

Abstract
Transporter-mediated glutamate uptake plays an essential role in shaping synaptic neurotransmission. The rapid removal of synaptically released glutamate ensures the high temporal dynamics characteristic of fast excitatory chemical neurotransmission and prevents the overexcitation of extrasynaptic NMDA receptors that have been implicated in synaptic plasticity impairments and cell death. Despite clear regional differences in plasticity and excitotoxic thresholds, few studies have compared extracellular glutamate dynamics across different brain regions and in response to a range of neural activity including plasticity-inducing stimuli. Here, we used the rapid extracellular fluorescent glutamate sensor iGluSnFR (intensity-based glutamate-sensing fluorescent reporter) and high-speed imaging (205 frames per second) to quantify relative differences in glutamate clearance rates over a wide range of presynaptic activity in situ in the hippocampus, cortex, and striatum of male C57/BL6NCrl mice. We found that the hippocampus was significantly more efficient than the cortex and striatum at clearing synaptically released glutamate and that this efficiency could be attributed, at least in part, to faster glutamate diffusion away from the release site. In addition, we found that pharmacological inhibition of GLT-1, the brain's most abundant glutamate transporter, slowed clearance rates to only a fraction (∼20–25%) of the effect induced by nonselective transporter blockade, regardless of the brain region and the duration of presynaptic activity. In all, our data reveal clear regional differences in glutamate dynamics after neural activity and suggest that non-GLT-1 transporters can make a large contribution to the rate of glutamate clearance in the hippocampus, cortex, and striatum.
SIGNIFICANCE STATEMENT Glutamate is the brain's most abundant neurotransmitter, and although essential for rapid cell–cell communication, too much glutamate can negatively impact cellular health. Extracellular glutamate levels are tightly regulated by membrane-bound transporters that rapidly remove the glutamate that is released during neural activity, thereby shaping both the spatial and temporal dynamics of excitatory neurotransmission. Using high-speed imaging of an optical sensor of extracellular glutamate, we show that glutamate dynamics vary widely from one brain region to the next and are highly dependent on the duration of synaptic activity. Our data demonstrate the heterogeneous nature of glutamate regulation in the brain and suggest that such regional differences can dramatically affect both the localization and duration of postsynaptic receptor activation during synaptic neurotransmission.
Keywords: GLT-1, glutamate, iGluSnFR, optogenetics, transporter
Introduction
Rapid excitatory synaptic transmission in the CNS relies heavily on efficient uptake mechanisms to clear the extracellular space of synaptically released glutamate. As glutamate cannot be degraded in the extracellular space, termination of glutamatergic neurotransmission is accomplished through a combination of diffusion and uptake through various sodium-dependent glutamate transporters, many of which are strategically located in close proximity to glutamate release sites (Rothstein et al., 1994; Danbolt, 2001). Despite the extremely organized morphology of tripartite synaptic structure (Perea et al., 2009), with the exception of electrophysiological recordings of synaptically activated glutamate transporter currents (STCs), the large majority of our knowledge of glutamate transporter function has been derived from biochemical uptake assays in homogenized tissue preparations. Whereas it is widely accepted that astrocytes mediate the bulk of glutamate uptake in the brain, the biochemical uptake assay was recently shown to dramatically overemphasize neuronal uptake at the expense of astrocytic uptake, highlighting the need to study glutamate dynamics and glutamate transporter function in situ (Petr et al., 2015). Moreover, regional differences in tortuosity (barriers to diffusion) and astrocyte-to-postsynaptic density distances have also been noted (Hrabětová, 2005; Chai et al., 2017); such differences are likely to dramatically influence the extracellular profile of glutamate during neural activity, yet such differences cannot be reflected in biochemical measures of glutamate uptake. Excessive amounts of extracellular glutamate may be detrimental to synaptic plasticity (Li et al., 2011) and can cause apoptotic cell death (Hardingham and Bading, 2010; Parsons and Raymond, 2014), and glutamate transporter dysfunction has been implicated in numerous neurodegenerative diseases and other CNS conditions (Maragakis and Rothstein, 2006; Parsons and Raymond, 2014; Phatnani and Maniatis, 2015). To gain a better understanding of how glutamate transporter dysfunction contributes to various disease states, it is imperative to first understand how these transporters regulate extracellular glutamate levels in the healthy brain in such a way that promotes efficient synaptic neurotransmission, robust synaptic plasticity, and neuroprotection.
The glutamate clearance requirements at a given excitatory synapse can vary tremendously, from action potential-independent quantal release to longer trains of sustained high-frequency firing in an afferent population. Using a fast and specific genetically encoded fluorescent sensor of extracellular glutamate (Marvin et al., 2013), we and others have demonstrated that the rate at which glutamate is cleared from the extracellular space is heavily influenced by presynaptic activity, in that uptake slows with increasing durations of burst activity (Armbruster et al., 2016; Parsons et al., 2016). This observation suggests that transporters may become overwhelmed when challenged with long durations of presynaptic activity. It has become widely accepted that glutamate transporter 1 (GLT-1) mediates the large majority of glutamate uptake in the brain, whereas the contribution from non-GLT-1 transporters is diminished in comparison. However, much of these data stem from biochemical uptake assays using exogenous glutamate, and little attention has been given to the relative importance of GLT-1 and non-GLT-1 transporters across the wide spectrum of the activity patterns experienced at excitatory synapses. Similarly, whether activity-dependent transporter-mediated uptake is similar from one brain region to the next is relatively unexplored, despite prior reports of regional differences in transporter expression (Furuta et al., 1997; Milton et al., 1997).
Here, we used a genetically encoded intensity-based glutamate-sensing fluorescent reporter (iGluSnFR) to visualize, in real time, the spatiotemporal extracellular dynamics of synaptically released glutamate over a wide range of presynaptic activity in the hippocampus, cortex, and striatum and used a pharmacological approach to quantify the relative importance of GLT-1 to total uptake. We found stark regional differences in the brain's ability to clear various glutamate challenges and provide evidence to suggest that non-GLT-1 transporters can make a substantial contribution to the overall glutamate clearance rate, particularly when GLT-1 is dysfunctional.
Materials and Methods
Animals.
All experiments were performed on acute brain slices obtained from male C57BL/6NCrl mice. Mice were ordered from Charles River at ∼3–4 weeks of age and were provided with a minimum of 2 d of acclimatization upon arriving at Memorial University's animal care facility. Mice were group housed in ventilated cage racks, were provided with standard chow and water ad libitum, and were maintained on a normal 12 h light/dark cycle. All experimental procedures were approved by Memorial University's Institutional Animal Care Committee and were performed in accordance with the guidelines set by the Canadian Council on Animal Care.
Stereotaxic surgery.
Male C57BL/6NCrl mice (4–6 weeks of age) were anesthetized by isoflurane inhalation (3%) and maintained with 1.5–2% isoflurane for the duration of the surgical procedure. Mice were secured within the ear bars of a standard stereotaxic apparatus (Stoelting), eye drops were used to lubricate the eyes throughout the procedure, and a subcutaneous 0.5 ml injection of 0.9% sterile saline was provided to help prevent dehydration. When unresponsive to toe-pinch, a small amount of fur above the scalp was cut with a pair of scissors, and a 0.1 ml bolus of 0.2% lidocaine was injected below the scalp. A small incision was then made in the scalp around bregma, and the underlying skull was exposed. A hand drill was used to carefully thin the skull at the desired coordinates from bregma, and a fine bent needle tip was used to peel back the last layer of skull to expose the underlying cortex while minimizing tissue damage. A Neuros 7002 Hamilton syringe was attached to an infusion pump (Pump 11 Elite Nanomite; Harvard Apparatus), which was then secured to the stereotaxic frame. A total volume of 1 μl of AAV1.hSyn.iGluSnFr.WPRE.SV40 (synapsin-iGluSnFR; Penn Vector Core, catalog #AV-1-PV2723; kindly provided by Dr. Loren L. Looger and Janelia Research Campus of the Howard Hughes Medical Institute, Ashburn, VA) was injected into the hippocampus, cortex, or striatum at an injection rate of 2 nl/s. The syringe was left in place for an additional 5 min after the injection. The following coordinates were used with respect to bregma: hippocampus, 2.6 mm posterior, 2.4 mm lateral (right), 1.2–1.4 mm ventral to brain surface; cortex, 0.7 mm anterior, 2.0 mm lateral (right), 0.6 mm ventral; striatum, 0.7 mm anterior, 2.0 mm lateral (right), 2.6 mm ventral to brain surface. The syringe was slowly withdrawn, the incision was sutured, and mice were given subcutaneous injections of 0.5 ml of 0.9% saline containing 2 mg/kg meloxicam before being placed on a heating pad for ∼30 min to accelerate recovery.
Slice preparation.
At 2–3 months of age (3–6 weeks after iGluSnFR injection), mice were anesthetized with isoflurane and decapitated, and the brain was quickly removed and immersed in ice-cold oxygenated (95% O2/5% CO2) slicing solution consisting of (in mm) 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 MgCl2, 0.5 CaCl2, and 10 glucose. Coronal brain slices (350 μm) containing the hippocampus, striatum, and cortex were obtained with a Leica VT1000 Vibratome. Slices were then placed in artificial CSF (ACSF) containing (in mm) 125 NaCl, 2.5 KCl, 25 NaHCO3, 1.25 NaH2PO4, 1 MgCl2, 2 CaCl2, and 10 glucose. Slices were recovered in oxygenated ACSF for 45 min at room temperature before experimentation.
Imaging and image analysis.
Slices were then transferred to the recording chamber, and a peristaltic pump (MP-II; Harvard Apparatus) was used to perfuse oxygenated ACSF at a flow rate of 2 ml/min. ACSF was heated to 32°C using an in-line heater and temperature controller (TC-344C; Harvard Apparatus). Glass stimulating electrodes were pulled using a Narishige PB-7 pipette puller to a resistance of 1–3 MΩ when filled with ACSF. Sections were viewed on an Olympus BX51 microscope with a 4x objective (0.28 NA). For hippocampal sections, the stimulating electrode was placed directly in the Schaffer collateral pathway within the stratum radiatum. For cortical sections, the stimulating electrode was placed in the deep layers of the somatosensory cortex. For striatal sections, the stimulation electrode was placed in the dorsal striatum. In all cases, the stimulating electrode was placed at a depth ∼50–100 μm below the slice surface. Clampex software (Molecular Devices) was used to send TTL triggers through the digital outputs of a Digidata 1550A (Molecular Devices) for precise control over an LED illumination source (Prior, Lumen 300), an EM-CCD camera (Andor, iXon Ultra 897), and an Iso-flex stimulus isolator (AMPI). Basal, unstimulated iGluSnFR expression was first measured in each slice using a constant LED power and exposure time. iGluSnFR responses to evoked neural activity were recorded with Andor Solis software, using 4 × 4 binning and an acquisition rate of 205 frames per second. Evoked iGluSnFR responses were averaged over three to five trials, with nonstimulus trials interleaved to control for any bleaching of the iGluSnFR signal during acquisition. The nonstimulus trials were averaged in ImageJ and subtracted from the average of the stimulus trials using the IOS and VSD signal processor plugin. The dynamics of extracellular glutamate dynamics within a given field were determined by calculating the average fluorescence intensity within a 10 × 10 pixel ROI (1 pixel at 4 × 4 binning = 16 μm) placed adjacent to the location of the stimulating electrode. Values for %ΔF/F were copied to GraphPad Prism, where decay tau was calculated from the response peak (in the case of short bursts of activity) or the end of stimulation (in the case of longer high-frequency trains) using a single-exponential nonlinear curve fit. To visually represent the time course of the response, we applied the “fire” heat map in ImageJ and used the “volume viewer” 3D plugin to display the response along the z (time) axis.
Pharmacology.
All the drugs used for these experiments were from Tocris Bioscience. Drugs used in the study and their concentrations are as follows: dihydrokainic acid (DHK), a competitive and selective GLT-1 blocker (EAAT-2; 300 μm); dl-threo-β-benzyloxyaspartic acid (dl-TBOA), a competitive and nonselective excitatory amino acid transporter blocker (10 and 100 μm); DNQX disodium salt, an AMPA/kainate receptor antagonist (20 μm); and d-AP-5, a selective NMDA receptor antagonist (50 μm).
Western blotting.
The hippocampus, cortex, and striatum were dissected out and homogenized in 200–400 μl each of lysis buffer containing protease/phosphatase inhibitors. Supernatant was collected from each brain region, and protein concentration was determined using BCA standards. Fifty micrograms of protein from each region were added to separate lanes of 10% SDS-PAGE gels for electrophoresis and transferred to nitrocellulose membranes. Primary antibodies for GLT-1 (E1, 1:1,000, Santa Cruz Biotechnology, mouse monoclonal) and actin (C4, 1:1,000, Santa Cruz Biotechnology, mouse monoclonal) were used, with actin used as a loading control. A goat anti-mouse IgG–HRP secondary antibody (sc-2005, 1:5,000, Santa Cruz Biotechnology, monoclonal) was then added to the membranes. Blots were developed using chemiluminescent HRP substrate (catalog #WBKLS0100, Lot No. 1712501, Millipore), and ImageJ was used to analyze and quantify the developed bands to determine GLT-1 expression.
Experimental design and statistics.
The statistical tests used included one-way ANOVA, one-way repeated-measures (RM) ANOVA, two-way ANOVA, two-way RM ANOVA, and linear regression. Post hoc tests included Tukey's, Bonferroni's, and Dunnett's tests. The statistical test used for each experiment is indicated in Results. P values of <0.05 were considered significant. Where indicated, N and n refer to the number of animals and slices used in each experiment, respectively.
Results
Characterization of glutamate clearance rates in the hippocampus, cortex, and striatum
The CA3–CA1 glutamate projection within the hippocampus is one of the most widely studied synaptic connections in the brain. We first examined activity-dependent regulation of extracellular glutamate dynamics within hippocampal circuitry by stimulating glutamate release at the CA3–CA1 synapse with a glass electrode placed in the Schaffer collateral pathway of iGluSnFR-injected mice (Fig. 1A). iGluSnFR embeds within the membrane with its glutamate-binding domain exposed to the extracellular surface (Marvin et al., 2013), and we used the synapsin promoter to restrict iGluSnFR expression to neurons. Thus, the fluorescent signals quantified in the present study represent relative differences in the amount and time course of synaptically released glutamate sensed at the extracellular neuronal surface. Glutamate release and clearance rates were visualized in real time by high-speed wide-field imaging (205 frames per second) of iGluSnFR transients in response to afferent stimulation of short bursts of activity (2, 5, and 10 pulses at 100 Hz) and longer trains of high-frequency stimulation (HFS; 50 and 100 pulses at 100 Hz). The relative magnitude of glutamate release was measured by the peak iGluSnFR response (Fig. 1B,D), and the clearance rate was measured by calculating the iGluSnFR decay tau immediately after the termination of the stimulation period (Fig. 1C,E), as described previously (Armbruster et al., 2016; Parsons et al., 2016). Not surprisingly, we found that increasing the number of pulses within the shorter bursts dramatically increased glutamate release (Fig. 1D; N = 10, n = 17, RM ANOVA, p < 0.001), with %ΔF/F values more than doubling as afferent stimulation increased from 2 to 10 pulses at 100 Hz. In contrast, glutamate clearance rates were comparatively stable over this stimulation range (Fig. 1E) despite the clear increase in response size. When longer trains of HFS were used, including 50 pulses at 100 Hz and the commonly used LTP induction protocol of 100 pulses at 100 Hz, glutamate clearance rates slowed over threefold (Fig. 1E; N = 10, n = 17, RM ANOVA, p < 0.001). These data demonstrate that glutamate clearance rates are more closely linked to the duration of presynaptic activity (i.e., short bursts vs HFS) than the amount of glutamate released. Indeed, within a given stimulation paradigm, we found no significant correlation between iGluSnFR %ΔF/F and decay tau, both for short bursts (Fig. 1F) and HFS (Fig. 1G). In addition, we found no significant correlation between decay tau and the total amount of glutamate released during longer stimulus trains, as measured by the area under the curve of iGluSnFR responses (50 pulses, r = 0.046, p = 0.864; 100 pulses, r = 0.120, p = 0.709).
Figure 1.
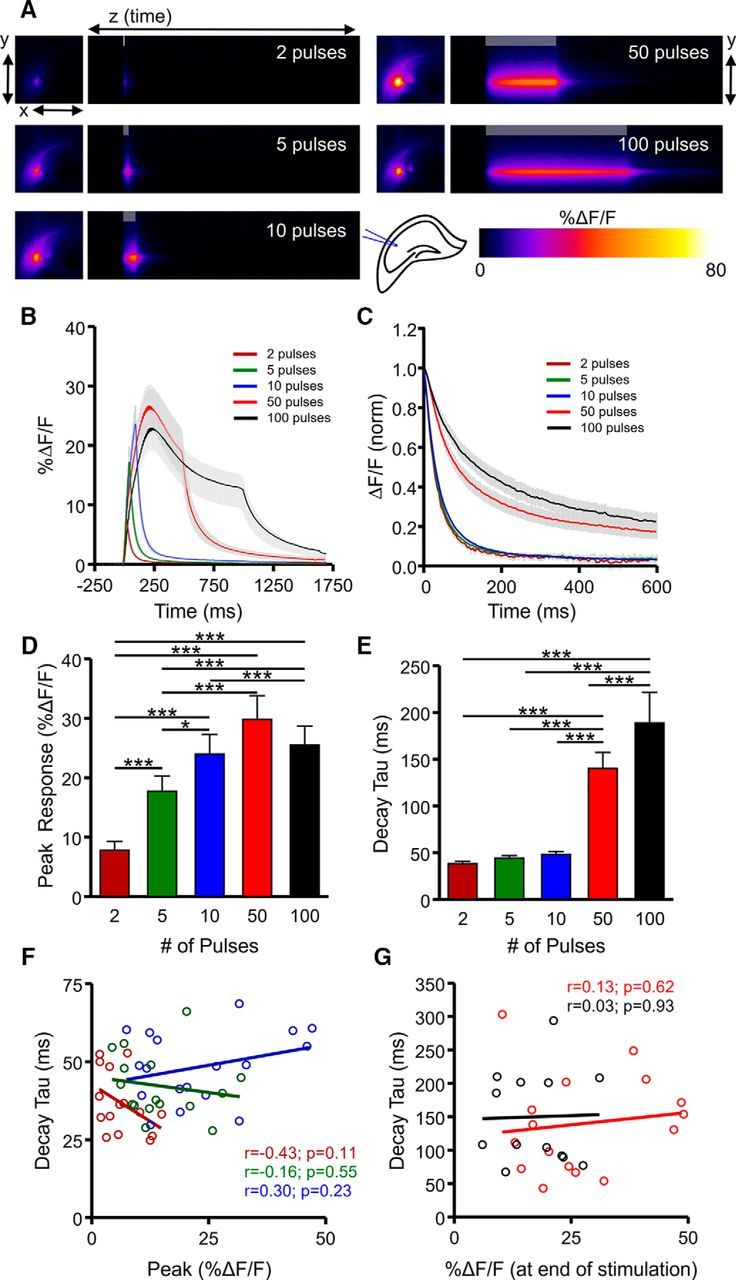
Characterization of extracellular glutamate dynamics in the hippocampus. A, Representative heatmaps of iGluSnFR responses after afferent stimulation (100 Hz). Peak responses are shown in the x–y plane (image size, 2 × 2 mm), and the y–z (time) plots show the kinetics of the response at a defined x-coordinate adjacent to the site of stimulation (image represents 2 s). The gray shaded area within the images denotes the onset and duration of afferent stimulation. B, Mean (±SEM) iGluSnFR response profiles to 2, 5, 10, 50, or 100 pulses of afferent stimulation at 100 Hz. C, Mean responses (±SEM) from B that were normalized to the peak value at the end of the stimulation. D–E, Grouped data showing mean (±SEM) iGluSnFR response peak (D) and decay tau (E). RM ANOVA with Tukey's post hoc results are indicated by *p < 0.05 and ***p < 0.001. F–G, Decay and %ΔF/F correlation of the iGluSnFR responses with a short burst of activity (F) and longer trains of high-frequency stimulation (G).
Transporter-mediated glutamate clearance is commonly quantified by a biochemical uptake assay, which requires tissue homogenization and therefore results in the loss of the local microenvironment of synaptic structure. It was recently demonstrated that astroglial ensheathment of the postsynaptic density can differ quite substantially between brain regions (Chai et al., 2017), and the profile and tortuosity of the extracellular space can also differ dramatically in a region-dependent manner (Hrabětová, 2005). Furthermore, as extracellular glutamate clearance rates may influence synaptic plasticity (Li et al., 2011) and neuronal vulnerability to excitotoxic cell death (Hardingham and Bading, 2010; Parsons and Raymond, 2014), it is important to understand whether the temporal profile of extracellular glutamate differs in a region-dependent manner. To this end, we repeated the above experiments in two other widely studied brain areas that contain dense glutamatergic afferents yet exhibit dramatically different plasticity responses to stimuli such as HFS: the cortex (specifically the deep layers of the somatosensory cortex) and the dorsal striatum. In the cortex (Fig. 2A), increasing the number of pulses dramatically increased the response size of iGluSnFR transients, in line with our expectations and our results obtained in the hippocampus (Fig. 2B,D; N = 8, n = 11, RM ANOVA, p < 0.001). During short bursts of presynaptic activity, the mean decay tau increased from 2 to 10 pulses, although post hoc significance was only observed when longer trains of HFS were applied (Fig. 2C,E; N = 8, n = 11, RM ANOVA, p < 0.001). In response to short bursts of activity, there was no significant correlation between iGluSnFR %ΔF/F and decay measurements (Fig. 2F) as seen in the hippocampus; however, a strong association was observed following HFS protocols in the cortex (Fig. 2G), with larger responses taking longer to clear from the extracellular space. We also found a significant positive correlation between decay tau and the total amount of glutamate released during longer stimulus trains, as measured by the area under the curve of iGluSnFR responses (50 pulses, r = 0.743, p = 0.009; 100 pulses, r = 0.842, p < 0.001).
Figure 2.
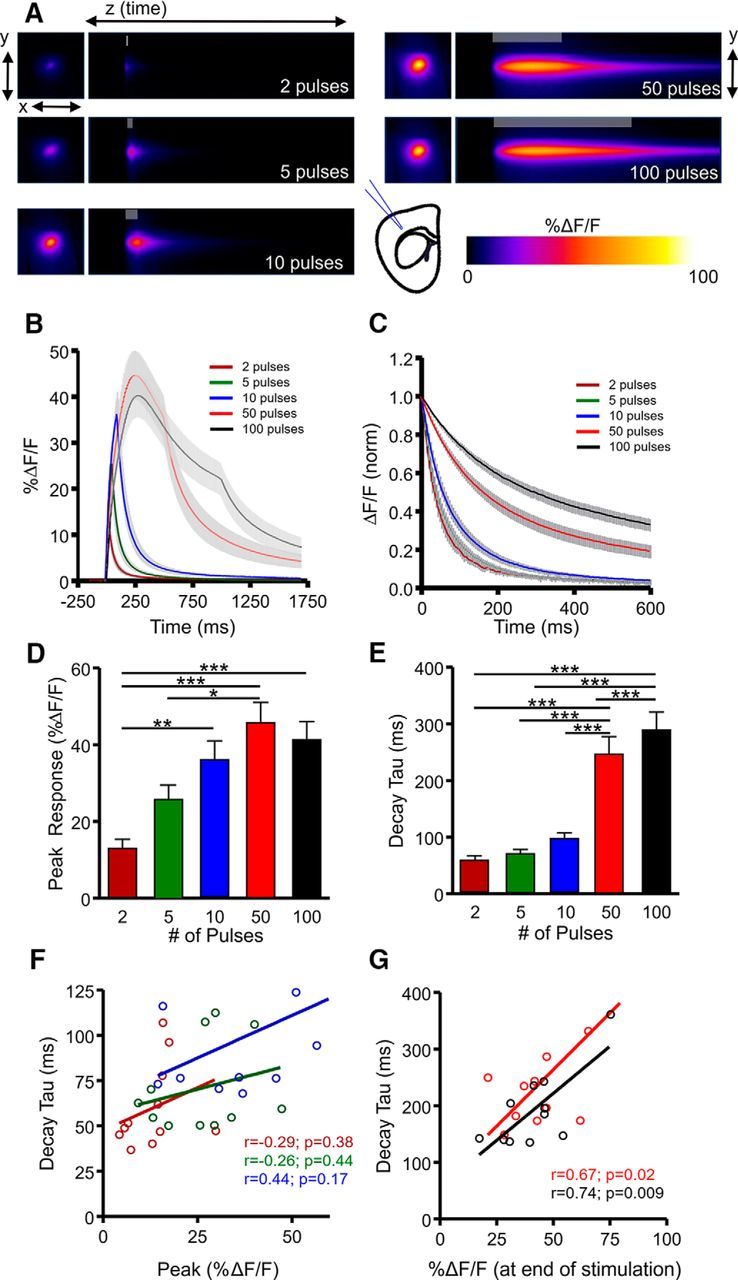
Characterization of extracellular glutamate dynamics in the cortex. A, Representative heatmaps of iGluSnFR responses after afferent stimulation (100 Hz). Peak responses are shown in the x–y plane (image size, 2 × 2 mm), and the y–z (time) plots show the kinetics of the response at a defined x-coordinate adjacent to the site of stimulation (image represents 2 s). The gray shaded area within the images denotes the onset and duration of afferent stimulation. B, Mean (±SEM) iGluSnFR response profiles to 2, 5, 10, 50, or 100 pulses of afferent stimulation at 100 Hz. C, Mean responses (±SEM) from B that were normalized to the peak value at the end of the stimulation. D–E, Grouped data showing mean (±SEM) iGluSnFR response peak (D) and decay tau (E). RM ANOVA with Tukey's post hoc results are indicated by *p < 0.05 and ***p < 0.001. F–G, Decay and %ΔF/F correlation of the iGluSnFR responses with a short burst of activity (F) and longer trains of high-frequency stimulation (G). **p < 0.01.
In the striatum (Fig. 3A), the amount of evoked glutamate release was also highly sensitive to the number of pulses of afferent stimulation, as expected (Fig. 3B,D; N = 4, n = 8, RM ANOVA, p < 0.001). The mean clearance rate increased as burst size increased from 2 to 10 pulses, but again, post hoc significance was only observed after longer trains of HFS (Fig. 3C,E; N = 4, n = 8, RM ANOVA, p < 0.001), similar to our observations in the cortex. Within a given stimulation paradigm, we observed no significant correlation between iGluSnFR %ΔF/F and decay taus for short bursts (Fig. 3F) or longer trains of HFS (Fig. 3G), suggesting that like the hippocampus, striatal clearance rates are more dependent on the duration of presynaptic activity than the amount of glutamate release per se. There was also no significant correlation between decay tau and the total amount of glutamate released during longer stimulus trains, as measured by the area under the curve of iGluSnFR responses (50 pulses, r = 0.108, p = 0.798; 100 pulses, r = 0.543, p = 0.164).
Figure 3.
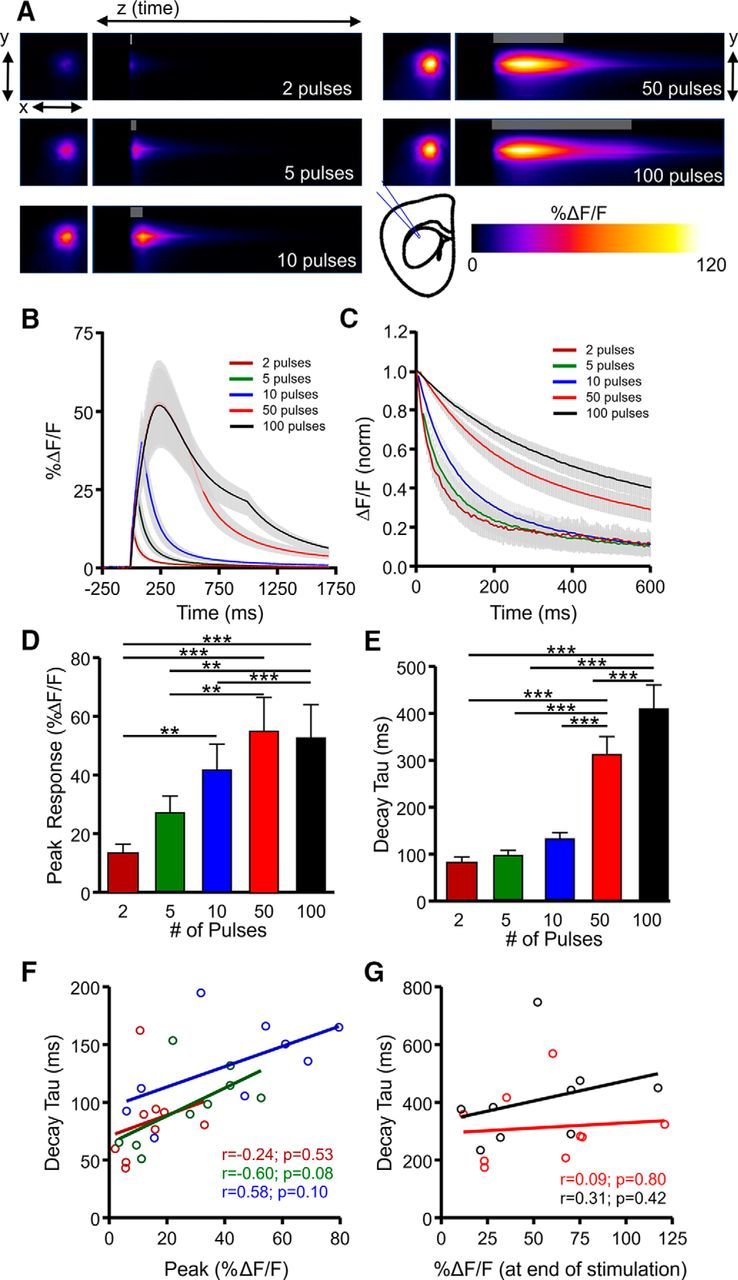
Characterization of extracellular glutamate dynamics in the striatum. A, Representative heatmaps of iGluSnFR responses after afferent stimulation (100 Hz). Peak responses are shown in the x–y plane (image size, 2 × 2 mm), and the y–z (time) plots show the kinetics of the response at a defined x-coordinate adjacent to the site of stimulation (image represents 2 s). The gray shaded area within the images denotes the onset and duration of afferent stimulation. B, Mean (±SEM) iGluSnFR response profiles to 2, 5, 10, 50, or 100 pulses of afferent stimulation at 100 Hz. C, Mean responses (±SEM) from B that were normalized to the peak value at the end of the stimulation. D–E, Grouped data showing mean (±SEM) iGluSnFR response peak (D) and decay tau (E). RM ANOVA with Tukey's post hoc results are indicated by **p < 0.01 and ***p < 0.001. F–G, Decay and %ΔF/F correlation of the iGluSnFR responses with a short burst of activity (F) and longer trains of high-frequency stimulation (G).
Direct comparison of glutamate dynamics in the hippocampus, cortex, and striatum
Although the general relationships between presynaptic activity and clearance rate appeared to be similar for the hippocampus, cortex, and striatum, interesting regional differences were revealed when we directly compared glutamate clearance rates in the three regions in response to shorter bursts of presynaptic activity and longer trains of HFS (Fig. 4). On direct comparison of the raw decay tau data, we found that the hippocampus was significantly more efficient than both the cortex and striatum in clearing glutamate released into the extracellular space during synaptic activity. With respect to short bursts, two-way RM ANOVA revealed clear regional effects, with the fastest mean clearance rates occurring in the hippocampus, the intermediate clearance rates in the cortex, and the slowest clearance rates in the striatum (Fig. 4A–E; hippocampus, N = 10, n = 17; cortex, N = 8, n = 11; striatum, N = 4, n = 8; two-way RM ANOVA; brain region, p < 0.001; number of pulses, p < 0.001; interaction, p < 0.001). Responses tended to be larger in the cortex and striatum compared with the hippocampus; however, the lower peaks in the hippocampus cannot explain the faster decay kinetics observed in this region as the same regional differences were observed when we compared the larger hippocampal responses with the smaller cortical and striatal responses such that the peaks matched (Fig. 4D). A significant interaction effect was also observed, reflecting the fact that mean clearance rates became gradually slower in the cortex and striatum as the number of pulses increased from 2 to 10, whereas the hippocampus was relatively unaffected by the same increase in burst duration. Indeed, the fold increase in iGluSnFR decay tau as the number of pulses within the burst increased from 2 to 10 was significantly greater for the cortex and the striatum compared with the hippocampus (Fig. 4F; hippocampus, N = 10, n = 17; cortex, N = 8, n = 11; striatum, N = 4, n = 8; ANOVA, p = 0.007). Similarly, during HFS, clear regional effects were observed (Fig. 4G; hippocampus, N = 10, n = 17; cortex, N = 8, n = 11; striatum, N = 4, n = 8; two-way RM ANOVA; brain region, p < 0.001; number of pulses, p < 0.001; interaction, p = 0.272), with mean clearance rates that were fastest in the hippocampus, slower in the cortex, and slowest in the striatum. Together, these data demonstrate that the hippocampus is more efficient than the cortex and striatum at clearing extracellular glutamate after a wide range of presynaptic activity and that transporters in the cortex and striatum appear to become overwhelmed before those in the hippocampus (Diamond and Jahr, 2000) after shorter bursts of activity.
Figure 4.
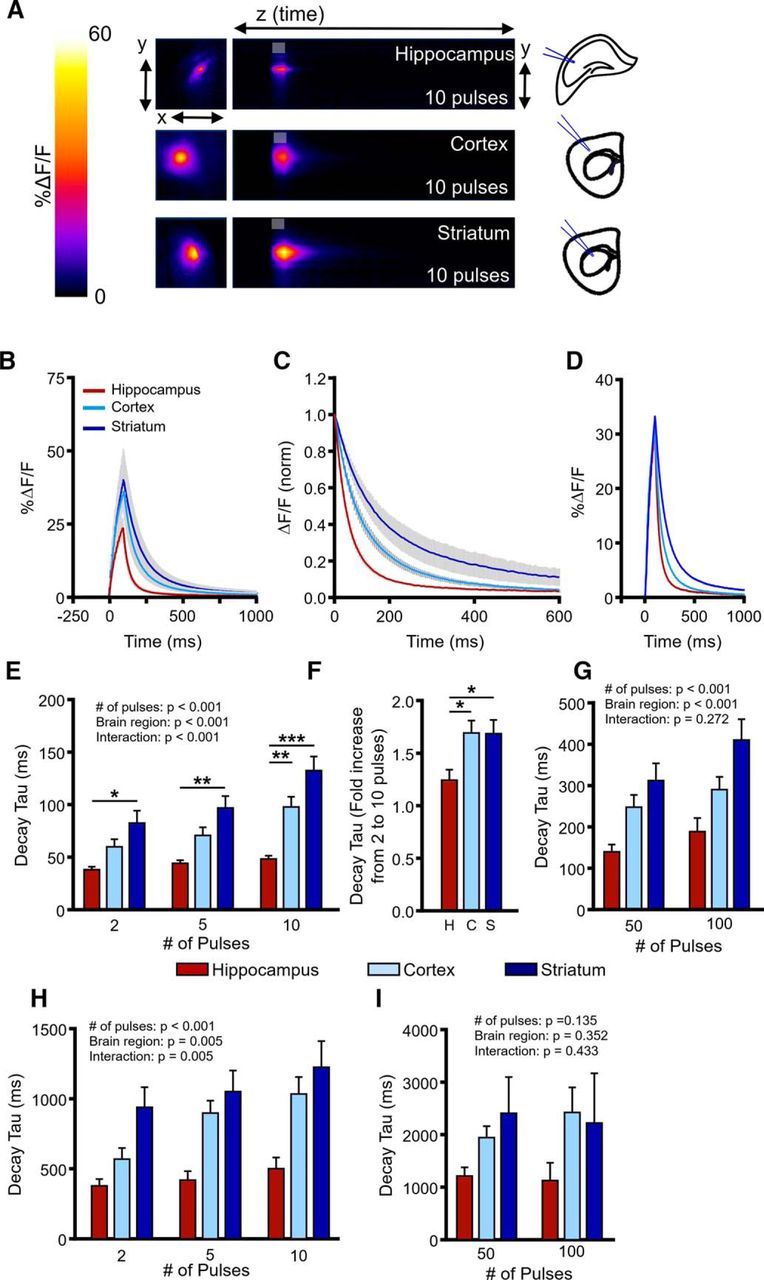
Regional differences in glutamate clearance capacity. A, Representative heatmaps of iGluSnFR responses in the hippocampus, cortex, and striatum after afferent stimulation (10 pulses at 100 Hz). Peak responses are shown in the x–y plane (image size, 2 × 2 mm), and the y–z (time) plots show the kinetics of the response at a defined x-coordinate adjacent to the site of stimulation (image represents 2 s). The gray shaded area within the images denotes the onset and duration of afferent stimulation. B, Mean (±SEM) iGluSnFR responses in the hippocampus, cortex, and striatum to 10 pulses at 100 Hz. C, Mean responses (±SEM) from B that were normalized to the peak value at the end of the stimulation. D, Raw responses (±SEM) from B were selected so that the average peak value was comparable between the three brain regions. E–G, Regional comparison of mean (±SEM) iGluSnFR decay tau values showing fastest glutamate clearance rates in the hippocampus, intermediate clearance rates in the cortex, and slow clearance rates in the striatum, for both shorter bursts of activity (E, F) and longer trains of high-frequency stimulation (G). H–I, Mean (±SEM) iGluSnFR decay tau values in the hippocampus, cortex, and striatum during glutamate transporter blockade with TBOA (100 μm) to quantify relative glutamate diffusion rates. *p < 0.05, **p < 0.01, ***p < 0.001.
Because glutamate is not enzymatically degraded in the extracellular space, the faster clearance rates observed in the hippocampus could be attributable to faster diffusion away from the release site and/or faster transporter-mediated uptake. Using integrative optical imaging and real-time iontophoresis, it has been previously suggested that tortuosity (i.e., barriers to diffusion) is low in the hippocampus compared with other brain areas (Hrabětová, 2005). Here, we quantified the relative glutamate diffusion rate of endogenously released glutamate by stimulating neural activity in the presence of the glutamate transporter blocker TBOA (100 μm) and monitored iGluSnFR transients as before. Both d-APV (50 μm) and DNQX (20 μm) were added to the bath during these experiments. Not surprisingly, TBOA had a dramatic effect on the rate at which glutamate was cleared from the extracellular space after synaptic activity; however, we still observed significant regional differences in iGluSnFR decay tau after short bursts of activity, with faster decay kinetics observed in the hippocampus compared with the cortex and striatum (Fig. 4H; hippocampus, N = 3, n = 7; cortex, N = 4, n = 6; striatum, N = 5, n = 9; two-way RM ANOVA; number of pulses, p < 0.001: brain region, p = 0.005; interaction, p = 0.005). These data are in agreement with diffusion studies (Hrabětová, 2005) and suggest that rapid extracellular diffusion can account, at least in part, for the efficient clearance rates observed in the hippocampus. Mean clearance rates in TBOA were also faster in the hippocampus after HFS, although the dataset was much more variable, particularly in the striatum, and no significant effect was observed (Fig. 4I; hippocampus, N = 3, n = 5; cortex, N = 5, n = 7; striatum, N = 5, n = 10; two-way RM ANOVA; number of pulses, p = 0.135; brain region, p = 0.352; interaction, p = 0.433).
To account for regional differences in the number of axons recruited during electrical stimulation, we varied the stimulation intensity from 25 to 250 μA while holding the stimulation duration constant (5 pulses at 100 Hz; Fig. 5). Not surprisingly, increasing the stimulus intensity resulted in larger iGluSnFR responses in all regions (Fig. 5A; hippocampus, N = 4, n = 7; cortex, N = 4, n = 9; striatum, N = 5, n = 11; two-way RM ANOVA; stimulus intensity, p < 0.001; brain region, p = 0.117; interaction, p = 0.060). As before, glutamate clearance rates were consistently the fastest in the hippocampus, intermediate in the cortex, and slowest in the striatum across all stimulation intensities tested (Fig. 5B; hippocampus, N = 4, n = 7; cortex, N = 4, n = 9; striatum, N = 5, n = 11; two-way RM ANOVA; stimulus intensity, p < 0.001; brain region, p < 0.001; interaction, p = 0.147). Stimulus intensity also had a small but consistent and significant effect on iGluSnFR decay tau; however, iGluSnFR response peaks were much more sensitive than decay to increasing stimulus strengths regardless of the brain region tested (Fig. 5C–E; hippocampus, N = 4, n = 7; two-way RM ANOVA; stimulus intensity, p < 0.001; peak vs decay, p < 0.001; interaction, p < 0.001; cortex, N = 4, n = 9; two-way RM ANOVA; stimulus intensity, p < 0.001; peak vs decay, p < 0.001; interaction, p < 0.001; striatum, N = 5, n = 11; two-way RM ANOVA; stimulus intensity, p < 0.001; peak vs decay, p < 0.001; interaction, p < 0.001). Therefore, recruiting more glutamate afferents with larger stimulus intensities dramatically enhances the size of the iGluSnFR response but has a relatively limited effect on decay kinetics. These data support the idea that clearance rates are more heavily influenced by the duration of afferent activity than the magnitude of release.
Figure 5.

Effect of stimulus intensity on extracellular glutamate dynamics. A, Mean (±SEM) iGluSnFR peaks in the hippocampus, cortex, and striatum in response to increasing the stimulus intensity (5 pulses, 100 Hz) from 25 to 250 μA. B, Mean (±SEM) iGluSnFR decay tau values in the hippocampus, cortex, and striatum in response to increasing the stimulus intensity (5 pulses, 100 Hz) from 25 to 250 μA. C–E, Comparison of the effect of stimulus intensity on iGluSnFR peak (solid bars) and decay tau (hatched bars) for the hippocampus (C), cortex (D), and striatum (E). Data were normalized to the response at 25 μA. All p values were obtained by two-way RM ANOVA.
Relative contributions of GLT-1 and non-GLT-1 transporters to glutamate clearance
Prior work used a pharmacological approach to quantify the functional contribution of GLT-1 relative to other excitatory amino acid transporters by measuring STCs before and after bath application of a saturating concentration of DHK (300 μm). The authors calculated a decay ratio, simply defined as the STC decay tau after transporter inhibition divided by the decay tau before transporter inhibition, and determined that the GLT-1 contribution to total uptake was greater in the hippocampus compared with the cortex, but only in the first postnatal week (Hanson et al., 2015). Here, we applied the same approach to the iGluSnFR transients to calculate a DHK (300 μm) decay ratio, representing the relative contribution of GLT-1 transporters to overall clearance kinetics, and also calculated a TBOA (100 μm) decay ratio, representing the relative contribution of all transporters to overall clearance kinetics. Note that the latter is not possible with STC recordings, as the STC is completely inhibited by this concentration of TBOA. In the hippocampus (Fig. 6), blocking GLT-1 resulted in an approximate twofold increase in both the size of evoked iGluSnFR responses (Fig. 6A,B,D) as well as the time to clear, regardless of the amount of presynaptic activity (Fig. 6A,C,E). We confirmed that 300 μm DHK was also a saturating concentration in our experimental setup (Fig. 6B, inset). The observed twofold increase in clearance rates after GLT-1 inhibition is similar to the decay ratio observed previously for STCs (Hanson et al., 2015). Interestingly, when we applied TBOA (100 μm) to block all transporter-mediated uptake, decay ratios were approximately fivefold larger than the decay ratios observed after DHK treatment (Fig. 6E; N = 3, n = 6; two-way RM ANOVA; treatment, p < 0.001; number of pulses, p = 0.397; interaction, p = 0.632), and TBOA had a slight but significant tendency to increase the size of the responses relative to DHK (Fig. 6D; N = 3, n = 6; two-way RM ANOVA; treatment, p = 0.037; number of pulses, p = 0.616; interaction, p = 0.698). For decay ratios, significant post hoc differences between the DHK and TBOA effects were observed for each stimulation paradigm tested (Fig. 6E; p < 0.001, Bonferroni's).
Figure 6.
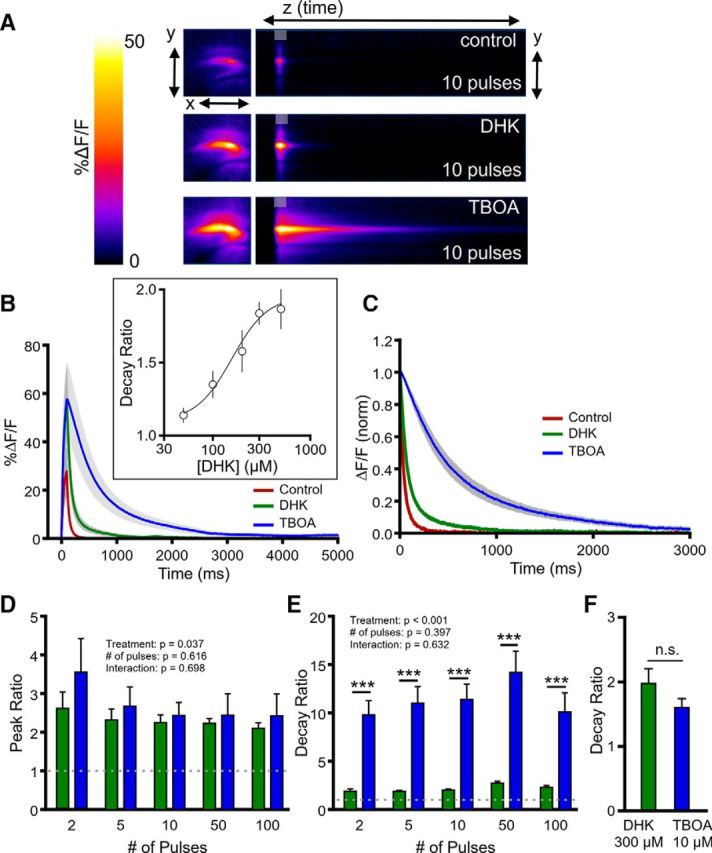
Comparison of the effects of selective GLT-1 inhibition and nonselective transporter inhibition on glutamate dynamics in the hippocampus. A, Representative heatmaps of hippocampal iGluSnFR responses after afferent stimulation (10 pulses at 100 Hz) in control conditions and after GLT-1 blockade with DHK (300 μm) and nonselective glutamate transporter blockade with TBOA (100 μm). Peak responses are shown in the x–y plane (image size, 2 × 2 mm), and the y–z (time) plots show the kinetics of the response at a defined x-coordinate adjacent to the site of stimulation (image represents 2 s). The gray shaded area within the images denotes the onset and duration of afferent stimulation. B, Mean (±SEM) iGluSnFR response profiles to 10 pulses of afferent stimulation at 100 Hz in control, DHK, and TBOA conditions. The inset shows a concentration–response curve for the DHK effect on the iGluSnFR decay ratio. C, Mean responses (±SEM) from B that were normalized to the peak value at the end of the stimulation. D–E, Grouped data showing mean (±SEM) DHK and TBOA peak ratios (D) and decay ratios (E) over a variety of stimulation paradigms. Peak and decay ratios indicate the fold effect of drug treatment over control levels. F, Mean (±SEM) decay ratios for 300 μm DHK and 10 μm TBOA. Two-way RM ANOVA with Bonferroni's post hoc results are indicated by ***p < 0.001. Unpaired t-test used in F. n.s. non-significant.
The dramatic difference between the decay ratio obtained in TBOA compared with DHK indicates either that non-GLT-1 transporters can contribute substantially to uptake capacity when GLT-1 is dysfunctional or that GLT-1 plays much less of a role in glutamate uptake than previously thought. To address the latter possibility, we compared the effect of saturating DHK (300 μm) with a concentration of TBOA (10 μm) previously shown to have the same effect as 300 μm DHK on STCs in the hippocampus (Diamond, 2005). We reasoned that if GLT-1 plays only a minor role in overall glutamate clearance, 10 μm TBOA, which blocks all glutamate transporter subtypes, should have a greater effect than 300 μm DHK on iGluSnFR decay kinetics. We found that the decay ratio was not significantly different between 10 μm TBOA and 300 μm DHK (Fig. 6F; DHK, N = 3, n = 4; TBOA, N = 4, n = 6; t test, p = 0.149), arguing against the interpretation that GLT-1 plays only a minor role in glutamate clearance. In all, our pharmacological data indicate that non-GLT-1 transporters contribute substantially to uptake capacity when GLT-1 is dysfunctional.
Next, we evaluated decay ratios induced by DHK (300 μm) and TBOA (100 μm) in the cortex. Interestingly, GLT-1 inhibition by DHK increased the peak of evoked glutamate transients (Fig. 7A,B,D), similar to our observation in the hippocampus; however, peaks were reduced back to baseline levels after the application of TBOA, and we observed a significant difference between DHK and TBOA on peak responses (Fig. 7D; N = 5, n = 7, two-way RM ANOVA; treatment, p < 0.001; number of pulses, p = 0.858; interaction, p = 0.999) regardless of the amount of presynaptic activity. Despite the larger responses after DHK application, we again found that TBOA had a much more profound effect on glutamate clearance, with DHK only slowing clearance rates to a fraction of the effect observed after TBOA (Fig. 7A,C,E; N = 5, n = 7, two-way RM ANOVA; treatment, p < 0.001; number of pulses, p = 0.147; interaction, p = 0.451).
Figure 7.
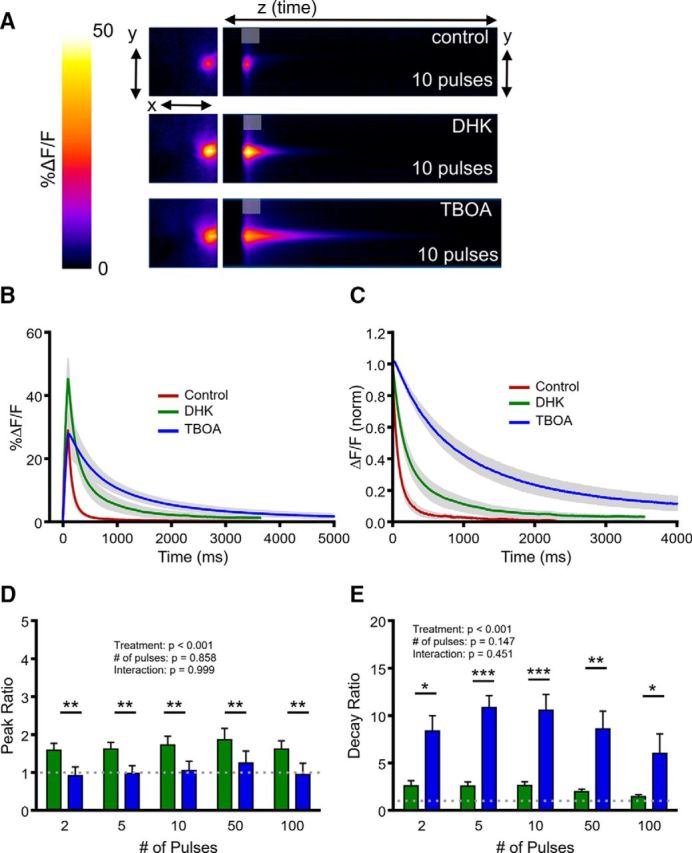
Comparison of the effects of selective GLT-1 inhibition and nonselective transporter inhibition on glutamate dynamics in the cortex. A, Representative heatmaps of cortical iGluSnFR responses after afferent stimulation (10 pulses at 100 Hz) in control conditions and after GLT-1 blockade with DHK (300 μm) and nonselective glutamate transporter blockade with TBOA (100 μm). Peak responses are shown in the x–y plane (image size, 2 × 2 mm), and the y–z (time) plots show the kinetics of the response at a defined x-coordinate adjacent to the site of stimulation (image represents 2 s). The gray shaded area within the images denotes the onset and duration of afferent stimulation. B, Mean (±SEM) iGluSnFR response profiles to 10 pulses of afferent stimulation at 100 Hz in control, DHK and TBOA conditions. C, Mean responses (±SEM) from B that were normalized to the peak value at the end of the stimulation. D–E, Grouped data showing mean (±SEM) DHK and TBOA peak ratios (D) and decay ratios (E) over a variety of stimulation paradigms. Peak and decay ratios indicate the fold effect of drug treatment over control levels. Two-way RM ANOVA with Bonferroni's post hoc results are indicated by *p < 0.05, **p < 0.01, and ***p < 0.001.
In the striatum, DHK and TBOA also had different effects on the amount of evoked glutamate release, with DHK facilitating release and TBOA returning iGluSnFR peaks to control or lower-than-control values (Fig. 8A,B,D; N = 6, n = 11, two-way RM ANOVA; treatment, p < 0.001; number of pulses, p = 0.232; interaction, p = 0.040). The significant interaction effect reflects the tendency for TBOA to suppress evoked glutamate release primarily at lower stimulus durations (Fig. 8D). Regardless of the peak response, we again found that DHK had a relatively small effect on clearance rates when compared with the effect of TBOA (Fig. 8A,C,E; N = 6, n = 11, two-way RM ANOVA; treatment, p < 0.001; number of pulses, p = 0.105; interaction, p = 0.379), similar to the results obtained in the hippocampus and cortex. Together, these data suggest a complex effect of transporter dysfunction on activity-dependent glutamate release and that a saturating concentration of the GLT-1 blocker DHK only slows glutamate clearance to ∼20–25% of that induced by nonselective transporter blockade with TBOA.
Figure 8.
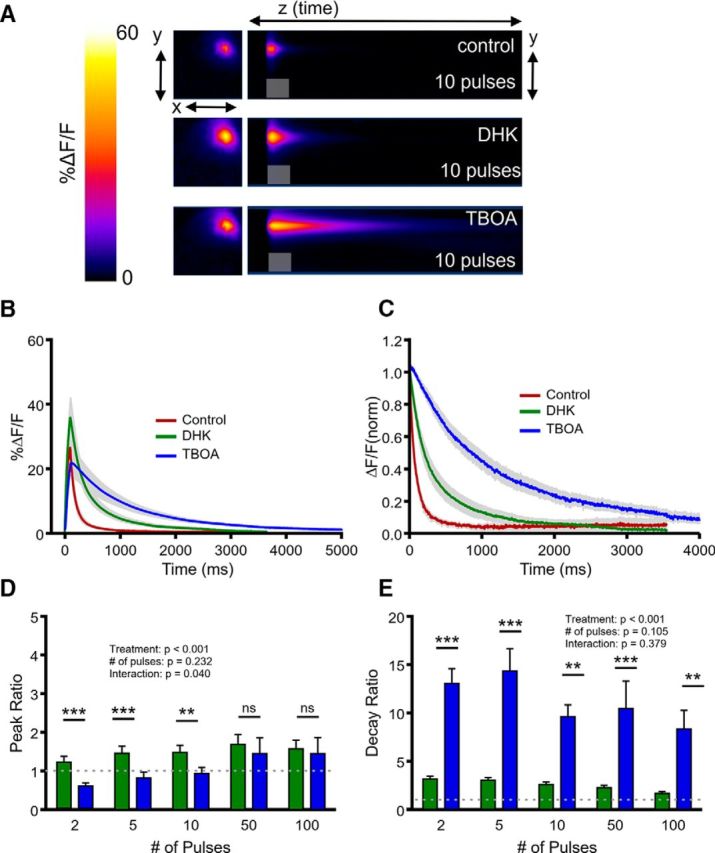
Comparison of the effects of selective GLT-1 inhibition and nonselective transporter inhibition on glutamate dynamics in the striatum. A, Representative heatmaps of striatal iGluSnFR responses after afferent stimulation (10 pulses at 100 Hz) in control conditions and after GLT-1 blockade with DHK (300 μm) and nonselective glutamate transporter blockade with TBOA (100 μm). Peak responses are shown in the x–y plane (image size, 2 × 2 mm), and the y–z (time) plots show the kinetics of the response at a defined x-coordinate adjacent to the site of stimulation (image represents 2 s). The gray shaded area within the images denotes the onset and duration of afferent stimulation. B, Mean (±SEM) iGluSnFR response profiles to 10 pulses of afferent stimulation at 100 Hz in control, DHK and TBOA conditions. C, Mean responses (±SEM) from B that were normalized to the peak value at the end of the stimulation. D–E, Grouped data showing mean (±SEM) DHK and TBOA peak ratios (D) and decay ratios (E) over a variety of stimulation paradigms. Peak and decay ratios indicate the fold effect of drug treatment over control levels. Two-way RM ANOVA with Bonferroni's post hoc results are indicated by **p < 0.01 and ***p < 0.001.
As DHK decay ratios were similar for the three regions tested, we asked whether GLT-1 expression levels were similar at the age at which our functional experiments were conducted (∼2 months). In agreement with our functional data, protein expression of GLT-1 was similar for the hippocampus, cortex, and striatum (Fig. 9; n = 9–11 per bar, ANOVA, p = 0.640), suggesting that the enhanced clearance rates in the hippocampus are likely not attributable to an increase in GLT-1 function or expression.
Figure 9.

GLT-1 expression levels are similar in the adult hippocampus, cortex, and striatum. A, Representative Western blot of GLT-1 expression in the cortex (Ctx), hippocampus (Hpc), and striatum (Str). B, Quantification of mean (±SEM) GLT-1 expression in the cortex (C), hippocampus (H), and striatum (S). ANOVA, p = 0.640.
Glutamate transporters are responsible for the rapid removal of synaptically released glutamate from the extracellular space but also work to keep ambient levels of extracellular glutamate low in the absence of substantial neural activity. By maintaining the ambient concentration of extracellular glutamate low, glutamate transporters ensure a high signal-to-noise ratio during synaptic neurotransmission (Danbolt, 2001). Therefore, we asked whether transporter inhibition affects ambient glutamate levels (as measured by raw, unstimulated iGluSnFR fluorescence; Parsons et al., 2016) differently in the hippocampus, cortex, and striatum. Experiments were performed in the presence of d-APV (50 μm) and DNQX (20 μm). Interestingly, DHK (300 μm) had very little effect on basal iGluSnFR values in all three regions (Fig. 10), suggesting that any rise in ambient glutamate induced by GLT-1 inhibition is too low to be detected by iGluSnFR. However, when TBOA was applied, clear elevations in basal iGluSnFR fluorescence were detected in the cortex and striatum (Fig. 10B,C; ANOVA, p < 0.001, and post hoc Dunnett's test of control vs TBOA, p < 0.01, for both the cortex and striatum), whereas the elevation observed in the hippocampus was modest in comparison (Fig. 10A; ANOVA, p = 0.027, and post hoc Dunnett's test of control vs TBOA, p > 0.05). Thus, similar to the clearance of evoked glutamate transients, non-GLT-1 transporters can help maintain low ambient levels of extracellular glutamate when GLT-1 is dysfunctional.
Figure 10.

Ambient glutamate levels increase after the application of TBOA but not DHK. A–C, Basal, unstimulated iGluSnFR fluorescence in control conditions and after GLT-1 inhibition with 300 μm DHK and nonselective glutamate transporter inhibition with 100 μm TBOA in the hippocampus (A), cortex (B), and striatum (C). One-way RM ANOVA significance was observed for all three regions. Dunnett's test post hoc significance is indicated by **p < 0.01.
Discussion
The time course of synaptically released extracellular glutamate is shaped by transporter-mediated uptake and the morphological properties of the extracellular space proximal to the release site (Danbolt, 2001; Thomas et al., 2011). It is important to understand how extracellular glutamate is regulated in the CNS, as excess extracellular glutamate can impair synaptic plasticity (Li et al., 2011) and promote excitotoxicity (Hardingham and Bading, 2010; Parsons and Raymond, 2014), and deficits in GLT-1-mediated uptake and the subsequent pathological accumulation of extracellular glutamate have been implicated in numerous conditions of the CNS including Alzheimer's disease, Huntington disease, and amyotrophic lateral sclerosis (Rothstein et al., 1995; Masliah et al., 1996; Liévens et al., 2001; Behrens et al., 2002; Howland et al., 2002; Miller et al., 2008; Huang et al., 2010; Scott et al., 2011). Here, we used wide-field iGluSnFR imaging (Marvin et al., 2013; Armbruster et al., 2016; Parsons et al., 2016) to characterize the real-time extracellular profiles of synaptically released glutamate in various brain regions and in response to a wide range of synaptic stimulation.
Regional differences in glutamate clearance
By quantifying the decay kinetics of STCs in acute hippocampal slices, it was previously demonstrated that glutamate transporters are not overwhelmed by stimulus trains up to 10 pulses at 100 Hz (Diamond and Jahr, 2000). We demonstrate a similar efficiency here in the hippocampus using iGluSnFR but show that this efficiency is not observed in the cortex and striatum. In the cortex and striatum, increasing the number of pulses in the stimulus burst from 2 to 10 (100 Hz) increased the glutamate clearance time approximately twofold, suggesting that transporters in these regions become overwhelmed earlier than those in the hippocampus. Furthermore, iGluSnFR decay rates were consistently faster in the hippocampus than in the cortex and striatum regardless of the amount of presynaptic activity, consistent with a recent observation of STCs at postnatal day 14, where cortical STC decay tau values were approximately twofold slower than the same values in the hippocampus (Hanson et al., 2015). Interestingly, a recent study elegantly demonstrated using serial electron microscopy that the distance between astrocytes and the postsynaptic density is shorter in the hippocampus compared with the striatum (Chai et al., 2017), which may account for the enhanced efficiency of transporter-mediated uptake in the hippocampus. In addition, prior integrative optical imaging and real-time iontophoresis experiments demonstrated that diffusion rates are faster in the hippocampus compared with the cortex (Hrabětová, 2005). In the present study, relative diffusion rates of synaptically released glutamate could be quantified by monitoring the iGluSnFR decay tau while blocking transporter-mediated uptake with 100 μm TBOA, and we found faster diffusion rates in the hippocampus compared with both the cortex and striatum, regardless of the amount of presynaptic activity. Together, these data demonstrate clear regional- and activity-dependent differences in the rate of glutamate clearance.
A recent study used both iGluSnFR and STC recordings to demonstrate that presynaptic activity influences glutamate clearance in the cortex (Armbruster et al., 2016). Here, we also observed a clear effect of presynaptic activity on the rate of glutamate clearance, with all tested regions showing a dramatic increase in the time required to clear extracellular glutamate after longer HFS trains of 50 or 100 pulses compared with shorter bursts of activity. As glutamate uptake is voltage dependent (Brew and Attwell, 1987), astrocyte depolarization during neural activity (Meeks and Mennerick, 2007) may account for the slow clearance rates observed after long stimulus trains. Furthermore, neural activity induces glial swelling and reduces the extracellular volume fraction, which can increase tortuosity and slow glutamate diffusion during long stimulus trains (Syková et al., 1999; Syková and Nicholson, 2008).
Effects of transporter inhibition on iGluSnFR responses
For the past two decades, it has been widely reported that GLT-1 is responsible for the large majority of glutamate uptake in the brain, with percentages of 90% or higher often being cited in manuscript introductions and review articles. Here, we found that although blocking GLT-1 with 300 μm DHK indeed prolonged the time course of evoked iGluSnFR transients, it produced only a fraction of the effect observed after 100 μm TBOA application. Interestingly, approximately half of the STC persists after a saturating concentration of DHK (Diamond and Jahr, 2000; Thomas et al., 2011), which is likely to represent current through GLAST transporters. GLAST is also abundant on astrocyte membranes (Danbolt, 2001) and exists at an approximate density of 3200 transporters/μm3 in the hippocampus stratum radiatum (Lehre and Danbolt, 1998). Our data here confirm that whereas GLT-1 may be the most abundant transporter in the brain, non-GLT-1 transporters can make large contributions to the rate of glutamate clearance, particularly when GLT-1 is dysfunctional.
iGluSnFR also represents a powerful tool to study relative alterations in evoked presynaptic glutamate release. In the hippocampus, transporter inhibition by either DHK or TBOA increased the response peak, in agreement with a previously reported increase in EPSC amplitude after transporter inhibition in the adult hippocampus (Diamond, 2005). On the other hand, although DHK tended to increase response peaks in the cortex and striatum, response sizes were reduced in these regions after TBOA application, also in agreement with a prior observation of a TBOA-induced reduction of striatal EPSCs (Milnerwood et al., 2010). Transporter inhibition in the hypothalamus increases extracellular glutamate and reduces presynaptic release by activating presynaptic group III metabotropic glutamate receptors (Oliet et al., 2001). Here, TBOA had the greatest effect on ambient glutamate levels in the striatum and cortex, the two regions that exhibited a TBOA-induced depression of evoked glutamate release. Indeed, presynaptic mGluR activation has been shown to inhibit excitatory transmission at the corticostriatal synapse (Pisani et al., 1997) and in the deep layers of the cortex (Bandrowski et al., 2003). Thus, our results are consistent with electrophysiological observations of transporter inhibitor effects on EPSCs and suggest that iGluSnFR can be used as a powerful tool to study glutamate transmission, both with respect to relative changes in release probability and extracellular clearance rates.
Quantifying glutamate clearance in situ: methodological considerations
STCs have been used to great effect to study glutamate dynamics in situ and provide the greatest temporal resolution of the available methods, particularly when the filtering properties are mathematically eliminated from the STC kinetics (Diamond, 2005). However, STCs are only able to sense glutamate at the surface of an individual astrocyte, are technically demanding, and are extremely difficult to elicit in brain regions that have a low to moderate density of glutamate afferents. Wide-field imaging of iGluSnFR complements STCs in that they monitor extracellular glutamate sensed at the surface of a population of neurons (synapsin promoter) or astrocytes (GFAP promoter) and can be easily performed in multiple brain regions. However, iGluSnFR itself acts as a glutamate buffer and, therefore, exhibits slower absolute decay tau values compared with STCs under similar conditions. Nonetheless, the regional differences in glutamate clearance detected in the present study were robust enough to be clearly detected using iGluSnFR imaging. In fact, despite the differences in raw iGluSnFR and STC decay tau values, GLT-1 blockade with 300 μm DHK increases both the iGluSnFR (this study) and the STC (Diamond and Jahr, 2000; Scimemi et al., 2009, 2013; Thomas et al., 2011; Hanson et al., 2015) decay tau approximately twofold. Although it is possible that the slower absolute iGluSnFR decay reflects iGluSnFR detection of glutamate concentrations too low to be detected by STCs, iGluSnFR affinity is in the micromolar range, and the %ΔF/F values obtained in the present study match the values obtained by exogenous applications of micromolar concentrations of glutamate in the acute slice preparation (Parsons et al., 2016). Simultaneous iGluSnFR and STC recordings will help distinguish the detection thresholds for each method during synaptic activity and is of interest for future studies.
Conclusion
The results of the present study demonstrate clear regional- and activity-dependent differences in the rate of glutamate clearance after synaptic activity and further highlight the importance of studying glutamate regulation in situ. The two major findings of the present study are as follows: (1) the hippocampus is more efficient at clearing extracellular glutamate compared with the cortex and striatum; and (2) GLT-1 inhibition by DHK only slowed clearance rates to a fraction of that induced by TBOA, suggesting non-GLT-1 transporters can make a substantial contribution to the glutamate clearance rate, particularly when GLT-1 is dysfunctional. The relative efficiency of glutamate clearance may play a role in dictating the type and magnitude of synaptic plasticity observed after long trains of HFS. For example, the same HFS that produces clear long-term potentiation in the hippocampus results in presynaptically mediated long-term depression in the striatum. It is possible that the efficient uptake/clearance of glutamate in the hippocampus limits perisynaptic/extrasynaptic mGluR activity, which is required for HFS-induced striatal long-term depression (Sung et al., 2001). Furthermore, slow clearance rates may play a role in regional vulnerability to excitotoxity. It is tempting to speculate that the slow clearance rates in the striatum can partially explain this region's vulnerability in Huntington disease, a devastating neurodegenerative disease in which glutamate toxicity and extrasynaptic NMDA receptor activation are major contributors to cell death (Okamoto et al., 2009; Milnerwood et al., 2010; Parsons and Raymond, 2014). Future research is required to fully understand the functional consequences of such dramatic regional differences in glutamate clearance, as the spatiotemporal dynamics of glutamate ultimately determines the location and types of glutamate receptors that are activated during neurotransmission.
Footnotes
This work was supported by a Natural Sciences and Engineering Research Council of Canada Discovery Grant. We thank Firoozeh Nafar for excellent technical services and the animal care staff at Memorial University for assistance with animal housing.
The authors declare no competing financial interests.
References
- Armbruster M, Hanson E, Dulla CG (2016) Glutamate clearance is locally modulated by presynaptic neuronal activity in the cerebral cortex. J Neurosci 36:10404–10415. 10.1523/JNEUROSCI.2066-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandrowski AE, Huguenard JR, Prince DA (2003) Baseline glutamate levels affect group I and II mGluRs in layer V pyramidal neurons of rat sensorimotor cortex. J Neurophysiol 89:1308–1316. 10.1152/jn.00644.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB (2002) Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the huntington mutation. Brain 125:1908–1922. 10.1093/brain/awf180 [DOI] [PubMed] [Google Scholar]
- Brew H, Attwell D (1987) Electrogenic glutamate uptake is a major current carrier in the membrane of axolotl retinal glial cells. Nature 327:707–709. 10.1038/327707a0 [DOI] [PubMed] [Google Scholar]
- Chai H, Diaz-Castro B, Shigetomi E, Monte E, Octeau JC, Yu X, Cohn W, Rajendran PS, Vondriska TM, Whitelegge JP, Coppola G, Khakh BS (2017) Neural circuit-specialized astrocytes: transcriptomic, proteomic, morphological, and functional evidence. Neuron 95:531–549.e9. 10.1016/j.neuron.2017.06.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danbolt NC. (2001) Glutamate uptake. Prog Neurobiol 65:1–105. 10.1016/S0301-0082(00)00067-8 [DOI] [PubMed] [Google Scholar]
- Diamond JS. (2005) Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci 25:2906–2916. 10.1523/JNEUROSCI.5125-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diamond JS, Jahr CE (2000) Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol 83:2835–2843. 10.1152/jn.2000.83.5.2835 [DOI] [PubMed] [Google Scholar]
- Furuta A, Rothstein JD, Martin LJ (1997) Glutamate transporter protein subtypes are expressed differentially during rat CNS development. J Neurosci 17:8363–8375. 10.1523/JNEUROSCI.17-21-08363.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson E, Armbruster M, Cantu D, Andresen L, Taylor A, Danbolt NC, Dulla CG (2015) Astrocytic glutamate uptake is slow and does not limit neuronal NMDA receptor activation in the neonatal neocortex. Glia 63:1784–1796. 10.1002/glia.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696. 10.1038/nrn2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howland DS, Liu J, She Y, Goad B, Maragakis NJ, Kim B, Erickson J, Kulik J, DeVito L, Psaltis G, DeGennaro LJ, Cleveland DW, Rothstein JD (2002) Focal loss of the glutamate transporter EAAT2 in a transgenic rat model of SOD1 mutant-mediated amyotrophic lateral sclerosis (ALS). Proc Natl Acad Sci U S A 99:1604–1609. 10.1073/pnas.032539299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabětová S. (2005) Extracellular diffusion is fast and isotropic in the stratum radiatum of hippocampal CA1 region in rat brain slices. Hippocampus 15:441–450. 10.1002/hipo.20068 [DOI] [PubMed] [Google Scholar]
- Huang K, Kang MH, Askew C, Kang R, Sanders SS, Wan J, Davis NG, Hayden MR (2010) Palmitoylation and function of glial glutamate transporter-1 is reduced in the YAC128 mouse model of huntington disease. Neurobiol Dis 40:207–215. 10.1016/j.nbd.2010.05.027 [DOI] [PubMed] [Google Scholar]
- Lehre KP, Danbolt NC (1998) The number of glutamate transporter subtype molecules at glutamatergic synapses: chemical and stereological quantification in young adult rat brain. J Neurosci 18:8751–8757. 10.1523/JNEUROSCI.18-21-08751.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Jin M, Koeglsperger T, Shepardson NE, Shankar GM, Selkoe DJ (2011) Soluble A oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J Neurosci 31:6627–6638. 10.1523/JNEUROSCI.0203-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liévens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP (2001) Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis 8:807–821. 10.1006/nbdi.2001.0430 [DOI] [PubMed] [Google Scholar]
- Maragakis NJ, Rothstein JD (2006) Mechanisms of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2:679–689. 10.1038/ncpneuro0355 [DOI] [PubMed] [Google Scholar]
- Marvin JS, Borghuis BG, Tian L, Cichon J, Harnett MT, Akerboom J, Gordus A, Renninger SL, Chen TW, Bargmann CI, Orger MB, Schreiter ER, Demb JB, Gan WB, Hires SA, Looger LL (2013) An optimized fluorescent probe for visualizing glutamate neurotransmission. Nat Methods 10:162–170. 10.1038/nmeth.2333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Alford M, DeTeresa R, Mallory M, Hansen L (1996) Deficient glutamate transport is associated with neurodegeneration in Alzheimer's disease. Ann Neurol 40:759–766. 10.1002/ana.410400512 [DOI] [PubMed] [Google Scholar]
- Meeks JP, Mennerick S (2007) Astrocyte membrane responses and potassium accumulation during neuronal activity. Hippocampus 17:1100–1108. 10.1002/hipo.20344 [DOI] [PubMed] [Google Scholar]
- Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, Kennedy RT, Rebec GV (2008) Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington's disease phenotype in the R6/2 mouse. Neuroscience 153:329–337. 10.1016/j.neuroscience.2008.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milnerwood AJ, Gladding CM, Pouladi MA, Kaufman AM, Hines RM, Boyd JD, Ko RW, Vasuta OC, Graham RK, Hayden MR, Murphy TH, Raymond LA (2010) Early increase in extrasynaptic NMDA receptor signaling and expression contributes to phenotype onset in Huntington's disease mice. Neuron 65:178–190. 10.1016/j.neuron.2010.01.008 [DOI] [PubMed] [Google Scholar]
- Milton ID, Banner SJ, Ince PG, Piggott NH, Fray AE, Thatcher N, Horne CH, Shaw PJ (1997) Expression of the glial glutamate transporter EAAT2 in the human CNS: an immunohistochemical study. Mol Brain Res 52:17–31. 10.1016/S0169-328X(97)00233-7 [DOI] [PubMed] [Google Scholar]
- Okamoto S, Pouladi MA, Talantova M, Yao D, Xia P, Ehrnhoefer DE, Zaidi R, Clemente A, Kaul M, Graham RK, Zhang D, Vincent Chen HS, Tong G, Hayden MR, Lipton SA (2009) Balance between synaptic versus extrasynaptic NMDA receptor activity influences inclusions and neurotoxicity of mutant huntingtin. Nat Med 15:1407–1413. 10.1038/nm.2056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SHR, Piet R, Poulain DA (2001) Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science 292:923–926. 10.1126/science.1059162 [DOI] [PubMed] [Google Scholar]
- Parsons MP, Raymond LA (2014) Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82:279–293. 10.1016/j.neuron.2014.03.030 [DOI] [PubMed] [Google Scholar]
- Parsons MP, Vanni MP, Woodard CL, Kang R, Murphy TH, Raymond LA (2016) Real-time imaging of glutamate clearance reveals normal striatal uptake in huntington disease mouse models. Nat Commun 7:11251. 10.1038/ncomms11251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Navarrete M, Araque A (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32:421–431. 10.1016/j.tins.2009.05.001 [DOI] [PubMed] [Google Scholar]
- Petr GT, Sun Y, Frederick NM, Zhou Y, Dhamne SC, Hameed MQ, Miranda C, Bedoya EA, Fischer KD, Armsen W, Wang J, Danbolt NC, Rotenberg A, Aoki CJ, Rosenberg PA (2015) Conditional deletion of the glutamate transporter GLT-1 reveals that astrocytic GLT-1 protects against fatal epilepsy while neuronal GLT-1 contributes significantly to glutamate uptake into synaptosomes. J Neurosci 35:5187–5201. 10.1523/JNEUROSCI.4255-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phatnani H, Maniatis T (2015) Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol 7:1–18. 10.1101/cshperspect.a020628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisani A, Calabresi P, Centonze D, Bernardi G (1997) Activation of group III metabotropic glutamate receptors depresses glutamatergic transmission at corticostriatal synapse. Neuropharmacology 36:845–851. 10.1016/S0028-3908(96)00177-3 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW (1994) Localization of neuronal and glial glutamate transporters. Neuron 13:713–725. 10.1016/0896-6273(94)90038-8 [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Van Kammen M, Levey AI, Martin LJ, Kuncl RW (1995) Selective loss of glial glutamate transporter GLT-1 in amyotrophic lateral sclerosis. Ann Neurol 38:73–84. 10.1002/ana.410380114 [DOI] [PubMed] [Google Scholar]
- Scimemi A, Tian H, Diamond JS (2009) Neuronal transporters regulate glutamate clearance, NMDA receptor activation, and synaptic plasticity in the hippocampus. J Neurosci 29:14581–14595. 10.1523/JNEUROSCI.4845-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, Meabon JS, Woltjer RL, Sullivan JM, Diamond JS, Cook DG (2013) Amyloid-β1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci 33:5312–5318. 10.1523/JNEUROSCI.5274-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott HA, Gebhardt FM, Mitrovic AD, Vandenberg RJ, Dodd PR (2011) Glutamate transporter variants reduce glutamate uptake in Alzheimer's disease. Neurobiol Aging 32:553.e1–11. 10.1016/j.neurobiolaging.2010.03.008 [DOI] [PubMed] [Google Scholar]
- Sung KW, Choi S, Lovinger DM (2001) Activation of group I mGluRs is necessary for induction of long-term depression at striatal synapses. J Neurophysiol 86:2405–2412. 10.1152/jn.2001.86.5.2405 [DOI] [PubMed] [Google Scholar]
- Syková E, Vargová L, Prokopová S, Simonová Z (1999) Glial swelling and astrogliosis produce diffusion barriers in the rat spinal cord. Glia 25:56–70. [DOI] [PubMed] [Google Scholar]
- Syková E, Nicholson C (2008) Diffusion in brain extracellular space. Physiol Rev 88:1277–1340. 10.1152/physrev.00027.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas CG, Tian H, Diamond JS (2011) The relative roles of diffusion and uptake in clearing synaptically released glutamate change during early postnatal development. J Neurosci 31:4743–4754. 10.1523/JNEUROSCI.5953-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]