

Abstract
Mitochondrial Ca2+ uptake is gated by the mitochondrial calcium uniplex, which is comprised of mitochondrial calcium uniporter (MCU), the Ca2+ pore-forming subunit of the complex, and its regulators. Ca2+ influx through MCU affects both mitochondrial function and movement in neurons, but its direct role in mitochondrial movement has not been explored. In this report, we show a link between MCU and Miro1, a membrane protein known to regulate mitochondrial movement. We find that MCU interacts with Miro1 through MCU's N-terminal domain, previously thought to be the mitochondrial targeting sequence. Our results show that the N-terminus of MCU has a transmembrane domain that traverses the outer mitochondrial membrane, which is dispensable for MCU localization into mitochondria. However, this domain is required for Miro1 interaction and is critical for Miro1 directed movement. Together, our findings reveal Miro1 as a new component of the MCU complex, and that MCU is an important regulator of mitochondrial transport.
SIGNIFICANCE STATEMENT Mitochondrial calcium level is critical for mitochondrial metabolic activity and mitochondrial transport in neurons. While it has been established that calcium influx into mitochondria is modulated by mitochondrial calcium uniporter (MCU) complex, how MCU regulates mitochondrial movement still remains unclear. Here, we discover that the N-terminus of MCU plays a different role than previously thought; it is not required for mitochondrial targeting but is essential for interaction with Miro1, an outer mitochondrial membrane protein important for mitochondrial movement. Furthermore, we show that MCU–Miro1 interaction is required to maintain mitochondrial transport. Our data identify that Miro1 is a novel component of the mitochondrial calcium uniplex and demonstrate that coupling between MCU and Miro1 as a novel mechanism modulating both mitochondrial Ca2+ uptake and mitochondrial transport.
Keywords: axon, MCU, mitochondria, transport
Introduction
Proper distribution and transport of mitochondria within extremely polarized regions of a neuron in response to physiological needs are essential for neuronal functions (Phillips et al., 1996; Ligon and Steward, 2000; Chada and Hollenbeck, 2003). Consequently, defects in transportation of mitochondria in neurons may create detrimental consequences and are linked to numerous neurological disorders (Ligon and Steward, 2000; Chada and Hollenbeck, 2003). It is also known that (1) Ca2+ plays a key role in regulating both metabolic capability of mitochondria and mitochondrial transport in neurons, and (2) mitochondrial calcium uniplex primarily facilitates Ca2+ uptake into the mitochondrial matrix (Deluca and Engstrom, 1961; Baughman et al., 2011; De Stefani et al., 2011). The core component of this uniplex, the mitochondrial calcium uniporter (MCU; Baughman et al., 2011; De Stefani et al., 2011), forms a Ca2+-specific pentameric pore within the inner mitochondrial membrane, along with its paralog MCUb (Raffaello et al., 2013; Oxenoid et al., 2016). MCU is regulated by its subunits, MICU1 (Perocchi et al., 2010; Petrungaro et al., 2015; Liu et al., 2016), MICU2 (Plovanich et al., 2013; Patron et al., 2014), and EMRE (Sancak et al., 2013; Vais et al., 2016), which establish a baseline threshold for Ca2+ sensitivity, ensuring that Ca2+ uptake only occurs during a significant increase in intracellular Ca2+ concentrations. These subunits are then assembled into the complex with MCUR1 (Chaudhuri et al., 2016; Tomar et al., 2016), which serves as a scaffolding factor. Although MCU's action in Ca2+ uptake is well established, its potential roles in regulating other Ca2+-dependent mitochondrial functions, such as mitochondrial transport, are not well understood.
The outer mitochondrial membrane protein Miro1, a Rho GTPase that contains two canonical EF-hands (Fransson et al., 2006), is known to modulate mitochondrial transport (Saotome et al., 2008; Macaskill et al., 2009; Wang and Schwarz, 2009; Chang et al., 2011; Pan et al., 2013). In mammals, Miro1 binds to TRAK1/2, which in turn binds to kinesin or dynein to promote microtubule-driven anterograde and retrograde movement, respectively (van Spronsen et al., 2013). In axons, mitochondrial movement is arrested upon a high pulse of Ca2+, due to EF-hand-mediated change in Miro1 configuration. Initial studies using Miro1 siRNA in neurons found a significant decrease in the overall number of moving mitochondria in axons (Macaskill et al., 2009), and overexpression of a Ca2+ insensitive mutant of Miro1 enhanced mitochondrial movement in the presence of increased Ca2+ levels (Saotome et al., 2008; Chang et al., 2011). However, tests using a Miro1 knock-out mouse model have reported conflicting results. An initial study reported only a modest decrease in retrograde movement (Nguyen et al., 2014), whereas a follow-up examining the difference between Miro1 and Miro2 knock-out phenotypes found that only the Miro1 knock-out mouse showed a dramatic decrease in axonal mitochondrial transport, making Miro1 the primary regulator of mitochondrial movement (López-Doménech et al., 2016). Both of these studies found that deletion of Miro1 produced severe neural defects.
Although MCU's action in Ca2+ uptake is well established, its potential roles in regulating mitochondrial transport are not well understood. Our previous work (Chang et al., 2011) suggested that MCU and Miro1 might work in the same biological pathway to simultaneously regulate mitochondrial functions and transport in axons. Overexpression of a Ca2+ insensitive mutant of Miro1 was sufficient to impair Ca2+ uptake into mitochondria, whereas pharmaceutical repression of MCU rescued Ca2+-induced arrest of mitochondrial movement (Chang et al., 2011). Further studies have confirmed that Miro plays a critical role in regulation of mitochondrial calcium, and controls interaction with the ER (Lee et al., 2016). However, the possibility of interaction between Miro1 and the inner mitochondrial components has not been examined. In this study, we have investigated the interaction between Miro1 and MCU, and the role of this interaction plays in mitochondrial transport and Ca2+ influx.
Materials and Methods
Animal care.
Animals were used in accordance with protocols approved by the Animal Care and Use Committees of UNIST. C57BL/6 mouse strain was purchased from Hyochang Science.
Coverslip preparation.
Glass-bottom dishes (Cellvis, D35-14-1.5-N) were prepared as follows for all microscopy experiments. First, dishes were immersed in 1 m HCl at 55°C for at least 4 h. Dishes were then washed thoroughly with dH2O, followed by 30 min washes of 50%, 70% and 100% ethanol. Dishes were then dried in a biosafety cabinet and exposed to UV radiation for at least 15 min for sterilization. Fifty micrograms per milliliter of poly-d-lysine (Thermo Fisher Scientific, P6407) were added to the glass portal in dishes, and were incubated in a 37°C and 5% CO2 cell culture incubator overnight. Dishes were washed the next day with autoclaved dH2O, dried in a biosafety cabinet, and kept in a sterile cell culture incubator until use. Coverslips (1.5H; Deckglaser, 0117520) were also prepared using an identical protocol.
Cell culture.
Primary neurons were dissected from E18 mouse embryos. Pregnant mouse was killed with CO2, followed by cervical dislocation. Embryos were removed and immediately placed in ice-cold HBSS (Thermo Fisher Scientific, 14065056). Hippocampi were dissected out of embryos, washed twice in Ca2+- and Mg2+-free HBSS, and incubated in EDTA-free trypsin (Thermo Fisher Scientific, 15050065) for 15 min. Hippocampi were washed in trituration media twice, and then vigorously pipetted up and down to dissociate the cells. The cell mixture was pipetted through a 40 μm cell filter (Falxon, 352340), and then plated onto glass-bottom dishes or coverslips at a density of 35,000 cells/cm2. Trituration media was replaced by neuronal maintenance media consisting of Neurobasal media (Thermo Fisher Scientific, 21103049), B27 (Thermo Fisher Scientific, 17504044), and GlutaMAX (Thermo Fisher Scientific, 35050061). HEK293 cells were obtained from the Korean Cell Line Bank. Neuro2A cells were obtained as a gift from Professor K. Kim at POSTECH (South Korea). Cell lines were maintained in DMEM (Invitrogen) supplemented with penicillin/streptomycin (ThermoFisher Scientific, 15070063) and 10% FBS (Millipore, Catalog #TMS-013-BKR), in a cell culture incubator kept at 5% CO2 and 37°C. Transfection was performed on all cell types using Lipofectamine 2000 (Invitrogen). For neurons, a total of 1.2 μg of plasmid DNA and 1 μl of Lipofectamine 2000, diluted in 100 μl Opti-MEM (ThermoFisher Scientific), was used. DNA/Lipofectamine mixture was incubated together at room temperature for 20 min. During this time, approximately half of the cell culture media was removed from the dish, and the transfection mixture was added to the cells. Transfection was allowed to occur for 3 h, at which point the media was removed, and a 50% mixture of old and new maintenance media was added back to the neurons. For other cell types, 2 μg of plasmid DNA and 2 μl of Lipofectamine 2000, diluted in 200 μl Opti-MEM, was used per dish. After 20 min of room temperature incubation, the mixture was added, without any follow-up washout step.
Mitochondrial extraction.
For mitochondrial extraction, HEK 293 cells were trypsinized for 5 min at 37°C and harvested by centrifugation at 500 × g at 4°C. Harvested cells were washed twice with 1 ml of PBS, and resuspended in 1 ml of ice-cold mitochondrial isolation buffer (200 mm mannitol, 70 mm sucrose, 10 mm HEPES pH7.4, 1 mm EGTA, and protease inhibitor cocktail). Cells were homogenized by 50–60 strokes with a douncer. Homogenized samples were centrifuged twice at 700 × g at 4°C to collect supernatant containing intact mitochondria. The supernatant was centrifuged for 10 min at 10,000 × g at 4°C. The resulting pellet is the mitochondrial fraction. The mitochondrial pellet was resuspended in 100 μl of the mitochondrial isolation buffer for further analysis. For proteinase K experiments, proteinase K (Roche, 3115828001) diluted 1:2000 in isolation buffer was added for 10 min on ice prior and washed twice with ice-cold isolation buffer, before adhering mitochondria to coverslips.
Plasmid preparation.
shMCU and shMiro1 vectors were obtained from Sigma-Aldrich (shMCU: TRCN0000251261; shMiro1: TRCN0000326200). Myc-Miro1 plasmid was obtained as a gift from Dr. Pontus Aspenstrom, Karolinska Institute, Stockholm, Sweden. MCU was cloned using silverscript III one-step RT-PCR kit (ThermoFisher Scientific, 12574018) in pFLAG-CMV6B (Sigma-Aldrich) using primers 5′-CTGAATTCATATGGCGGCCGCCGCAGGTAG-3′ and 5′-GCTGGATCCTCATTCCTTTTCTCCGATCTG-3′. MCU was then subcloned into C-Flag (Addgene, 20011) and sfGFP-N1 (Addgene, 54737). TOMM20-Myc was cloned from TOMM20-sfGFP, which was subcloned from mEos3.2-TOMM20-N-10 (Addgene, 57483). sfGFP was removed and Myc tag was added using the primers 5′-TCCGAGGAGGACCTGACCGGTGCGGCCGCGACTCTAGATCA-3′ and 5′-GATCAGCTTCTGCTCAGGATCCCCGCTACCGCCTTCCACATC-3′.
Immunocytochemistry.
Mitochondria and cells were stained using identical procedures. Isolated mitochondria were pipetted onto coverslips and allowed to adhere for 20 min. Mitochondria were fixed using 4% paraformaldehyde and 0.3% glutaraldehyde for 15 min. Free amines were quenched using 1 mg/ml NaBH4 for 10 min. Coverslips were permeabilized with 0.3% Triton X for 10 min, blocked in 1% BSA and 0.05% sodium azide for at least 1 h. Coverslips were inverted and stained in blocking buffer overnight at 4°C using the following antibodies: 1:500 anti-FLAG (Sigma-Aldrich, F7425), 1:400 anti-Myc (Santa Cruz Biotechnologies, 9E10), 1:200 anti-TOMM20 (Abcam, ab56783), and 1:400 cytochrome C (BD PharMingen, Catalog #556433). After staining, coverslips were washed three times with PBST (0.1% Tween 20). Secondary staining was performed with Invitrogen Alexa antibodies (1:400) in blocking buffer for 2 h. Coverslips were washed three times with PBST, and mounted using Prolong Gold mounting reagent (ThermoFisher Scientific, P36934). For examination of the role of calcium on MCU processing, cells were transfected and incubated for 8 h, followed by addition of 2 μm ionomycin for 10 min, immediately followed by fixation. For endogenous MCU staining, 14 DIV primary neurons were fixed in −20°C methanol for 30 min, followed by normal staining procedures. We confirmed the knock-down of MCU in primary neurons using imaging analyses and found that shMCU decreased MCU level to 30% of control. Although the transfection rate for primary neurons was low (<1% per dish), imaging analyses allow us to selectively examine transfected neurons.
Coimmunoprecipitation and Western blotting.
Fourteen hours after transfection cells are collected and lysed in 400 μl of RIPA buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 0.05% C24H39NaO4, 0.1% SDS, 1% Triton-X, and protease inhibitor cocktail). Protein concentrations were measured using the Bradford assay (Bio-Rad). One milligram of proteins was rotated head-over-tail with 0.3 μl of anti-Flag antibody (F7425, Sigma-Aldrich) or 1 μl of anti-Myc antibody (9E10, Santa Cruz Biotechnology) overnight at 4°C. The protein-Ab mixtures were rotated head-over-tail with the protein A/G agarose beads (ThermoFisher Scientific, 20421) for 4 h at 4°C. The beads were washed with 1 ml of the RIPA buffer and centrifuged for 5 min at 5000 × g at 4°C. The supernatant was removed after centrifugation. This washing step was repeated five times. For elution, 10 μl of RIPA buffer, 7 μl of 4× SDS loading dye, and 3 μl of 1 m DTT was added into washed beads. Immunoprecipitated proteins are eluted from beads by heating at 95°C for 15 min.
For Western blot analysis, proteins were separated on 12% SDS-gel and transferred onto a PVDF membrane (IPVH00010, Millipore). After blocking the membrane with 5% skim milk in TBST (0.01% Tween 20 in TBS) for 1 h at room temperature, the membrane was incubated with primary antibodies (anti-Flag Ab, 1:3000; Anti-Myc Ab, 1:1000) overnight at 4°C. The membrane was washed 3 times with TBST for 10 min and incubated with the secondary antibody for 90 min at room temperature. The membrane was washed five times with TBST for 10 min at room temperature. Imaging is performed using ImageQuant LAS 4000 (GE Healthcare).
Confocal microscopy.
All confocal images were taken using a Zeiss LSM 780 confocal microscope. For live cell imaging, 37°C was maintained using a (heating instrument), and axial stability was maintained using the Zeiss Definite Focus z-correction hardware. Velocity measurements were taken using a C-Apochromat 20× dry objective, and calcium measurements were taken using a C-Apochromat 40× water-immersion objective. All immunocytochemistry images were taken using a Plan-Apochromat 63×/1.4 oil-immersion objective (Zeiss, 420782-9900-000) at ∼21°C. For FRET experiment preparation, AlexaFluor 488 served as the donor, and AlexaFluor 555 served as the acceptor. Individual fixed mitochondria were identified, and acceptor photo-bleached using a 561 nm laser over the course of 20 frames. For the FRET region-of-interest, only the area occupied by the outer mitochondrial membrane was used. All FRET efficiencies were calculated and normalized using the Zeiss FRET analysis software package.
Calcium measurements.
For calcium measurements, Mito-GEM-GECO, cytoplasmic R-GECO, and the vectors of interest were transfected into Neuro2A cells at least 2 d before experiments. Calcium measurements were made using a C-Apochromat 40×/1.2 Water Corr M27 objective (Zeiss, 421767-9970-000), at a frame of 1 frame every 7 s. Baseline calcium measurements were made for at least 20 frames before and following the addition of 2 μm ionomycin. The subsequently induced calcium spike was confirmed using cytoplasmic R-GECO. For calcium measurements, the ratio of the blue and green emissions of GEM-GECO was used to measure the calcium levels of mitochondria over time. Data values were collected in ImageJ using the multiple positions plugin. Values collected from the cells in each dish was averaged, and counted as a single replica to form the response curve. All experiments were performed on at least 3 separate days and cell passages. To calculate the relative change in calcium, the values in the frame 20 frames preceding the addition of ionomycin (resting values) considered the initial value and were used to set a baseline for the effect of ionomycin-induced calcium spikes.
Velocity measurements.
Velocity distributions were calculated as previously described (Niescier et al., 2016). Primary hippocampal neurons were transfected at 4 DIV mito-dendra2 and the respective vector to be tested. Cells were maintained until 13–17 DIV, at which point they were used for experiments. Only one neuron per glass-bottom dish was imaged, and each dataset was collected from two to five independent cultures.
To obtain mitochondrial velocities, mitochondria in the cell body were photo-converted using a 405 nm laser, to make this subpopulation of mitochondria convert to the RFP form of dendra2. A 708 μm × 708 μm field-of-view was captured using a Plan-Apochromat 20×/0.8 objective (Zeiss, 420650-9901-000) for 60 min, at a rate of 1 frame every 15 s. Data were imported into Fiji software, where the movement of the photo-converted mitochondria was traced over the course of the time lapse, providing a map of the axonal branches. A kymograph of axon segments >200 μm from the cell body was drawn, and mitochondrial velocity was calculated based on the slope of the line of mitochondria as they travel within the kymograph. A velocity distribution histogram of these data was generated using MATLAB, and the resulting histogram was fitted into our derivation of the Fokker–Planck equation using Microsoft Excel. Kolmogorov–Smirnov test was performed using the Real Stats Microsoft Excel plugin.
Statistics.
Tukey's, Dunnett's, and Fisher's LSD ANOVA, paired Student's t test, and two-tailed Student's t test was performed using GraphPad Prism.
Results
Physiological roles of MCU
We hypothesized that Miro1 and MCU play a key role in regulating mitochondrial movement in axons (Saotome et al., 2008; Chang et al., 2011). Previously, we established a new analytical method for examining mitochondrial velocity in axons by transfecting neurons with mito-dendra2, a photo-switchable fluorescent protein (Chudakov et al., 2007), and generating a velocity distribution of photo-converted mitochondria as they leave the cell body (Niescier et al., 2016). A modified Fokker–Planck equation is then fit to the distribution (Niescier et al., 2016). The velocity distribution of mitochondria movement in the entire axon provides a better representation of mitochondrial transport in axons, by eliminating bias toward axon areas with minimal movement activity. As predicted, reduction of MCU significantly reduced the number of mitochondria leaving the cell body over the course of 1 h in primary neurons (Fig. 1A–C). MCU knockdown significantly reduced the overall velocity of mitochondria, causing mitochondria to prematurely halt movement (Fig. 1B,D,E).
Figure 1.
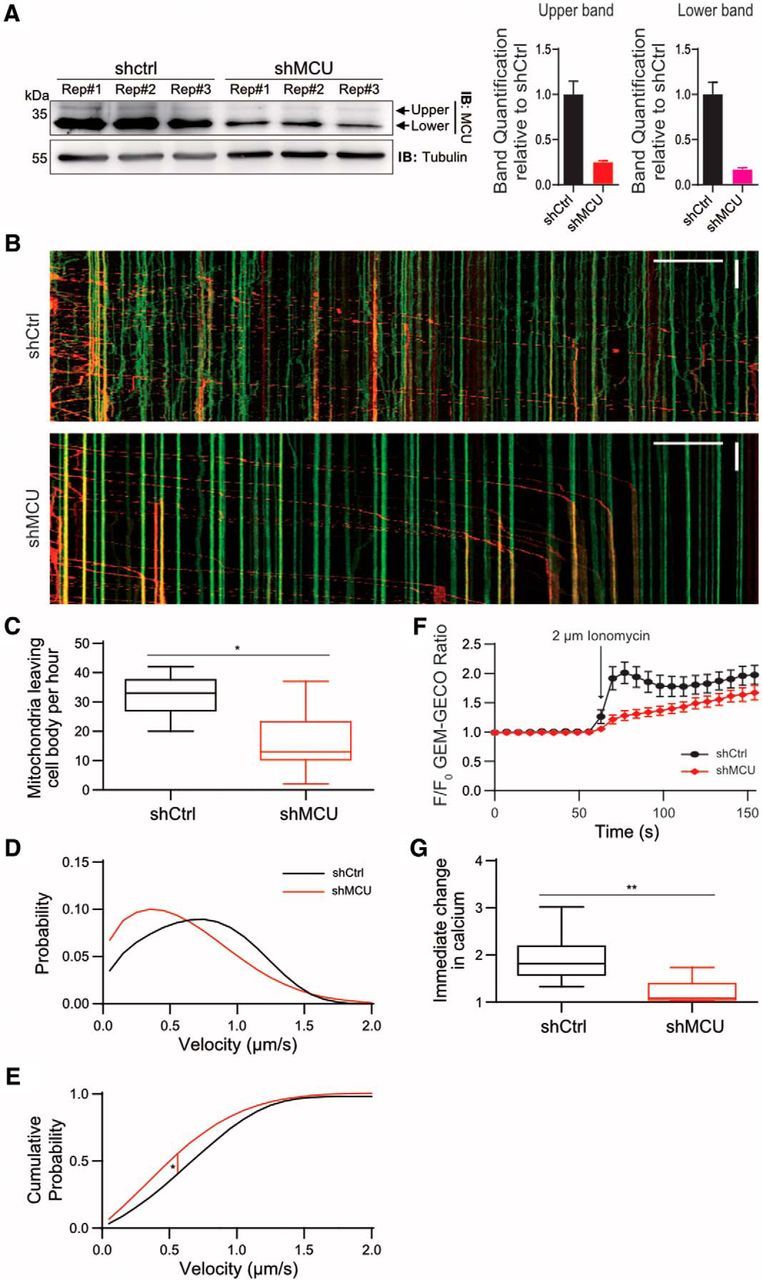
MCU and Miro1 regulate axonal mitochondrial transport and calcium influx. A, Neuro2A cells were transfected with either shctrl or shMCU in triplicate. Both the native upper and lower band was normalized to tubulin, and quantified relative to shctrl. B, Kymograph of anterograde mitochondrial movement in control and MCU knockdown conditions. Mitochondria expressing Mito-dendra in the cell body was photoconverted to red color to allow visualization of mitochondrial movement originating from the cell body. Green signals mark mitochondria already present in the axon. C, Number of mito-dendra2 photoconverted mitochondria leaving the cell body over the course of 1 h. *p < 0.002, one-way ANOVA uncorrected Fisher's LSD. Tukey's box plot profile is shown. D, Anterograde velocity distribution of axonal mitochondria, fitted to a Fokker–Planck equation. E, Cumulative probability graph derived from the data in B. *p < 2E−05, tested for statistical significance by Kolmogorov–Smirnov test. N = shctrl: 9 neurons, shMCU: 12, shMiro1: 7. F, Analyses of Ca2+ influx into mitochondria using Neuro2A cells. Cells expressing Mito-GEM-GECO, R-GECO, and either shctrl or shMCU were exposed to 2 μm ionomycin. G, Quantification of the Mito-GEM-GECO ratio the first frame after ionomycin was added to cells. **p < 0.0005, one-way ANOVA Dunnett's multiple-comparisons test. Tukey's box plot profile is shown.
Next, we tested whether MCU modulates mitochondrial Ca2+ uptake. Neuro 2A cells with reduced levels of MCU were transfected with the genetically encoded Ca2+ indicators, R-GECO and mito-GEM-GECO (Zhao et al., 2011). Baseline Ca2+ measurements were made for at least 100 s before and following the addition of 2 μm ionomycin, a Ca2+ ionophore. At low concentrations, this ionophore has been previously established to selectively increase the plasma membrane's permeability to Ca2+, while not permeabilizing mitochondria (Mallilankaraman et al., 2012). Ca2+ elevation following ionomycin treatment was confirmed using cytoplasmic R-GECO, and mitochondrial Ca2+ uptake was monitored using mito-GEM-GECO (Fig. 1E–G). As expected and previously shown (Baughman et al., 2011; De Stefani et al., 2011), we found that MCU knockdown reduced mitochondrial Ca2+ uptake (Fig. 1F,G).
MCU and Miro1 interact biochemically
Our result showing that MCU regulates mitochondrial velocity suggest that this protein and Miro1 may exist in a single regulatory complex. To test this hypothesis, we first performed an immunoprecipitation assay. Because MCU and Miro1 are found in the both the outer and inner mitochondrial membrane, we used stringent RIPA buffer to completely solubilize the membranes, and provide an environment that would promote conformation of the transmembrane helices in solution (Montserret et al., 2000). Analysis of HEK cells transfected with MCU-Flag and Myc-Miro1 confirmed the presence of a biochemical relationship between these two proteins (Fig. 2A). We also detected that Myc-Miro1 and MCU-Flag were colocalized in axons of primary hippocampal neurons (Fig. 2B). Interestingly, we noted that MCU migrated as two bands on the Western blot. Furthermore, Miro1 displayed a significant bias toward coimmunoprecipitating with the upper 40 kDa band of MCU (Fig. 2A). It is understood that MCU possesses a cleavable N-terminal domain, which supposedly guides its localization to mitochondria. However, Miro1's preferential binding to the 40 kDa band suggests that this N-terminal domain may have a different role. Thus, we investigated whether MCU's N-terminal domain is indeed required for MCU mitochondrial localization, or whether it plays a different role. Previous analysis using MITOPROT software (Claros and Vincens, 1996) predicted a likely mitochondrial targeting sequence (MTS) in this domain (p = 0.976). However, we found that the recently developed targeting sequence software MitoFates (Fukasawa et al., 2015) does not predict the presence of an N-terminal targeting sequence in MCU (p = 0.253; Fig. 2C). The N-terminal domain of MCU lacks the putative positively charged amphipathic α helix that marks a mitochondrial targeting sequence (von Heijne, 1986). To test this experimentally, if the N-terminal domain plays a role in mitochondrial localization, we expressed a mutant of MCU without the putative N-terminal MTS [MCU(Δ2–57)-Flag; Martell et al., 2012] in primary neurons (Fig. 2D). MCU(Δ2–57)-Flag still exhibited colocalization with Miro1 in axons, suggesting that this N-terminal domain is dispensable for mitochondrial localization. We also constructed a vector that only has the N-terminal domain of MCU fused to superfolder GFP [MCU(1–57)-sfGFP] to test its capability to guide a protein to mitochondria. Transfection of this vector into HEK cells and primary hippocampal neurons revealed that sfGFP is located in the cytoplasm rather than mitochondria (Fig. 2E,F), suggesting that the N-terminal domain of MCU is not sufficient to direct MCU localization into mitochondria. Together, these indicate that Miro1 preferentially interacts with the 40 kDa size of MCU and the N-terminal domain of MCU does not function as MTS.
Figure 2.
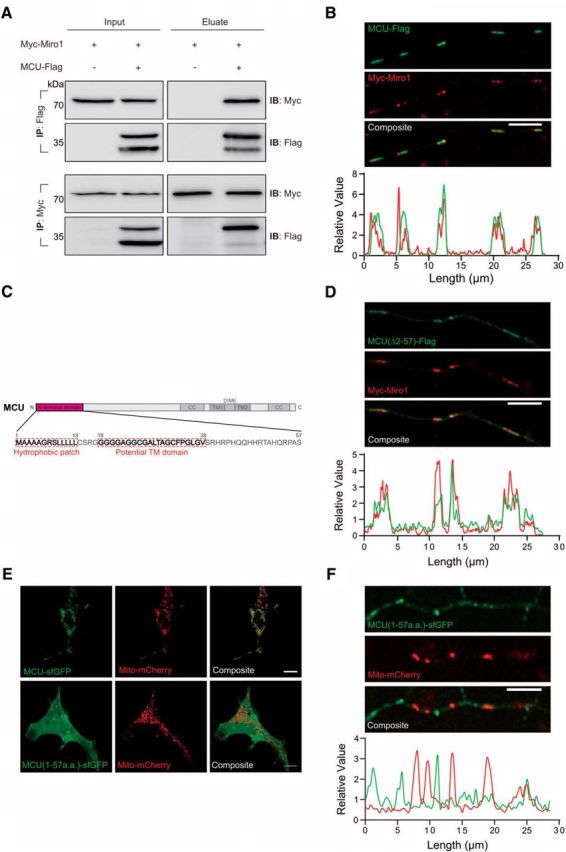
MCU interacts with Miro1. A, MCU-Flag and Myc-Miro1 were coimmunoprecipitated using Flag or Myc antibody, and then blotted with indicated antibody, respectively. MCU-Flag and Miro-1 show interaction, but Myc-Miro1 preferentially immunoprecipitates unprocessed MCU. B, Images of MCU-Flag with Myc-Miro1 in axons of hippocampal primary neurons. Scale bar, 5 μm. The graph below shows the profile plot of the axon above, demonstrating colocalization of MCU-Flag with Myc-Miro1. C, Domain map of MCU, showing a predicted transmembrane domain and hydrophobic patch in the putative MTS. D, MCU(Δ2–57)-Flag is sufficient to colocalize with Myc-Miro1 in primary axons. Scale bar, 5 μm. E, F, The MTS of MCU coupled to sfGFP (MCU(1–57)-sfGFP) is not sufficient to direct localization of sfGFP to mitochondria in HEK cells and primary hippocampal axons. Scale bar, 10 μm.
If the N-terminus of MCU is not required for mitochondrial localization, is it required for interaction with Miro? To explore this possibility, we performed immunoprecipitation analysis using HEK cell extracts containing either MCU-Flag or MCU(Δ2–57)-Flag, and Myc-Miro1. MCU-Flag or MCU(Δ2–57)-Flag was immunoprecipitated using Flag antibody, and interaction with Miro1 was detected by Myc antibody. Figure 3A clearly demonstrated that deletion of MCU's N-terminus eliminated the interaction between MCU and Miro1, confirming that the N-terminus of MCU is essential for interaction with Miro1.
Figure 3.
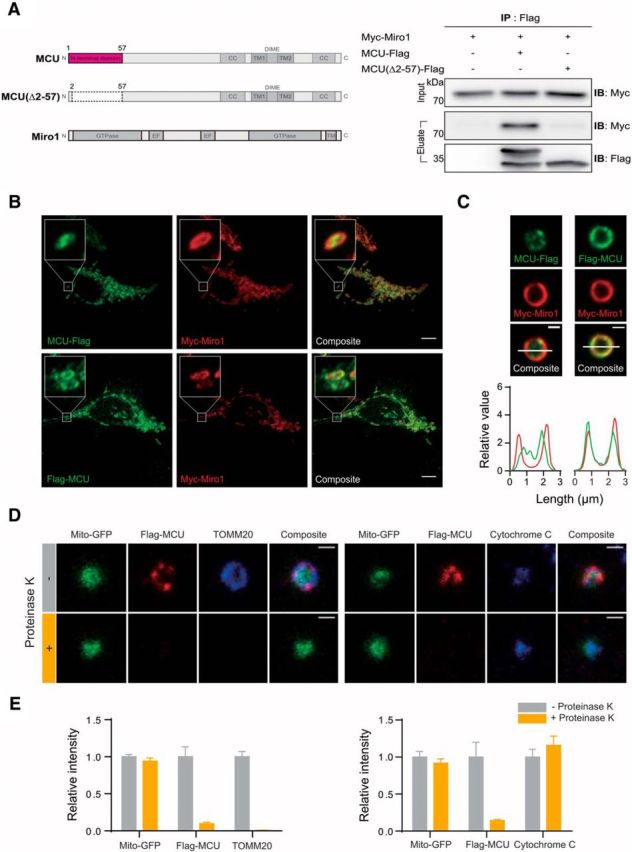
The N-terminus of MCU is located on the cytoplasmic side of the outer mitochondrial membrane. A, Coimmunoprecipitation analysis of MCU(Δ2–57)-Flag and Myc-Miro1. MCU(Δ2–57)-Flag was unable to produce the unprocessed 40 kDa band of MCU, and was also unable to coimmunoprecipitate Myc-Miro1. B, C, Colocalization of MCU-Flag and Flag-MCU with Myc-Miro1 in Neuro2A cells. Isolated mitochondria from HEK cells overexpressing Myc-Miro1 and either MCU-Flag or Flag-MCU, showing that Flag-MCU epitope resides in the outer mitochondrial membrane. Scale bar, 1 μm. D, Treatment of mitochondria isolated from HEK cells, and transfected with Flag-MCU, with proteinase K. Proteinase K is able to digest the Flag and TOMM20 signal, while leaving the cytochrome C signal intact. Scale bar, 1 μm. E, Quantification of samples in D. N = 96 mitochondria for TOMM20 control, 51 for TOMM20 proteinase K, 43 for Cytochome C control, 82 for cytochrome C proteinase K.
An issue with the possibility of an interaction between Miro1 and MCU is that they are outer and inner mitochondrial proteins, respectively, and the current putative topology of these proteins suggests that there are few possible areas that could host such an interaction. To better investigate interaction between Miro1 and MCU in mitochondria, we attached a Flag tag to either the N-terminus or the C-terminus to MCU, and examined its localization. Flag-MCU and MCU-Flag are both present in mitochondria, but showed subtle difference in their relative distribution within mitochondria and colocalization with Myc-Miro1 (Fig. 3B). To better delineate protein localization within mitochondria subregions, we extracted mitochondria from HEK cells. Extracted mitochondria lose their putative oblong shape and turn into flat spheres (Ishihara et al., 2004), but the outer mitochondrial membrane remains intact and is resolvable from other mitochondrial components. Consistent with a previous report (Martell et al., 2012), MCU-Flag signal localized within the internal area of the mitochondrion and did not well overlap with Miro1 location, confirming that the C-terminus of MCU is located inside the mitochondrial matrix. In contrast, Flag-MCU showed excellent colocalization with Myc-Miro1, showing that the N-terminus of MCU is stably localized to the outer mitochondrial membrane (Fig. 3C).
Although the N-terminus of MCU appears to colocalize with the outer mitochondrial membrane, it is unclear whether it is present on the inside or outside of this membrane. Topology analysis shows a potential transmembrane domain at the 18–38 position of MCU, as well as a hydrophobic patch at the 1–13 position (Fig. 2C), providing a plausible area for MCU to interact with this area. To further delineate whether MCU's N-terminus is located in the outer mitochondrial membrane or present in the inter membrane space of mitochondria, we treated extracted mitochondria with proteinase K, which selectively digests components located on the cytoplasmic side of the outer mitochondrial membrane, but does not act on proteins in the inter membrane space or the matrix (Fig. 3D). TOMM20 was efficiently digested by proteinase K treatment, whereas cytochrome C (located within the inter membrane space) and Mito-GFP were not affected (Fig. 3D,E). We also found that the Flag-MCU signal was completely eliminated upon treatment of proteinase K (Fig. 3D,E), confirming that the N-terminus of Flag-MCU is located on the cytoplasmic side of the outer mitochondrial membrane. This model is consistent with the presence of a transmembrane domain within the N-terminus region as predicted by TMBASE (Hofmann and Stoffel, 1993), as well as a hydrophobic patch at the tip of the domain (Fig. 2C). Note that we also measured the Feret's minimum diameter of our extracted mitochondria, which examines the smallest possible diameter of a particle in an image, to ensure that there is no abnormal swelling of mitochondria following different treatment conditions. Our analysis of Mito-GFP signal showed that this diameter of mitochondria was an average of 1.1 μm in all conditions. This is in agreement with manually measured diameter of intact mitochondria imaged with TEM (Franko et al., 2013), though the reduced resolution found with confocal microscopy compared with TEM can give mitochondria a slightly larger appearance.
Interacting domains between MCU and Miro1
Having demonstrated that the N-terminus of MCU is facing the cytosolic side of mitochondria and is required for its interaction with Miro1, we next explored the possibility that other functional domains of MCU also modulate its interaction with Miro1. We tested the possibility that the dime motif of MCU regulates MCU–Miro1 interaction, as the dime motif is predicted to project into the mitochondrial inter membrane space and is important for Ca2+ pore formation (Baughman et al., 2011; Fig. 4A). To this end, we altered several amino acids in the dime motif (257EYSW260 to 257AAAA260 or 261DIME264 to 261AAAA264). However, these mutant MCU proteins were still able to find to Miro1, suggesting that the dime motif is not involved in MCU–Miro1 interaction (Fig. 4A). Next, we examined the regions on Miro1 required for its interaction with MCU. We prepared two different constructs: (1) Myc-Miro1 lacking transmembrane domain and (2) Myc-Miro1 without the last 3 aa (616KQR618; Fig. 4B). Immunoprecipitation assay using Flag antibody and detection with Myc antibody revealed that Miro1 transmembrane domain is required for its interaction with MCU.
Figure 4.
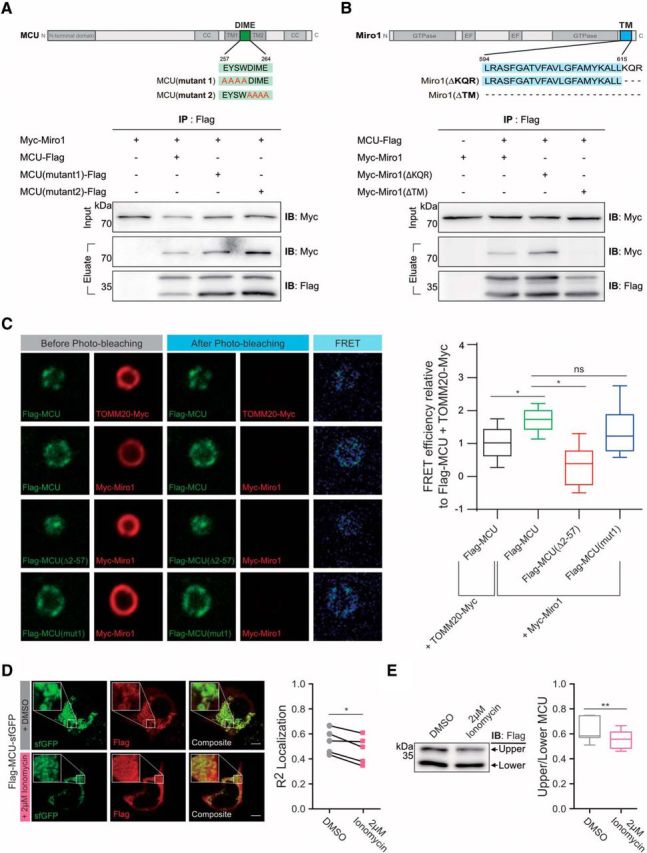
Domain mapping of the interaction between MCU and Miro1. A, Mutations on the dime motif of MCU (257EYSW260 to 257AAAA260 or 261DIME264 to 261AAAA264) did not affect the interaction between MCU and Miro1. B, MCU did not interact with Myc-Miro1 without transmembrane domain, but still immunoprecipitated Myc-Miro1 lacking 3 aa (616KQR618). C, FRET analysis of Myc-Miro1 and Flag-MCU showed a significantly higher FRET efficiency compared with that of TOMM20-Myc and Flag-MCU. Mutation on the dime motif of MCU (257EYSW260 to 257AAAA260) did not affect Myc-Miro1 binding, while elimination of MCU's N-terminal domain [Flag-MCU(Δ2–57)] or adding Ca2+ significantly reduced interaction with Myc-Miro1. N = Flag-MCU + TOMM20-Myc: 72 mitochondria. Flag-MCU + Myc-Miro1: 78 mitochondria, Flag-MCU (257EYSW260 to 257AAAA260) + Myc-Miro1: 25 mitochondria, Flag-MCU(Δ2–57) + Myc-Miro1: 23 mitochondria. Scale bar, 1 μm. *p < 0.0001. Tukey's box plot profile is shown. D, Ca2+ elevation alters MCU–Miro1 interaction. MCU expression vector dually tagged in the N-terminus (Flag) and the C-terminus (sfGFP), respectively, was prepared and used to investigate localization of these tags after treating cells with 2 μm ionomycin for 10 min. The R2 correlation between Flag and sfGFP was compared between paired samples with or without addition of 2 μm ionomycin. N = 5 experiments, *p = 0.0171. E, HEK cells transfected with MCU-Flag were treated with 2 μm ionomycin as shown above and then blotted for Flag. Graph shows the ratio of the full-length MCU band (top band) to the processed MCU (bottom band). N = 7 experiments, **p = 0.0094. Tukey's boxplot profile is shown.
To verify interaction between MCU and Miro1 in the mitochondria, we performed FRET using acceptor-photobleaching with isolated mitochondria (Fig. 4C). Myc-Miro1 and Flag-MCU have a significantly higher FRET efficiency compared with that of TOMM20-Myc and Flag-MCU, supporting the claim that Miro1 interacts with MCU. Furthermore, consistent with results shown in Figure 4A, the dime motif mutations did not affect the MCU–Miro1 interaction in isolated mitochondria (Fig. 4C). In contrast, deletion of the N-terminus of MCU significantly reduced the FRET signal (Fig. 4C).
Having demonstrated that MCU's N-terminus interacts with Miro1, we next asked whether Ca2+ elevation alters MCU–Miro1 interaction. MCU is present in two different forms, and Miro1 preferentially binds to the 40 kDa size of MCU in mitochondria (Fig. 2A). Thus, we hypothesized that high cytosolic Ca2+ levels would stimulate cleavage of the N-terminal domain of MCU, disrupting MCU–Miro1 interaction to facilitate uptake of Ca2+ into mitochondria through MCU. To test this hypothesis, we prepared the MCU expression vector dually tagged in the N-terminus (Flag) and the C-terminus (sfGFP). Next, we investigated the location of these tags before and after treatment with 2 μm ionomycin for 10 min (Fig. 4D). Figure 4D demonstrates that both Flag and GFP tags showed clear colocalization in mitochondria before ionomycin treatment. Strikingly, the N-terminus Flag tag was distributed to the cytoplasm, whereas C-terminal sfGFP signal remained in mitochondria after ionomycin treatment, implying that Ca2+ elevation initiates cleavage of the N-terminus of MCU to abrogate its interaction with Miro1. Furthermore, the intensity of MCU bands on the Western blot showed a decrease in the amount of unprocessed MCU following exposure to ionomycin (Fig. 4E). Together, these data support a model in which Ca2+ elevation triggers proteolytic cleavage of MCU, thus disrupting interaction between MCU and Miro1.
Miro1–MCU interaction is essential for mitochondrial transport in axons
Although MCU and Miro1 have been shown to be essential for both mitochondrial transport and Ca2+ influx (Fig. 1), it is not known what role their interaction plays in these functions. To examine the possibility of a functional relationship, we tested whether abnormal mitochondrial movement and Ca2+ influx in cells expressing shMCU can be restored by expressing either MCU-Flag or MCU(Δ2–57)-Flag (Fig. 5A). MCU has been shown to integrate normally into the uniplex complex with or without the presence of the N-terminus in artificial membranes (De Stefani et al., 2011), and our results show that MCU(Δ2–57)-Flag integrates into axonal mitochondria (Fig. 2D), similar to wild-type MCU-Flag (Fig. 2B) but does not interact with Miro1. Whereas expression of MCU-Flag was able to rescue the reduced velocity caused by MCU reduction, overexpression of MCU(Δ2–57)-Flag remained significantly slower compared with control (Fig. 5B,C). MCUΔ2–57-Flag was also unable to rescue the total number of mobile mitochondria (Fig. 6D). Conversely, MCU-Flag or MCUΔ2–57-Flag expression was able to rescue Ca2+ influx to that of control (Figs. 5C–D). Together, these results imply that the N-terminal domain of MCU that interacts with Miro1 is required for mitochondrial transport; however, the interaction does not play a role in stimulating uniplex activity of Ca2+ influx into mitochondria.
Figure 5.
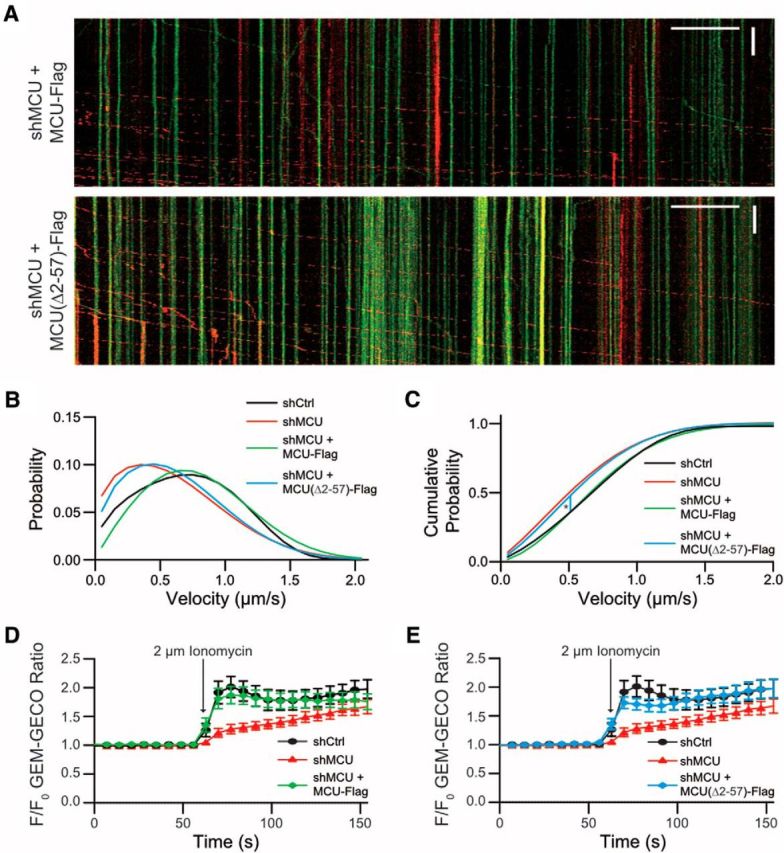
MCU(Δ2–57) is able to rescue calcium influx, but not mitochondrial movement. A, Number of mito-dendra2 switched mitochondria leaving the cell body over the course of 1 h. *p < 0.02 one-way ANOVA, uncorrected Fisher's LSD test. Tukey's box plot profile is shown. B, Velocity distribution of axonal mitochondria fitted to a derived Fokker–Planck equation. C, Cumulative probability graph derived from the data in B. *p < 0.0002, tested for statistical significance by Kolmogorov–Smirnov test. N = shctrl: 9 neurons, shMCU: 12, shMCU + MCU(Δ2–57)-Flag: 7, shMCU + MCU-Flag: 5. D–F, Analysis of Ca2+ influx into mitochondria using Neuro2A cells. Cells expressing Mito-GEM-GECO, R-GECO, and either shctrl, shMCU, shMCU + MCU-Flag (D), or shMCU, shMCU + MCU(Δ2–57)-Flag (E) were exposed to 2 μm ionomycin. Note that the same control was used as in Figure 1, and statistics were all performed as a single ANOVA.
Figure 6.

MCU-Miro1 interaction prevents MCU's Ca2+ influx capability. A, The mutant MCU (7R to A and 8S to A) binds to Miro1, but it cannot be cleaved. B, C, Cells containing the mutant MCU showed that Ca2+ influx was significantly reduced after ionomycin treatment. *p < 0.01, ****p < 0.0001. One-way ANOVA Dunnett's multiple-comparisons test. Tukey's box plot profile is shown. N = shctrl: 12 neurons, shMCU: 12, shMCU + MCU-Flag: 11, shMCU + MCU(Δ2–57)-Flag: 12, shMCU + MCU(mut1)-Flag: 8, shMCU + MCU(R7A,S8A)-Flag: 12. D, Neurons expressing the mutant MCU showed extremely small number of mitochondria leaving the cell body, which made difficult to measure proper mitochondrial velocity in axon. *p < 0.01, ****p < 0.0001. One-way ANOVA Dunnett's multiple-comparisons test. Tukey's box plot profile is shown. N = shctrl: 9 neurons, shMCU: 12, shMCU + MCU-Flag: 8, shMCU + MCU(Δ2–57)-Flag: 5, shMCU + MCU(R7A,S8A)-Flag: 5.
To further investigate the importance of the MCU amino acids within the membrane, we generated a mutant MCU that has mutations on two different amino acids in the N-terminus (7R to A and 8S to A), because it is known that arginine in the membrane side of a protein plays an important role by carrying charge (Hristova and Wimley, 2011). Because this mutant MCU cannot be cleaved (Fig. 6A), we used it to address the functional significance of MCU cleavage: whether it is important for Ca2+ influx or mitochondrial movement. Cells containing the mutant MCU (MCUR7AS8A) displayed significantly reduced Ca2+ influx following ionomycin treatment (Fig. 6B,C), suggesting that the cleavage and disassociation of MCU and Miro1 is required for efficient Ca2+ influx through MCU. We also noticed that neurons expressing the mutant MCU (MCUR7AS8A) showed dramatically reduced number of mitochondria leaving the cell body (Fig. 6D), which made it difficult to measure proper mitochondrial transport in axon. It is plausible that the MCU mutant acts as a dominant-negative form that interferes with the normal MCU–Miro1 interaction required for mitochondrial movement.
Discussion
Here, we provide evidence that MCU plays critical roles in regulating mitochondrial Ca2+ influx and axonal transport. We have shown that MCU interacts with Miro1, a protein located in the outer mitochondrial membrane (Fransson et al., 2006), through an alternative use of its putative N-terminal mitochondrial targeting domain. This N-terminal domain of MCU is neither sufficient nor necessary for its localization. We report that MCU and Miro1 interaction is required for efficient mitochondrial transport in axons, but not for mitochondrial Ca2+ influx. We also show that the N-terminus of MCU can be cleaved, and its cleavage is required for mitochondrial Ca2+ influx.
An important issue in this study is the endogenous expression level of the larger interacting form of MCU, which we propose is responsible for regulation of mitochondrial transport, compared with the transport machinery of mitochondria. Although it is true that the majority of this protein is in the cleaved formation, we do not believe that this excludes this form from having a biological purpose regarding transport. MCU forms a pentameric pore to conduct calcium influx (Oxenoid et al., 2016), and our model only requires a single MCU protein out of five to remain unprocessed. In addition, the majority of mitochondria in a typical neuron are stationary, and only 20–30% of mitochondria are mobile (Chang et al., 2011; Nguyen et al., 2014; López-Doménech et al., 2016), meaning that the majority of the MCU in a given neuron will be in its processed form. The total number of mobile mitochondria also decreases as the neuron matures (Chang and Reynolds, 2006; Faits et al., 2016), which would result in a lowered presence of the larger MCU form. Altogether, we find that the unprocessed form's reduced presence does not exclude it from being a functional domain. Another issue that arises is the quantity of MCU compared with the transport machinery in mitochondria. Although the total number of motors on a typical mammalian mitochondrion remains unknown, a study using giant amoeba Reticulomyxa found 1–4 motors per mitochondrion in vivo (Ashkin et al., 1990). However, it should be noted that these mitochondria are much smaller compared with those found in mammalian tissue, and it is likely that this number underestimates what is found in other organisms. Interestingly, MCU particles found using super-resolution imaging techniques have also revealed a surprisingly low number of channels per mitochondrion (De La Fuente et al., 2016), a number that may correlate with the number of motor complexes present on the outer mitochondrial membrane of a moving mitochondrion.
We propose that (1) MCU–miro1 interaction maintains mitochondrial transport in axons; (2) cytoplasmic Ca2+ elevation triggers the cleavage of the N-terminus of MCU, leading to dissociation of MCU–Miro interaction; 3) disrupting the interaction between MCU and Miro1 negatively impacts mitochondrial movement but promotes efficient Ca2+ entry into mitochondrial matrix. The role of Ca2+ in the cleavage of MCU's N-terminal domain, as well as the role of the Miro1 yeast ortholog gem1 on interaction with the ER (Kornmann et al., 2011), also alludes that these proteins may have a more intertwined role in Ca2+ regulation. It will be interesting to examine in the future mechanisms regulating cleavage of MCU, identify the enzyme responsible for the cleavage of MCU, as well as understand how a disruption in MCU and Miro1 interaction promotes Ca2+ entry. In summary, we have found that Miro1 is a novel component of the mitochondrial Ca2+ uniplex, and demonstrate that coupling between MCU and Miro1 is a novel mechanism modulating mitochondrial transport.
Footnotes
This work was supported by Samsung Science and Technology Foundation (SSTF-BA1301003) and the Leading Research Program, National Research Foundation of Korea Grant funded by the Korea government (MEST; 2016R1A3B1905982).
The authors declare no competing financial interests.
References
- Ashkin A, Schütze K, Dziedzic JM, Euteneuer U, Schliwa M (1990) Force generation of organelle transport measured in vivo by an infrared laser trap. Nature 348:346–348. 10.1038/348346a0 [DOI] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476:341–345. 10.1038/nature10234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chada SR, Hollenbeck PJ (2003) Mitochondrial movement and positioning in axons: the role of growth factor signaling. J Exp Biol 206:1985–1992. 10.1242/jeb.00263 [DOI] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ (2006) Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience 141:727–736. 10.1016/j.neuroscience.2006.01.034 [DOI] [PubMed] [Google Scholar]
- Chang KT, Niescier RF, Min KT (2011) Mitochondrial matrix Ca2+ as an intrinsic signal regulating mitochondrial motility in axons. Proc Natl Acad Sci U S A 108:15456–15461. 10.1073/pnas.1106862108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Artiga DJ, Abiria SA, Clapham DE (2016) Mitochondrial calcium uniporter regulator 1 (MCUR1) regulates the calcium threshold for the mitochondrial permeability transition. Proc Natl Acad Sci U S A 113:E1872–E1880. 10.1073/pnas.1602264113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chudakov DM, Lukyanov S, Lukyanov KA (2007) Tracking intracellular protein movements using photoswitchable fluorescent proteins PS-CFP2 and Dendra2. Nat Protoc 2:2024–2032. 10.1038/nprot.2007.291 [DOI] [PubMed] [Google Scholar]
- Claros MG, Vincens P (1996) Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur J Biochem 241:779–786. 10.1111/j.1432-1033.1996.00779.x [DOI] [PubMed] [Google Scholar]
- De La Fuente S, Fernandez-Sanz C, Vail C, Agra EJ, Holmstrom K, Sun J, Mishra J, Williams D, Finkel T, Murphy E, Joseph SK, Sheu SS, Csordás G (2016) Strategic positioning and biased activity of the mitochondrial calcium uniporter in cardiac muscle. J Biol Chem 291:23343–23362. 10.1074/jbc.M116.755496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deluca HF, Engstrom GW (1961) Calcium uptake by rat kidney mitochondria. Proc Natl Acad Sci U S A 47:1744–1750. 10.1073/pnas.47.11.1744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, Rizzuto R (2011) A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476:336–340. 10.1038/nature10230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faits MC, Zhang C, Soto F, Kerschensteiner D (2016) Dendritic mitochondria reach stable positions during circuit development. eLife 5:e11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franko A, Baris OR, Bergschneider E, von Toerne C, Hauck SM, Aichler M, Walch AK, Wurst W, Wiesner RJ, Johnston IC, de Angelis MH (2013) Efficient isolation of pure and functional mitochondria from mouse tissues using automated tissue disruption and enrichment with anti-TOM22 magnetic beads. PloS One 8:e82392. 10.1371/journal.pone.0082392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransson S, Ruusala A, Aspenström P (2006) The atypical rho GTPases miro-1 and miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun 344:500–510. 10.1016/j.bbrc.2006.03.163 [DOI] [PubMed] [Google Scholar]
- Fukasawa Y, Tsuji J, Fu SC, Tomii K, Horton P, Imai K (2015) MitoFates: improved prediction of mitochondrial targeting sequences and their cleavage sites. Mol Cell Proteomics 14:1113–1126. 10.1074/mcp.M114.043083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann K, Stoffel W (1993) TMBASE: a database of membrane spanning protein segments. Biol Chem Hoppe-Seyler 374:166. [Google Scholar]
- Hristova K, Wimley WC (2011) A look at arginine in membranes. J Membr Biol 239:49–56. 10.1007/s00232-010-9323-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara N, Eura Y, Mihara K (2004) Mitofusin 1 and 2 play distinct roles in mitochondrial fusion reactions via GTPase activity. J Cell Sci 117:6535–6546. 10.1242/jcs.01565 [DOI] [PubMed] [Google Scholar]
- Kornmann B, Osman C, Walter P (2011) The conserved GTPase Gem1 regulates endoplasmic reticulum-mitochondria connections. Proc Natl Acad Sci U S A 108:14151–14156. 10.1073/pnas.1111314108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Lee KS, Huh S, Liu S, Lee DY, Hong SH, Yu K, Lu B (2016) Polo kinase phosphorylates miro to control ER-mitochondria contact sites and mitochondrial Ca2+ homeostasis in neural stem cell development. Dev Cell 37:174–189. 10.1016/j.devcel.2016.03.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon LA, Steward O (2000) Movement of mitochondria in the axons and dendrites of cultured hippocampal neurons. J Comp Neurol 427:340–350. [DOI] [PubMed] [Google Scholar]
- Liu JC, Liu J, Holmström KM, Menazza S, Parks RJ, Fergusson MM, Yu ZX, Springer DA, Halsey C, Liu C, Murphy E, Finkel T (2016) MICU1 serves as a molecular gatekeeper to prevent in vivo mitochondrial calcium overload. Cell Rep 16:1561–1573. 10.1016/j.celrep.2016.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Doménech G, Higgs NF, Vaccaro V, Roš H, Arancibia-Cárcamo IL, MacAskill AF, Kittler JT (2016) Loss of dendritic complexity precedes neurodegeneration in a mouse model with disrupted mitochondrial distribution in mature dendrites. Cell Rep 17:317–327. 10.1016/j.celrep.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT (2009) Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61:541–555. 10.1016/j.neuron.2009.01.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallilankaraman K, Cárdenas C, Doonan PJ, Chandramoorthy HC, Irrinki KM, Golenár T, Csordás G, Madireddi P, Yang J, Müller M, Miller R, Kolesar JE, Molgó J, Kaufman B, Hajnóczky G, Foskett JK, Madesh M (2012) MCUR1 is an essential component of mitochondrial Ca2+ uptake that regulates cellular metabolism. Nat Cell Biol 14:1336–1343. 10.1038/ncb2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martell JD, Deerinck TJ, Sancak Y, Poulos TL, Mootha VK, Sosinsky GE, Ellisman MH, Ting AY (2012) Engineered ascorbate peroxidase as a genetically encoded reporter for electron microscopy. Nat Biotechnol 30:1143–1148. 10.1038/nbt.2375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montserret R, McLeish MJ, Böckmann A, Geourjon C, Penin F (2000) Involvement of electrostatic interactions in the mechanism of peptide folding induced by sodium dodecyl sulfate binding. Biochemistry 39:8362–8373. 10.1021/bi000208x [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Oh SS, Weaver D, Lewandowska A, Maxfield D, Schuler MH, Smith NK, Macfarlane J, Saunders G, Palmer CA, Debattisti V, Koshiba T, Pulst S, Feldman EL, Hajnóczky G, Shaw JM (2014) Loss of Miro1-directed mitochondrial movement results in a novel murine model for neuron disease. Proc Natl Acad Sci U S A 111:E3631–E3640. 10.1073/pnas.1402449111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niescier RF, Kwak SK, Joo SH, Chang KT, Min KT (2016) Dynamics of mitochondrial transport in axons. Front Cell Neurosci 10:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenoid K, Dong Y, Cao C, Cui T, Sancak Y, Markhard AL, Grabarek Z, Kong L, Liu Z, Ouyang B, Cong Y, Mootha VK, Chou JJ (2016) Architecture of the mitochondrial calcium uniporter. Nature 533:269–273. 10.1038/nature17656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X, Liu J, Nguyen T, Liu C, Sun J, Teng Y, Fergusson MM, Rovira II, Allen M, Springer DA, Aponte AM, Gucek M, Balaban RS, Murphy E, Finkel T (2013) The physiological role of mitochondrial calcium revealed by mice lacking the mitochondrial calcium uniporter. Nat Cell Biol 15:1464–1472. 10.1038/ncb2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, Rizzuto R (2014) MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 53:726–737. 10.1016/j.molcel.2014.01.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK (2010) MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature 467:291–296. 10.1038/nature09358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrungaro C, Zimmermann KM, Küttner V, Fischer M, Dengjel J, Bogeski I, Riemer J (2015) The Ca2+-dependent release of the Mia40-induced MICU1-MICU2 dimer from MCU regulates mitochondrial Ca2+ uptake. Cell Metab 22:721–733. 10.1016/j.cmet.2015.08.019 [DOI] [PubMed] [Google Scholar]
- Phillips JM, Hollenbeck JR, Ilgen DR (1996) Prevalence and prediction of positive discrepancy creation: examining a discrepancy between two self-regulation theories. J Appl Psychol 81:498–511. 10.1037/0021-9010.81.5.498 [DOI] [PubMed] [Google Scholar]
- Plovanich M, Bogorad RL, Sancak Y, Kamer KJ, Strittmatter L, Li AA, Girgis HS, Kuchimanchi S, De Groot J, Speciner L, Taneja N, Oshea J, Koteliansky V, Mootha VK (2013) MICU2, a paralog of MICU1, resides within the mitochondrial uniporter complex to regulate calcium handling. PLoS One 8:e55785. 10.1371/journal.pone.0055785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raffaello A, De Stefani D, Sabbadin D, Teardo E, Merli G, Picard A, Checchetto V, Moro S, Szabò I, Rizzuto R (2013) The mitochondrial calcium uniporter is a multimer that can include a dominant-negative pore-forming subunit. EMBO J 32:2362–2376. 10.1038/emboj.2013.157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancak Y, Markhard AL, Kitami T, Kovács-Bogdán E, Kamer KJ, Udeshi ND, Carr SA, Chaudhuri D, Clapham DE, Li AA, Calvo SE, Goldberger O, Mootha VK (2013) EMRE is an essential component of the mitochondrial calcium uniporter complex. Science 342:1379–1382. 10.1126/science.1242993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnóczky G (2008) Bidirectional Ca2+-dependent control of mitochondrial dynamics by the miro GTPase. Proc Natl Acad Sci U S A 105:20728–20733. 10.1073/pnas.0808953105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomar D, Dong Z, Shanmughapriya S, Koch DA, Thomas T, Hoffman NE, Timbalia SA, Goldman SJ, Breves SL, Corbally DP, Nemani N, Fairweather JP, Cutri AR, Zhang X, Song J, Jaña F, Huang J, Barrero C, Rabinowitz JE, Luongo TS, et al. (2016) MCUR1 is a scaffold factor for the MCU complex function and promotes mitochondrial bioenergetics. Cell Rep 15:1673–1685. 10.1016/j.celrep.2016.04.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vais H, Mallilankaraman K, Mak DD, Hoff H, Payne R, Tanis JE, Foskett JK (2016) EMRE is a matrix Ca2+ sensor that governs gatekeeping of the mitochondrial Ca2+ uniporter. Cell Rep 14:403–410. 10.1016/j.celrep.2015.12.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC (2013) TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77:485–502. 10.1016/j.neuron.2012.11.027 [DOI] [PubMed] [Google Scholar]
- von Heijne G. (1986) Mitochondrial targeting sequences may form amphiphilic helices. EMBO J 5:1335–1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Schwarz TL (2009) The mechanism of Ca2+-dependent regulation of kinesin-mediated mitochondrial motility. Cell 136:163–174. 10.1016/j.cell.2008.11.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Araki S, Wu J, Teramoto T, Chang YF, Nakano M, Abdelfattah AS, Fujiwara M, Ishihara T, Nagai T, Campbell RE (2011) An expanded palette of genetically encoded Ca2+ indicators. Science 333:1888–1891. 10.1126/science.1208592 [DOI] [PMC free article] [PubMed] [Google Scholar]