

Abstract
Functional neuronal connectivity requires proper neuronal morphogenesis and its dysregulation causes neurodevelopmental diseases. Transforming growth factor-β (TGF-β) family cytokines play pivotal roles in development, but little is known about their contribution to morphological development of neurons. Here we show that the Smad-dependent canonical signaling of TGF-β family cytokines negatively regulates neuronal morphogenesis during brain development. Mechanistically, activated Smads form a complex with transcriptional repressor TG-interacting factor (TGIF), and downregulate the expression of a neuronal polarity regulator, collapsin response mediator protein 2. We also demonstrate that TGF-β family signaling inhibits neurite elongation of human induced pluripotent stem cell-derived neurons. Furthermore, the expression of TGF-β receptor 1, Smad4, or TGIF, which have mutations found in patients with neurodevelopmental disorders, disrupted neuronal morphogenesis in both mouse (male and female) and human (female) neurons. Together, these findings suggest that the regulation of neuronal morphogenesis by an evolutionarily conserved function of TGF-β signaling is involved in the pathogenesis of neurodevelopmental diseases.
SIGNIFICANCE STATEMENT Canonical transforming growth factor-β (TGF-β) signaling plays a crucial role in multiple organ development, including brain, and mutations in components of the signaling pathway associated with several human developmental disorders. In this study, we found that Smads/TG-interacting factor-dependent canonical TGF-β signaling regulates neuronal morphogenesis through the suppression of collapsin response mediator protein-2 (CRMP2) expression during brain development, and that function of this signaling is evolutionarily conserved in the mammalian brain. Mutations in canonical TGF-β signaling factors identified in patients with neurodevelopmental disorders disrupt the morphological development of neurons. Thus, our results suggest that proper control of TGF-β/Smads/CRMP2 signaling pathways is critical for the precise execution of neuronal morphogenesis, whose impairment eventually results in neurodevelopmental disorders.
Keywords: canonical TGF-β signaling, CRMP2, neurodevelopmental diseases, neuronal morphogenesis, TGIF
Introduction
Higher brain function relies on the exquisite architecture of neural circuits that are established during development. Functional neural networks are orchestrated through the proper growth of axons to their targets and the formation of complex dendrite branches that integrate multiple synaptic inputs. Defects in neuronal morphogenesis lead to neurological diseases including mental retardation, autism spectrum disorders, and psychiatric diseases (Ramocki and Zoghbi, 2008; Walsh and Engle, 2010).
Transforming growth factor-β (TGF-β) signaling controls a variety of biological processes, including cell proliferation, differentiation, apoptosis, and tissue patterning (Massagué et al., 2000). The TGF-β superfamily comprises TGF-βs, bone morphogenetic proteins (BMPs), growth and differentiation factors (GDFs), activins, and Nodal. TGF-β family ligands bind to their cognate type I and type II serine/threonine kinase receptors and induce transphosphorylation of the type I receptors. Activated type I receptors in turn phosphorylate pathway-restricted transcription factor Smads (R-Smads), which then form a complex with common partner Smad4. The resultant Smad complexes translocate into the nucleus, where they modulate the transcription of target genes through interaction with transcriptional cofactors (Derynck and Zhang, 2003; Shi and Massagué, 2003). There also exist Smad-independent noncanonical TGF-β signaling pathways, containing molecules such as p38 mitogen-activated protein kinase, TGF-β-activated kinase 1, TNF receptor associated factor 4 (TRAF4) and TRAF6 (Derynck and Zhang, 2003; Zhang, 2009); however, their precise biological contributions to TGF-β signaling are much less clear compared with the canonical pathway.
Smad-dependent canonical TGF-β signaling is critical for multiple aspects of neurodevelopment, including adult neurogenesis and neuroprotection (König et al., 2005; Liu and Niswander, 2005; Ageta et al., 2008; Colak et al., 2008). It has been shown that TGF-β receptor 1 [TGFβR1 (or ALK5)]-dependent TGF-β signaling promotes the maturation of newborn neurons in the adult hippocampus (He et al., 2014). Moreover, mutations in signaling pathway components, such as TGFβR1, TGFβR2, Smad4, and TG-interacting factor (TGIF), are associated with several human developmental disorders characterized by cognitive abnormality and mental retardation (Gripp et al., 2000; Loeys et al., 2005; Le Goff et al., 2011), suggesting that canonical TGF-β signaling plays a crucial role in neuronal functions and development. However, its functional role in neuronal morphogenesis, especially during brain development, and how defects of canonical TGF-β signaling lead to neurodevelopmental disorders, are not fully elucidated.
Proper control of microtubule and actin cytoskeleton is fundamental for neuronal development and maintenance (Conde and Cáceres, 2009). The family of collapsin response mediator proteins (CRMPs) includes molecules critical for neurite development. CRMPs comprise the five cytosolic proteins CRMP1–5, and all CRMPs bind to tubulin. CRMP1, CRMP2, CRMP3, and CRMP4 show 75% sequence similarity with each other, whereas CRMP5 shares only 50% similarity (Quach et al., 2015). All CRMPs can be phosphorylated and are abundantly expressed in the developing and adult nervous systems (Wang and Strittmatter, 1996; Bretin et al., 2005). CRMP2 [also known as DPYSL2 (dihydropyriminidase-like 2)] is the first identified member of the CRMP family and has been studied most extensively. It binds to α- and β-tubulin heterodimers and enhances microtubule assembly, thereby promoting axon specification and formation (Inagaki et al., 2001; Fukata et al., 2002). CRMP2 also binds to actin to control cytoskeletal dynamics (Arimura et al., 2005). In addition, phosphorylation by glycogen synthase kinase 3β negatively regulates the activity of CRMP2 (Yoshimura et al., 2005). Although a recent study has reported that BMP–Smad signaling suppresses CRMP2 expression in neuronal progenitor cells in the developing brain (Sun et al., 2010b), the mechanisms that regulate the expression of CRMP2 to define the morphological development of neurons have yet to be elucidated.
In the present study, using mouse embryonic hippocampal neurons, we first show that the activation of canonical TGF-β superfamily signaling impairs neuronal morphogenesis. Smads bind to the Crmp2 promoter and repress it upon TGF-β signal activation in neurons. We further found that TGIF mediates this Smad-dependent suppression of Crmp2 expression. Similar to the case of mouse neurons, we also show that TGF-β signaling negatively regulates the morphological development of neurons established from human fibroblast-derived induced pluripotent stem cells (hiPSCs), suggesting an evolutionarily conserved function for TGF-β signaling between mouse and human. Notably, the expression of TGFβR1, Smad4, or TGIF, which have mutations found in patients with neurodevelopmental disorders, impaired morphological maturation of both mouse and human neurons.
Materials and Methods
Animals.
All aspects of animal care and treatment were performed according to the guidelines of the Experimental Animal Care Committee of Kyushu University (Fukuoka, Japan). Timed-pregnant ICR mice (Japan SLC) were used in this study.
Cell culture.
Hippocampal neurons were isolated from embryonic day 17 (E17) mouse embryos (male and female) according to a previously described protocol (Irie et al., 2016). In brief, hippocampal neurons were dissociated with papain at 37°C for 20 min and triturated by gentle pipetting. Dissociated cells were plated on poly-l-lysine (catalog #P2636, Sigma-Aldrich)-coated culture dishes with α-MEM (catalog #12571-063, Invitrogen) containing 5% fetal bovine serum (FBS) and 0.6% glucose. When neurons had adhered (after 3–4 h), the medium was replaced with maintenance medium (Neurobasal medium; catalog #21103-049, Invitrogen) supplemented with 20 μl/ml B-27 (catalog #17504-44, Invitrogen) and 0.5 mm GlutaMax (catalog #35050-061, Invitrogen). Neurons were treated with 50 ng/ml TGF-β1 (catalog #240-B, R&D Systems), 50 ng/ml BMP2 (catalog #355-BM, R&D Systems), 50 ng/ml BMP4 (catalog #314-BP, R&D Systems), 50 ng/ml BMP7 (catalog #354-BP, R&D Systems), 5 ng/ml SB431542 (catalog #13031, Cayman Chemical), and 50 ng/ml Noggin (catalog #6057-NG, R&D Systems). Half-volume medium changes were performed every 3 d. Cytosine β-d-arabinofuranoside hydrochloride (catalog #C1768, Sigma-Aldrich) was added 1 d after plating to eliminate proliferating undifferentiated and glial cells.
Neural stem/precursor cells (NS/PCs) were isolated from E14 mouse embryo (male and female) telencephalons. The telencephalons were triturated with Hank's balanced salt solution (catalog #H2387, Sigma-Aldrich) by gentle pipetting. Dissociated cells were cultured for 4 d before being used in an experiment in N2 medium [DMEM/F12 (catalog #124000-24, Invitrogen), 25 μg/ml insulin (catalog #097-06474, Wako), 100 μg/ml apo-transferrin (catalog #34401-55, Nacalai Tesque), 16 μg/ml putrescine dihydrochloride (catalog #P5780, Sigma-Aldrich), 30 μm sodium selenite (catalog #S5261, Sigma-Aldrich), 20 μm progesterone (P0130, Sigma-Aldrich)], supplemented with 10 ng/ml bFGF (catalog #100-18B, PeproTech), on culture dishes coated with poly-l-ornithine (catalog #P3655, Sigma-Aldrich) and fibronectin (catalog #F4759, Sigma-Aldrich).
Female human NSPCs (hNS/PCs; a gift from Dr. H. Okano, Keio University, Tokyo, Japan; Andoh-Noda et al., 2015) were plated onto poly-l-ornithine- and laminin-coated (Corning) plates in maintenance medium (DMEM/F12, 1 μl/ml B27, 10 ng/ml bFGF and 10 ng/ml EGF; PeproTech). The cells were passaged at a ratio of 1:3 every third or fourth day using TrypLE (catalog #12563011, Invitrogen). To induce neuronal differentiation, cells were plated onto Matrigel (Corning)-coated dishes in maintenance medium lacking both EGF and bFGF. Half of the medium was changed every 2 d. The study protocol was reviewed and approved by the Research Ethics Committees of Kyushu University (Approval No. 27–59) and Keio University (Approval No. 20080016).
Constructs.
Lentivirus vectors used to express short-hairpin RNAs (shRNAs; pLLX), and FLAG-tagged TGFβR1, TGFβR2, TGFβR1-D400G, Smad1, DN-Smad1, Smad2, Smad4, hSmad4, hSmad4-I500T, CRMP2, TGIF, and TGIF-S162F (pLEMPRA) were provided by Dr. Z. Zhou (University of Pennsylvania School of Medicine) and Dr. M.E. (Harvard Medical School) Greenberg. pLLX and pLEMPRA are dual-promoter lentivirus vectors constructed by inserting the U6 promoter-driven shRNA cassette 5′ into the ubiquitin-C promoter in the FUIGW plasmid (Lois et al., 2002; Zhou et al., 2006). Empty vector was used as a negative control in knock-down and overexpression experiments. shRNAs against Smad1, Smad4, Crmp2, and Tgif were designed to target mouse genes with the following sequences: shSmad1, 5′-ACAGCCGAGTAACTGCGTCACCATT-3′; shSmad4-1, 5′-GGATGAGTACGTTCACGAC-3′; shSmad4-2, 5′-GTAATCGCGCATCAACGGA-3′; shCRMP2-1, 5′-ACTCCTTCCTCGTGTACAT-3′; shCRMP2-2, 5′-GCCTATTGGCTGCCTTTGA-3′; shTGIF-1, 5′-ATCTGGACCAAGTACGAAT-3′; and shTGIF-2, 5′-AGACACACCTGTCCACACT-3′.
Lentivirus production.
Lentiviruses were produced as described previously (Tsujimura et al., 2015). Briefly, lentivirus particles were constructed by cotransfecting HEK293T cells with the lentivirus constructs pCMV-VSV-G-RSV-Rev and pCAG-HIVgp using polyethyleneimine (catalog #23966-1, Polysciences). The culture supernatants were collected 48 h after transfection, and virus was introduced into neurons and hNS/PCs by adding these supernatants to the culture media.
Immunocytochemistry.
Cells were fixed at the indicated days in vitro (DIV) with 4% paraformaldehyde in PBS and washed with PBS. They were then permeabilized and blocked with blocking buffer (3% FBS and 0.1% Triton X-100 in PBS) at room temperature (RT) and incubated with primary antibodies diluted in blocking buffer at RT for 3 h. The following primary antibodies were used in this study: rabbit anti-microtubule-associated protein 2 (MAP2; 1:500; catalog #AB5622, Millipore; You et al., 2015); guinea pig anti-MAP2 (1:1000; catalog #188-004, Synaptic Systems; Pribiag and Stellwagen, 2013); mouse anti-Tau1 (1:500; catalog #MAB3420, Millipore; van Beuningen et al., 2015); chick anti-GFP (1:1000; catalog #GFP-1020, Aves Laboratories; Taylor et al., 2010); chick anti-RFP (1:500; catalog #PM005, MBL; Okamoto et al., 2016); and Smad4 (catalog #9515, Cell Signaling Technology; Park et al., 2012). After being washed with PBS, the cells were incubated for 1 h at RT with the following corresponding secondary antibodies: CF488 donkey anti-chick IgG (H+L), highly cross-adsorbed (1:500; Biotium); CF555 donkey anti-mouse IgG (H+L), highly cross-adsorbed (1:500; Biotium); CF555 donkey anti-rabbit IgG (H+L), highly cross-adsorbed (1:500; Biotium); and CF647 donkey anti-guinea pig IgG (H+L), highly cross-adsorbed (1:500; Biotium). Hoechst 33258 (1:500; catalog #04928-92, Nacalai Tesque) was used for nuclear staining. After a final washing with PBS, they were mounted on glass slides with Immu-Mount (Thermo Fisher Scientific), and images were taken using a Leica AF600 fluorescence microscope.
Immunohistochemistry.
Sections were washed with PBS, permeabilized and blocked with blocking buffer at RT, and incubated overnight at 4°C with primary antibody solution. After being washed with PBS, the sections were incubated for 2 h at RT with secondary antibody solution. After being washed with PBS, the sections were mounted on glass slides. Fluorescence images were acquired using a Zeiss LSM 700 confocal microscope.
In utero electroporation.
To evaluate dendritic growth in vivo, in utero electroporation was performed on E14 mice embryos (male and female), as described previously (Irie et al., 2016). Briefly, plasmid DNA [1.0 μg/μl (axon development) or 0.1 μg/μl (dendrite development) in PBS containing 0.1% Fast Green] was injected into the lateral ventricle of the embryonic brain from outside the uterus. Fifty millisecond electric pulses of 45 V were delivered five times at intervals of 950 ms using a model CUY21 Single Cell Electroporator (Nepa Gene). Glass micropipettes were prepared using a P-1000IVF Micropipette Puller (Sutter Instrument). Animals were perfused with 4% paraformaldehyde at postnatal day 1 (P1; axon development) or P10 (dendrite development).
Morphological analysis.
Morphological assay was performed as previously described (Irie et al., 2016). To analyze axon development in vitro, hippocampal neurons were subjected to immunocytochemistry using anti-Tau-1 and anti-GFP antibody at 3DIV. Neurites with a strong Tau-1 signal at their proximal end were counted as axons. Axon length and branch number were measured using ImageJ. For analysis of axon development in vivo, brain sections were subjected to immunohistochemistry with anti-GFP and anti-RFP antibodies. Axon lengths were measured by taking the edge of the GFP and tdTomato colabeled region as the starting point using ImageJ.
For analysis of dendrite development in vitro, hippocampal neurons were subjected to immunostaining with antibodies against MAP2 and GFP at 6DIV. Dendrites were defined as MAP2-positive neurites. Sholl analysis and quantification of dendritic length and branch numbers were performed using ImageJ software. For Sholl analysis, concentric circles having 10 μm increments in radius were defined from the center of the cell body. The number of MAP2-positive dendrites crossing each circle was counted. For analysis of dendrite development in vivo, brain sections were subjected to immunohistochemistry with anti-GFP antibody. Total dendritic length and branch numbers of GFP-positive neurons in layer 4 were measured using ImageJ. For Sholl analysis, concentric circles having 10 μm increments in radius were defined from the center of cell body. The number of GFP-positive dendrites crossing each circle was counted.
qRT-PCR analysis.
Total RNAs were isolated with TRIzol (catalog #15596-018, Invitrogen) and subjected to reverse transcription with the SuperScript VILO cDNA Synthesis Kit (catalog #11754250, Invitrogen) according to the manufacturer instructions. qRT-PCR was performed using a KAPA SYBR Fast qPCR Kit (catalog #KK4602, Kapa Biosystems) with ROX as the reference dye (Thermo Fisher Scientific) with the StepOne Real-Time PCR System (Applied Biosystems). Expression levels of each gene were normalized to GAPDH and calculated relative to the control. Primers were as follows: CRMP2, 5′-TATTCCACGCATCACGAGCGA-3′ (forward), 5′-GGTCTTCACCCCTCCTGGTA-3′ (reverse); Smad4, 5′-GGCCGTGGCAGGGAACA-3′ (forward), 5′-CTGCAGAGCTCGGTGAAGGTGAAT-3′ (reverse); CRMP1, 5′-ACACGGCCAGTGATGTGAG-3′ (forward), 5′-AGGAGCCGGTCACTCTGG-3′ (reverse); CRMP3, 5′-TCACACTCGGACTTCCTAAGC-3′ (forward), 5′-CTTTGATGAGAAGGCGGTCG-3′ (reverse); TGIF, 5′-GATTCTGCGAGACTGGCTGT-3′ (forward), 5′-CAGTTACAGACCTGTAGTGTGG-3′ (reverse); and GAPDH, 5′-ACCACAGTCCATGCCATCAC-3′ (forward), 5′-TCCACCACCCTGTTGCTGTA-3′ (reverse).
Western blot analysis.
Whole-cell extracts were separated by SDS-PAGE and transferred onto PVDF membranes (Millipore). After completion of the transfer, the membrane was cut into separate proteins according to molecular mass, and then probed with antibodies against CRMP2 (60–80 kDa; catalog #9393, Cell Signaling Technology), Smad1 (58–60 kDa; catalog #9743, Cell Signaling Technology), Smad2 (60 kDa; catalog #5339, Cell Signaling Technology), phosphorylated (p)-Smad1/5 (60 kDa; catalog #9516, Cell Signaling Technology), and p-Smad2 (60 kDa; catalog #3108, Cell Signaling Technology), with GAPDH (37 kDa; catalog #MAB374, Millipore) as an internal control. Immunoreactive bands were detected by enhanced chemiluminescence using ECL Prime Western Blotting Detection Reagent (GE Healthcare). After the first detection, the membrane was stripped, if necessary, using stripping solution (catalog #05364-55, Nacalai Tesque), and then incubated with other primary antibodies.
Chromatin immunoprecipitation assay.
A chromatin immunoprecipitation (ChIP) assay was performed as previously described (Noguchi et al., 2016). Cells were cross-linked in medium containing 1% formaldehyde for 10 min, neutralized with 1.5 m glycine for 10 min, and then washed with cold PBS. The cells were sonicated using a Sonifer 450 (Branson) in SDS-lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.0) until the DNA fragments were 200–500 bp in length. Immunoprecipitation was performed at 4°C overnight with 2 μg of an antibody against p-Smad1/5 (catalog #9516, Cell Signaling Technology) or p-Smad2 (catalog #3108, Cell Signaling Technology), or histone H3-Lys27 acetylation (H3K27Ac; catalog #CMA309, Millipore). Immunocomplexes were then incubated with 20 μl of anti-mouse or anti-rabbit IgG Dynabead magnetic beads for at least 6 h. The beads were washed successively with the following four buffers: low-salt buffer (0.1% SDS, 1% Triton X-100, 0.1% NaDOC, 1 mm EDTA, 50 mm Tris-HCl, pH 8.1, and 150 mm NaCl); high-salt buffer (0.1% SDS, 1% Triton X-100, 0.1% NaDOC, 1 mm EDTA, and 50 mm Tris-HCl, pH 8.1, 500 mm NaCl); and LiCl buffer (0.25 m LiCl, 0.5% NP-40, 0.5% NaDOC, 1 mm EDTA, and 10 mm Tris-HCl, pH 8.0). They were then washed twice with TE buffer (10 mm Tris-HCl and 1 mm EDTA, pH 8.0). The cross-linking in immunocomplexes and input samples was reversed with direct elution buffer (0.5% SDS, 5 mm EDTA, and 10 mm Tris-HCl, pH 8.0, 300 mm NaCl) at 65°C overnight. The DNA was further treated with RNase at 37°C for 30 min and then incubated with proteinase K (Nacalai Tesque) at 65°C for 1 h. The DNA was purified by phenol-chloroform extraction followed by ethanol precipitation. The DNA pellet was dissolved in 20 μl of H2O and used as a template for PCR or quantitative PCR. Primers were as follows: p-Smad1/5 and p-Smad2, primerI: 5′-CTCCATTGTGGCCTGCATTG-3′ (forward), 5′-GCATATCCCACGATTCTGACCA-3′ (reverse); p-Smad1/5 and p-Smad2, primerII: 5′-ACCTGAAGATTTCCGCAGTCC-3′ (forward), 5′-CATGGGTCACAATCACAGGTTC-3′ (reverse); and H3K27Ac: 5′-TACAGCGCCTACCTAATGGC-3′ (forward), 5′-TGCCTCATAACCCTCCCTCA-3′ (reverse).
Luciferase reporter assay.
Hippocampal neurons treated with TGF-β1 and BMP2 were transfected with a reporter construct harboring the Crmp2 promoter, using PEI (Sigma-Aldrich). After transfection, the cells were incubated for 3 d and were lysed with Reporter Lysis Buffer. Luciferase activity of the lysates was measured with the Dual-Glo Luciferase Assay System (Promega) according to the manufacturer protocol. Firefly luciferase activity was determined in three independent transfections and normalized by comparison with the Renilla luciferase activity of the internal control.
Experimental design and statistical analysis.
Statistical analyses were performed with Student's t test (for two-group comparisons) and one-way ANOVA, followed by Tukey's multiple-comparison tests, as appropriate (for multiple groups comparison), using Prism 7 (GraphPad Software). All data are presented as the mean ± SEM. p Values <0.05 were considered significant. The sample size was similar to those reported in previous publications (Tsujimura et al., 2015; Irie et al., 2016; Noguchi et al., 2016) and is indicated in the relevant figure legends. No statistical method was used to predetermine sample size.
Results
TGF-β family cytokines impair dendritic and axonal growth/branching of mouse hippocampal neurons
Canonical TGF-β signaling controls the development of diverse organs including brain. However, our understanding of the roles of this signaling pathway in the morphological development of neurons is sparse. To explore such roles, we exposed primary-cultured mouse hippocampal neurons to TGF-β family cytokines and assessed axonal and dendritic morphology. We found that total dendritic length and dendritic branch numbers were decreased in neurons treated with TGF-β1, BMP2, and BMP4 compared with control in a dose-dependent manner [Figs. 1A–C (Fig. 1B: TGF-β1, p = 0.0199; BMP2, p = 0.0073; BMP4, p = 0.0135; ANOVA; Fig. 1C: TGF-β1, p = 0.0055; BMP2, p = 0.0004; BMP4, p = 0.0043; ANOVA), 2A–D (Fig. 2A: TGF-β1 20, p = 0.0356; TGF-β1 50, p = 0.0063; TGF-β1 125, p = 0.0019; ANOVA; Fig. 2B: TGF-β1 20, p = 0.007; TGF-β1 50, p = 0.0013; TGF-β1 125, p = 0.0001; ANOVA; Fig. 2C: BMP2 20, p = 0.0198; BMP2 50, p = 0.0006; BMP2 125, p = 0.0002; ANOVA; Fig. 2D: BMP2 20, p = 0.0322; BMP2 50, p = 0.0013; BMP2 125, p = 0.0005; ANOVA)]. We also found that TGF-β1 and BMP2 regulate dendrite development additively rather than synergistically [Fig. 2E–H (Fig. 2E: TGF-β1 20, p = 0.0262; BMP2 20, p = 0.0476; ANOVA; Fig. 2F: TGF-β1 20, p = 0.0055; BMP2 20, p = 0.0107; ANOVA; Fig. 2G: TGF-β1 50, p > 0.9999; BMP2 50, p = 0.994; ANOVA; Fig. 2H: TGF-β1 50, p = 0.9853; BMP2 50, p = 0.9923; ANOVA)]. We further observed decreased dendritic complexity in TGF-β1-, BMP2-, and BMP4-treated neurons, as assessed by Sholl analysis (Fig. 1D). Consistent with previous studies suggesting that BMP7 promotes dendritic growth through a noncanonical pathway (Lee-Hoeflich et al., 2004), dendritic branch numbers increased after BMP7 treatment, although dendritic length was not affected [Fig. 1B,C (Fig. 1B: BMP7, p = 0.682; ANOVA; Fig. 1C; BMP7, p = 0.0015; ANOVA]. On the other hand, treatment with the TGFβR1 inhibitor SB431542 and a BMP antagonist Noggin increased dendritic length, numbers, and complexity [Fig. 1E–H (Fig. 1F: SB431542, p = 0.0002; Noggin, p = 0.0021; ANOVA; Fig. 1G: SB431542, p = 0.0014; Noggin, p = 0.0068; ANOVA)]. In addition to dendritogenesis, we found that total axonal length and branch numbers were reduced by treatment with TGF-β1, BMP2, and BMP4 [Fig. 1I–K (Fig. 1J: TGF-β1, p = 0.0003; BMP2, p = 0.0003; BMP4; p = 0.0002; ANOVA; Fig. 1K: TGF-β1, p = 0.0079; BMP2, p = 0.0037; BMP4, p = 0.0118; ANOVA)].
Figure 1.
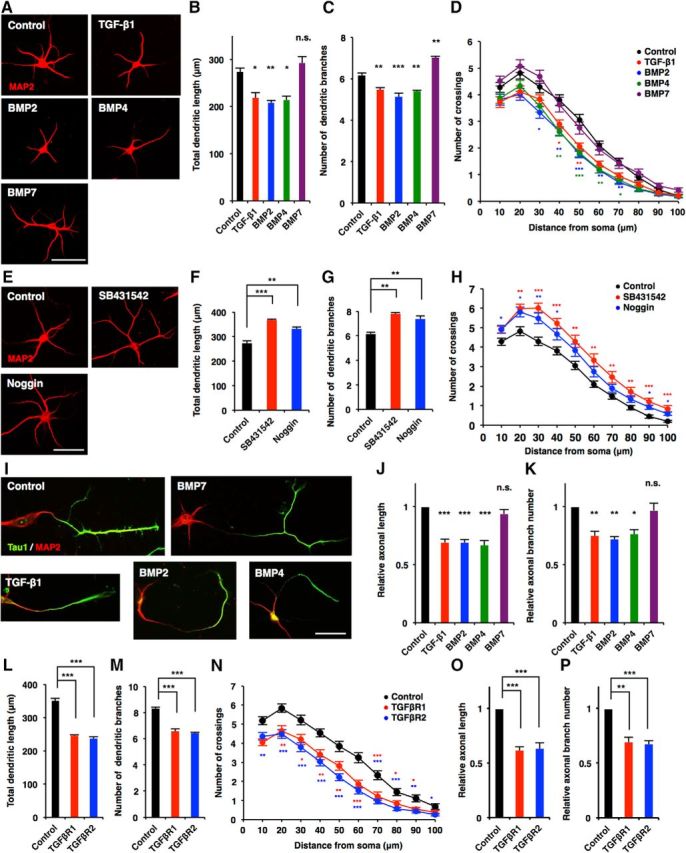
TGF-β family signals negatively regulate neuronal development in hippocampal neurons. A, Representative images of MAP2 (red) staining of 6DIV hippocampal neurons treated with BSA (control), TGF-β1, BMP2, BMP4, and BMP7. Scale bar, 50 μm. B, C, Quantification of total dendritic length (B) and branch numbers (C) of A. D, Quantification of dendrite complexity by Sholl analysis of 3DIV hippocampal neurons treated with BSA, TGF-β1, BMP2, BMP4, and BMP7. E, Representative images of MAP2 (red) staining of 6DIV hippocampal neurons treated with BSA (control), SB431542, and Noggin. Scale bar, 50 μm. F, G, Quantification of total dendritic length (F) and branch numbers (G) in E. H, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons treated with BSA, SB431542, and Noggin. I, Representative images of Tau1 (green) and MAP2 (red) staining of 3DIV hippocampal neurons treated with BSA, TGF-β1, BMP2, BMP4, and BMP7. Scale bar, 50 μm. J, K, Quantification of total axon length (J) and axon branch numbers (K) of 3DIV hippocampal neurons treated with BSA, TGF-β1, BMP2, BMP4, and BMP7 immunostained with antibodies against Tau1. Total length and branch numbers of Tau1-positive axons were measured. L, M, Quantification of total dendritic length (L) and branch numbers (M) of 6DIV hippocampal neurons infected with lentiviruses expressing GFP alone (control) or GFP together with TGFβR1 or TGFβR2. N, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing TGFβR1 or TGFβR2. O, P, Quantification of total axon length (O) and axon branch numbers (P) of 3DIV hippocampal neurons infected with lentiviruses expressing TGFβR1 or TGFβR2. Data are presented as the mean ± SEM. p > 0.05 (n.s.); *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
Figure 2.

TGF-β1 and BMP2 additively suppress neuronal development in hippocampal neurons in a dose-dependent manner. A–D, Hippocampal neurons were treated with 20, 50, or 125 ng/ml TGF-β1 (A, B) or BMP2 (C, D). Quantification of total dendritic length (A, C) and branch numbers (B, D). E, F, Hippocampal neurons were treated with 20 ng/ml TGF-β1 or BMP2 or with 20 ng/ml TGF-β1 and BMP2. G, H, Hippocampal neurons were treated with 50 ng/ml TGF-β1 or BMP2 or with 50 ng/ml TGF-β1 and BMP2. Quantification of total dendritic length (E, G) and branch numbers (F, H). Data are presented as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
Since TGF-β signaling is mediated through the activation of the TGFβR complex, which is composed of type I and type II receptors, we next investigated the effect of TGFβR forced expression on the morphological maturation of neurons. Overexpression of TGFβR1 (type I) and TGFβR2 (type II) significantly suppressed dendritic and axonal morphogenesis [Fig. 1L–P (Fig. 1L: TGFβR1, p = 0.0003; TGFβR2, p = 0.0002; ANOVA; Fig. 1M: TGFβR1, p < 0.0001; TGFβR2, p < 0.0001; ANOVA; Fig. 1O: TGFβR1, p = 0.0008; TGFβR2, p = 0.001; ANOVA; Fig. 1P: TGFβR1, p = 0.0013; TGFβR2, p = 0.0001; ANOVA)]. Together, these results suggest that these TGF-β family members, except for BMP7, inhibit morphological development of cultured hippocampal neurons.
Downstream transcription factor Smads are responsible for TGF-β family-induced suppression of dendritic and axonal growth/branching in cultured hippocampal neurons
Canonical TGF-β family signaling is mediated by downstream signal transducer Smad transcription factors. Pathway-restricted R-Smads are activated by particular TGF-β members (e.g., TGF-β1 activates Smad2 and Smad3), while BMPs activate Smad1, Smad5, and Smad8. To confirm that Smads are indeed activated in neurons treated with TGF-β family cytokines, we immunostained hippocampal neurons treated with BSA, TGF-β1, BMP2, BMP4, or BMP7 using antibodies against p-Smads (p-Smad2 for TGF-β1 and p-Smad1/5 for BMPs). We detected a p-Smad2 signal in neurons treated with TGF-β1 for 12 h (Fig. 3A). Although we also clearly detected a p-Smad1/5 signal in BMP2- and BMP4-treated neurons, the signal was faint in BMP7-treated neurons (Fig. 3B). When we analyzed Smad1/5 activation by Western blot, the BMP2-activated Smad1/5 signal was dramatically increased by 12 h, whereas little or no activation was observed in BMP7-treated neurons (Fig. 3C). It has been reported that conditional knockout of TGFβR1 and overexpression of a constitutively active form of TGFβR1 (caTGFβR1) impaired and stimulated, respectively, dendrite growth in the adult hippocampus in vivo (He et al., 2014), which is opposite to what we observed in the overexpression of TGFβR1. We therefore generated lentiviral vectors coexpressing GFP and caTGFβR1 (Wieser et al., 1995) and infected hippocampal neurons with the viruses. We first confirmed that canonical TGF-β signaling was activated in the caTGFβR1-expressing virus-infected neurons by assessing Smad2 phosphorylation, and detected a p-Smad2 signal in the nucleus of the neurons (Fig. 3D). Overexpression of caTGFβR1 in hippocampal neurons also decreased total dendritic length and branch numbers compared with control [Fig. 3F–H (Fig. 3G: caTGFβR1, p = 0.0056; ANOVA; Fig. 3H: caTGFβR1, p = 0.0079; ANOVA)]. Furthermore, overexpression of the constitutively active form of BMP receptor type IA (caBMPR1A; Akiyama et al., 1997) in hippocampal neurons induced phosphorylation of Smad1/5 and diminished total dendritic length and branch numbers compared with control [Fig. 3E–H (Fig. 3G: caBMPR1A, p = 0.0026; ANOVA; Fig. 3H: caBMPR1A, p = 0.0052; ANOVA)].
Figure 3.

Canonical TGF-β and BMP signaling pathways suppress dendrite development in hippocampal neurons. A, Representative images of p-Smad2 (red) and MAP2 (cyan) staining of hippocampal neurons treated with BSA and TGF-β1. Scale bar, 20 μm. B, Representative images of p-Smad1/5 (red) and MAP2 (cyan) staining of hippocampal neurons treated with BSA, BMP2, BMP4, and BMP7. Scale bar, 20 μm. C, Western blot analysis of p-Smad1/5 expression in neurons treated with BMP2 and BMP7 for 4, 8, and 12 h. D, E, Representative images of GFP (green) and p-Smad2 (cyan; D) or p-Smad1/5 (cyan; E) staining of 6DIV hippocampal neurons infected with lentiviruses expressing caTGFβR1 or caBMPR1A. Scale bar, 30 μm. F, Representative images of GFP (green) and MAP2 (red) staining of 6DIV hippocampal neurons infected with lentiviruses expressing caTGFβR1 or caBMPR1A. Scale bar, 50 μm. G, H, Quantification of total dendritic length (G) and branch numbers (H) in F. Data are presented as the mean ± SEM. **p < 0.01 by one-way ANOVA andTukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
We next investigated the expression pattern of Smad1 and Smad2 during the development of cultured hippocampal neurons, and found that Smad1 and Smad2 expression gradually decreased over time (Fig. 4A,B). We then examined the functional effects of Smads on neuronal morphological development by gain-of-function and loss-of-function experiments. Overexpression of Smad1, Smad2, and Smad4 in hippocampal neurons suppressed total dendritic length, branch numbers, and complexity compared with control [Fig. 4C–F (Fig. 4D: Smad1, p < 0.0001; Smad2, p < 0.0001; Smad4, p < 0.0001; ANOVA; Fig. 4E: Smad1, p = 0.0024; Smad2, p = 0.0348; Smad4, p = 0.0257; ANOVA)]. For the loss-of-function experiment, we generated lentiviral vectors coexpressing GFP and a specific shRNA against Smad1 (shSmad1) or Smad4 (shSmad4-1 and shSmad4-2; Fig. 4I,J). In contrast to the results obtained with overexpression, the knockdown of Smad1 and Smad4 promoted dendrite development [Fig. 4K–M,P–R (Fig. 4K: shSmad4-1, p = 0.0041; shSmad4-2, p = 0.0056; ANOVA; Fig. 4L: shSmad4-1, p = 0.0035; shSmad4-2, p = 0.0031; ANOVA; Fig. 4P: shSmad1, p = 0.0018; ANOVA; Fig. 4Q: shSmad1, p = 0.009; ANOVA)]. Likewise, the expression of a dominant-negative (DN) form of Smad1 (Yoshiura et al., 2007) promoted dendrite development (Fig. 4P–R (Fig. 4P: DN-Smad1, p = 0.0002; ANOVA; Fig. 4Q: DN-Smad1, p = 0.0026; ANOVA)]. Furthermore, similar effects of Smads were observed regarding axonal growth [Fig. 4G,H,N,O,S,T (Fig. 4G: Smad1, p < 0.0001; Smad2, p < 0.0001; Smad4, p < 0.0001; ANOVA; Fig. 4H: Smad1, p < 0.0001; Smad2, p < 0.0001; Smad4, p < 0.0001; ANOVA; Fig. 4N: shSmad4-1, p = 0.0089; shSmad4-2, p = 0.0096; ANOVA; Fig. 4O: shSmad4-1, p = 0.0224; shSmad4-2, p = 0.0074; ANOVA; Fig. 4S: shSmad1, p = 0.0002; DN-Smad1, p = 0.0003; ANOVA; Fig. 4T: shSmad1, p = 0.0002; DN-Smad1, p = 0.0006; ANOVA)].
Figure 4.
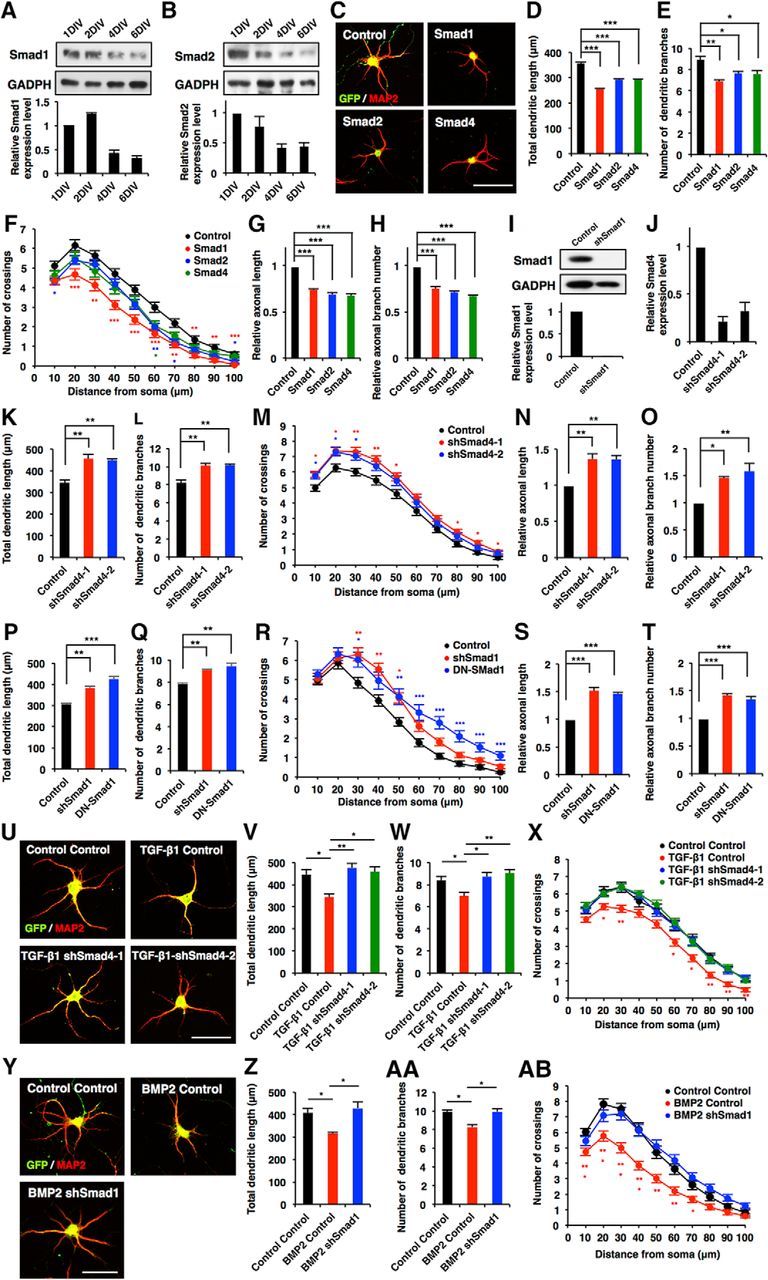
Canonical Smad-dependent signaling pathways suppress morphological development in hippocampal neurons. A, B, Western blot analysis of Smad1 and Smad2 expression in developing cultured hippocampal neurons. C, Representative images of GFP (green) and MAP2 (red) staining of 6DIV hippocampal neurons infected with lentiviruses expressing Smad1, Smad2, or Smad4. Scale bar, 50 μm. D, E, Quantification of total dendritic length (D) and branch numbers (E) of 6DIV hippocampal neurons infected with lentiviruses expressing Smad1, Smad2, or Smad4. F, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing Smad1, Smad2, or Smad4. G, H, Quantification of total axon length (G) and axon branch numbers (H) of 3DIV hippocampal neurons infected with lentiviruses expressing Smad1, Smad2, or Smad4. I, Hippocampal neurons were infected with lentiviruses expressing shRNA against Smad1. The expression of Smad1 protein was measured by Western blotting using an anti-Smad1 antibody. J, Validation of the effect of Smad4 shRNAs. qRT-PCR analysis of Smad4 levels in cultured hippocampal neurons infected with lentiviruses expressing shSmad4-1 or shSmad4-2. K, L, Quantification of total dendritic length (K) and branch numbers (L) of 6DIV hippocampal neurons infected with lentiviruses expressing shSmad4-1 and shSmad4-2. M, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing shSmad4-1 and shSmad4-2. N, O, Quantification of total axon length (N) and axon branch numbers (O) of 3DIV hippocampal neurons infected with lentiviruses expressing shSmad4-1 and shSmad4-2. P, Q, Hippocampal neurons were infected with lentiviruses expressing shSmad1 and DN-Smad1: quantification of total dendritic length (P) and branch numbers (Q). R, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing shSmad1 and DN-Smad1. S, T, Quantification of total axon length (S) and axon branch number (T) of 3DIV hippocampal neurons infected with lentiviruses expressing shSmad1 and DN-Smad1. U, Representative images of GFP and MAP2 staining of 6DIV hippocampal neurons treated with TGF-β1, and infected with lentiviruses expressing control shRNA, shSmad4-1 or shSmad4-2. Scale bar, 50 μm. V, W, Quantification of total dendritic length (V) and branch numbers (W) of 6DIV hippocampal neurons treated with TGF-β1, and infected with lentiviruses expressing control or shSmad4-1 and shSmad4-2. X, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons treated with TGF-β1, and infected with lentiviruses expressing control or shSmad4-1 and shSmad4-2. Y, Representative images of GFP (green) and MAP2 (red) staining of 6DIV hippocampal neurons treated with BMP2, and infected with lentiviruses expressing shSmad1. Scale bar, 50 μm. Z, AA, Quantification of total dendritic length (Z) and branch numbers (AA) of 6DIV hippocampal neurons treated with BMP2, and infected with lentiviruses expressing control or shSmad1. AB, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons treated with BMP2, and infected with lentiviruses expressing control or shSmad1. Data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
To determine whether Smads are indeed acting downstream of TGF-β signaling in neuronal morphogenesis, we investigated the consequence of Smad4 loss of function in TGF-β1-treated neurons. Knocking down Smad4 abolished the effect of TGF-β1 on dendritic development [Fig. 4U–X (Fig. 4V: TGF-β1 Control, p = 0.0193; TGF-β1 shSmad4-1, p = 0.006; TGF-β1 shSmad4-2, p = 0.0127; ANOVA; Fig. 4W: TGF-β1 Control, p = 0.0443; TGF-β1 shSmad4-1, p = 0.0217; TGF-β1 shSmad4-2, p = 0.0079; ANOVA)]. Similarly, knockdown of Smad1 cancelled the effect of BMP2 on dendritic development [Fig. 4Y,AB (Fig. 4Z: BMP2 Control, p = 0.0439; BMP2 shSmad1, p = 0.0204; ANOVA; Fig. 4AA: BMP2 Control, p = 0.01; BMP2 shSmad1, p = 0.0109; ANOVA)]. Together, these observations suggest that Smad-dependent canonical TGF-β signaling suppresses the morphological maturation of hippocampal neurons.
Canonical TGF-β signaling represses CRMP2 expression
Previous studies have shown that CRMP2 regulates various phases of neuronal development including polarization, axonal formation (Inagaki et al., 2001; Nishimura et al., 2003), and dendritic growth (Zhang et al., 2016). However, the mechanisms that regulate Crmp2 expression for neuronal maturation remain poorly understood. Since BMP signaling has been reported to downregulate CRMP2 expression in ES cells (Fei et al., 2010) and neuronal progenitor cells in the developing brain (Sun et al., 2010b), we hypothesized that canonical TGF-β signaling might also control the expression of CRMP2 in differentiated neurons. To test this idea, we investigated the expression pattern of CRMP2 in maturating hippocampal neurons. Opposite to the expression patterns of Smads, CRMP2 expression gradually increased as the neurons matured (Fig. 5A,B). It has been reported that CRMP2 has two isoforms (64 and 75 kDa; Yuasa-Kawada et al., 2003), and an immunoblot using anti-CRMP2 antibody detected two bands corresponding to these isoforms when CRMP2 expression was high (Fig. 5A, lanes 4DIV and 6DIV). We also observed a small but significant reduction of Crmp2 expression level upon TGF-β1 and BMP2 stimulation in the hippocampal neurons, whereas mRNA levels of two other CRMP family members, Crmp1 and Crmp3, were unchanged [Fig. 5C–E (Fig. 5C: TGF-β1, p = 0.0194; BMP2, p = 0.0248; ANOVA; Fig. 5D: TGF-β1, p = 0.7491; BMP2, p = 0.9887; ANOVA; Fig. 5E: TGF-β1, p = 0.9840; BMP2, p = 0.9893; ANOVA)]. TGF-β1 and BMP2 treatment also decreased the expression level of CRMP2 protein [Fig. 5F–I (Fig. 5G: p < 0.0001, unpaired t test; Fig. 5I: p = 0.0032; unpaired t test]. To examine whether phosphorylated Smads bind to the Crmp2 promoter region, we made two types of qPCR primer that recognize either the Crmp2 promoter (Primer I) or the 3′UTR (Primer II) region and performed a ChIP assay (Fig. 5J). This revealed that Smad2 and Smad1/5 binding to the promoter, but not to the 3′UTR region of Crmp2, increased in response to TGF-β1 and BMP2 stimulation, respectively [Fig. 5K–N (Fig. 5K: p = 0.0138, unpaired t test; Fig. 5L: p = 0.3092, unpaired t test; Fig. 5M: p = 0.016, unpaired t test; Fig. 5N: p = 0.4131, unpaired t test)]. Moreover, we performed a luciferase assay using a reporter construct harboring the Crmp2 promoter, and found that promoter activity was suppressed by stimulation with TGF-β1 and BMP2 [Fig. 5O,P (Fig. 5P: TGF-β1, p = 0.0255; BMP2, p = 0.0181; ANOVA)]. Together, these results suggest that canonical TGF-β signaling represses CRMP2 expression at the transcription level in neurons.
Figure 5.

Canonical TGF-β/BMP signaling represses expression of CRMP2 at the transcription level in hippocampal neurons. A, Western blot analysis of CRMP2 expression in developing cultured hippocampal neurons at 1DIV, 2DIV, 4DIV, and 6DIV. B, Quantification of CRMP2 expression levels in A. C–E, qRT-PCR analysis of Crmp2, Crmp1, and Crmp3 levels in cultured hippocampal neurons. The cells were treated with TGF-β1 or BMP2 for 12 h. F, H, Western blot analysis of CRMP2 expression in cultured hippocampal neurons. The cells were treated with TGF-β1 or BMP2 for 12 h. G, I, Quantification of CRMP2 expression levels in F and H. J, Diagram of the Crmp2 promoter and 3′UTR region. PrimerI and primerII recognize the Crmp2 promoter and 3′UTR region, respectively. K–N, ChIP-qPCR analysis of p-Smad2 (K, L) or p-Smad1/5 (M, N) binding to the Crmp2 promoter or 3′UTR in cultured hippocampal neurons. The cells were treated with TGF-β1 or BMP2 for 1 h. O, Diagram of the luciferase construct containing the Crmp2 promoter region and TSS. P, Quantification of luciferase activity of hippocampal neurons treated with BSA, TGF-β1, and BMP2, and transfected with the luciferase construct. Data are presented as the mean ± SEM. G, K–N, *p < 0.05, **p < 0.01, ***p < 0.001 by Student's t test. p > 0.05 (n.s.). C–E, P, *p < 0.05 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
In light of the above finding, we next performed gain-of-function and loss-of-function experiments for CRMP2 and evaluated dendritic development. We first validated the knockdown efficiency of shRNAs against Crmp2 (shCRMP2-1 and shCRMP2-2) for the loss-of-function experiments (Fig. 6A,B). Overexpression of CRMP2 in hippocampal neurons increased total dendritic length, branch numbers, and complexity compared with control, whereas knockdown of Crmp2 had the opposite effect [Fig. 6C–F (Fig. 6D: CRMP2, p = 0.0007; shCRMP2-1, p = 0.0081; shCRMP2-2, p = 0.0192; ANOVA; Fig. 6E: CRMP2, p = 0.0001; shCRMP2-1, p = 0.0002; shCRMP2-2, p = 0.0006; ANOVA)]. We further confirmed that total axonal length and branch numbers were increased by CRMP2 expression [Fig. 6G,H (Fig. 6G: CRMP2, p < 0.0001; shCRMP2-1, p = 0.0007; shCRMP2-2, p = 0.002; ANOVA; Fig. 6H: CRMP2, p < 0.0001; shCRMP2-1, p = 0.0004; shCRMP2-2, p < 0.0001; ANOVA)]. If the downregulation of CRMP2 is responsible for the TGF-β family-induced suppression of neuronal morphogenesis, forced expression of CRMP2 should abolish the effect of TGF-β family stimulation. To test this prediction, we expressed CRMP2 in TGF-β family-treated neurons and found that CRMP2 expression indeed rescued the decrease in total dendritic length, branch numbers, and complexity induced by TGF-β1 and BMP2 treatment [Fig. 6I–P (Fig. 6J: TGF-β1 Control, p = 0.0013; TGF-β1 CRMP2, p = 0.0016; ANOVA; Fig. 6K: TGF-β1 Control, p = 0.0065; TGF-β1 CRMP2, p = 0.0134; ANOVA; Fig. 6N: BMP2 Control, p = 0.0221; BMP2 CRMP2, p = 0.003; ANOVA; Fig. 6O: BMP2 Control, p = 0.009; BMP2 CRMP2, p = 0.0005; ANOVA)]. We also found that CRMP2 expression abolishes the effects of Smad1 and Smad4 overexpression on dendritic development [Fig. 6Q–X (Fig. 6R: Smad4 Control, p = 0.0455; Smad4 CRMP2, p = 0.0123; ANOVA; Fig. 6S: Smad4 Control, p = 0.0067; Smad4 CRMP2, p = 0.0042; ANOVA; Fig. 6V: Smad1 Control, p = 0.0433; Smad1 CRMP2, p = 0.0394; ANOVA; Fig. 6W: Smad1 Control, p = 0.0411; Smad1 CRMP2, p = 0.0327; ANOVA)]. These findings indicate that the downregulation of CRMP2 accounts for the TGF-β signaling-induced impairment of neuronal morphogenesis.
Figure 6.
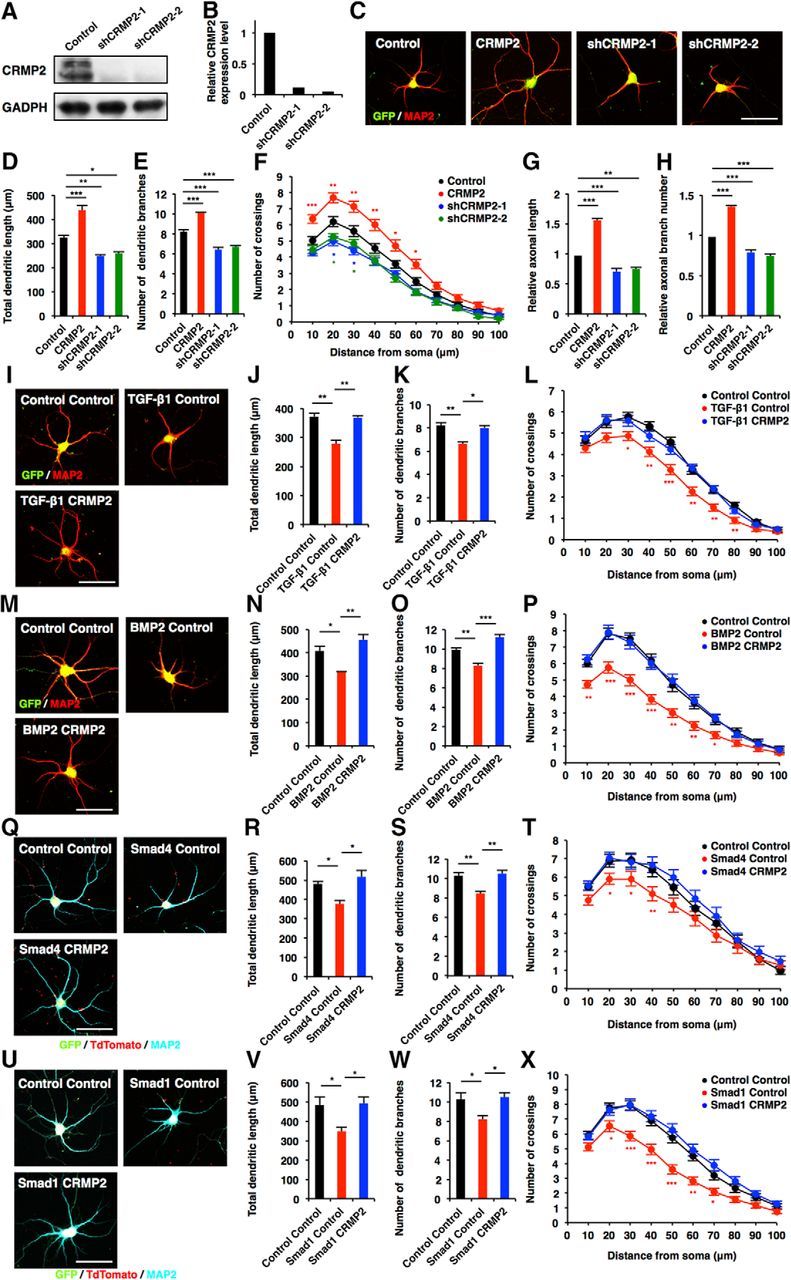
Canonical TGF-β/BMP signaling suppresses morphological development in hippocampal neurons through the downregulation of CRMP2. A, Hippocampal neurons were infected with lentiviruses expressing shRNAs against Crmp2. Expression of CRMP2 protein was measured by Western blotting using an anti-CRMP2 antibody. B, Quantification of CRMP2 protein level in A. C, Representative images of GFP (green) and MAP2 (red) staining of 6DIV hippocampal neurons infected with lentiviruses expressing CRMP2, shCRMP2-1, or shCRMP2-2. Scale bar, 50 μm. D, E, Quantification of total dendritic length (D) and branch numbers (E) of 6DIV hippocampal neurons infected with lentiviruses expressing CRMP2, shCRMP2-1, or shCRMP2-2. F, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing CRMP2, shCRMP2-1, or shCRMP2-2. G, H, Quantification of total axon length (G) and axon branch numbers (H) of 3DIV hippocampal neurons infected with lentiviruses expressing shCRMP2-1 or shCRMP2-2. I, Representative image of GFP and MAP2 staining of 6DIV hippocampal neurons treated with TGF-β1, and infected with lentiviruses expressing CRMP2. Scale bar, 50 μm. J, K, Quantification of total dendritic length (J) and branch numbers (K) of 6DIV hippocampal neurons treated with TGF-β1, and infected with lentiviruses expressing control or CRMP2. L, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons treated with TGF-β1, and infected with lentiviruses expressing control or CRMP2. M, Representative images of GFP and MAP2 staining of 6DIV hippocampal neurons treated with BMP2, and infected with lentiviruses expressing CRMP2. Scale bar, 50 μm. N, O, Quantification of total dendritic length (N) and branch numbers (O) of 6DIV hippocampal neurons treated with BMP2, and infected with lentiviruses expressing CRMP2. P, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons treated with BMP2, and infected with lentiviruses expressing CRMP2. Q, Representative images of GFP, tdTomato (red), and MAP2 (cyan) staining of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad4 and CRMP2. Scale bar, 50 μm. R, S, Quantification of total dendritic length (R) and branch numbers (S) of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad4 and CRMP2. T, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad4 and CRMP2. U, Representative images of GFP, tdTomato, and MAP2 staining of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad1 and CRMP2. Scale bar, 50 μm. V, W, Quantification of total dendritic length (V) and branch numbers (W) of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad1 and CRMP2. X, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad1 and CRMP2. Data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
TGIF is the key molecule for TGF-β signaling in the regulation of dendritic and axonal growth/branching of neurons
TGIF, a homeodomain-containing transcription factor, has been identified as a Smad-interacting corepressor (Wotton et al., 1999). TGIF regulates normal neuronal development (Knepper et al., 2006), and mutations in TGIF are associated with the genetic disease holoprosencephaly (HPE), which is the most common defect of the developing forebrain in humans (Gripp et al., 2000). Therefore, we reasoned that TGIF should be a candidate factor for mediating TGF-β-induced repression of Crmp2 expression. We first examined the expression levels of Tgif in NS/PCs and neurons, and detected Tgif expression in both cell types, although the level was lower in neurons than in NS/PCs (Fig. 7A). In addition, when we expressed TGIF in neurons, Crmp2 expression was decreased (Fig. 7B, p = 0.0048, unpaired t test). It has been reported that TGIF interacts specifically with Smad1 and Smad2 and recruits a complex of corepressors, including C-terminal binding protein (CtBP), mSin3, and histone deacetylases (HDACs; Wotton et al., 1999, 2001). We then examined whether the treatment of neurons with TGF-β1 decreased H3K27ac levels in the Crmp2 promoter, and found that this was the case (Fig. 7C, p = 0.0039, unpaired t test).
Figure 7.
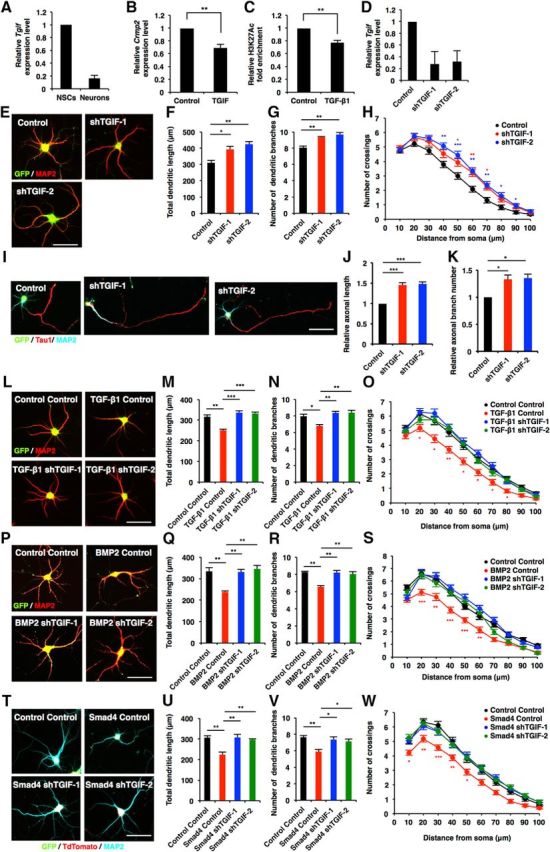
TGIF is key transcriptional regulator for TGF-β/Smad signaling in the regulation of neuronal morphogenesis. A, qRT-PCR analysis of Tgif expression levels in E14 NS/PCs and E17 neurons. B, qRT-PCR analysis of Crmp2 expression levels in cultured hippocampal neurons infected with lentiviruses expressing TGIF. C, ChIP-qPCR analysis of H3K27Ac binding to the Crmp2 promoter in cultured hippocampal neurons. The cells were treated with TGF-β1 for 12 h. D, Validation of TGIF shRNAs. qRT-PCR analysis of Tgif expression levels in cultured hippocampal neurons infected with lentiviruses expressing shRNAs against Tgif. E, Representative images of GFP (green) and MAP2 (red) staining of 6DIV hippocampal neurons infected with lentiviruses expressing shTGIF-1 or shTGIF-2. Scale bar, 50 μm. F, G, Quantification of total dendritic length (F) and branch numbers (G) of 6DIV hippocampal neurons infected with lentiviruses expressing shTGIF-1 or shTGIF-2. H, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing shTGIF-1 or shTGIF-2. I, Representative images of GFP, Tau1 (red), and MAP2 (cyan) staining of 3DIV hippocampal neurons infected with lentiviruses expressing shTGIF-1 or shTGIF-2. Scale bar, 50 μm. J, K, Quantification of total axon length (J) and axon branch numbers (K) of 3DIV hippocampal neurons infected with lentiviruses expressing shTGIF-1 or shTGIF-2. L, P, Representative images of GFP and MAP2 staining of 6DIV hippocampal neurons treated with TGF-β1 or BMP2, and infected with lentiviruses expressing shTGIF-1 or shTGIF-2. Scale bar, 50 μm. M, N, Q, R, Quantification of total dendritic length (M, Q) and branch numbers (N, R) of 6DIV hippocampal neurons treated with TGF-β1 or BMP2, and infected with lentiviruses expressing control, shTGIF-1, or shTGIF-2. O, S, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons treated with TGF-β1 or BMP2, and infected with lentiviruses expressing control or shTGIF-1 or shTGIF-2. T, Representative images of GFP, tdTomato (red), and MAP2 (cyan) staining of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad4 and either shTGIF-1 or shTGIF-2. Scale bar, 50 μm. U, V, Quantification of total dendritic length (U) and branch numbers (V) of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad4 and either shTGIF-1 or shTGIF-2. W, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons coinfected with lentiviruses expressing Smad4 and either shTGIF-1 or shTGIF-2. Data are presented as the mean ± SEM. B, C, **p < 0.01 by Student's t test. F, G, J, K, M, N, Q, R, U, V, *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
To investigate the role of TGIF in neuronal morphogenesis, we performed a loss-of-function experiment using lentiviruses expressing specific shRNAs against Tgif (shTGIF-1 and shTGIF-2; Fig. 7D). Knockdown of Tgif elevated total dendritic length, branch numbers, and the complexity of neurons compared with control [Fig. 7E–H (Fig. 7F: shTGIF-1, p = 0.027; shTGIF-2, p = 0.0064; ANOVA; Fig. 7G: shTGIF-1, p = 0.058; shTGIF-2, p = 0.0018; ANOVA)]. Furthermore, a similar effect of Tgif knockdown was observed on axonal growth [Fig. 7I–K (Fig. 7J: shTGIF-1, p = 0.0008; shTGIF-2, p = 0.0006; ANOVA; Fig. 7K: shTGIF-1, p = 0.0242; shTGIF-2, p = 0.0176; ANOVA)]. To determine whether TGIF acts downstream of TGF-β signaling in neuronal morphogenesis, we examined the effect of TGIF loss-of-function in TGF-β1- and BMP2-treated neurons. The reduction of TGIF expression eliminated the effect of TGF-β1 and BMP2 on dendritic development [Fig. 7L–S (Fig. 7M: TGF-β1 control, p = 0.0022; TGF-β1 shTGIF-1, p = 0.0003; TGF-β1 shTGIF-2, p = 0.0005; ANOVA; Fig. 7N: TGF-β1 control, p = 0.0211; TGF-β1 shTGIF-1, p = 0.0043; TGF-β1 shTGIF-2, p = 0.0036; ANOVA; Fig. 7Q: BMP2 control, p = 0.0065; BMP2 shTGIF-1, p = 0.0077; BMP2 shTGIF-2, p = 0.0032; ANOVA; Fig. 7R: BMP2 control, p = 0.0012; BMP2 shTGIF-1, p = 0.0016; BMP2 shTGIF-2, p = 0.0032; ANOVA)]. Moreover, we found that knocking down Tgif expression abolished the effects of Smad4 overexpression on dendritic development [Fig. 7T–W (Fig. 7U: Smad4 control, p = 0.0056; Smad4 shTGIF-1, p = 0.005; Smad4 shTGIF-2, p = 0.0097; ANOVA; Fig. 7V: Smad4 control, p = 0.0067; Smad4 shTGIF-1, p = 0.0178; Smad4 shTGIF-2, p = 0.0291; ANOVA)]. Thus, these observations suggest that TGIF is a key factor for TGF-β signaling in the regulation of morphological maturation of neurons.
Activation of TGF-β signaling impairs dendritic and axonal elaboration in vivo
To understand the role of TGF-β and BMP signaling under physiological conditions, we next evaluated these signaling activities in P1 hippocampus by immunostaining using anti-p-Smad1/5 or -p-Smad2 antibodies together with those against Sox2 (an NS/PC marker), NeuN (a mature neuron marker), and MAP2 (which is expressed from earlier stages than NeuN during neuronal differentiation). p-Smad1/5 and p-Smad2 signals were observed primarily in Sox2-positive NS/PCs, but were barely detectable in NeuN-positive mature neurons (Fig. 8A,B). However, we could observe p-Smad1/5 and p-Smad2 signals in some fraction of MAP2-positive cells (Fig. 8C,D). These results indicate that activation of Smads is gradually reduced during the course of neuronal differentiation and maturation.
Figure 8.

Activation of Smads gradually decreases during neuronal differentiation and maturation in vivo. A, B, Representative images of Sox2 (green), NeuN (blue) and p-Smad1/5 (red; A) or p-Smad2 (red; B) staining of P1 hippocampus. The boxed areas are enlarged in the right panels. Arrowheads, Representative cells double-positive for Sox2 and p-Smad1/5 (A) or Sox2 and p-Smad2 (B); arrows, p-Smad1/5 (A) or p-Smad2 (B) single-positive cells. p-Smads and NeuN signals are mutually exclusive. Scale bar, 100 μm. C, D, Representative images of MAP2 (blue) and p-Smad1/5 (red; C) or p-Smad2 (red; D) staining of P1 hippocampus. Arrowheads, Representative cells double-positive for MAP2 and p-Smad1/5 (C) or Sox2 and p-Smad2 (D); arrows, p-Smad1/5 (C) or p-Smad2 (D) single-positive cells. Subpopulations of MAP2-positive cells are p-Smad1/5-positive or p-Smad2-positive. Scale bar, 100 μm.
Next, to investigate whether canonical TGF-β signaling regulates morphological maturation of neurons in vivo, we performed in utero electroporation with vectors coexpressing GFP and TGFβR1 into E14.5 mouse telencephalon, and analyzed dendritic morphology at P10. GFP+ neurons expressing TGFβR1 showed significantly reduced dendritic length, branch numbers, and complexity compared with control neurons [Fig. 9A–E (Fig. 9C: TGFβR1, p = 0.0002; ANOVA; Fig. 9D: TGFβR1, p = 0.0076; ANOVA)]. We then examined the effects of two downstream effectors of TGF-β signaling, Smad4 and TGIF, on neuronal morphogenesis in vivo. The expression of both Smad4 and TGIF impaired the development of neuronal dendrites [Fig. 9A–E (Fig. 9C: Smad4, p < 0.0001; TGIF, p = 0.0148; ANOVA; Fig. 9D: Smad4, p < 0.0001; TGIF, p = 0.0107; ANOVA)].
Figure 9.

TGIF-mediated canonical TGF-β signaling downregulates neuronal morphogenesis in vivo. A, Representative images of electroporated neurons, stained with anti-GFP antibodies. Mouse embryos were electroporated with plasmids expressing only GFP (control, green), or GFP together with TGFβR1, Smad4, and TGIF, by in utero electroporation at E14 and killed at P10. Scale bar, 50 μm. B, Representative tracing images of neurons electroporated with the constructs as in A. Scale bar, 50 μm. C, D, Quantification of total dendrite length (C) and dendrite branch numbers (D) in B. Number of cells analyzed: control, 18 neurons; TGFβR1, 12 neurons; Smad4, 13 neurons; TGIF, 16 neurons from three brains. E, Quantification of dendrite complexity by Sholl analysis of neurons in B. Data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test.
We further examined whether canonical TGF-β signal-related factors also regulate axonal growth in vivo. To do this, we cointroduced CRMP2 (CRMP2-GFP) and Smad1 (Smad1-tdTomato) constructs into telencephalon at E14.5 by in utero electroporation, and measured the axon length of GFP and tdTomato colabeled neurons at P1. Control GFP and tdTomato double-labeled axons extended to the contralateral side of the cortex; however, this was impaired by the expression of Smad1 [Fig. 10A,B (Fig. 10B: Control Smad1, p = 0.0082; CRMP2 Smad1, p = 0.0076; ANOVA)]. Remarkably, the coexpression of CRMP2 with Smad1 restored the impaired axonal elongation induced by Smad1 expression. Thus, as in the case of cultured neurons in vitro, canonical TGF-β signaling suppresses the morphological development of neurons in the developing mouse cortex in vivo.
Figure 10.

CRMP2 restores Smad1-induced impairment in axonal elongation of neurons in vivo. A, Representative images of electroporated brain sections stained with anti-GFP and tdTomato antibodies. Mouse embryos were coelectroporated with plasmids expressing GFP and tdTomato (N = 4 mouse brains), GFP and Smad1 (N = 4 mouse brains), or CRMP2 and Smad1 (N = 4 mouse brains) by in utero electroporation at E14 and killed at P1. Axons were measured by taking the edge of the GFP and tdTomato colabeled region as the starting point (white line). The arrowheads indicate the end of the axonal tract. Scale bar, 500 μm. B, Quantification of axonal length in A. Data are presented as the mean ± SEM. **p < 0.01 by one-way ANOVA, Tukey's post-test.
TGIF-mediated canonical TGF-β signaling suppresses neurite elongation of hiPSC-derived neurons
iPSC technology enables us to undertake comparative studies between human and other animal species including rodents (Takahashi et al., 2007; Marchetto et al., 2013). Using this technology, we next investigated whether the effects of canonical TGF-β signaling that we found here in mouse neurons could be replicated in human neurons. hiPSC-derived neurons were cultured in the presence of TGF-β1, BMP2, or Noggin. Neurite extension was significantly inhibited by TGF-β1 and BMP2 treatment, whereas treatment with Noggin markedly promoted neurite growth of hiPSC-derived neurons [Fig. 11A,B (Fig. 11B: TGF-β1, p = 0.0055; BMP2, p = 0.0003; Noggin, p = 0.0116; ANOVA)]. We next expressed hTGFβR1 in hiPSC-derived neurons, and found that it repressed neurite elongation [Fig. 11C,D (Fig. 11D: TGFβR1, p = 0.0064; ANOVA)]. In addition, the overexpression of caTGFβR1 and caBMPR1A in hiPSC-derived neurons repressed neurite elongation [Fig. 11C,D (Fig. 11D: caTGFβR1, p = 0.0004; caBMPR1A, p = 0.0003; ANOVA)]. We also observed inhibition of neurite growth following the expression of hSmad4 and hTGIF in hiPSC-derived neurons [Fig. 11E,F (Fig. 11F: hSmad4, p = 0.0004; hTGIF, p = 0.0006; ANOVA)]. These findings are consistent with the results in mouse neurons, and indicate that the negative effects of canonical TGF-β signaling on the morphological development of neurons are evolutionarily conserved between mouse and human.
Figure 11.

TGIF-mediated canonical TGF-β signaling downregulates the morphological development of hiPSC-derived neurons. A, Representative images of GFP (green) staining of 7DIV neurons differentiated from hNS/PCs treated with BSA as a control, TGF-β1, BMP2, and Noggin. Scale bar, 50 μm. B, Quantification of total neurite length in A. C, Representative images of GFP staining of 7DIV neurons differentiated from hNS/PCs infected with lentiviruses expressing hTGFβR1, caTGFβR1, and caBMPR1A. Scale bar, 50 μm. D, Quantification of total neurite length in C. E, Representative images of GFP staining of 7DIV neurons differentiated from hNS/PCs infected with lentiviruses expressing hSmad4 or hTGIF. Scale bar, 50 μm. F, Quantification of total neurite length in E. Data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
Expression of mutants found in patients with neurodevelopmental disorders disrupt neuronal morphogenesis
Since it has been demonstrated that mutations in components of canonical TGF-β signaling are associated with neurodevelopmental disorders characterized by cognitive deficits (Loeys et al., 2005) and, in the present study, that canonical TGF-β signaling controls morphological maturation in human neurons, we next asked whether the dysregulation of neuronal morphogenesis by canonical TGF-β signaling might underlie the pathology of these disorders. To this end, we examined the functional consequence of mutations found in patients with neurodevelopmental disorders in the regulation of neuronal morphogenesis. An aspartate (D) to glycine (G) mutation at amino acid residue 400 (D400G) of hTGFβR1 is observed in patients with multiple developmental anomalies (Loeys et al., 2005). When we expressed the hTGFβR1-D400G mutant in mouse hippocampal neurons, it robustly inhibited dendritic development more severely than wild-type hTGFβR1 [Fig. 12A–D (Fig. 12B: hTGFβR1, p < 0.0001; hTGFβR1-D400G, p = 0.0076; ANOVA; Fig. 12C: hTGFβR1, p = 0.0005; hTGFβR1-D400G, p = 0.0014; ANOVA)], and this held true for hiPSC-derived neurons [Fig. 13A,B (Fig. 13B: hTGFβR1, p < 0.0001; hTGFβR1-D400G, p = 0.0034; ANOVA)]. We next expressed wild-type hSmad4 and the hSmad4-isoleucine (I) 500 threonine (T) mutant, which is found in Myhre syndrome (Le Goff et al., 2011), in mouse hippocampal neurons, and evaluated dendritic morphology. Although we found that hSmad4-I500T was expressed at a level similar to that of endogenous Smad4 protein in hSmad4-I500T-expressing, virus-infected hippocampal neurons [Fig. 12E,F (Fig. 12F: p = 0.0005, unpaired t test)], the expression of this mutant enhanced dendritic growth, whereas wild-type hSmad4 expression inhibited it [Fig. 12G–J (Fig. 12H: hSmad4, p = 0.0022; hSmad4-I500T, p < 0.0001; ANOVA; Fig. 12I: hSmad4, p = 0.0115; hSmad4-I500T, p = 0.0007; ANOVA)], suggesting that Smad4-I500T acts as a dominant negative form in the regulation of the morphological maturation of neurons. Furthermore, we assessed the function of the hTGIF mutant harboring serine (S) to phenylalanine (F) substitution at amino acid 162 (hTGIF-S162F), which is found in HPE patients (Gripp et al., 2000) and in mouse hippocampal neurons. In contrast to the inhibitory effects of wild-type hTGIF on dendritic morphology, the expression of hTGIF-S162F significantly promoted dendritic growth [Fig. 12K–N (Fig. 12L: hTGIF, p = 0.0011; hTGIF-S162F, p < 0.0001; ANOVA; Fig. 12M: hTGIF, p = 0.015; hTGIF-S162F, p = 0.0007; ANOVA)]. We also expressed hSmad4-I500T and hTGIF-S162F in hiPSC-derived neurons and assessed neurite morphology, and found that they enhanced neurite elongation [Fig. 13C–F (Fig. 13D: hSmad4, p = 0.0361; hSmad4-I500T, p = 0.0027; ANOVA; Fig. 13F: hTGIF, p = 0.0374; hTGIF-S162F, p = 0.0007; ANOVA)]. Together, these results imply that the dysregulation of neuronal morphogenesis by canonical TGF-β signaling may contribute to the pathology of particular neurodevelopmental disorders.
Figure 12.

Mutations in canonical TGF-β signaling factors disrupt neuronal morphogenesis. A, Representative images of GFP (green) and MAP2 (red) staining of 6DIV hippocampal neurons infected with lentiviruses expressing hTGFβR1 or hTGFβR1-D400G. Scale bar, 50 μm. B, C, Quantification of total dendritic length (B) and branch numbers (C) of 6DIV hippocampal neurons infected with lentiviruses expressing hTGFβR1 or hTGFβR1-D400G. D, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing hTGFβR1 or hTGFβR1-D400G. E, Representative images of Smad4 (cyan) and GFP (green) staining of 6DIV hippocampal neurons infected with lentivirus expressing hSmad4-I500T. Scale bar, 30 μm. F, Quantification of hSmad4-I500T expression. hSmad4-I500T signal intensity in neuronal soma infected with control virus was divided by GFP signal intensity, and this value was used for normalization of the value obtained with hSmad4-I500T-expressing, virus-infected cells. G, Representative images of GFP and MAP2 staining of 6DIV hippocampal neurons infected with lentiviruses expressing hSmad4 or hSmad4-I500T. Scale bar, 50 μm. H, I, Quantification of total dendritic length (H) and branch numbers (I) of 6DIV hippocampal neurons infected with lentiviruses expressing hSmad4 or hSmad4-I500T. J, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing hSmad4 or hSmad4-I500T. K, Representative images of GFP and MAP2 staining of 6DIV hippocampal neurons infected with lentiviruses expressing hTGIF or hTGIF-S162F. Scale bar, 50 μm. L, M, Quantification of total dendritic length (L) and branch numbers (M) of 6DIV hippocampal neurons infected with lentiviruses expressing hTGIF or hTGIF-S162F. N, Quantification of dendrite complexity by Sholl analysis of 6DIV hippocampal neurons infected with lentiviruses expressing hTGIF or hTGIF-S162F. Data are presented as the mean ± SEM. F, ***p < 0.001 by Student's t test. B, C, H, I, L, M, *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
Figure 13.

Mutations of TGFβR1, Smad4, and TGIF abnormally regulate neuronal morphogenesis of iPSC-derived human neurons. A, C, E, Representative images of GFP staining of 7DIV neurons differentiated from hNS/PCs infected with lentiviruses expressing hTGFβR1, hTGFβR1-D400G, hSmad4, hSmad4-I500T, hTGIF, or hTGIF-S162F. Scale bars, 50 μm. B, D, F, Quantification of total neurite length in A, C, and E. Data are presented as the mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 by one-way ANOVA, Tukey's post-test. N = 3 independent experiments; at least 50 neurons were analyzed in each experiment.
Discussion
TGF-β superfamily signaling is one of several pleiotropic regulatory signal transduction mechanisms that play pivotal roles in embryonic development and the maintenance of adult homeostasis. Canonical TGF-β signaling controls a diverse array of cellular processes and tissue development. However, the functional role of canonical TGF-β signaling in the regulation of neuronal morphogenesis has not been previously defined. Our present findings strongly indicate that canonical TGF-β signaling negatively regulates neuronal morphogenesis. We have shown that treatment with TGF-β1, BMP2, and BMP4 suppressed neurite morphogenesis. In addition, overexpression of Smad1, Smad2, and Smad4 inhibited both axonal and dendritic growth. On the contrary, knockdown of Smad1 and Smad4 enhanced neurite development and alleviated the decreased axon/dendrite growth induced by TGF-β1 and BMP2 treatments. These results suggest that TGF-β signaling during neurite morphogenesis depends on Smads, which are critical signal transducers downstream of TGF-β signals. A previous study showed that TGFβR1 (Alk5)-dependent TGF-β signaling promotes the maturation of newborn neurons and that physiological TGF-β signaling is active in NeuN-positive mature neurons in the adult hippocampus (He et al., 2014). However, in our experimental setting using embryonic hippocampal neurons and newly generated human iPSC-derived neurons, TGFβR1 (Alk5)-dependent canonical TGF-β signaling negatively regulated the morphological development of neurons. Although we do not currently have a specific explanation of why canonical TGF-β signaling exerts these opposite functions in neuronal morphogenesis, it may be that post-translational modifications of Smads differ between distinct developmental stages and among neuronal subtypes, since modifications of Smads such as acetylation are also known to affect Smad functions (Gaarenstroom and Hill, 2014). Another previous study, using E18 hippocampal neurons, also showed that GDF5 promotes dendrite growth by activating the Smad signaling pathway (Osório et al., 2013), a finding that appears to conflict with our observations here using neurons from E17 hippocampus. Although GDF5 is known to activate the same Smads (1, 5, and 8) as BMP2 and BMP4, the developmental stage difference (E17 and E18) of neurons may have affected the consequence of GDF5 and these BMPs. For example, it is known that during the middle embryonic period, BMP signaling initially promotes neurogenesis by pushing progenitors toward a neuronal fate (Bond et al., 2012). However, BMP signaling then switches from being a proneuronal to a proastroglial cue and enhances astroglial differentiation in the late embryonic stage. Moreover, TGF-β signaling negatively regulates self-renewal of midbrain neuroepithelial stem cells, although it induces the proliferation of mesenchymal stem cells (Sakaki-Yumoto et al., 2013). These studies imply that TGF-β and BMP signaling effects are diverse and depend on the cell type, as well as the microenvironment, developmental stage, and physiological state of the cells. Further mechanistic studies on TGF-β and BMP signal transduction and the cross talk with other signaling pathways will provide a better understanding of the roles of TGF-β and BMP signaling in the regulation of axon and dendrite formation.
Neuronal morphology is defined by microtubule and actin cytoskeletal dynamics (Conde and Cáceres, 2009). Increasing evidence suggests that CRMPs control neuronal morphogenesis by regulating mictrotubule and actin cytoskeleton dynamics (Quach et al., 2015); however, the regulatory mechanisms of the expression of CRMPs in neurons remains largely unknown. In this study, our analyses revealed that Smad-dependent TGF-β/BMP signaling downregulates CRMP2 expression in neurons. Consistent with the results of a previous report in neuronal progenitor cells (Sun et al., 2010b), we demonstrated that Smads bind to the Crmp2 promoter in neurons in response to TGF-β1 and BMP2 stimulations. In addition, the expression of CRMP2 ameliorated the negative effect of TGF-β1/BMP2 stimulation and Smad expression on neuronal development. These results indicate that CRMP2 downregulation by TGF-β/Smad signaling is important for TGF-β/Smad signaling to exert their negative effect on neuronal morphogenesis. How, then, do Smads repress Crmp2 expression? A previous study (Wotton et al., 2001) reported that TGIF recruits a repressor complex, including CtBP, mSin3, and HDACs, to Smad target genes through its interaction with Smads. In support of this observation, we have shown that TGIF clearly suppresses Crmp2 expression and that knockdown of TGIF restored the impaired neuronal morphogenesis induced by TGF-β1/BMP2 treatment and Smad4 expression. Furthermore, by analyzing public ChIP sequence datasets (Estarás et al., 2012; Willer et al., 2015; Yoon et al., 2015), we found that TGIF and Smads bind to the Crmp2 promoter region [−700 to −150 bp from the transcription start site (TSS)], a sequence that includes the fragment in which we detected Smads binding in response to TGF-β1 and BMP2 stimulation in the ChIP assay. Although further investigation will be required to elucidate the precise mechanism, these findings nevertheless strongly suggest that Smads suppress Crmp2 expression through TGIF-mediated epigenetic gene silencing (Fig. 14).
Figure 14.

Schematic representation of the effect of canonical TGF-β family signaling on the morphological development of neurons. TGF-β family cytokines, such as TGF-β and BMP2/4, bind and activate its receptors. Smads are subsequently activated by the receptors and are translocated into nucleus. Activated Smads then form a complex with TGIF in nucleus and downregulate the Crmp2 expression, which result in the suppression of morphological development in neurons. We found that TGF-β/Smads–TGIF/CRMP2 signaling pathways are conserved between mouse and human.
Previous work has shown that BMP7 enhances the dendrite growth of neurons in a noncanonical manner (Lein et al., 1995; Lee-Hoeflich et al., 2004). Consistent with these studies, we did not observe a negative effect of BMP7 on dendrite development in cultured hippocampal neurons. In addition, we observed very little, if any, phosphorylation of Smads by BMP7 stimulation. Although we do not currently have any likely explanation for why BMP7 exerts a different effect on Smads activation from other TGF-β members, we speculate that BMP7 preferentially activates noncanonical TGF-β/BMP signaling pathway molecules such as LIMK through type II BMPR. Further investigations will be required to explain these distinct effects among the members of TGF-β superfamily.
In several previous studies, in situ hybridization and immunohistochemical experiments have demonstrated that TGF-β ligands are expressed in the mouse CNS during development (Heine et al., 1987; Mecha et al., 2008), and that TGF-β receptors are highly expressed in mouse embryonic hippocampus (Tomoda et al., 1996). In the developing mouse brain, moreover, BMPs are expressed in certain regions of the telencephalon, including the medial walls of the lateral ventricles, which correspond to the regions of prospective hippocampus and choroid plexus (Furuta et al., 1997). BMP receptors have been also shown to be expressed in the lateral ventricular zone of the embryonic mouse brain (Panchision et al., 2001). These studies thus strongly suggest that TGF-β and BMP signaling affect brain development. In this study, we detected p-Smad1/5 and p-Smad2 in Sox2-positive NS/PCs but not in NeuN-positive mature neurons. However, we could also observe p-Smad1/5 and p-Smad2 signals in a subpopulation of MAP2-positive cells that contain relatively immature neurons compared with NeuN-positive cells. These results suggested that the activation of Smads gradually decreases during neuronal differentiation and maturation. Because we have shown in this study that TGF-β and BMP signaling suppress dendritic and axonal growth/branching, it is plausible that the gradual reduction of Smad activation enables hippocampal neurons to acquire the ability to execute neurite morphogenesis efficiently when they need to do so during brain development.
Mutations in components of canonical TGF-β signaling such as hTGFβR1, hSmad4, and hTGIF have been linked to several human developmental disorders characterized by mental retardation and cognitive abnormality (Gripp et al., 2000; Loeys et al., 2005; Le Goff et al., 2011). However, how the dysregulation of canonical TGF-β signaling causes neurodevelopmental disorders remains unknown. In this study, we have demonstrated that mutants of hTGFβR1, hSmad4, and hTGIF show detrimental effects on neuronal morphogenesis. We expressed the hSmad4-I500T mutant in mouse hippocampal neurons. Although we found that hSmad4-I500T was expressed at a level similar to that of the endogenous Smad4 protein in these neurons, the expression of the mutant enhanced dendritic growth, whereas wild-type hSmad4 expression inhibited it, suggesting that Smad4-I500T acts as a DN form in the regulation of morphological maturation of neurons. In analysis of the function of TGIF mutation, we used the hTGIF-S162F mutant. This mutation is in the HDAC and Smad binding domain of TGIF. A previous immunoprecipitation study (Gripp et al., 2000) showed that the interaction between TGIF-S162F and HDAC1 or Smad2 was weaker than that with wild-type TGIF. Therefore, it seems likely that the overexpression of TGIF-S162F interferes with the binding of HDAC and Smads and then promotes dendrite development. Although functional experiments involving the hTGFβR1-D400G mutant were not reported in the previous article, increased intensity of nuclear phosphorylated Smad2 and expression of TGF-β signaling target genes have been observed in tissues derived from individuals with mutations in TGFβR1 and TGFβR2 (Loeys et al., 2005), which is indicative of increased TGF-β signaling (gain of function) in vivo. Taking these previous data together with our finding that overexpression of the TGFβR1 mutant resulted in an effect that was stronger than the wild type, we infer that the TGFβR1 mutant acts in a hypermorphic (gain-of-function) manner. Our experiments with human variants suggest that both excessive and insufficient TGF-β signaling activity cause abnormal morphological development of neurons, which in turn leads to impaired neuronal network formation. Moreover, these findings provide a plausible explanation for how the dysregulation of canonical TGF-β signaling results in neurodevelopmental disorders, and suggest that precise activity control of TGF-β signaling is crucial for proper neuronal development.
Recent evidence has suggested that TGF-β signaling is also associated with psychiatric disorders. TGF-β signaling molecules such as TGF-β2 and TGFβR1 have been reported to be upregulated in the brains of patients with schizophrenia and bipolar disorder (Benes et al., 2007); another study has shown that forebrain-specific Smad4 knock-out mice exhibit schizophrenia-like phenotypes (Sun et al., 2010a). In addition, there is increasing evidence for altered CRMP expression in psychiatric diseases including schizophrenia and mood disorders (Blouin et al., 1998; Nakata et al., 2003; Quach et al., 2015). Since altered neurite morphology is known to contribute to various psychiatric disorders (Rosoklija et al., 2000; Soetanto et al., 2010; Kulkarni and Firestein, 2012), it is conceivable that the dysregulation of TGF-β/Smads/CRMP2 signaling pathways contributes to the pathogenesis of psychiatric disorders.
We have extended this knowledge by revealing the negative effects of canonical TGF-β signaling on neurite morphogenesis in hiPSC-derived neurons. In addition, mutations of canonical TGF-β signaling components found in patients have been shown to abnormally regulate neuritogenesis in human neurons. Given that the canonical TGF-β/BMP signal critically affects morphogenesis in both mouse and human neurons, we propose that the proper control of this evolutionarily conserved signaling pathway is very important for the perfect execution of neuronal morphogenesis, whose impairment eventually results in neurodevelopmental disorders.
Footnotes
This work was funded by the Japan Society for the Promotion of Science (JSPS) KAKENHI 17H01390; an Intramural Research Grant 27–7 for Neurological and Psychiatric Disorders of the National Center of Neurology and Psychiatry to K.N.; JSPS KAKENHI 16K18391 to K.T.; and JSPS KAKENHI 16J03827 to H.N. We thank T. Imamura and S. Katada for valuable discussion; M.E. Greenberg, Z. Zhou, and H. Okano for sharing reagents and cells; I. Smith for editing the manuscript; and all members of the Laboratory of Molecular Neuroscience, Department of Stem Cell Biology and Medicine, Kyushu University. We also thank the Research Support Center, Research Center for Human Disease Modeling, Kyushu University Graduate School of Medical Sciences for technical assistance.
The authors declare no competing financial interests.
References
- Ageta H, Murayama A, Migishima R, Kida S, Tsuchida K, Yokoyama M, Inokuchi K (2008) Activin in the brain modulates anxiety-related behavior and adult neurogenesis. PLoS One 3:e1869. 10.1371/journal.pone.0001869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyama S, Katagiri T, Namiki M, Yamaji N, Yamamoto N, Miyama K, Shibuya H, Ueno N, Wozney JM, Suda T (1997) Constitutively active BMP type I receptors transduce BMP-2 signals without the ligand in C2C12 myoblasts. Exp Cell Res 235:362–369. 10.1006/excr.1997.3680 [DOI] [PubMed] [Google Scholar]
- Andoh-Noda T, Akamatsu W, Miyake K, Matsumoto T, Yamaguchi R, Sanosaka T, Okada Y, Kobayashi T, Ohyama M, Nakashima K, Kurosawa H, Kubota T, Okano H (2015) Differentiation of multipotent neural stem cells derived from rett syndrome patients is biased toward the astrocytic lineage. Mol Brain 8:31. 10.1186/s13041-015-0121-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimura N, Ménager C, Kawano Y, Yoshimura T, Kawabata S, Hattori A, Fukata Y, Amano M, Goshima Y, Inagaki M, Morone N, Usukura J, Kaibuchi K (2005) Phosphorylation by rho kinase regulates CRMP-2 activity in growth cones. Mol Cell Biol 25:9973–9984. 10.1128/MCB.25.22.9973-9984.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benes FM, Lim B, Matzilevich D, Walsh JP, Subburaju S, Minns M (2007) Regulation of the GABA cell phenotype in hippocampus of schizophrenics and bipolars. Proc Natl Acad Sci U S A 104:10164–10169. 10.1073/pnas.0703806104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin JL, Dombroski BA, Nath SK, Lasseter VK, Wolyniec PS, Nestadt G, Thornquist M, Ullrich G, McGrath J, Kasch L, Lamacz M, Thomas MG, Gehrig C, Radhakrishna U, Snyder SE, Balk KG, Neufeld K, Swartz KL, DeMarchi N, Papadimitriou GN, et al. (1998) Schizophrenia susceptibility loci on chromosomes 13q32 and 8p21. Nat Genet 20:70–73. 10.1038/1734 [DOI] [PubMed] [Google Scholar]
- Bond AM, Bhalala OG, Kessler JA (2012) The dynamic role of bone morphogenetic proteins in neural stem cell fate and maturation. Dev Neurobiol 72:1068–1084. 10.1002/dneu.22022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bretin S, Reibel S, Charrier E, Maus-Moatti M, Auvergnon N, Thevenoux A, Glowinski J, Rogemond V, Prémont J, Honnorat J, Gauchy C (2005) Differential expression of CRMP1, CRMP2A, CRMP2B, and CRMP5 in axons or dendrites of distinct neurons in the mouse brain. J Comp Neurol 486:1–17. 10.1002/cne.20465 [DOI] [PubMed] [Google Scholar]
- Colak D, Mori T, Brill MS, Pfeifer A, Falk S, Deng C, Monteiro R, Mummery C, Sommer L, Götz M (2008) Adult neurogenesis requires Smad4-mediated bone morphogenic protein signaling in stem cells. J Neurosci 28:434–446. 10.1523/JNEUROSCI.4374-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde C, Cáceres A (2009) Microtubule assembly, organization and dynamics in axons and dendrites. Nat Rev Neurosci 10:319–332. 10.1038/nrn2631 [DOI] [PubMed] [Google Scholar]
- Derynck R, Zhang YE (2003) Smad-dependent and smad-independent pathways in TGF-beta family signalling. Nature 425:577–584. 10.1038/nature02006 [DOI] [PubMed] [Google Scholar]
- Estarás C, Akizu N, García A, Beltrán S, de la Cruz X, Martínez-Balbás MA (2012) Genome-wide analysis reveals that Smad3 and JMJD3 HDM co-activate the neural developmental program. Development 139:2681–2691. 10.1242/dev.078345 [DOI] [PubMed] [Google Scholar]
- Fei T, Xia K, Li Z, Zhou B, Zhu S, Chen H, Zhang J, Chen Z, Xiao H, Han JDJ, Chen YG (2010) Genome-wide mapping of SMAD target genes reveals the role of BMP signaling in embryonic stem cell fate determination. Genome Res 20:36–44. 10.1101/gr.092114.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Itoh TJ, Kimura T, Ménager C, Nishimura T, Shiromizu T, Watanabe H, Inagaki N, Iwamatsu A, Hotani H, Kaibuchi K (2002) CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat Cell Biol 4:583–591. 10.1038/ncb825 [DOI] [PubMed] [Google Scholar]
- Furuta Y, Piston DW, Hogan BL (1997) Bone morphogenetic proteins (BMPs) as regulators of dorsal forebrain development. Development 124:2203–2212. [DOI] [PubMed] [Google Scholar]
- Gaarenstroom T, Hill CS (2014) TGF-signaling to chromatin: how smads regulate transcription during selfrenewal and differentiation. Semin Cell Dev Biol 32:107–118. 10.1016/j.semcdb.2014.01.009 [DOI] [PubMed] [Google Scholar]
- Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massagué J, Muenke M, Elledge SJ (2000) Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet 25:205–208. 10.1038/76074 [DOI] [PubMed] [Google Scholar]
- He Y, Zhang H, Yung A, Villeda SA, Jaeger PA, Olayiwola O, Fainberg N, Wyss-Coray T (2014) ALK5-dependent TGF-β signaling is a major determinant of late-stage adult neurogenesis. Nat Neurosci 17:943–952. 10.1038/nn.3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heine U, Munoz EF, Flanders KC, Ellingsworth LR, Lam HY, Thompson NL, Roberts AB, Sporn MB (1987) Role of transforming growth factor-beta in the development of the mouse embryo. J Cell Biol 105:2861–2876. 10.1083/jcb.105.6.2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Chihara K, Arimura N, Ménager C, Kawano Y, Matsuo N, Nishimura T, Amano M, Kaibuchi K (2001) CRMP-2 induces axons in cultured hippocampal neurons. Nat Neurosci 4:781–782. 10.1038/90476 [DOI] [PubMed] [Google Scholar]
- Irie K, Tsujimura K, Nakashima H, Nakashima K (2016) MicroRNA-214 promotes dendritic development by targeting the schizophrenia-associated gene quaking (Qki). J Biol Chem 291:13891–13904. 10.1074/jbc.M115.705749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knepper JL, James AC, Ming JE (2006) TGIF, a gene associated with human brain defects, regulates neuronal development. Dev Dyn 235:1482–1490. 10.1002/dvdy.20725 [DOI] [PubMed] [Google Scholar]
- König HG, Kögel D, Rami A, Prehn JH (2005) TGF-beta 1 activates two distinct type 1 receptors in neurons: implications for neuronal NF-kappa B signaling. J Cell Biol 168:1077–1086. 10.1083/jcb.200407027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni VA, Firestein BL (2012) The dendritic tree and brain disorders. Mol Cell Neurosci 50:10–20. 10.1016/j.mcn.2012.03.005 [DOI] [PubMed] [Google Scholar]
- Lee-Hoeflich ST, Causing CG, Podkowa M, Zhao X, Wrana JL, Attisano L (2004) Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J 23:4792–4801. 10.1038/sj.emboj.7600418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, Afenjar A, Destrée A, di Rocco M, Héron D, Jacquemont S, Marlin S, Simon M, Tolmie J, Verloes A, Casanova JL, Munnich A, Cormier-Daire V (2011) Mutations at a single codon in mad homology 2 domain of SMAD4 cause myhre syndrome. Nat Genet 44:85–88. 10.1038/ng.1016 [DOI] [PubMed] [Google Scholar]
- Lein P, Johnson M, Guo X, Rueger D, Higgins D (1995) Osteogenic protein-1 induces dendritic growth in rat sympathetic neurons. Neuron 15:597–605. 10.1016/0896-6273(95)90148-5 [DOI] [PubMed] [Google Scholar]
- Liu A, Niswander LA (2005) Bone morphogenetic protein signalling and vertebrate nervous system development. Nat Rev Neurosci 6:945–954. 10.1038/nrn1805 [DOI] [PubMed] [Google Scholar]
- Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, et al. (2005) A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet 37:275–281. 10.1038/ng1511 [DOI] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, Baltimore D (2002) Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295:868–872. 10.1126/science.1067081 [DOI] [PubMed] [Google Scholar]
- Marchetto MCN, Narvaiza I, Denli AM, Benner C, Lazzarini TA, Nathanson JL, Paquola ACM, Desai KN, Herai RH, Weitzman MD, Yeo GW, Muotri AR, Gage FH (2013) Differential L1 regulation in pluripotent stem cells of humans and apes. Nature 503:525–529. 10.1038/nature12686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Blain SW, Lo RS (2000) TGFbeta signaling in growth control, cancer, and heritable disorders. Cell 103:295–309. 10.1016/S0092-8674(00)00121-5 [DOI] [PubMed] [Google Scholar]
- Mecha M, Rabadán MA, Peña-Melián A, Valencia M, Mondéjar T, Blanco MJ (2008) Expression of TGF-betas in the embryonic nervous system: analysis of interbalance between isoforms. Dev Dyn 237:1709–1717. 10.1002/dvdy.21558 [DOI] [PubMed] [Google Scholar]
- Nakata K, Ujike H, Sakai A, Takaki M, Imamura T, Tanaka Y, Kuroda S (2003) The human dihydropyrimidinase-related protein 2 gene on chromosome 8p21 is associated with paranoid-type schizophrenia. Biol Psychiatry 53:571–576. 10.1016/S0006-3223(02)01729-8 [DOI] [PubMed] [Google Scholar]
- Nishimura T, Fukata Y, Kato K, Yamaguchi T, Matsuura Y, Kamiguchi H, Kaibuchi K (2003) CRMP-2 regulates polarized numb-mediated endocytosis for axon growth. Nat Cell Biol 5:819–826. 10.1038/ncb1039 [DOI] [PubMed] [Google Scholar]
- Noguchi H, Murao N, Kimura A, Matsuda T, Namihira M, Nakashima K (2016) DNA methyltransferase 1 is indispensable for development of the hippocampal dentate gyrus. J Neurosci 36:6050–6068. 10.1523/JNEUROSCI.0512-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto M, Miyata T, Konno D, Ueda HR, Kasukawa T, Hashimoto M, Matsuzaki F, Kawaguchi A (2016) Cell-cycle-independent transitions in temporal identity of mammalian neural progenitor cells. Nat Commun 7:11349. 10.1038/ncomms11349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osório C, Chacón PJ, Kisiswa L, White M, Wyatt S, Rodríguez-Tébar A, Davies AM (2013) Growth differentiation factor 5 is a key physiological regulator of dendrite growth during development. Development 140:4751–4762. 10.1242/dev.101378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panchision DM, Pickel JM, Studer L, Lee SH, Turner PA, Hazel TG, McKay RD (2001) Sequential actions of BMP receptors control neural precursor cell production and fate. Genes Dev 15:2094–2110. 10.1101/gad.894701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SO, Kumar M, Gupta S (2012) TGF-β and iron differently alter HBV replication in human hepatocytes through TGF-β/BMP signaling and cellular microRNA expression. PLoS One 7:e39276. 10.1371/journal.pone.0039276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pribiag H, Stellwagen D (2013) TNF-α downregulates inhibitory neurotransmission through protein phosphatase 1-dependent trafficking of GABAA receptors. J Neurosci 33:15879–15893. 10.1523/JNEUROSCI.0530-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quach TT, Honnorat J, Kolattukudy PE, Khanna R, Duchemin AM (2015) CRMPs: critical molecules for neurite morphogenesis and neuropsychiatric diseases. Mol Psychiatry 20:1037–1045. 10.1038/mp.2015.77 [DOI] [PubMed] [Google Scholar]
- Ramocki MB, Zoghbi HY (2008) Failure of neuronal homeostasis results in common neuropsychiatric phenotypes. Nature 455:912–918. 10.1038/nature07457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosoklija G, Toomayan G, Ellis SP, Keilp J, Mann JJ, Latov N, Hays AP, Dwork AJ (2000) Structural abnormalities of subicular dendrites in subjects with schizophrenia and mood disorders: preliminary findings. Arch Gen Psychiatry 57:349–356. 10.1001/archpsyc.57.4.349 [DOI] [PubMed] [Google Scholar]
- Sakaki-Yumoto M, Katsuno Y, Derynck R (2013) TGF-β family signaling in stem cells. Biochim Biophys Acta 1830:2280–2296. 10.1016/j.bbagen.2012.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massagué J (2003) Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113:685–700. 10.1016/S0092-8674(03)00432-X [DOI] [PubMed] [Google Scholar]
- Soetanto A, Wilson RS, Talbot K, Un A, Schneider JA, Sobiesk M, Kelly J, Leurgans S, Bennett DA, Arnold SE (2010) Association of anxiety and depression with microtubule-associated protein 2- and synaptopodin-immunolabeled dendrite and spine densities in hippocampal CA3 of older humans. Arch Gen Psychiatry 67:448–457. 10.1001/archgenpsychiatry.2010.48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Gewirtz JC, Bofenkamp L, Wickham RJ, Ge H, O'Connor MB (2010a) Canonical TGF-β signaling is required for the balance of excitatory/inhibitory transmission within the hippocampus and prepulse inhibition of acoustic startle. J Neurosci 30:6025–6035. 10.1523/JNEUROSCI.0789-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Fei T, Yang T, Zhang F, Chen YG, Li H, Xu Z (2010b) The suppression of CRMP2 expression by bone morphogenetic protein (BMP)-SMAD gradient signaling controls multiple stages of neuronal development. J Biol Chem 285:39039–39050. 10.1074/jbc.M110.168351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S (2007) Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131:861–872. 10.1016/j.cell.2007.11.019 [DOI] [PubMed] [Google Scholar]
- Taylor AM, Dieterich DC, Ito HT, Kim SA, Schuman EM (2010) Microfluidic local perfusion chambers for the visualization and manipulation of synapses. Neuron 66:57–68. 10.1016/j.neuron.2010.03.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomoda T, Shirasawa T, Yahagi YI, Ishii K, Takagi H, Furiya Y, Arai KI, Mori H, Muramatsu MA (1996) Transforming growth factor-beta is a survival factor for neonate cortical neurons: coincident expression of type I receptors in developing cerebral cortices. Dev Biol 179:79–90. 10.1006/dbio.1996.0242 [DOI] [PubMed] [Google Scholar]
- Tsujimura K, Irie K, Nakashima H, Egashira Y, Fukao Y, Fujiwara M, Itoh M, Uesaka M, Imamura T, Nakahata Y, Yamashita Y, Abe T, Takamori S, Nakashima K (2015) miR-199a links MeCP2 with mTOR signaling and its dysregulation leads to rett syndrome phenotypes. Cell Rep 12:1887–1901. 10.1016/j.celrep.2015.08.028 [DOI] [PubMed] [Google Scholar]
- van Beuningen SFB, Will L, Harterink M, Chazeau A, van Battum EY, Frias CP, Franker MAM, Katrukha EA, Stucchi R, Vocking K, Antunes AT, Slenders L, Doulkeridou S, Sillevis Smitt P, Altelaar AFM, Post JA, Akhmanova A, Pasterkamp RJ, Kapitein LC, de Graaff E, et al. (2015) TRIM46 controls neuronal polarity and axon specification by driving the formation of parallel microtubule arrays. Neuron 88:1208–1226. 10.1016/j.neuron.2015.11.012 [DOI] [PubMed] [Google Scholar]
- Walsh CA, Engle EC (2010) Allelic diversity in human developmental neurogenetics: insights into biology and disease. Neuron 68:245–253. 10.1016/j.neuron.2010.09.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LH, Strittmatter SM (1996) A family of rat CRMP genes is differentially expressed in the nervous system. J Neurosci 16:6197–6207. 10.1523/JNEUROSCI.16-19-06197.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser R, Wrana JL, Massagué J (1995) GS domain mutations that constitutively activate T beta R-I, the downstream signaling component in the TGF-beta receptor complex. EMBO J 14:2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willer A, Jakobsen JS, Ohlsson E, Rapin N, Waage J, Billing M, Bullinger L, Karlsson S, Porse BT (2015) TGIF1 is a negative regulator of MLL-rearranged acute myeloid leukemia. Leukemia 29:1018–1031. 10.1038/leu.2014.307 [DOI] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massagué J (1999) A smad transcriptional corepressor. Cell 97:29–39. 10.1016/S0092-8674(00)80712-6 [DOI] [PubMed] [Google Scholar]
- Wotton D, Knoepfler PS, Laherty CD, Eisenman RN, Massagué J (2001) The smad transcriptional corepressor TGIF recruits mSin3. Cell Growth Differ 12:457–463. [PubMed] [Google Scholar]
- Yoon SJ, Foley JW, Baker JC (2015) HEB associates with PRC2 and SMAD2/3 to regulate developmental fates. Nat Commun 6:6546. 10.1038/ncomms7546 [DOI] [PubMed] [Google Scholar]
- Yoshimura T, Kawano Y, Arimura N, Kawabata S, Kikuchi A, Kaibuchi K (2005) GSK-3beta regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120:137–149. 10.1016/j.cell.2004.11.012 [DOI] [PubMed] [Google Scholar]
- Yoshiura S, Ohtsuka T, Takenaka Y, Nagahara H, Yoshikawa K, Kageyama R (2007) Ultradian oscillations of stat, Smad, and Hes1 expression in response to serum. Proc Natl Acad Sci U S A 104:11292–11297. 10.1073/pnas.0701837104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You X, Vlatkovic I, Babic A, Will T, Epstein I, Tushev G, Akbalik G, Wang M, Glock C, Quedenau C, Wang X, Hou J, Liu H, Sun W, Sambandan S, Chen T, Schuman EM, Chen W (2015) Neural circular RNAs are derived from synaptic genes and regulated by development and plasticity. Nat Neurosci 18:603–610. 10.1038/nn.3975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa-Kawada J, Suzuki R, Kano F, Ohkawara T, Murata M, Noda M (2003) Axonal morphogenesis controlled by antagonistic roles of two CRMP subtypes in microtubule organization. Eur J Neurosci 17:2329–2343. 10.1046/j.1460-9568.2003.02664.x [DOI] [PubMed] [Google Scholar]
- Zhang H, Kang E, Wang Y, Yang C, Yu H, Wang Q, Chen Z, Zhang C, Christian KM, Song H, Ming G-L, Xu Z (2016) Brain-specific Crmp2 deletion leads to neuronal development deficits and behavioural impairments in mice. Nat Commun 7:0. 10.1038/ncomms11773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang YE. (2009) Non-Smad pathways in TGF-beta signaling. Cell Res 19:128–139. 10.1038/cr.2008.328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Hong EJ, Cohen S, Zhao WN, Ho HY, Schmidt L, Chen WG, Lin Y, Savner E, Griffith EC, Hu L, Steen JA, Weitz CJ, Greenberg ME (2006) Brain-specific phosphorylation of MeCP2 regulates activity-dependent bdnf transcription, dendritic growth, and spine maturation. Neuron 52:255–269. 10.1016/j.neuron.2006.09.037 [DOI] [PMC free article] [PubMed] [Google Scholar]