC, p = 0.0463; scrambled TTX vs scrambled BICC, p = 0.0004; USP8_A DMSO vs USP8_A TTX, p = 0.8546; USP8_A DMSO vs USP8_A BICC, p = 0.9759; USP8_A TTX vs USP8_A BICC, p = 0.8644; USP8_B DMSO vs USP8_B TTX, p = 0.5462; USP8_B DMSO vs USP8_B BICC, p = 0.8134; USP8_B TTX vs USP8_B BICC, p = 0.4643; Puncta intensity between shRNA treatment p = #: scrambled DMSO vs USP8_A DMSO, p = 0.0161; scrambled DMSO vs USP8_A TTX, p = 0.0023; scrambled DMSO vs USP8_A BICC, p = 0.0094; scrambled DMSO vs USP8_B DMSO, p = 0.0249; scrambled DMSO vs USP8_B TTX, p = 0.0902; scrambled DMSO vs USP8_B BICC, p = 0.0304; puncta size within drug treatment p = *: one-way ANOVA: F = 7.330, p < 0.0001; post hoc Student's t test scrambled DMSO vs scrambled TTX, p = 0.0197; scrambled DMSO vs scrambled BICC, p = 0.0293; scrambled TTX vs scrambled BICC, p = 0.0004; USP8_A DMSO vs USP8_A TTX, p = 0.6818; USP8_A DMSO vs USP8_A BICC, p = 0.4774; USP8_A TTX vs USP8_A BICC, p = 0.6699; USP8_B DMSO vs USP8_B TTX, p = 0.3103; USP8_B DMSO vs USP8_B BICC, p = 0.9049; USP8_B TTX vs USP8_B BICC, p = 0.3346; puncta size between shRNA treatment p = #: scrambled DMSO vs USP8_A DMSO, p = 0.0123; scrambled DMSO vs USP8_A TTX, p = 0.0119; scrambled DMSO vs USP8_A BICC, p = 0.0488; scrambled DMSO vs USP8_B DMSO, p = 0.0288; scrambled DMSO vs USP8_B TTX, p = 0.2267; scrambled DMSO vs USP8_B BICC, p = 0.0406. ##p < 0.01. B, Knockdown of USP8 has no change on overall PSD-95 puncta fluorescence intensity or puncta size, as shown previously (Fig. 4A) and PSD-95 in USP8 knockdown neurons still responds synaptic activity modulation (*). n = 3 independent cultures, ∼8 neurons quantified for each condition per culture. Puncta intensity within drug treatment: one-way ANOVA: F = 15.45, p < 0.0001; post hoc Student's t test scrambled DMSO vs scrambled TTX, p = 0.0026; scrambled DMSO vs scrambled BICC, p = 0.0057; scrambled TTX vs scrambled BICC, p < 0.0001; USP8_A DMSO vs USP8_A TTX, p < 0.0001; USP8_A DMSO vs USP8_A BICC, p = 0.0338; USP8_A TTX vs USP8_A BICC, p < 0.0001; USP8_B DMSO vs USP8_B TTX, p = 0.0013; USP8_B DMSO vs USP8_B BICC, p = 0.0032; USP8_B TTX vs USP8_B BICC, p < 0.0001; puncta size within drug treatment: one-way ANOVA: F = 4.753, p < 0.0001; post hoc Student's t test scrambled DMSO vs scrambled TTX, p = 0.0231; scrambled DMSO vs scrambled BICC, p = 0.2250; scrambled TTX vs scrambled BICC, p = 0.0005; USP8_A DMSO vs USP8_A TTX, p = 0.0091; USP8_A DMSO vs USP8_A BICC, p = 0.6029; USP8_A TTX vs USP8_A BICC, p = 0.0002; USP8_B DMSO vs USP8_B TTX, p = 0.0162; USP8_B DMSO vs USP8_B BICC, p = 0.7856; USP8_B TTX vs USP8_B BICC, p = 0.0092. C, Representative images. Scale bar, 5 μm. *p < 0.05, **p < 0.01, ***p < 0.001.
Tandem ubiquitin binding domain pulldown.
Ubiquitination of proteins was determined using a magnetic TUBE pulldown assay. TUBEs recognize tetra-ubiquitin in the nanomolar range and have been demonstrated to bind to all seven linkage types. For pulldowns, cells were treated with 1 μm MG132 for 3–5 h before lysis. Cells were lysed (50 mm Tris, pH 7.5, 150 mm NaCl, 1 mm EDTA, 1% NP-40, 10% glycerol) with protease, phosphatase, and DUB inhibitors and lysates were clarified by centrifugation for 10 min at 14,000 × g at 4°C. Input samples were removed before adding 40 μl of magnetic-TUBE slurry to 1 ml of lysate. Inputs and pulldowns were rotated overnight at 4°C. Beads were then washed three times with TBST (20 mm Tris, pH 8.0, 150 mm NaCl, 0.1% Tween-20), and samples were resolved with SDS-PAGE, where input lanes contain 1–5% of protein lysate and were analyzed by immunoblotting.
In vitro direct deubiquitination assay.
HA-tagged ubiquitin was cotransfected along with SHANK1, SHANK2, or SHANK3 in six-well plates of HEK293 cells using Lipofectamine 2000 for 48 h. Cell were treated with 1 μm MG132 for 3–5 h before lysis. Cells were lysed in RIPA buffer with protease, phosphatase, and DUB inhibitors, and then heated to 85°C for 10 min to kill any remaining DUB activity and to disrupt any protein–protein interactions. Lysates were then cooled to 4°C and normalized to total protein concentration. Lysates were precleared with protein A/G beads for 20 min rotating at 4°C. Beads were removed, and lysates were incubated overnight at 4°C with 2 μg of PAN SHANK antibody. The following day, immune complexes were precipitated with protein A/G beads for 2 h at 4°C followed by washing with RIPA buffer three times and then reconstituting in 100 μl of DUB buffer (10 mm Tris-HCl, pH 8, 1 mm dithiothreitol, 1 mm EDTA). Next, immunoprecipitated lysates were incubated with vehicle, 5 μm purified recombinant USP8, or 5 μm purified recombinant USP8 plus a 40 mm N-ethylmaleimide DUB inhibitor rotating for 37°C for 90 min. Beads were again washed three times and then incubated in sample loading buffer for 10 min at 100°C before loading on SDS-PAGE and Western blotting for SHANK and HA-ubiquitin.
Experimental design and statistical analysis.
All statistical analyses were performed using GraphPad Prism 6 software. A p value <0.05 was regarded as statistically significant, and specific statistical tests along with p values are reported within each figure legend. All data are displayed as the mean with SEM, and significance is represented with asterisks (*) or hashtags (#) within the figures. One-way ANOVA with post hoc Student's t tests were used to analyze the quantification of the DUB screen in Figure 1D–F, and I, and Figure 2, A and B. All imaging data were collected and quantified by a single experimenter blinded to condition or treatment, and data were analyzed from three to five independent primary cultures with ∼10 neurons images per culture (n = number of neurons). One-way ANOVA with post hoc Student's t tests were used to analyze the quantification of USP8 overexpression or knockdown in Figure 1I, Figure 2, C, E, G, and H, Figure 3, A, C, and F, Figure 4A, and Figure 6, A and B. The cumulative frequency was analyzed with Kolmogorov–Smirnov tests, shown in Figure 3G. A repeated-measures ANOVA with post hoc Student's t tests were used to analyze the TUBE pulldown assays in Figure 5, B and D.
Figure 5.
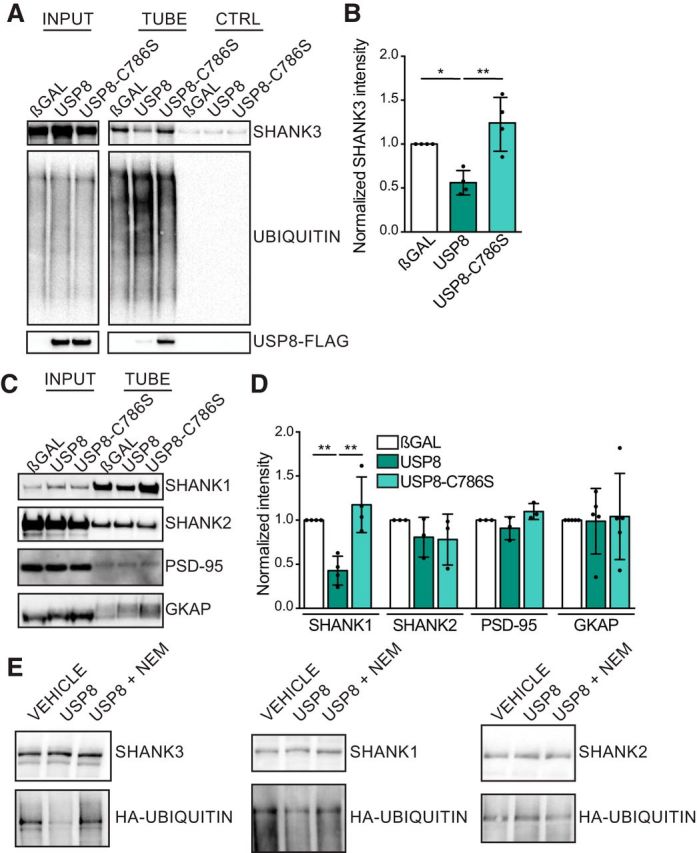
USP8 regulates the ubiquitination of SHANK3 and SHANK1. A, Representative immunoblots from assays that used TUBEs coupled to magnetic beads to pull down polyubiquitinated proteins. Stable SHANK3-expressing cells were transfected with βGAL control, USP8, or USP8–C786S (enzymatic dead) and were treated with the proteasome inhibitor MG132 for 3 h before lysis and pulldown. B, Intensity of SHANK3 immunoblot bands from pulldown lanes were quantified. Overexpression of USP8 leads to a decrease in the level of polyubiquitinated SHANK3 relative to βGAL control or USP8–C786S. n = 4 assays from independent cultures. One-way ANOVA: F = 12.63, p = 0.0024; βGAL vs USP8, p = 0.0264; βGAL vs C786S, p = 0.2388; USP8 vs C786S, p = 0.0020. C, Representative immunoblots of TUBE pulldown assays from HEK293 lysates transiently transfected with SHANK1, SHANK2, PSD-95, or GKAP along with βGAL, USP8, or USP8–C786S. D, Quantification of TUBE pulldown assays from C. SHANK1 was less ubiquitinated when USP8 was overexpressed relative to βGAL control or USP8–C786S. SHANK2, PSD-95, and GKAP polyubiquitination were unaffected by USP8 overexpression. n = 3–5 assays from independent cultures. SHANK1: one-way ANOVA: F = 14.53, p = 0.0015; βGAL vs USP8, p = 0.0085; βGAL vs C786S, p = 0.4814; USP8 vs C786S, p = 0.0016; SHANK2: one-way ANOVA: F = 0.9666, p = 0.4326; PSD-95: one-way ANOVA: F = 3.235, p = 0.5189; GKAP: one-way ANOVA: F = 0.03123, p = 0.9693. E, Ubiquitinated SHANK proteins were immunoprecipitated from HEK293 cells overexpressing SHANK1, SHANK2, or SHANK3 together with HA-ubiquitin and treated with MG132. Immunoprecipitated SHANK1, SHANK2, or SHANK3 was incubated with vehicle, recombinant USP8, or recombinant USP8 plus the DUB inhibitor NEM in an in vitro deubiquitination assay. HA-ubiquitin signal on SHANK3 and SHANK1 was reduced by recombinant USP8. *p < 0.05, **p < 0.01.
Results
Identification of USP8 as a putative SHANK3 DUB using an unbiased overexpression screen
To confirm that SHANK3 is regulated by the ubiquitin-proteasome in neurons, we treated cultured dissociated rat cortical neurons at DIV21 for 3 h with the proteasome inhibitor MG132. Compared with vehicle control, MG132 increased the level of endogenous SHANK3 protein by ∼25%, as measured by the immunofluorescence intensity of SHANK3 puncta (Fig 1A).
By deubiquitinating their substrates, DUBs should protect substrates from proteasome degradation and lead to higher steady-state levels of those substrates. To identify specific DUBs that might regulate the ubiquitination of SHANK3, we performed an unbiased screen of DUBs for their ability to boost SHANK3 protein levels in a stable HEK293 cell line engineered to overexpress SHANK3. SHANK3 protein expression increased over time following MG132 treatment, confirming it can be degraded by the proteasome in this cell line (Fig 1B). From a FLAG-tagged human DUB cDNA library containing all 114 known human DUBs, we found that seven DUBs (USP3, UCHL1, USP22, HDAC6, ATXN3, JOSD1, USP8) consistently and significantly increased SHANK3 protein expression in this stable cell line, compared with βGAL control (Fig 1C,D). The degree of elevation of SHANK3 induced by the coexpression of these seven DUBs was comparable to MG132 treatment (Fig 1C,D).
To determine whether these seven DUBs regulate SHANK3 protein expression in neurons, we transfected USP3, UCHL1, USP22, HDAC6, ATXN3, JOSD1, or USP8 individually in DIV16 rat dissociated hippocampal neurons along with SHANK3-TdTomato and GFP. As expected, SHANK3-TdTomato localized in a punctate synaptic pattern along dendrites (Fig. 1G). Only cotransfection of USP8 resulted in a significant increase in SHANK3-TdTomato intensity, whereas the other six DUBs tested had no significant effect (Fig 1E). Enhancing SHANK3 protein levels is known to increase dendritic spine number and promote synapse stability (Roussignol et al., 2005; Durand et al., 2012). Indeed, we found that the overexpression of USP8 also led to an ∼60% increase in dendritic spine density in transfected neurons compared with βGAL control, perhaps due to the increase in SHANK3 protein expression (Fig 1F). To determine whether these seven DUBs can act on endogenous SHANK3, we overexpressed each DUB individually in DIV16 hippocampal neurons and immunostained for SHANK3. We found that only the overexpression of USP8 resulted in a significant increase in endogenous SHANK3 protein expression (Fig. 2A), as well as a 45% increase in dendritic spine density (Fig. 2B). These results are consistent with a role for USP8 in deubiquitinating SHANK3 and preventing its degradation via the proteasome.
To test whether the effects of USP8 overexpression and proteasome inhibition might occlude each other, we combined USP8 overexpression with proteasome inhibitor treatment (USP8+MG132) in SHANK3 stable HEK293 cells. This combination did not elevate SHANK3 protein levels more than either USP8 or MG132 alone (Fig. 1H,I). The lack of additivity of the effects of USP8 and MG132 on SHANK3 protein levels is consistent with the idea that USP8 overexpression is acting via the inhibition of UPS degradation of SHANK3. USP8 overexpression was associated with a slight but nonsignificant decrease in SHANK3 mRNA (Fig. 1J), excluding the possibility that the elevation of SHANK3 protein levels upon USP8 overexpression is due to increased transcription of SHANK3.
USP8 regulates SHANK3 and SHANK1 protein expression and dendritic spine density in hippocampal neurons
To determine whether USP8 DUB activity is important for the increase in SHANK3 protein levels, we made a USP8 “dead” enzyme by mutating the catalytic cysteine residue to a serine (USP8-C786S). This mutation has previously been shown to kill the enzyme activity while preserving the overall structure and folding of USP8 (Wu et al., 2004; Mizuno et al., 2005; Avvakumov et al., 2006). In contrast to wild-type (WT) USP8, the overexpression of USP8-C786S did not change SHANK3 levels in the SHANK3 stable HEK293 cell line (Fig 2C,D) and did not significantly change SHANK3-TdTomato intensity or dendritic spine density in hippocampal neurons (Fig 2E,F). Thus, DUB activity is necessary for the positive effects of USP8 on SHANK3 protein levels and dendritic spine density. To investigate whether USP8 DUB activity is important for regulating endogenous SHANK3, we transfected USP8 or USP8-C786S in DIV15 hippocampal neurons and quantified endogenous SHANK3 intensity using immunofluorescence 24 h later. Compared with βGAL control, the overexpression of USP8 increased the endogenous protein levels of SHANK3 by approximately twofold as measured by immunofluorescence intensity of SHANK3 and led to an ∼45% increase in spine density. These changes were not observed with overexpression of the catalytically dead mutant USP8-C786S (Fig. 2G–I), further supporting the hypothesis that USP8 deubiquitinates endogenous SHANK3 and prevents its proteasome degradation.
We next examined the effect of the loss of function of individual DUBs in primary hippocampal neurons. Loss of DUB function should result in a reduction of substrate levels due to enhanced ubiquitination and targeting of the substrate to the proteasome and subsequent degradation. We used Accell siRNAs to knock down each DUB for 8 d in mature primary hippocampal cultures and quantified total SHANK3 levels by immunoblotting. Among the seven DUBs tested, only siRNA knockdown of the DUB USP8, or the knockdown of SHANK3 itself, resulted in a significant loss of SHANK3 protein levels (Fig. 3A,B). These findings further support the hypothesis that USP8 plays an important role in deubiquitinating SHANK3. Additionally, in the stable HEK293 SHANK3-expressing cells, 60% knockdown of the endogenous human USP8 with siRNA resulted in an ∼50% reduction in SHANK3 protein levels (Fig 3C).
To further investigate the impact of loss of USP8 on endogenous SHANK3 in rat hippocampal neurons, we used shRNAs targeting rat USP8. Two of four shRNA-expressing constructs tested (shRNA_A and shRNA_B) were effective in knocking down USP8 protein expression when transiently cotransfected with rat myc-tagged USP8 cDNA in heterologous HEK293 cells (Fig. 3D). Relative to a scrambled shRNA control, shRNA_A and shRNA_B resulted in 90% and 65% loss of USP8 protein after 72 h, respectively (Fig 3D). We tested the knockdown of USP8 for 3 d in DIV17 hippocampal neurons using the shRNA_A, shRNA_B, or scrambled shRNA control. shRNA-transfected neurons were identified by GFP that was expressed from the same vector. We used ImageJ software to identify SHANK3 puncta along a segment of transfected dendrite and quantified SHANK3 intensity per puncta. Knockdown of USP8 resulted in an ∼50% reduction in the density of SHANK3 puncta and an ∼30% decrease in SHANK3 immunofluorescence intensity and size of the puncta that remain (Fig 3E,F). The decrease in size and intensity of SHANK3 puncta is also indicated by a significant leftward shift of the cumulative frequency graphs of individual puncta size and density (Fig 3G).
Beyond SHANK3, we sought to determine whether USP8 knockdown affected other PSD proteins using immunofluorescence to quantify the endogenous levels of SHANK1, SHANK2, PSD-95, GKAP, and HOMER1A. As predicted from the reduction of spine density (and of SHANK3 puncta density), the density of puncta of each of these PSD proteins along dendrites was reduced (though this did not reach significance with HOMER1A), presumably correlating with the loss of synapses in USP8 knockdown neurons (Fig 4A,B). However, when quantifying the intensity and size of individual immunofluorescence puncta, only SHANK1 puncta intensity and puncta size were impacted by the knockdown of USP8 (Fig 4A,B). The size and fluorescence intensity of SHANK2, PSD-95, GKAP, and HOMER1A puncta were unchanged with USP8 knockdown. Together, these results suggest that USP8 controls the protein levels of SHANK1, as well as SHANK3, at excitatory synapses. Depletion of SHANK1 and SHANK3 proteins by USP8 knockdown could account for an overall loss of synapse density.
USP8 is a DUB for SHANK3 and SHANK1
To determine more directly whether USP8 can act as a DUB on SHANK, we used a TUBE pulldown assay to quantify the ubiquitination of proteins. TUBEs have a nanomolar affinity for polyubiquitinated proteins and can be conjugated to magnetic beads to pull down polyubiquitinated species from total cell lysates. In SHANK3-expressing stable cell lines transfected with USP8, steady-state SHANK3 protein levels were increased in input lysates, compared with the same cell line transfected with βGAL control or the USP8 catalytically dead mutant USP8-C786S (Fig 5A; but see Fig 2C,D). However, the level of polyubiquitinated SHANK3 in USP8-overexpressing cells was significantly reduced, as measured by TUBE pulldown from the same lysates (Fig 5A,B). These results are consistent with a role for USP8 in deubiquitinating SHANK3 to enhance protein stability. The total levels of USP8 protein were similar for USP8 WT and USP8-C786S, as observed in the input lanes (Fig 5A); however, we noted that a larger fraction of USP8-C786S is ubiquitinated than for WT USP8 (Fig 5A), which is perhaps explicable by the self-deubiquitination activity of WT USP8 (Mizuno et al., 2005).
Since the knockdown of USP8 also decreased SHANK1 expression in neurons (Fig 4A,B), we tested whether USP8 could deubiquitinate SHANK1 in the same TUBE assay. Similar to SHANK3, SHANK1 protein expression increased with the coexpression of USP8 (Fig 5C, input lanes). As measured by TUBE pulldown assay, SHANK1 was less polyubiquitinated with coexpression of USP8, compared with βGAL control or USP8-C786S (Fig 5C,D, TUBE lanes), suggesting that USP8 can deubiquitinate SHANK1 as well as SHANK3. In similar experiments, WT USP8 coexpression had no effect on steady-state levels or polyubiquitination of SHANK2 (Fig 5C,D), which is consistent with our finding in neurons that the knockdown of USP8 reduced the synaptic levels of SHANK1 and SHANK3, but not SHANK2 (Figs. 3E,F, 4A,B). By TUBE pulldown, we found that PSD-95 was weakly ubiquitinated, which is consistent with previous studies (Colledge et al., 2003; Ehlers, 2003; Na et al., 2012), and the small amount that was ubiquitinated was not influenced by USP8 coexpression (Fig 5C,D). Finally, we probed whether USP8 could deubiquitinate GKAP, an important SHANK binding protein whose ubiquitination is known to play an important role at synapses (Hung et al., 2010; Shin et al., 2012). We found that the levels of GKAP were unchanged among USP8, USP8-C786S, and βGAL control in both input lanes and TUBE pulldowns (Fig 5C,D). Thus, USP8 shows selective DUB activity toward SHANK1 and SHANK3, among the PSD proteins tested.
To determine whether USP8 can directly deubiquitinate SHANK3 and SHANK1, we performed an in vitro deubiquitination assay using purified recombinant USP8 enzyme and immunopurified ubiquitinated SHANK proteins. We generated ubiquitinated SHANK proteins by overexpressing SHANK1, SHANK2, or SHANK3 in HEK293 cells along with HA-ubiquitin. Cells were treated with MG132 to enhance substrate ubiquitination, and lysates were heated to 85°C to kill any enzymatic activity and disrupt protein–protein interactions. SHANK1, SHANK2, or SHANK3 were immunoprecipitated and then incubated with recombinant USP8. As measured by the HA–ubiquitin signal, we found that SHANK3 and SHANK1 were deubiquitinated in the presence of recombinant USP8 in vitro, but SHANK2 was unaffected (Fig. 5E). Furthermore, the deubiquitination of SHANK3 and SHANK1 by USP8 was blocked when DUB inhibitor NEM was included in the assay. These biochemical data indicate that USP8 can act directly as a DUB for SHANK proteins, with apparent specificity for SHANK3 and SHANK1.
Loss of USP8 blocks the regulation of SHANK3 by synaptic activity
Prolonged inhibition of synaptic activity with TTX increases the protein levels of SHANK, while the stimulation of synaptic activity with BICC leads to a reduction in SHANK (Ehlers, 2003; Hung et al., 2010; Tao-Cheng et al., 2010; Shin et al., 2012). To test whether deubiquitination of SHANK3 via USP8 is important for these changes, we knocked down USP8 in mature hippocampal neuron cultures and then treated them with TTX, BICC, or DMSO vehicle control for 24 h. In control neurons transfected with a scrambled shRNA construct, we found that TTX treatment led to an ∼50% increase in SHANK3 puncta immunofluorescence intensity and 40% increase in SHANK3 puncta size, whereas BICC caused an ∼20–25% reduction in SHANK3 puncta immunofluorescence intensity and puncta size (Fig 6A,C). Knockdown of USP8 with shRNA_A or shRNA_B reduced the baseline intensity and size of SHANK3 puncta (Fig 6A,C), as seen before (Fig. 3E,F). Furthermore, knockdown of USP8 prevented the upregulation of SHANK3 puncta size and puncta intensity induced by TTX, and occluded the downregulation of SHANK3 puncta size and puncta intensity induced by BICC (Fig 6A,C). Unlike SHANK3, the postsynaptic levels of PSD-95, measured by puncta size and immunofluorescence intensity, were unaffected by shRNA_A or shRNA_B in basal conditions; nor were the upregulation and downregulation of PSD-95 in response to TTX and BICC impacted by USP8 knockdown (Fig 6B,C). Thus, USP8 plays a specific role in the modulation of SHANK3 ubiquitination and protein levels in response to synaptic activity.
Discussion
SHANK3 is a member of the SHANK family of postsynaptic scaffolding proteins that coordinates the recruitment of receptors and cytoskeletal regulators to influence structure and signaling at the excitatory synapse (Sheng and Kim, 2000). In humans, haploinsufficiency of SHANK3 due to small deletions or balanced translocations have been identified in patients with neurodevelopmental and intellectual disability (Wong et al., 1997; Anderlid et al., 2002; Bonaglia et al., 2006), and the loss of SHANK3 is described as the genetic cause of Phelan–McDermid syndrome, which is characterized by developmental delay and autistic behaviors among other symptoms (Wilson et al., 2003). Additionally, mutations in the SHANK3 gene are estimated to contribute to ∼1% of ASDs (Durand et al., 2007; Gauthier et al., 2009; Boccuto et al., 2013; Leblond et al., 2014) and may also play a role in schizophrenia (Failla et al., 2007; Choi et al., 2015; de Sena Cortabitarte et al., 2017). In mice, multiple groups have found that the deletion or mutation of Shank3 causes significant changes to the composition of synaptic proteins, alters synapse number and morphology, and impairs animal behavior (Bozdagi et al., 2010; Peça et al., 2011; Wang et al., 2011; Yang et al., 2012; Mei et al., 2016). Furthermore, the overexpression of Shank3 causes manic-like behaviors and abnormal excitatory–inhibitory balance in mice (Roussignol et al., 2005; Han et al., 2013). These data highlight the importance of precisely controlled levels of SHANK3 protein for proper synapse development and brain function.
In neurons, we and others have shown that SHANK3 and its family members are highly ubiquitinated, and that this ubiquitination is an important mechanism to control activity-dependent protein turnover and morphological changes at the synapse (Figs. 1, 5, 6; Ehlers, 2003; Bingol and Schuman, 2006; Shin et al., 2012). Identifying the DUB that deubiquitinates SHANK3 is an important step, as manipulation of this enzyme could enhance the protein stability of SHANK3, potentially increasing steady-state SHANK3 levels and perhaps ameliorating the symptoms in patients with haploinsufficiency of SHANK3 or ASD (Mei et al., 2016; Selimbeyoglu et al., 2017; Siegel et al., 2017). From screening a library of >100 DUBs, we identified USP8 as a DUB that can remove ubiquitin from SHANK3 and enhance SHANK3 protein expression in cultured neurons. Previous publications have found that the loss of SHANK3 in mice or cultured neurons leads to an overall loss of synapses and reductions in the levels of multiple PSD proteins (Bozdagi et al., 2010; Peça et al., 2011; Wang et al., 2011; Yang et al., 2012; Reim et al., 2017). Here we show that loss of USP8 leads to decreased dendritic spine density, while the overexpression of USP8 causes an increase in spine density. We hypothesize that by regulating the ubiquitination and proteasomal degradation of SHANK3 and its family member SHANK1, USP8 controls SHANK1/SHANK3 stability and dendritic spine density.
In non-neuronal cells, USP8 is reported to act at multiple stages in the endosomal sorting complexes required for transport (ESCRT) pathway. USP8 promotes the recycling of cell surface receptors, such as epidermal growth factor receptor (EGFR), back to the plasma membrane or enhances their degradation depending on when and where it deubiquitinates its substrate along the recycling pathway (Mizuno et al., 2005; Niendorf et al., 2007; Wright et al., 2011). Furthermore, USP8 interacts with the proline-rich SH3 domain of the ESCRT0 protein STAM2 via two noncanonical SH3-binding domains (Kato et al., 2000; Berry et al., 2002). SHANK3 also contains an SH3 domain, which may act as a potential site of interaction with USP8 (Naisbitt et al., 1999; Tu et al., 1999). In fact, USP8 has previously been identified as a putative SHANK3 binder via immunoprecipitation from mouse brain overexpressing Shank3-GFP (Han et al., 2013) and is enriched in dendritic spines of primary cultured neurons (Scudder et al., 2014).
UPS-driven protein degradation contributes to PSD remodeling in response to synaptic activity (Bingol and Sheng, 2011). Therefore, we wanted to determine whether modulating the deubiquitination status of SHANK3 via USP8 changes its response to synaptic activity. We found that the loss of USP8 leads to decreased SHANK3 protein levels and occludes the bidirectional changes in SHANK3 in response to synaptic activity. However, the loss of USP8 does not change the response of PSD-95 to synaptic activity, suggesting the specificity of USP8 for SHANK3. Previous publications have found that the overexpression of USP8 increases synaptic strength and reduces AMPAR ubiquitination, while the loss of USP8 decreases synaptic strength and increases AMPAR ubiquitination (Scudder et al., 2014). This study additionally reported that the overexpression of USP8 prevents the loss of surface AMPAR following heightened synaptic activity and contributes to the downscaling of synaptic strength (Scudder et al., 2014).
Our results suggest a model where USP8 deubiquitinates SHANK3 following chronic blockage of synaptic activity, preventing degradation via the proteasome and increasing synaptic SHANK3 protein levels. In contrast, following chronic synaptic activity, SHANK3 is no longer deubiquitinated by USP8 and is subsequently degraded. Previous publications have found that total USP8 protein levels are downregulated following chronic synaptic activity, suggesting a mechanism for decreased deubiquitination of USP8 substrates, such as SHANK3 (Scudder et al., 2014). Together with our results, these studies highlight the critical role for USP8 in controlling the levels of synaptic proteins, such as SHANK3, SHANK1, and AMPAR, in response to synaptic activity modulation. Future work should determine functionally how USP8 is modulated in response to synaptic activity and how it interacts with its substrates.
Footnotes
We thank Eunjoon Kim for providing DNA constructs, Baris Bingol, and Christopher Wall for providing materials and technical advice on biochemical experiments; Christine Kim for proving technical assistance; and Casper Hoogenraad, Borislav Dejanovic, Daniel Fleck, and Annette Vogl for critical discussions on experimental design and results.
The authors are full time employees of Genentech, a member of the Roche Group.
References
- Anderlid BM, Schoumans J, Annerén G, Tapia-Paez I, Dumanski J, Blennow E, Nordenskjöld M (2002) FISH-mapping of a 100-kb terminal 22q13 deletion. Hum Genet 110:439–443. 10.1007/s00439-002-0713-7 [DOI] [PubMed] [Google Scholar]
- Araki Y, Zeng M, Zhang M, Huganir RL (2015) Rapid dispersion of SynGAP from synaptic spines triggers AMPA receptor insertion and spine enlargement during LTP. Neuron 85:173–189. 10.1016/j.neuron.2014.12.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avvakumov GV, Walker JR, Xue S, Finerty PJ Jr, Mackenzie F, Newman EM, Dhe-Paganon S (2006) Amino-terminal dimerization, NRDP1-rhodanese interaction, and inhibited catalytic domain conformation of the ubiquitin-specific protease 8 (USP8). J Biol Chem 281:38061–38070. 10.1074/jbc.M606704200 [DOI] [PubMed] [Google Scholar]
- Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, Endris V, Roberts W, Szatmari P, Pinto D, Bonin M, Riess A, Engels H, Sprengel R, Scherer SW, Rappold GA (2010) Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet 42:489–491. 10.1038/ng.589 [DOI] [PubMed] [Google Scholar]
- Berkel S, Tang W, Treviño M, Vogt M, Obenhaus HA, Gass P, Scherer SW, Sprengel R, Schratt G, Rappold GA (2012) Inherited and de novo SHANK2 variants associated with autism spectrum disorder impair neuronal morphogenesis and physiology. Hum Mol Genet 21:344–357. 10.1093/hmg/ddr470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry DM, Nash P, Liu SK, Pawson T, McGlade CJ (2002) A high-affinity arg-X-X-lys SH3 binding motif confers specificity for the interaction between gads and SLP-76 in T cell signaling. Curr Biol 12:1336–1341. 10.1016/S0960-9822(02)01038-2 [DOI] [PubMed] [Google Scholar]
- Bhattacharyya S, Yu H, Mim C, Matouschek A (2014) Regulated protein turnover: snapshots of the proteasome in action. Nat Rev Mol Cell Biol 15:122–133. 10.1038/nrm3741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Schuman EM (2005) Synaptic protein degradation by the ubiquitin proteasome system. Curr Opin Neurobiol 15:536–541. 10.1016/j.conb.2005.08.016 [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM (2006) Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature 441:1144–1148. 10.1038/nature04769 [DOI] [PubMed] [Google Scholar]
- Bingol B, Sheng M (2011) Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron 69:22–32. 10.1016/j.neuron.2010.11.006 [DOI] [PubMed] [Google Scholar]
- Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M (2014) The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510:370–375. 10.1038/nature13418 [DOI] [PubMed] [Google Scholar]
- Boccuto L, Lauri M, Sarasua SM, Skinner CD, Buccella D, Dwivedi A, Orteschi D, Collins JS, Zollino M, Visconti P, Dupont B, Tiziano D, Schroer RJ, Neri G, Stevenson RE, Gurrieri F, Schwartz CE (2013) Prevalence of SHANK3 variants in patients with different subtypes of autism spectrum disorders. Eur J Hum Genet 21:310–316. 10.1038/ejhg.2012.175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonaglia MC, Giorda R, Mani E, Aceti G, Anderlid BM, Baroncini A, Pramparo T, Zuffardi O (2006) Identification of a recurrent breakpoint within the SHANK3 gene in the 22q13.3 deletion syndrome. J Med Genet 43:822–828. 10.1136/jmg.2005.038604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozdagi O, Sakurai T, Papapetrou D, Wang X, Dickstein DL, Takahashi N, Kajiwara Y, Yang M, Katz AM, Scattoni ML, Harris MJ, Saxena R, Silverman JL, Crawley JN, Zhou Q, Hof PR, Buxbaum JD (2010) Haploinsufficiency of the autism-associated Shank3 gene leads to deficits in synaptic function, social interaction, and social communication. Mol Autism 1:15. 10.1186/2040-2392-1-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi SY, Pang K, Kim JY, Ryu JR, Kang H, Liu Z, Kim WK, Sun W, Kim H, Han K (2015) Post-transcriptional regulation of SHANK3 expression by microRNAs related to multiple neuropsychiatric disorders. Mol Brain 8:74. 10.1186/s13041-015-0165-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colledge M, Snyder EM, Crozier RA, Soderling JA, Jin Y, Langeberg LK, Lu H, Bear MF, Scott JD (2003) Ubiquitination regulates PSD-95 degradation and AMPA receptor surface expression. Neuron 40:595–607. 10.1016/S0896-6273(03)00687-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Sena Cortabitarte A, Degenhardt F, Strohmaier J, Lang M, Weiss B, Roeth R, Giegling I, Heilmann-Heimbach S, Hofmann A, Rujescu D, Fischer C, Rietschel M, Nöthen MM, Rappold GA, Berkel S (2017) Investigation of SHANK3 in schizophrenia. Am J Med Genet B Neuropsychiatr Genet 174:390–398. 10.1002/ajmg.b.32528 [DOI] [PubMed] [Google Scholar]
- Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, Nygren G, Rastam M, Gillberg IC, Anckarsäter H, Sponheim E, Goubran-Botros H, Delorme R, Chabane N, Mouren-Simeoni MC, de Mas P, Bieth E, Rogé B, Héron D, Burglen L, et al. (2007) Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet 39:25–27. 10.1038/ng1933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, Montcouquiol M, Sans N (2012) SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry 17:71–84. 10.1038/mp.2011.57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. (2003) Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci 6:231–242. 10.1038/nn1013 [DOI] [PubMed] [Google Scholar]
- Failla P, Romano C, Alberti A, Vasta A, Buono S, Castiglia L, Luciano D, Di Benedetto D, Fichera M, Galesi O (2007) Schizophrenia in a patient with subtelomeric duplication of chromosome 22q. Clin Genet 71:599–601. 10.1111/j.1399-0004.2007.00819.x [DOI] [PubMed] [Google Scholar]
- Gauthier J, Spiegelman D, Piton A, Lafrenière RG, Laurent S, St-Onge J, Lapointe L, Hamdan FF, Cossette P, Mottron L, Fombonne E, Joober R, Marineau C, Drapeau P, Rouleau GA (2009) Novel de novo SHANK3 mutation in autistic patients. Am J Med Genet B Neuropsychiatr Genet 150B:421–424. 10.1002/ajmg.b.30822 [DOI] [PubMed] [Google Scholar]
- Guilmatre A, Huguet G, Delorme R, Bourgeron T (2014) The emerging role of SHANK genes in neuropsychiatric disorders. Carbonetto S, Bourgeron T, eds. Dev Neurobiol 74:113–122. 10.1002/dneu.22128 [DOI] [PubMed] [Google Scholar]
- Han K, Holder JL Jr, Schaaf CP, Lu H, Chen H, Kang H, Tang J, Wu Z, Hao S, Cheung SW, Yu P, Sun H, Breman AM, Patel A, Lu HC, Zoghbi HY (2013) SHANK3 overexpression causes manic-like behaviour with unique pharmacogenetic properties. Nature 503:72–77. 10.1038/nature12630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung AY, Sung CC, Brito IL, Sheng M (2010) Degradation of postsynaptic scaffold GKAP and regulation of dendritic spine morphology by the TRIM3 ubiquitin ligase in rat hippocampal neurons. PLoS One 5:e9842. 10.1371/journal.pone.0009842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Ehlers MD (2013) Modeling autism by SHANK gene mutations in mice. Neuron 78:8–27. 10.1016/j.neuron.2013.03.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Miyazawa K, Kitamura N (2000) A deubiquitinating enzyme UBPY interacts with the src homology 3 domain of hrs-binding protein via a novel binding motif PX(V/I)(D/N)RXXKP. J Biol Chem 275:37481–37487. 10.1074/jbc.M007251200 [DOI] [PubMed] [Google Scholar]
- Leblond CS, Nava C, Polge A, Gauthier J, Huguet G, Lumbroso S, Giuliano F, Stordeur C, Depienne C, Mouzat K, Pinto D, Howe J, Lemière N, Durand CM, Guibert J, Ey E, Toro R, Peyre H, Mathieu A, Amsellem F, et al. (2014) Meta-analysis of SHANK mutations in autism spectrum disorders: a gradient of severity in cognitive impairments. PLoS Genet 10:e1004580. 10.1371/journal.pgen.1004580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PC, Dodart JC, Aron L, Finley LW, Bronson RT, Haigis MC, Yankner BA, Harper JW (2013) Altered social behavior and neuronal development in mice lacking the Uba6-Use1 ubiquitin transfer system. Mol Cell 50:172–184. 10.1016/j.molcel.2013.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacGillavry HD, Kerr JM, Kassner J, Frost NA, Blanpied TA (2016) Shank-cortactin interactions control actin dynamics to maintain flexibility of neuronal spines and synapses. Eur J Neurosci 43:179–193. 10.1111/ejn.13129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429:761–766. 10.1038/nature02617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei Y, Monteiro P, Zhou Y, Kim JA, Gao X, Fu Z, Feng G (2016) Adult restoration of Shank3 expression rescues selective autistic-like phenotypes. Nature 530:481–484. 10.1038/nature16971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno E, Iura T, Mukai A, Yoshimori T, Kitamura N, Komada M (2005) Regulation of epidermal growth factor receptor down-regulation by UBPY-mediated deubiquitination at endosomes. Mol Biol Cell 16:5163–5174. 10.1091/mbc.e05-06-0560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na CH, Jones DR, Yang Y, Wang X, Xu Y, Peng J (2012) Synaptic protein ubiquitination in rat brain revealed by antibody-based ubiquitome analysis. J Proteome Res 11:4722–4732. 10.1021/pr300536k [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nägerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T (2004) Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron 44:759–767. 10.1016/j.neuron.2004.11.016 [DOI] [PubMed] [Google Scholar]
- Naisbitt S, Kim E, Tu JC, Xiao B, Sala C, Valtschanoff J, Weinberg RJ, Worley PF, Sheng M (1999) Shank, a novel family of postsynaptic density proteins that binds to the NMDA receptor/PSD-95/GKAP complex and cortactin. Neuron 23:569–582. 10.1016/S0896-6273(00)80809-0 [DOI] [PubMed] [Google Scholar]
- Niendorf S, Oksche A, Kisser A, Löhler J, Prinz M, Schorle H, Feller S, Lewitzky M, Horak I, Knobeloch KP (2007) Essential role of ubiquitin-specific protease 8 for receptor tyrosine kinase stability and endocytic trafficking in vivo. Mol Cell Biol 27:5029–5039. 10.1128/MCB.01566-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nijman SM, Luna-Vargas MP, Velds A, Brummelkamp TR, Dirac AM, Sixma TK, Bernards R (2005) A genomic and functional inventory of deubiquitinating enzymes. Cell 123:773–786. 10.1016/j.cell.2005.11.007 [DOI] [PubMed] [Google Scholar]
- Peça J, Feliciano C, Ting JT, Wang W, Wells MF, Venkatraman TN, Lascola CD, Fu Z, Feng G (2011) Shank3 mutant mice display autistic-like behaviours and striatal dysfunction. Nature 472:437–442. 10.1038/nature09965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim D, Distler U, Halbedl S, Verpelli C, Sala C, Bockmann J, Tenzer S, Boeckers TM, Schmeisser MJ (2017) Proteomic analysis of post-synaptic density fractions from Shank3 mutant mice reveals brain region specific changes relevant to autism spectrum disorder. Front Mol Neurosci 10:26. 10.3389/fnmol.2017.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussignol G, Ango F, Romorini S, Tu JC, Sala C, Worley PF, Bockaert J, Fagni L (2005) Shank expression is sufficient to induce functional dendritic spine synapses in aspiny neurons. J Neurosci 25:3560–3570. 10.1523/JNEUROSCI.4354-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sala C, Piëch V, Wilson NR, Passafaro M, Liu G, Sheng M (2001) Regulation of dendritic spine morphology and synaptic function by shank and homer. Neuron 31:115–130. 10.1016/S0896-6273(01)00339-7 [DOI] [PubMed] [Google Scholar]
- Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S, O'Connor I, Russell C, Drmic IE, Hamdan FF, Michaud JL, Endris V, Roeth R, Delorme R, Huguet G, Leboyer M, Rastam M, Gillberg C, Lathrop M, Stavropoulos DJ, et al. (2012) SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet 90:879–887. 10.1016/j.ajhg.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmeisser MJ. (2015) Translational neurobiology in shank mutant mice- model systems for neuropsychiatric disorders. Ann Anat 200:115–117. 10.1016/j.aanat.2015.03.006 [DOI] [PubMed] [Google Scholar]
- Scudder SL, Goo MS, Cartier AE, Molteni A, Schwarz LA, Wright R, Patrick GN (2014) Synaptic strength is bidirectionally controlled by opposing activity-dependent regulation of Nedd4–1 and USP8. J Neurosci 34:16637–16649. 10.1523/JNEUROSCI.2452-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimbeyoglu A, Kim CK, Inoue M, Lee SY, Hong ASO, Kauvar I, Ramakrishnan C, Fenno LE, Davidson TJ, Wright M, Deisseroth K (2017) Modulation of prefrontal cortex excitation/inhibition balance rescues social behavior in CNTNAP2-deficient mice. Sci Transl Med 9:eaah6733. 10.1126/scitranslmed.aah6733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC (2007) The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem 76:823–847. 10.1146/annurev.biochem.76.060805.160029 [DOI] [PubMed] [Google Scholar]
- Sheng M, Kim E (2000) The Shank family of scaffold proteins. J Cell Sci 113:1851–1856. [DOI] [PubMed] [Google Scholar]
- Shin SM, Zhang N, Hansen J, Gerges NZ, Pak DT, Sheng M, Lee SH (2012) GKAP orchestrates activity-dependent postsynaptic protein remodeling and homeostatic scaling. Nat Neurosci 15:1655–1666. 10.1038/nn.3259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JJ, Chitwood RA, Ding JM, Payne C, Taylor W, Gray R, Zemelman BV, Johnston D (2017) Prefrontal cortex dysfunction in fragile X mice depends on the continued absence of fragile X mental retardation protein in the adult brain. J Neurosci 37:7305–7317. 10.1523/JNEUROSCI.0571-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swatek KN, Komander D (2016) Ubiquitin modifications. Cell Res 26:399–422. 10.1038/cr.2016.39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng JH, Dosemeci A, Gallant PE, Smith C, Reese T (2010) Activity induced changes in the distribution of shanks at hippocampal synapses. Neuroscience 168:11–17. 10.1016/j.neuroscience.2010.03.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu JC, Xiao B, Naisbitt S, Yuan JP, Petralia RS, Brakeman P, Doan A, Aakalu VK, Lanahan AA, Sheng M, Worley PF (1999) Coupling of mGluR/Homer and PSD-95 complexes by the shank family of postsynaptic density proteins. Neuron 23:583–592. 10.1016/S0896-6273(00)80810-7 [DOI] [PubMed] [Google Scholar]
- Wang X, McCoy PA, Rodriguiz RM, Pan Y, Je HS, Roberts AC, Kim CJ, Berrios J, Colvin JS, Bousquet-Moore D, Lorenzo I, Wu G, Weinberg RJ, Ehlers MD, Philpot BD, Beaudet AL, Wetsel WC, Jiang YH (2011) Synaptic dysfunction and abnormal behaviors in mice lacking major isoforms of Shank3. Hum Mol Genet 20:3093–3108. 10.1093/hmg/ddr212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson HL, Wong AC, Shaw SR, Tse WY, Stapleton GA, Phelan MC, Hu S, Marshall J, McDermid HE (2003) Molecular characterisation of the 22q13 deletion syndrome supports the role of haploinsufficiency of SHANK3/PROSAP2 in the major neurological symptoms. J Med Genet 40:575–584. 10.1136/jmg.40.8.575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AC, Ning Y, Flint J, Clark K, Dumanski JP, Ledbetter DH, McDermid HE (1997) Molecular characterization of a 130-kb terminal microdeletion at 22q in a child with mild mental retardation. Am J Hum Genet 60:113–120. [PMC free article] [PubMed] [Google Scholar]
- Wright MH, Berlin I, Nash PD (2011) Regulation of endocytic sorting by ESCRT-DUB-mediated deubiquitination. Cell Biochem Biophys 60:39–46. 10.1007/s12013-011-9181-9 [DOI] [PubMed] [Google Scholar]
- Wu X, Yen L, Irwin L, Sweeney C, Carraway KL 3rd (2004) Stabilization of the E3 ubiquitin ligase Nrdp1 by the deubiquitinating enzyme USP8. Mol Cell Biol 24:7748–7757. 10.1128/MCB.24.17.7748-7757.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Bozdagi O, Scattoni ML, Wöhr M, Roullet FI, Katz AM, Abrams DN, Kalikhman D, Simon H, Woldeyohannes L, Zhang JY, Harris MJ, Saxena R, Silverman JL, Buxbaum JD, Crawley JN (2012) Reduced excitatory neurotransmission and mild autism-relevant phenotypes in adolescent Shank3-null mutant mice. J Neurosci 32:6525–6541. 10.1523/JNEUROSCI.6107-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]