

Abstract
Synaptotagmin-1 (Syt1) binds Ca2+ through its tandem C2 domains (C2A and C2B) and triggers Ca2+-dependent neurotransmitter release. Here, we show that snt-1, the homolog of mammalian Syt1, functions as the Ca2+ sensor for both tonic and evoked neurotransmitter release at the Caenorhabditis elegans neuromuscular junction. Mutations that disrupt Ca2+ binding in double C2 domains of SNT-1 significantly impaired tonic release, whereas disrupting Ca2+ binding in a single C2 domain had no effect, indicating that the Ca2+ binding of the two C2 domains is functionally redundant for tonic release. Stimulus-evoked release was significantly reduced in snt-1 mutants, with prolonged release latency as well as faster rise and decay kinetics. Unlike tonic release, evoked release was triggered by Ca2+ binding solely to the C2B domain. Moreover, we showed that SNT-1 plays an essential role in the priming process in different subpopulations of synaptic vesicles with tight or loose coupling to Ca2+ entry.
SIGNIFICANCE STATEMENT We showed that SNT-1 in Caenorhabditis elegans regulates evoked neurotransmitter release through Ca2+ binding to its C2B domain in a similar way to Syt1 in the mouse CNS and the fly neuromuscular junction. However, the largely decreased tonic release in snt-1 mutants argues SNT-1 has a clamping function. Indeed, Ca2+-binding mutations in the C2 domains in SNT-1 significantly reduced the frequency of the miniature EPSC, indicating that SNT-1 also acts as a Ca2+ sensor for tonic release. Therefore, revealing the differential mechanisms between invertebrates and vertebrates will provide significant insights into our understanding how synaptic vesicle fusion is regulated.
Keywords: C. elegans, calcium sensor, evoked release, neuromuscular junction, tonic release, synaptotagmin
Introduction
Synaptotagmins are a large family of membrane proteins with multifunctional double C2 domains (C2A and C2B). Over 15 synaptotagmin isoforms have been identified in mammals, several of which have been shown to be important for synaptic vesicle (SV) exocytosis (Brose et al., 1992; Xu et al., 2007; Weber et al., 2014). Synaptotagmin-1 (Syt1), one of the most important members of the family, has been demonstrated to act as the major Ca2+ sensor of neurotransmitter release at many types of synapses (Geppert et al., 1994; Yoshihara and Littleton, 2002; Lee et al., 2013; Südhof, 2013). Each C2 domain of Syt1 contains five Ca2+-binding residues (aspartates; Nishiki and Augustine, 2004a). Upon binding Ca2+, the C2 domains insert into the plasma membrane, and promote membrane fusion. The C2 domains have been shown to interact with the SV fusion apparatus, such as syntaxin/synaptosomal-associated protein of 25 kDa (SNAP-25) dimer, to regulate SNARE-mediated membrane fusion (Rickman et al., 2004).
In cultured hippocampal or cortical neurons which lack Syt1, Ca2+-dependent evoked synchronous release is reduced, whereas slow asynchronous release is enhanced (Nishiki and Augustine, 2004b). Similar results have been observed at Drosophila neuromuscular junction (NMJ) in Syt1 null mutants (Lee et al., 2013). Although both the C2A and C2B domains contain Ca2+-binding residues and have similar topology, the Ca2+ binding by the latter domain is critical for evoked synchronous release (Littleton et al., 2001; Shin et al., 2003). Moreover, Syt2, the closest homolog of Syt1, exhibits a distinct expression pattern in mouse brain but exerts a similar function to Syt1 in triggering evoked synchronous release (Kochubey and Schneggenburger, 2011). Previous studies have reported that the asynchronous release requires another synaptotagmin isoform, synaptotagmin-7 (Syt7), which functions as an additional Ca2+ sensor (Weber et al., 2014; Bacaj et al., 2015).
However, the function of Syt1 in spontaneous release remains puzzling. Differing from its role in evoked release, Syt1 suppresses spontaneous release, as evidenced by the increased frequency of miniature EPSCs (mEPSCs) and IPSCs (mIPSCs) in cultured hippocampal or cortical neurons lacking Syt1 (Xu et al., 2009; H. Bai et al., 2016). Similar results have also been observed at the NMJ of Drosophila Syt1 null mutants (Lee et al., 2013). Several studies have hypothesized that Syt1 clamps another Ca2+ sensor which is activated in Syt1 knock-out neurons. In contrast to these studies, distinct roles of Syt1 in regulating spontaneous release have also been reported, with Syt1 knock-out being shown to result in a 40% decrease in spontaneous release at precalyceal synapses at the postnatal day 2 (P2) stage (Kochubey et al., 2016). In autapses, spontaneous release was unaffected in Syt1 knock-out mice (Geppert et al., 1994). Collectively, these studies suggest that the Syt1 may have differential roles for spontaneous release at different synapses.
The nematode Caenorhabditis elegans has been widely used to study the molecular and cellular mechanisms of synaptic transmission (Richmond et al., 1999; Martin et al., 2011). However, the major Ca2+ sensor for SV fusion in C. elegans has not yet been fully described. Synaptotagmin-1 in the worm is encoded by the snt-1 gene (Nonet et al., 1993). A previous study found that SV release at the neuromuscular junction of snt-1 mutants was significantly decreased (Yu et al., 2013). SNT-1 was also shown to regulate both the synaptic localization of Rab3-GAP on the SV membrane and repetitive synaptic release (Cheng et al., 2015). However, despite these findings, how synaptic transmission is regulated by Ca2+ binding to the C2 domains in SNT-1 is still to be determined.
In the present study, we investigated the function of SNT-1 in regulating Ca2+-dependent SV release at the C. elegans NMJ. We found that Ca2+ binding to the C2 domains in SNT-1 is an essential role in SV fusion. We also demonstrate that the priming of SVs with loose coupling to Ca2+ entry is nearly eliminated in snt-1 mutants, whereas the priming of SVs with tight coupling to Ca2+ entry is partially mediated by SNT-1.
Materials and Methods
Strains.
Strain maintenance and genetic manipulation were performed as described previously. Animals were cultivated at room temperature on nematode growth medium (NGM) agar plates seeded with OP50 bacteria. On the day before experiments, L4 larval stage animals were transferred to fresh plates seeded with OP50 bacteria for all the electrophysiological recordings. The following strains were used: wild-type, N2 Bristol; NM204, snt-1(md290) II; ZTH403, hztEx15001 [Psnb-1::SNT-1;snt-1(md290)I]; ZTH386, hztEx386 [Psnb-1::SNT-1ΔC2A;snt-1(md290)I]; ZTH385, hztEx385 [Psnb-1::SNT-1ΔC2B;snt-1(md290)I]; ZTH412, hztEx412 [Psnb-1::SNT-1C2A D3,4N;snt-1(md290)I]; ZTH413, hztEx413 [Psnb-1::SNT-1C2B D3,4N;snt-1(md290)I]; ZTH254, hztEx254 [Psnb-1::SNT-1C2AB D3,4N;snt-1(md290)I]; ZTH283, hztEx283 [Psnb-1::SNT-1C2A D1-5N;snt-1(md290)I]; ZTH282, hztEx282 [Psnb-1::SNT-1C2B D1-5N;snt-1(md290)I]; ZTH343, hztEx343 [Psnb-1::SNT-1C2AB D1-5N;snt-1(md290)I]; ZTH389, hztEx389 [Psnb-1::Syt1;snt-1(md290)I]; ZTH366, hztEx366 [Punc-129::SNT-1::GFP]; ZTH380, hztEx380 [Punc-129::SNT-1::mApple]; ZTH10, hztEX10 [Psnb-1::UNC-13L;unc-13(s69)]; ZTH11, hztEX11 [Psnb-1::UNC-13S;unc-13(s69)]; ZTH359, hztEX359 [Psnb-1::UNC-13L;unc-13(s69);snt-1(md290)]; ZTH373, hztEX373 [Psnb-1::UNC-13S;unc-13(s69);snt-1(md290)]; JSD95, tauIs44 [Punc129::mCherry::Rab3].
Constructs.
A 1.3 kb cDNA corresponding to snt-1a was amplified by PCR from the N2 cDNA library and inserted into MCSII (multiple cloning site II) of the JB6 vector between the KpnI and NotI sites.
The snb-1 promoter was inserted into MCSI (multiple cloning site I) between the SphI and BamHI sites. For the imaging strains, the snb-1 promoter was replaced by the unc-129 promoter using the same restriction sites. GFP/mApple was inserted after the C-terminal of SNT-1 between the NotI and MluI sites.
Transgenes and germline transformation.
Transgenic strains were isolated by microinjection of various plasmids using either Pmyo-2::NLS-GFP (KP#1106) or Pmyo-2::NLS-mCherry (KP#1480) as the coinjection marker. Integrated transgenes were obtained by UV irradiation of strains carrying extrachromosomal arrays. All integrated transgenes were outcrossed at least seven times.
Locomotion and behavior assays.
Worm tracking and analysis were performed as described previously (Dittman and Kaplan, 2008), with minor modifications. Young adult animals were washed with a drop of PBS and then transferred to fresh NGM plates with no bacterial lawn (30 worms per plate). Worm movement recordings were started 10 min after the worms were removed from food. A 1 min digital video of each plate was captured at a 1 Hz frame rate by a Qimaging camera on an Olympus MVX10 microscope using Micro-Manager 1.4.22. Average speed was determined for each animal using ImageJ plugin wrMTrch (http://www.phage.dk/plugins/wrmtrck.html).
Fluorescence imaging.
Animals were immobilized on 2% agarose pads with 30 mm levamisole. Fluorescence imaging was performed on a spinning-disk confocal system (3i Yokogawa W1 SDC) controlled by Slidebook 6.0 software. Animals were imaged with an Olympus 100× 1.4 numerical aperture Plan Apochromat objective. Z series of optical sections were acquired at 0.13 μm steps. Images were deconvolved with Huygens Professional version 16.10 (Scientific Volume Imaging) and then processed to yield maximum intensity projections using ImageJ 1.51n (W. Rasband, NIH, Bethesda, MD).
Protein purification.
The wild-type and mutant SNT-1 C2AB domains were cloned into pET28a at the SacI and XhoI sites for expression with an N-terminal His tag. The proteins were expressed in Escherichia coli BL21(DE3) cells overnight at 20°C and purified using Talon affinity resin, and elution with 50 mm HEPES, pH 7.5, 150 mm NaCl, and 200 mm imidazole. The proteins were then gel filtered using a Superdex200 FPLC column into 50 mm HEPES, pH 7.5, 150 mm NaCl, and 2 mm DTT.
Isothermal titration calorimetry.
Isothermal titration calorimetry (ITC) experiments were performed using a MicroCal PEAQ-ITC instrument. Experiments were performed at 25°C in 50 mm HEPES, pH 7.5, 150 mm NaCl, and 2 mm DTT, with 2 mm CaCl2 in the syringe injected into 50 μm proteins in the cell. The binding parameters were derived from a single-site binding model using MicroCal PEAQ-ITC analysis software. Due to limited protein concentrations, we did not attempt to analyze the data for the presence of multiple binding sites, and the stoichiometry was not well defined.
Electrophysiology.
Electrophysiology was conducted on dissected C. elegans as described previously (Hu et al., 2012). Worms were superfused in an extracellular solution containing the following (in mm): 127 NaCl, 5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 20 glucose, 1 CaCl2, and 4 MgCl2, bubbled with 5% CO2, 95% O2 at 20°C. Whole-cell recordings were carried out at −60 mV for all EPSCs, including mEPSCs, evoked EPSCs, and sucrose-evoked responses. The internal solution contained the following (in mm): 105 CH3O3SCs, 10 CsCl, 15 CsF, 4 MgCl2, 5 EGTA, 0.25 CaCl2, 10 HEPES, and 4 Na2ATP, adjusted to pH 7.2 using CsOH. Stimulus-evoked EPSCs were obtained by placing a borosilicate pipette (5–10 μm) near the ventral nerve cord (one muscle distance from the recording pipette) and applying a 0.4 ms, 85μA square pulse. Sucrose-evoked release was triggered by a 5s application of 0.5 m sucrose dissolved in normal bath solution. The pipette containing hypertonic sucrose was placed at the end of the patched muscle cell.
Data acquisition and statistical analysis.
All recordings were obtained using a HEKA EPC10 double amplifier filtered at 2 kHz, and analyzed by built-in programs in Igor 7 software (WaveMetrics). Each set of data represents the mean ± SEM of an indicated number (n) of animals. Statistical significance was determined using one-way ANOVA followed by Dunnett's test to control for multiple comparisons.
Results
The C2 domains are indispensable for the normal function of SNT-1
Animals lacking snt-1 display a strong locomotion defect (Fig. 1A,B), suggesting an important role of snt-1 in the nervous system. Neuronal expression of snt-1 (under the snb-1 promoter) fully rescued the locomotion speed to a wild-type level (Fig. 1A,B, SNT-1C2AB). Protein sequence analysis showed that snt-1 in C. elegans has a highly conserved C2 domain compared to Syt1 in mouse and fly (Fig. 1-1). The C2A domain has 70% identity to mouse Syt1 and 71% identity to fly Syt1, and the C2B domain has 83% identity to mouse Syt1 and 84% identity to fly Syt1 (Fig. 1-1). To determine the importance of the C2 domains, we made truncated SNT-1 lacking either the C2A or the C2B domain (SNT-1ΔC2A and SNT-1ΔC2B). Expression of the truncated SNT-1 failed to rescue the locomotion defect (Fig. 1A,B), indicating that both C2 domains are indispensable for the normal function of SNT-1.
Figure 1.

The C2 domains are essential for the normal function of SNT-1 (for sequence alignment of SNT-1 and Syt1, see Figure 1-1). A, Representative locomotory trajectories of 10 animals for the wild type, snt-1(md290), and snt-1 expressing an snt-1 rescue cDNA under the pan-neuronal promoter. The starting points of each trajectory have been aligned for clarity. B, Quantification of the average locomotion speed for the indicated genotypes or transgenes. C–E, Synaptic localization of the constructs used in B. Data are means ± SEM. ###p < 0.001 compared to wild type; ***p < 0.001 compared to SNT-1C2AB rescue. n.s., Nonsignificant compared to wild type. Significance tests were performed using one-way ANOVA followed by Dunnett's test. The number of worms analyzed for each genotype is indicated in the bar graph. Scale bar, 5 m.
The sequence alignment of SNT-1 and Syt1. A, Amino acid sequence alignment for C. elegans SNT-1, mouse Syt1, and Drosophila Syt1 using the online protein similarity analysis (Clustal Omega). The transmembrane domain (TM), C2A domain, and C2B domain are indicated in light green, light blue, and light red, respectively. All five aspartates (D1 to D5) in each C2 domain are indicated. B, Analysis of sequence identify for the TM domain, C2A domain, and C2B domain in the three representative model organisms (w, worm; f, fly; m, mouse). Download Figure 1-1, TIF file (2.1MB, tif)
One possibility for the loss of function of SNT-1ΔC2A and SNT-1ΔC2B could be the mislocalization of snt-1 in synapses. To test this, we first made a GFP-tagged fusion protein of SNT-1, which allowed us to demonstrate good colocalization of SNT-1::GFP with mCherry::Rab3, a SV marker (Fig. 1C). Colocalization of SNT-1ΔC2A::GFP or SNT-1ΔC2B::GFP with Rab3 was also observed (Fig. 1D,E), indicating that the loss of the C2A or the C2B domain does not affect the synaptic localization of snt-1.
Tonic release is severely impaired in snt-1 mutants
To directly assess the role of snt-1 in synaptic transmission, we first recorded mEPSCs from the body wall muscles of adult worms. As the C. elegans NMJ exhibits graded synaptic transmission and endogenous neural activity continuously drives neurotransmitter release (Q. Liu et al., 2009), the mEPSCs are better designated as “tonic release.” The mEPSCs were recorded in bath solution containing 1 mm Ca2+, to avoid high Ca2+-induced burst release (P. Liu et al., 2013). Despite the different characteristics of the synapses of C. elegans and mammals, tonic release and spontaneous release share many similarities. Both display Ca2+-dependent properties (Xu et al., 2009; H. Liu et al., 2018), and the molecular mechanisms that regulate tonic and spontaneous release are also similar in many cases. For example, SNAP-25 knock-out in the mouse NMJ leads to the complete elimination of evoked neurotransmitter release but leaves spontaneous release unaltered (Washbourne et al., 2002), with the same phenotype being observed in C. elegans ric-4/SNAP-25 mutants in which tonic release is normal, whereas evoked release is nearly abolished (Q. Liu et al., 2005; H. Liu et al., 2018). A loss of cpx-1/complexin, a SNARE binding protein, causes a dramatic increase in the mEPSCs at the worm NMJ, as well as in mouse hippocampal neurons (Maximov et al., 2009; Martin et al., 2011). These results suggest that tonic and spontaneous release are likely to be regulated by similar molecular mechanisms.
Our results showed that the tonic release was dramatically reduced in snt-1 mutants (Fig. 2A,B), differing from the observations in the mouse CNS and the fly NMJ, in which spontaneous release is increased in Syt1 knock-out neurons (Xu et al., 2009; Pang et al., 2011; Lee et al., 2013; H. Bai et al., 2016; Afuwape et al., 2017; Bouhours et al., 2017). To test whether the command interneuron activity can affect the tonic release at the NMJ, the entire head region was cut off after a standard dissection (Fig. 2-1A). Our results showed that the mEPSCs recorded from the head-cut animals were indistinguishable from those recorded in intact worms (Fig. 2-1B,C). In addition, we did not observe a further decrease in the mEPSC frequency in the snt-1 mutants following removal of the head. Therefore, the baseline tonic release at the NMJ synapses is unlikely to be affected by the upstream endogenous neuronal activity. Apart from a decrease in mEPSC frequency, a larger mEPSC amplitude was seen in snt-1 mutants (Fig. 2C,D). Neuronal expression of snt-1 cDNA fully rescued the mEPSC frequency and amplitude defects in snt-1 mutants. The mEPSC amplitudes in SNT-1 rescue animals were actually slightly smaller than those in wild-type animals, suggesting that overexpression of SNT-1 can reverse the effect on mEPSC size (Fig. 2C,D). Interestingly, a similar result was also observed at the Drosophila NMJ, where overexpression of Syt1 caused a reduction in SV size and a smaller miniature excitatory junction potential (mEJP) amplitude (Lee et al., 2013). Thus, it is likely that the larger mEPSC amplitude in snt-1 mutants is due to an increase in SV size.
Figure 2.

SNT-1 is required for tonic release. Miniature EPSCs were recorded from the body wall muscle of adult wild-type worms, snt-1 mutants, and snt-1 mutants rescued with the indicated transgenic constructs. It should be noted that tonic release is mainly triggered by endogenous motor neuron activity (Figure 2-1). A, Representative traces of mEPSCs. B, C, Summary of the mean frequency and amplitude of the mEPSCs for each genotype or transgene. ###p < 0.001 compared to wild type; ***p < 0.001 compared to SNT-1C2AB rescue. n.s., Nonsignificant compared to SNT-1C2AB rescue. Significance tests were performed using one-way ANOVA followed by Dunnett's test. D, Cumulative probability distributions of mEPSC amplitudes for the indicated genotypes or transgenes. E, F, Example traces and quantification of mEPSC frequency in wild-type and snt-1 mutants recorded at different [Ca2+]e. Data are means ± SEM. *p < 0.05; **p < 0.01. The number of worms analyzed for each genotype is indicated in the bar graphs. ##p < 0.01.
The tonic release at the NMJ is not affected by blocking the upstream command interneuron activity. A, Schematic depiction of head removal after a standard dissection. B, Representative traces of mEPSCs from intact and head-cut animals. C, Summary of the mean frequency and amplitude of the mEPSCs. Data are mean ± SEM (n.s., non-significant when compared to intact animals; student’s-t test). Download Figure 2-1, TIF file (633.1KB, tif)
Our findings are inconsistent with those in mouse central synapses and fly NMJ in which spontaneous release is dramatically increased in Syt1-deficient neurons. It has been proposed that this increased spontaneous release is mediated by another unknown Ca2+ sensor, as evidenced by the extracellular Ca2+ concentration-dependent enhancement of mEPSC frequency (Xu et al., 2009; Dai et al., 2015). To determine whether another Ca2+ sensor was mediating the remaining spontaneous release in snt-1 mutants, we analyzed the mEPSCs under different extracellular Ca2+ conditions. Our results showed that there were no significant changes in the mEPSC frequency in a bath solution containing 2 mm or 3 mm Ca2+ (Fig. 2E,F). Thus, the remaining spontaneous release in snt-1 mutants is likely triggered by other regulatory mechanisms.
SNT-1 is required for stimulus-evoked neurotransmitter release
In vertebrates, Syt1 serves as a Ca2+ sensor and is required for action potential-triggered synchronous neurotransmitter release (Brose et al., 1992; Geppert et al., 1994; Nishiki and Augustine, 2004b; Nagy et al., 2006; Schonn et al., 2008; Bacaj et al., 2015; Kochubey et al., 2016). We therefore asked whether SNT-1 regulates this release form in a similar way to Syt1. To address this, stimulus-evoked EPSCs were recorded at the NMJ. It should be noted that the motor neurons in C. elegans do not fire action potentials due to the lack of sodium channels. To open the voltage-gated Ca2+ channels and allow Ca2+ influx, we applied an electric stimulus onto the ventral nerve cord to depolarize the neurons. This experimental assay has been proven to be an effective way to trigger evoked neurotransmitter release (Richmond et al., 1999; Madison et al., 2005; Martin et al., 2011). We found that the evoked EPSCs were significantly impaired in amplitude and integrated charge transfer in snt-1 mutants (Fig. 3A–C), suggesting that SNT-1 regulates evoked release in a similar fashion to Syt1. The evoked EPSC defect was rescued by expressing wild-type snt-1 cDNA in the nervous system (Fig. 3A–C). Mouse Syt1 cDNA was also introduced into the nervous system in snt-1 mutants to further confirm that SNT-1 functions similarly to Syt1 in regulating evoked neurotransmitter release. Our results showed that the evoked EPSCs in snt-1 mutants were rescued to a level similar to those in wild-type SNT-1 rescue animals (Fig. 3A–C), revealing a conserved function of synaptotagmin-1 in the worm. Unexpectedly, the remaining evoked release in snt-1 mutants differed from that in the mouse CNS, which exhibits asynchronous release in Syt1 knock-out neurons, or that in the fly NMJ, in which the evoked EJP is completely eliminated (Bacaj et al., 2013; Lee et al., 2013). In fact, the amplitude of the evoked EPSCs was reduced by only 50% compared to the wild type, implying that another synaptotagmin isoform may also be required for evoked release (Fig. 3-1).
Figure 3.

The C2 domains are essential for stimulus-evoked neurotransmitter release. Electric stimulus–evoked EPSCs were recorded from the body wall muscle of the indicated genotypes or transgenes. A, Representative traces of evoked EPSCs. B, C, Quantifications of the mean amplitude and charge transfer of evoked EPSCs. The synaptotagmin isoforms that possibly mediate the remaining evoked release in snt-1 mutants are shown in Figure 3-1. ###p < 0.001 compared to wild type; **p < 0.01, ***p < 0.001 compared to SNT-1C2AB rescue. n.s., Nonsignificant compared to SNT-1C2AB rescue. Significance tests were performed using one-way ANOVA followed by Dunnett's test. D, E, Representative traces and quantification graph of sucrose-evoked responses from wild-type and snt-1 mutants. The recording conditions were the same as those for evoked EPSCs. The total released quanta were analyzed during the 5s sucrose puff. The number of worms analyzed for each genotype is indicated in the bar graphs. ***p < 0.001 compared to wild type. The significance test was performed using Student's t test. Data are means ± SEM. The number of worms analyzed for each genotype is indicated in the bar graphs.
The sequence alignment of SNT-1 and SNT-2/3. A, Amino acid sequence alignment for C. elegans SNT-1 and two other synaptotagmin isoforms, SNT-2 and SNT-3 (Clustal Omega). The third and fourth aspartates in the C2 domains are indicated. Download Figure 3-1, TIF file (468.4KB, tif)
To confirm the role of the C2 domains in synaptic transmission, we recorded both mEPSCs and evoked EPSCs in SNT-1ΔC2A and SNT-1ΔC2B rescue animals. Our results revealed that the synaptic transmission in these animals was indistinguishable from that in the snt-1 mutants (Figs. 2A–C, 3A–C), consistent with our locomotion assay observation. Together, these results demonstrate that SNT-1 plays an essential role in synaptic transmission.
The readily releasable pool of SVs is reduced in snt-1 mutants
The decrease in evoked EPSCs could result from a defect in SV priming. To test this, we performed hypertonic sucrose experiments to measure the size of readily releasable pool (RRP) of SVs. The number of released quanta within a 5 s puff of sucrose was analyzed. Our results showed that the sucrose-evoked quantal release in snt-1 mutants was significantly reduced compared to that in wild-type animals (Fig. 3D,E), indicating a role of SNT-1 in SV priming. Similar results have also been observed at the fly NMJ where the sucrose-evoked release is nearly eliminated in Syt1 mutants (Yoshihara et al., 2010). A significant reduction in the number of SVs near the active zone has also been found in electron microscopy experiments in both the worm and fly NMJ in the absence of snt-1 or Syt1 (Lee et al., 2013; Yu et al., 2013), supporting the role of SNT-1 in SV priming. Interestingly, there was also a reduction in the total numbers of vesicles (density) at synapses in snt-1 mutants (Yu et al., 2013). Consequently, the ratio of the docked vesicles to the total vesicles was unaltered, suggesting a normal function of SNT-1 in SV docking. In mouse cultured hippocampal neurons, the docked vesicle number in Syt1 null mutants is also comparable to that in wild-type animals (Geppert et al., 1994). Thus, far it is still unclear why synaptotagmin-1 has differential roles in SV priming in the worm/fly and mouse.
Ca2+ binding to the C2 domains of SNT-1 is essential for tonic and evoked release
Each C2 domain in Syt1 contains five aspartate residues that bind Ca2+ (Nishiki and Augustine, 2004a). Sequence analysis showed that these aspartates are perfectly conserved in the C2 domains of SNT-1 in C. elegans (Figure 1-1, light blue and light red). Given that the most important feature of Syt1 as the major Ca2+ sensor is that its C2 domains bind Ca2+ and regulate Ca2+-dependent neurotransmitter release, we asked whether the Ca2+ binding of the C2 domains in SNT-1 accounts for the synaptic transmission in C. elegans. To test this, we created a series of mutations of the Ca2+-binding sites (aspartate to asparagine, D to N) in a single C2 domain or both C2 domains, including the third and fourth aspartates (SNT-1C2A D3,4N, SNT-1C2B D3,4N, and SNT-1C2AB D3,4N), as previous studies have demonstrated that these two binding sites are critical for the Syt1 function at the fly NMJ (Lee et al., 2013), as well as all five aspartates in the C2 domain (SNT-1C2A D1-5N, SNT-1C2B D1-5N, SNT-1C2AB D1-5N). These mutations were introduced into transgenic constructs and expressed in an snt-1 null mutant background under a pan-neuronal promoter, snb-1.
To analyze the function of these mutations in synaptic transmission, we first determined whether they localized correctly at synapses. Colocalization was compared between GFP-tagged mutated proteins and Rab3. As shown in Figure 4, the majority of these proteins exhibited good synaptic localization (Fig. 4A–E). However, we failed to detect GFP fluorescence in SNT-1C2AB D1-5N transgenic animals. These results have been repeated in three independent transgenic lines, indicating that the proteins in these animals are unstable and they therefore could not be used for subsequent functional analysis.
Figure 4.

Expression and localization of the mutated SNT-1 constructs. SNT-1 transgenic constructs containing DN mutations in the C2 domain were expressed in D-type cholinergic motor neurons (under the unc-129 promoter). A–E, The colocalization of these constructs (GFP tagged) with Rab3 (mCherry tagged) was compared. F, The construct with all five aspartate mutations in the double C2 domains could not be detected, suggesting that the protein is unstable. Scale bar, 5 μm.
We next asked whether the D/N mutations in the C2 domains disrupt the Ca2+ binding. ITC experiments were performed to compare the Ca2+ binding properties between wild-type C2 domains (C2AB) and mutated C2 domains with D3,4N mutations (C2ABD3,4N). Our results showed that the wild-type protein bound Ca2+ ions with an affinity (Kd) of 15.6 ± 2 μm (SD; n = 2) and an exothermic binding enthalpy (ΔH) of −2.8 ± 0.3 kcal/mol at 25°C (Fig. 5). In contrast, the D3,4N mutations in C2AB abolished the interaction with Ca2+ under the conditions tested (Fig. 5). Our results therefore indicate that SNT-1 in the worm binds Ca2+ similarly to Syt1 in mammals.
Figure 5.
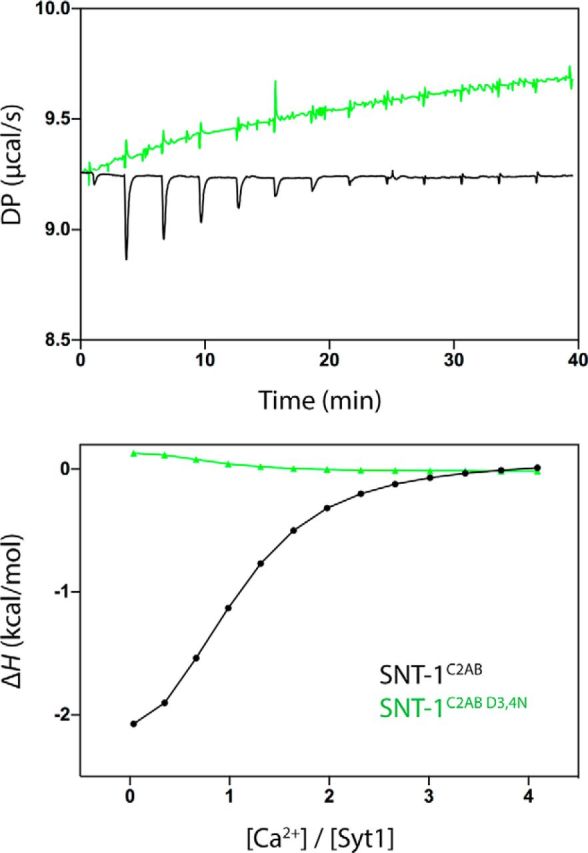
Binding of SNT-1 wild-type and mutant C2AB domains to Ca2+ by ITC. The top panel shows raw ITC data, and the bottom panel shows integrated normalized data. DP, Differential power.
To characterize the functions of these mutations, we analyzed both tonic and evoked neurotransmitter release in all transgenic animals. This revealed that the mEPSC frequency in SNT-1C2A D3,4N and SNT-1C2B D3,4N rescue animals was unaltered compared to that in the SNT-1C2AB rescue worms. However, the mEPSC frequency was significantly reduced in SNT-1C2AB D3,4N rescue animals (Fig. 6A–C). Thus, these results indicate that C2A and C2B function redundantly in triggering tonic release. In addition, the mEPSC amplitude was also significantly increased in SNT-1C2AB rescue animals but not in SNT-1C2A D3,4N or SNT-1C2B D3,4N rescue animals (Fig. 6C,D). Different from the tonic release, the evoked EPSCs were decreased in SNT-1C2B D3,4N rescue animals compared to those in SNT-1C2AB rescue animals, whereas no changes were observed in SNT-1C2A D3,4N rescue animals (Fig. 6F–H). These results are consistent with previous reports that evoked release is determined by Ca2+ binding to the C2B domain, but not the C2A domain, of Syt1 (Nishiki and Augustine, 2004a; Lee et al., 2013). To examine whether other aspartates in the C2 domains (D1, D2, and D5) are playing any roles in synaptic transmission, we measured mEPSCs and evoked EPSCs in SNT-1C2A D1-5N, and SNT-1C2B D1-5N transgenic animals. We found that expression of SNT-1C2A D1-5N or SNT-1C2B D1-5N in snt-1 null mutants fully rescued the tonic release (Fig. 6A–C), similar to that in SNT-1C2A D3,4N and SNT-1C2B D3,4N rescue animals. These results again demonstrate that the two C2 domains in SNT-1 function redundantly in triggering tonic release. The evoked EPSCs in the SNT-1C2B D1-5N transgenic animals were indistinguishable from those in SNT-1C2B D3,4N rescue animals and unaltered in SNT-1C2A D1-5N rescue animals (Fig. 6F–H), confirming that the C2B domain is essential for evoked neurotransmitter release.
Figure 6.

Effects of the DN mutations in the C2 domain on synaptic transmission. A, Representative traces of mEPSCs. B, C, Summary of the mean frequency and amplitude of the mEPSCs for each genotype or transgene. D, E, Cumulative probability distributions of mEPSC amplitudes for the indicated genotypes or transgenes. F, Representative traces of evoked EPSCs. G, H, Quantifications of the mean amplitude and charge transfer of evoked EPSCs in F. I, J, Representative traces and quantifications of the sucrose-activated responses from the wild-type and SNT-1C2B D3,4N rescue. Data are means ± SEM. **p < 0.01; ***p < 0.001 compared to SNT-1C2AB rescue. n.s., Nonsignificant compared to SNT-1C2AB rescue. The significance test was performed using Student's t test. The number of worms analyzed for each genotype is indicated in the bar graphs.
Similarly, mutating the third and fourth aspartates in the C2B domain of Syt1 is sufficient to disrupt the evoked release at the fly NMJ (Lee et al., 2013), indicating that calcium liganding to these two sites is essential for evoked release. In mouse hippocampal neurons, the evoked fast release was completely suppressed when the third aspartate in the C2B domain of Syt1 was mutated (Geppert et al., 1994). Thus, our results demonstrate that the C2 domain of SNT-1 regulates the evoked release in a similar fashion to Syt1.
Evoked neurotransmitter release is determined by Ca2+ sensing ability of C2B
The smaller RRP in snt-1 mutants raised another question regarding whether the reduction in evoked EPSCs in SNT-1C2B D3,4N rescue animals arises from a decrease in the RRP but not a change in Ca2+ sensing during fusion. To address this question, we analyzed the sucrose-evoked quantal release from SNT-1C2B D3,4N animals. As shown in Figure 6, I and J, the quantal number in the SNT-1C2B D3,4N rescue animals was comparable to that in wild-type worms. These results are consistent with those observed at the fly NMJ in which D3/4N mutations in both C2 domains lead to impairments in spontaneous and evoked release but no change in the number of SVs near the active zone (Lee et al., 2013). Taken together, our data demonstrate that SNT-1 in C. elegans regulates both tonic and evoked neurotransmitter release by acting as a Ca2+ sensor.
The kinetics of SV release are altered in snt-1 mutants
In addition to the total amount of neurotransmitter released, another determinant of synaptic transmission is the time course of release. Changes in release kinetics have significant effects on synaptic efficiency and plasticity (Schweizer and Augustine, 1998; Sabatini and Regehr, 1999). The kinetics of release are approximately described by three major parameters: delay, rise time, and decay. The onset of vesicle fusion was strikingly delayed in snt-1 mutants (Fig. 7A–C), indicating a role of SNT-1 in regulating the stimulus response time of SV release. It should be noted that the delay in snt-1 mutants is not due to a defect in endocytosis, as we found that the response time of the EPSCs in mutants lacking unc-41/Stonin or unc-57/endophilin, two proteins that are known to be essential for endocytosis (Phillips et al., 2000, 2010; Jung et al., 2007; J. Bai et al., 2010), was indistinguishable from that in wild-type animals (data not shown). The total charge transfer kinetics, EPSC rise time, and EPSC decay time all displayed faster kinetics in synapses lacking SNT-1 compared to wild type (Fig. 7B,D,E). Notably, these findings are different from those obtained in mouse Syt1 knock-out neurons, which exhibit slow and long-lasting asynchronous release (Nishiki and Augustine, 2004a, b; Schonn et al., 2008). Despite the longer latency between stimulation and release, the fast kinetics of exocytosis observed in snt-1 mutants suggests that this is phasic rather than delayed release. One possible model for the delayed fast release is that the SVs are still located near calcium channels in the absence of SNT-1, but are less sensitive to Ca2+.
Figure 7.

Changes in release kinetics in snt-1 mutants. The release kinetics of the evoked EPSCs were analyzed from the indicated genotypes or transgenes. A, Representative traces of evoked EPSCs from wild-type and snt-1 mutants. A magnified view of the boxed area shows the release delay in snt-1 mutants. B, The normalized charge transfer of the traces in A shows a delayed but faster release in snt-1 mutants. C–E, The kinetic parameters including delay, rise time, and decay are summarized. Data are means ± SEM. ##p < 0.01, ###p < 0.001 compared to wild type; *p < 0.05, **p < 0.01, ***p < 0.001 compared to SNT-1C2AB rescue. n.s., Nonsignificant compared to SNT-1C2AB rescue. Significance tests were performed using one-way ANOVA followed by Dunnett's test. The number of worms analyzed for each genotype is indicated in the bar graphs.
We next analyzed the impact of Ca2+ binding to the C2 domains on release kinetics. The synaptic delay was significantly prolonged in SNT-1C2A D1-5N and SNT-1C2B D1–5N rescue animals, indicating that calcium must bind to both C2A and C2B to trigger short latency release (Fig. 7C). For example, although mutations of the third and fourth aspartates in either C2 domain had no effects on synaptic delay, we observed a significantly longer latency in SNT-1C2AB D3,4N rescue (Fig. 7C). The fact that the delay in SNT-1C2AB D1-5N rescue was still shorter than that in the snt-1 mutants indicates that some other functions of SNT-1 are independent of calcium binding. In the case of the rise time, we observed significant changes in animals with SNT-1C2AB D3,4N transgenes. All other transgenes carrying D/N mutations exhibit a decreased trend in rise time but did not reach statistical significance (Fig. 7D). The evoked decay was mainly determined by Ca2+ binding to the C2B but not the C2A domain, consistent with the result that Ca2+ binding to the C2B domain is essential for evoked release (Fig. 6F).
Priming of slow release is abolished in the absence of SNT-1
Our previous studies demonstrated that evoked release at the C. elegans NMJ consists of fast and slow components, which represent the fusion of distinct subpopulations of SVs with tight or loose coupling to Ca2+ entry. UNC-13L and UNC-13S, the two isoforms of the priming factor unc-13, have been found to determine the coupling of SVs to Ca2+ entry, thereby mediating fast and slow release (Hu et al., 2013). The fast rise time and decay of the evoked EPSCs in snt-1 mutants led to the hypothesis that the slow component is eliminated in the absence of SNT-1. If this is true, we would expect that the slow release in unc-13 null mutants rescued by UNC-13S would be abolished by the removal of SNT-1. As expected, the evoked slow release was nearly abrogated by inactivation of SNT-1 in UNC-13S rescue animals (UNC-13S;snt-1; Fig. 8A–C). The tonic release was also completely eliminated in those animals (Fig. 8G–I). These results indicate that the synaptic transmission mediated by UNC-13S is completely dependent on SNT-1.
Figure 8.
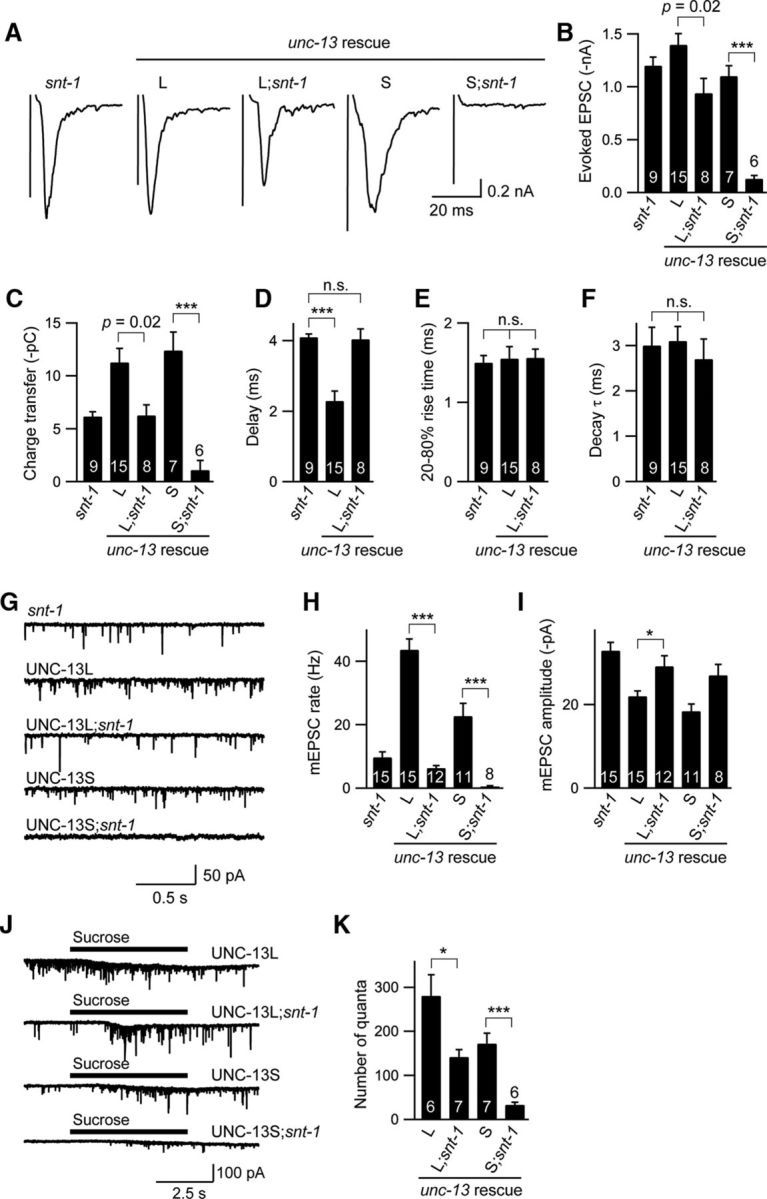
Effects of SNT-1 on the fast and slow evoked release mediated by UNC-13L and UNC-13S. A–C, Representative traces of the evoked EPSCs of indicated genotypes (A) and summaries of the amplitude (B) and charge transfer (C) are shown. D–F, Comparison of the kinetics (delay, rise time, and decay) from snt-1, UNC-13L rescue, and UNC-13L;snt-1 worms. G–I, Example traces of the mEPSCs (G) and summaries of the mean frequencies (H) and amplitudes of the mEPSCs (I) from the same strains as in A. J, K, Hypotonic sucrose-evoked currents (J) and quantifications of the quantal release during the sucrose puff (K) are shown. Data are means ± SEM. *p < 0.05; ***p < 0.001. n.s., Nonsignificant. Significance tests were performed using one-way ANOVA followed by Dunnett's test. The number of worms analyzed for each genotype is indicated in the bar graphs.
We then asked whether SNT-1 is required for the evoked fast release. To address this, we analyzed evoked EPSCs in UNC-13L;snt-1 animals. Our results showed that the lack of SNT-1 significantly diminished both the amplitude and charge transfer of the evoked EPSCs in UNC-13L rescue animals (Fig. 8A–C), suggesting a role of SNT-1 in fast release as well. By analyzing the release kinetics, we found that the fast release in UNC-13L rescue animals was significantly delayed, whereas the rise time and decay remained unchanged (Fig. 8D–F). These results demonstrate that SNT-1 also plays an essential role in the release of SVs with tight coupling to Ca2+ entry, particularly in regulating the response time of synaptic release to stimulus. Our data also imply that multiple Ca2+ sensors are involved in the regulation of fast release.
The complete loss of evoked slow release in UNC-13MR;snt-1 animals could result from the role of SNT-1 in vesicle priming rather than Ca2+ sensing in the fusion step. We therefore next analyzed the priming of the slow vesicle pool in the absence of SNT-1. As shown in Figure 8, J and K, hypertonic sucrose-evoked quantal release was nearly eliminated in UNC-13S;snt-1 animals compared to that in UNC-13S animals, demonstrating a crucial role of SNT-1 in priming the slow release component. In contrast, the quantal number decreased by 40% in UNC-13S;snt-1 animals compared to that in UNC-13L rescue worms. Given that the D3,4N mutations in the C2B domain (SNT-1C2B D3,4N) caused a loss of evoked slow release (Figs. 6F–H, 7D,E) but did not affect the RRP (Fig. 8), our data therefore suggest that the slow vesicle component possibly uses SNT-1 as its only Ca2+ sensor for Ca2+-triggered release.
Discussion
Ca2+ binding to C2 domains and tonic release
To date, it has been reported that Syt1 suppresses spontaneous release in many types of vertebrate neuron (Xu et al., 2009; Lee et al., 2013). Other neurons express Syt2 as their major Ca2+ sensor, where it has a very similar function to Syt1 (Kochubey et al., 2016). The increase in spontaneous release observed in Syt1 or Syt2 knock-out neurons suggests that a second unknown Ca2+ sensor is activated, with our results indicating that the worm may lack such a Ca2+ sensor. Mutating the Ca2+ binding sites in both the C2 domains of SNT-1 led to a significant decrease in tonic release (Fig. 2). Interestingly, disrupting Ca2+ binding in a single C2 domain did not affect this release. These results indicate that Ca2+ binding to a single C2 domain is sufficient to trigger tonic release at a wild-type level. A similar result has also been observed at the fly NMJ, where blocking Ca2+ binding in a single C2 domain of Syt1 fails to alter spontaneous release, whereas blocking Ca2+ binding in both C2 domains causes a dramatic increase in spontaneous release (Lee et al., 2013). Although only one C2 domain is required to bind Ca2+, the two C2 domains must coexist, as the truncated form of SNT-1 lacking either C2A or C2B failed to rescue the mEPSC defect (Fig. 2A,B). One possible explanation is that a single C2 domain can still interact with the SNARE complex or phospholipids without Ca2+, thereby maintaining the functional structure of SNT-1.
A recent study showed that the calyx of Held uses distinct synaptotagmins as Ca2+ sensors at different developmental stages (Kochubey et al., 2016). At mature calyx synapses, Syt2 is the Ca2+ sensor that suppresses spontaneous release, inhibits asynchronous release, and triggers synchronous release, similar to Syt1 at hippocampal synapses, whereas at immature calyx synapses, Syt1 is used to trigger synchronous release. However, Syt1 at this latter stage lacks a clamping function and cannot suppress spontaneous release. One explanation for this observation is that the P2 neurons do not express the Ca2+ sensor that has been proposed to maintain the high frequency of spontaneous release in Syt1 knock-out hippocampal or cortical neurons. Moreover, single isolated hippocampal neurons lacking Syt1 display normal wild-type mEPSCs (Geppert et al., 1994). These results indicate that Syt1 has distinct roles in spontaneous release at different types of synapses, probably due to the use of distinct synaptic fusion machinery or differential synaptic organization. Some evidence, however, argues that the fusion machinery in C. elegans is different from that in the mouse and fly. A loss of function of the SNARE-binding protein, complexin, causes a massive increase in spontaneous or tonic release in the mouse, fly, and worm (Huntwork and Littleton, 2007; Maximov et al., 2009; Martin et al., 2011). Moreover, complexin exhibits a similar role in controlling spontaneous and evoked release separately in the worm and mouse (Martin et al., 2011; Kaeser-Woo et al., 2012). Together, these results suggest that SV fusion in all three species is controlled by conserved fusion machinery. Thus, the key issue for understanding the different results in the worm is to identify the unknown Ca2+ sensor that is suppressed by Syt1 in the mouse and fly, and to determine whether this sensor is expressed at the worm NMJ.
It is still unclear what causes the larger mEPSC amplitude in snt-1 mutants. One possibility is that this defect results from a role of SNT-1 in endocytosis, as we observed very similar larger mEPCs in mutants lacking unc-41/Stonin (data not shown), a potential binding partner of SNT-1. In addition, the mouse stoning 2 and Drosophila STNB (Stoned B) have been shown to bind to the C2 domains of Syt1 (Phillips et al., 2000, 2010; Jung et al., 2007). A previous study showed that UNC-41 protein is expressed at endocytic sites and colocalizes with SNT-1 (Mullen et al., 2012). Based on these results, it is likely that a defect in endocytosis leads to the larger mEPSC amplitude observed in snt-1 mutants. Interestingly, the increased mEPSC amplitude in SNT-1C2AB D3,4N rescue animals suggests that Ca2+ binding to the C2 domains at least partially determines the vesicular size (Fig. 6; Lee et al., 2013). However, we cannot exclude the possibility that other regulatory mechanisms are also involved in this process.
Ca2+ binding to C2 domains and evoked neurotransmitter release
Previous studies have demonstrated that evoked synchronous release is mainly determined by Ca2+ binding to the C2B domain of Syt1 (Geppert et al., 1994; Nishiki and Augustine, 2004b; Lee et al., 2013). Similarly, we have found that SNT-1 also regulates evoked release via Ca2+ binding to C2B. Although sufficient to trigger spontaneous release, the binding of Ca2+ to C2A makes no contribution to evoked release. Thus far, all the results obtained from mouse, fly, and C. elegans support the notion that synaptotagmin-1 regulates evoked neurotransmitter release through Ca2+ binding to the C2B domain. These findings demonstrate that the tonic and evoked release are differentially regulated by SNT-1-mediated Ca2+ binding to C2 domains. In fact, a similar result has also been observed in the calyx of Held, in which the increased mEPSC frequency in Syt2 knock-out neurons was completely rescued by Syt2 cDNA carrying a mutation of the third aspartate in C2B, whereas the same construct failed to rescue the evoked EPSCs (Kochubey and Schneggenburger, 2011).
Our results suggest that another Ca2+ sensor is required to trigger the remaining evoked fast release in snt-1 mutants. Similarly, it has been shown that evoked synchronous release is triggered by two distinct Ca2+ sensors at mouse calyx synapses (Kochubey et al., 2016). Syt2 knock-out at the P7 stage led to a significant decrease in evoked fast release, and the remaining fast release was nearly abolished in Syt1–Syt2 double knock-out neurons, demonstrating that fast release is triggered by both Syt1 and Syt2. In another study, Syt1 and Syt2 were shown to be functionally redundant in regulating evoked release in parvalbumin interneurons (Bouhours et al., 2017). Based on these results, it is likely that the evoked release at the worm NMJ is triggered by two or more distinct Ca2+ sensors. Several other synaptotagmin isoforms are expressed in C. elegans, and two of these display a synaptotagmin-1-like sequence (snt-2 and snt-3; Figs. 1-1, 2-1, respectively). However, their involvement in synaptic transmission has not yet been characterized, and will form the basis of future investigations.
Although Ca2+ binding to the C2A domain is not required for evoked release, mutations of the Ca2+ binding site in this domain caused a significant delay similar to that caused by Ca2+-binding mutations in the C2B domain, indicating that the C2 domain binding Ca2+ is required to maintain a quick response of SVs to stimulation. Moreover, the delay observed in the truncated SNT-1 rescue animals lacking the C2A or C2B domain was longer than that in animals carrying Ca2+ binding mutations in SNT-1, suggesting that other mechanisms in the C2 domains regulate the release delay (Fig. 7C). It has been reported that Syt1 can enhance the rate of SNARE-dependent fusion of liposomes in vitro (Gaffaney et al., 2008), raising the possibility that the interaction of SNT-1 with the SNARE complex may regulate the delay.
Evoked asynchronous release at the worm NMJ
Apart from its role in evoked synchronous release, Syt1 in the mouse CNS suppresses asynchronous release, characterized by a long-lasting release duration (>100 ms). As a consequence, the evoked asynchronous release is much smaller than the synchronous release in wild-type neurons, and is enhanced in Syt1 knock-out neurons (Nishiki and Augustine, 2004b; Kochubey et al., 2016). The evoked EPSCs in snt-1 mutants, however, exhibit shorter release duration than in wild-type animals. The evoked responses end in around 10 ms in snt-1 mutants and 20 ms in wild-type animals (Fig. 7A), suggesting that the remaining release in the mutants is not asynchronous-like release. Interestingly, recent studies in the calyx of Held and parvalbumin interneurons showed that evoked fast release is mediated by two distinct Ca2+ sensors, Syt1 and Syt2 (Kochubey et al., 2016; Bouhours et al., 2017). Therefore, it is likely that the worm NMJ requires another synaptotagmin as a Ca2+ sensor for evoked release. This unknown sensor may suppress the asynchronous release in snt-1 mutants.
Role of SNT-1 for different kinetic release components
The identity of the Ca2+ sensor(s) for distinct SV populations remains largely unclear (Gracheva et al., 2006; Hammarlund et al., 2007). Here we show that SNT-1 is involved in the priming of both evoked fast and slow release at the worm NMJ. Priming of slow release relies on SNT-1, whereas priming of fast release requires SNT-1 and another unknown Ca2+ sensor. In fact, the fast rise time and decay in snt-1 mutants mainly result from the loss of the slow release. In addition, the evoked EPSCs in transgenic animals carrying C2B mutations (SNT-1C2B D3,4N, SNT-1C2AB D3,4N, or SNT-1C2B D1-5N) exhibit similar amplitude, charge transfer, rise time, and decay to those in the snt-1 mutants (Fig. 7), indicating that the release of the slow component is determined by Ca2+ binding to the C2B domain in SNT-1. The normal RRP of SVs but fast evoked EPSCs in SNT-1C2B D3,4N animals indicates that the evoked slow release is using SNT-1 as its major Ca2+ sensor.
Footnotes
This work was supported by Australia Research Council Project Grant DP160100849 (Z.H.), National Health and Medical Research Council (NHMRC) Grants APP1122351 (Z.H.) and APP1058734 (B.M.C.), NARSAD Young Investigator Grant 24980 (Z.H.), and NHMRC Senior Research Fellowship APP1136021 (B.M.C.). We thank Dr. Jeremy Dittman for providing the mCherry::Rab3 imaging line, and Dr. Rowan Tweedale for critically reading this manuscript. The imaging was performed at the Queensland Brain Institute's Advanced Microscopy Facility.
The authors declare no competing financial interests.
References
- Afuwape OA, Wasser CR, Schikorski T, Kavalali ET (2017) Synaptic vesicle pool-specific modification of neurotransmitter release by intravesicular free radical generation. J Physiol 595:1223–1238. 10.1113/JP273115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, Südhof TC (2013) Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80:947–959. 10.1016/j.neuron.2013.10.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Burré J, Malenka RC, Liu X, Südhof TC (2015) Synaptotagmin-1 and -7 Are redundantly essential for maintaining the capacity of the readily-releasable pool of synaptic vesicles. PLoS Biol 13:e1002267. 10.1371/journal.pbio.1002267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H, Xue R, Bao H, Zhang L, Yethiraj A, Cui Q, Chapman ER (2016) Different states of synaptotagmin regulate evoked versus spontaneous release. Nat Commun 7:10971. 10.1038/ncomms10971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai J, Hu Z, Dittman JS, Pym EC, Kaplan JM (2010) Endophilin functions as a membrane-bending molecule and is delivered to endocytic zones by exocytosis. Cell 143:430–441. 10.1016/j.cell.2010.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhours B, Gjoni E, Kochubey O, Schneggenburger R (2017) Synaptotagmin2 (Syt2) drives fast release redundantly with Syt1 at the output synapses of parvalbumin-expressing inhibitory neurons. J Neurosci 37:4604–4617. 10.1523/JNEUROSCI.3736-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Südhof TC, Jahn R (1992) Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256:1021–1025. 10.1126/science.1589771 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Wang J, Wang Y, Ding M (2015) Synaptotagmin 1 directs repetitive release by coupling vesicle exocytosis to the Rab3 cycle. eLife 4 e05118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai J, Chen P, Tian H, Sun J (2015) Spontaneous vesicle release is not tightly coupled to voltage-gated calcium channel-mediated Ca2+ influx and is triggered by a Ca2+ sensor other than synaptotagmin-2 at the juvenile mice calyx of held synapses. J Neurosci 35:9632–9637. 10.1523/JNEUROSCI.0457-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittman JS, Kaplan JM (2008) Behavioral impact of neurotransmitter-activated G-protein-coupled receptors: muscarinic and GABAB receptors regulate Caenorhabditis elegans locomotion. J Neurosci 28:7104–7112. 10.1523/JNEUROSCI.0378-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffaney JD, Dunning FM, Wang Z, Hui E, Chapman ER (2008) Synaptotagmin C2B domain regulates Ca2+-triggered fusion in vitro: critical residues revealed by scanning alanine mutagenesis. J Biol Chem 283:31763–31775. 10.1074/jbc.M803355200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC (1994) Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79:717–727. 10.1016/0092-8674(94)90556-8 [DOI] [PubMed] [Google Scholar]
- Gracheva EO, Burdina AO, Holgado AM, Berthelot-Grosjean M, Ackley BD, Hadwiger G, Nonet ML, Weimer RM, Richmond JE (2006) Tomosyn inhibits synaptic vesicle priming in Caenorhabditis elegans. PLoS Biol 4:e261. 10.1371/journal.pbio.0040261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarlund M, Palfreyman MT, Watanabe S, Olsen S, Jorgensen EM (2007) Open syntaxin docks synaptic vesicles. PLoS Biol 5:e198. 10.1371/journal.pbio.0050198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Hom S, Kudze T, Tong XJ, Choi S, Aramuni G, Zhang W, Kaplan JM (2012) Neurexin and neuroligin mediate retrograde synaptic inhibition in C. elegans. Science 337:980–984. 10.1126/science.1224896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z, Tong XJ, Kaplan JM (2013) UNC-13L, UNC-13S, and tomosyn form a protein code for fast and slow neurotransmitter release in Caenorhabditis elegans. eLife 2:e00967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntwork S, Littleton JT (2007) A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci 10:1235–1237. 10.1038/nn1980 [DOI] [PubMed] [Google Scholar]
- Jung N, Wienisch M, Gu M, Rand JB, Müller SL, Krause G, Jorgensen EM, Klingauf J, Haucke V (2007) Molecular basis of synaptic vesicle cargo recognition by the endocytic sorting adaptor stonin 2. J Cell Biol 179:1497–1510. 10.1083/jcb.200708107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser-Woo YJ, Yang X, Südhof TC (2012) C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J Neurosci 32:2877–2885. 10.1523/JNEUROSCI.3360-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochubey O, Schneggenburger R (2011) Synaptotagmin increases the dynamic range of synapses by driving Ca2+-evoked release and by clamping a near-linear remaining Ca2+ sensor. Neuron 69:736–748. 10.1016/j.neuron.2011.01.013 [DOI] [PubMed] [Google Scholar]
- Kochubey O, Babai N, Schneggenburger R (2016) A synaptotagmin isoform switch during the development of an identified CNS synapse. Neuron 91:1183. [DOI] [PubMed] [Google Scholar]
- Lee J, Guan Z, Akbergenova Y, Littleton JT (2013) Genetic analysis of synaptotagmin C2 domain specificity in regulating spontaneous and evoked neurotransmitter release. J Neurosci 33:187–200. 10.1523/JNEUROSCI.3214-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton JT, Bai J, Vyas B, Desai R, Baltus AE, Garment MB, Carlson SD, Ganetzky B, Chapman ER (2001) Synaptotagmin mutants reveal essential functions for the C2B domain in Ca2+-triggered fusion and recycling of synaptic vesicles in vivo. J Neurosci 21:1421–1433. 10.1523/JNEUROSCI.21-05-01421.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Li L, Wang W, Gong J, Yang X, Hu Z (2018) Spontaneous vesicle fusion is differentially regulated at cholinergic and GABAergic synapses. Cell Rep 22:2334–2345. 10.1016/j.celrep.2018.02.023 [DOI] [PubMed] [Google Scholar]
- Liu P, Chen B, Wang ZW (2013) Postsynaptic current bursts instruct action potential firing at a graded synapse. Nat Commun 4:1911. 10.1038/ncomms2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Chen B, Yankova M, Morest DK, Maryon E, Hand AR, Nonet ML, Wang ZW (2005) Presynaptic ryanodine receptors are required for normal quantal size at the Caenorhabditis elegans neuromuscular junction. J Neurosci 25:6745–6754. 10.1523/JNEUROSCI.1730-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Hollopeter G, Jorgensen EM (2009) Graded synaptic transmission at the Caenorhabditis elegans neuromuscular junction. Proc Natl Acad Sci U S A 106:10823–10828. 10.1073/pnas.0903570106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison JM, Nurrish S, Kaplan JM (2005) UNC-13 interaction with syntaxin is required for synaptic transmission. Curr Biol 15:2236–2242. 10.1016/j.cub.2005.10.049 [DOI] [PubMed] [Google Scholar]
- Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS (2011) Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol 21:97–105. 10.1016/j.cub.2010.12.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Südhof TC (2009) Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science 323:516–521. 10.1126/science.1166505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen GP, Grundahl KM, Gu M, Watanabe S, Hobson RJ, Crowell JA, McManus JR, Mathews EA, Jorgensen EM, Rand JB (2012) UNC-41/stonin functions with AP2 to recycle synaptic vesicles in Caenorhabditis elegans. PLoS One 7:e40095. 10.1371/journal.pone.0040095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Kim JH, Pang ZP, Matti U, Rettig J, Südhof TC, Sørensen JB (2006) Different effects on fast exocytosis induced by synaptotagmin 1 and 2 isoforms and abundance but not by phosphorylation. J Neurosci 26:632–643. 10.1523/JNEUROSCI.2589-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ (2004a) Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci 24:8542–8550. 10.1523/JNEUROSCI.2545-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ (2004b) Synaptotagmin I synchronizes transmitter release in mouse hippocampal neurons. J Neurosci 24:6127–6132. 10.1523/JNEUROSCI.1563-04.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML, Grundahl K, Meyer BJ, Rand JB (1993) Synaptic function is impaired but not eliminated in C. elegans mutants lacking synaptotagmin. Cell 73:1291–1305. 10.1016/0092-8674(93)90357-V [DOI] [PubMed] [Google Scholar]
- Pang ZP, Bacaj T, Yang X, Zhou P, Xu W, Südhof TC (2011) Doc2 supports spontaneous synaptic transmission by a Ca2+-independent mechanism. Neuron 70:244–251. 10.1016/j.neuron.2011.03.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AM, Smith M, Ramaswami M, Kelly LE (2000) The products of the Drosophila stoned locus interact with synaptic vesicles via synaptotagmin. J Neurosci 20:8254–8261. 10.1523/JNEUROSCI.20-22-08254.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips AM, Ramaswami M, Kelly LE (2010) Stoned. Traffic 11:16–24. 10.1111/j.1600-0854.2009.00999.x [DOI] [PubMed] [Google Scholar]
- Richmond JE, Davis WS, Jorgensen EM (1999) UNC-13 is required for synaptic vesicle fusion in C. elegans. Nat Neurosci 2:959–964. 10.1038/14755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickman C, Archer DA, Meunier FA, Craxton M, Fukuda M, Burgoyne RD, Davletov B (2004) Synaptotagmin interaction with the syntaxin/SNAP-25 dimer is mediated by an evolutionarily conserved motif and is sensitive to inositol hexakisphosphate. J Biol Chem 279:12574–12579. 10.1074/jbc.M310710200 [DOI] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG (1999) Timing of synaptic transmission. Annu Rev Physiol 61:521–542. 10.1146/annurev.physiol.61.1.521 [DOI] [PubMed] [Google Scholar]
- Schonn JS, Maximov A, Lao Y, Südhof TC, Sørensen JB (2008) Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proc Natl Acad Sci U S A 105:3998–4003. 10.1073/pnas.0712373105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer FE, Augustine GJ (1998) SNARE proteins and the timing of neurotransmitter release. Mol Psychiatry 3:293–297. 10.1038/sj.mp.4000422 [DOI] [PubMed] [Google Scholar]
- Shin OH, Rhee JS, Tang J, Sugita S, Rosenmund C, Südhof TC (2003) Sr2+ binding to the Ca2+ binding site of the synaptotagmin 1 C2B domain triggers fast exocytosis without stimulating SNARE interactions. Neuron 37:99–108. 10.1016/S0896-6273(02)01145-5 [DOI] [PubMed] [Google Scholar]
- Südhof TC. (2013) A molecular machine for neurotransmitter release: synaptotagmin and beyond. Nat Med 19:1227–1231. 10.1038/nm.3338 [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Benditó G, Molnár Z, Becher MW, Valenzuela CF, Partridge LD, Wilson MC (2002) Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5:19–26. 10.1038/nn783 [DOI] [PubMed] [Google Scholar]
- Weber JP, Toft-Bertelsen TL, Mohrmann R, Delgado-Martinez I, Sørensen JB (2014) Synaptotagmin-7 is an asynchronous calcium sensor for synaptic transmission in neurons expressing SNAP-23. PLoS One 9:e114033. 10.1371/journal.pone.0114033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mashimo T, Südhof TC (2007) Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54:567–581. 10.1016/j.neuron.2007.05.004 [DOI] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, Südhof TC (2009) Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci 12:759–766. 10.1038/nn.2320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT (2002) Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron 36:897–908. 10.1016/S0896-6273(02)01065-6 [DOI] [PubMed] [Google Scholar]
- Yoshihara M, Guan Z, Littleton JT (2010) Differential regulation of synchronous versus asynchronous neurotransmitter release by the C2 domains of synaptotagmin 1. Proc Natl Acad Sci U S A 107:14869–14874. 10.1073/pnas.1000606107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu SC, Klosterman SM, Martin AA, Gracheva EO, Richmond JE (2013) Differential roles for snapin and synaptotagmin in the synaptic vesicle cycle. PLoS One 8:e57842. 10.1371/journal.pone.0057842 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The sequence alignment of SNT-1 and Syt1. A, Amino acid sequence alignment for C. elegans SNT-1, mouse Syt1, and Drosophila Syt1 using the online protein similarity analysis (Clustal Omega). The transmembrane domain (TM), C2A domain, and C2B domain are indicated in light green, light blue, and light red, respectively. All five aspartates (D1 to D5) in each C2 domain are indicated. B, Analysis of sequence identify for the TM domain, C2A domain, and C2B domain in the three representative model organisms (w, worm; f, fly; m, mouse). Download Figure 1-1, TIF file (2.1MB, tif)
The tonic release at the NMJ is not affected by blocking the upstream command interneuron activity. A, Schematic depiction of head removal after a standard dissection. B, Representative traces of mEPSCs from intact and head-cut animals. C, Summary of the mean frequency and amplitude of the mEPSCs. Data are mean ± SEM (n.s., non-significant when compared to intact animals; student’s-t test). Download Figure 2-1, TIF file (633.1KB, tif)
The sequence alignment of SNT-1 and SNT-2/3. A, Amino acid sequence alignment for C. elegans SNT-1 and two other synaptotagmin isoforms, SNT-2 and SNT-3 (Clustal Omega). The third and fourth aspartates in the C2 domains are indicated. Download Figure 3-1, TIF file (468.4KB, tif)