

Abstract
Exposure of the developing brain to toxins, drugs, or deleterious endogenous compounds during the perinatal period can trigger alterations in cell division, migration, differentiation, and synaptogenesis, leading to lifelong neurological impairment. The brain is protected by cellular barriers acting through multiple mechanisms, some of which are still poorly explored. We used a combination of enzymatic assays, live tissue fluorescence microscopy, and an in vitro cellular model of the blood–CSF barrier to investigate an enzymatic detoxification pathway in the developing male and female rat brain. We show that during the early postnatal period the choroid plexus epithelium forming the blood–CSF barrier and the ependymal cell layer bordering the ventricles harbor a high detoxifying capacity that involves glutathione S-transferases. Using a functional knock-down rat model for choroidal glutathione conjugation, we demonstrate that already in neonates, this metabolic pathway efficiently prevents the penetration of blood-borne reactive compounds into CSF. The versatility of the protective mechanism results from the multiplicity of the glutathione S-transferase isoenzymes, which are differently expressed between the choroidal epithelium and the ependyma. The various isoenzymes display differential substrate specificities, which greatly widen the spectrum of molecules that can be inactivated by this pathway. In conclusion, the blood–CSF barrier and the ependyma are identified as key cellular structures in the CNS to protect the brain fluid environment from different chemical classes of potentially toxic compounds during the postnatal period. This metabolic neuroprotective function of brain interfaces ought to compensate for the liver postnatal immaturity.
SIGNIFICANCE STATEMENT Brain homeostasis requires a stable and controlled internal environment. Defective brain protection during the perinatal period can lead to lifelong neurological impairment. We demonstrate that the choroid plexus forming the blood–CSF barrier is a key player in the protection of the developing brain. Glutathione-dependent enzymatic metabolism in the choroidal epithelium inactivates a broad spectrum of noxious compounds, efficiently preventing their penetration into the CSF. A second line of detoxification is located in the ependyma separating the CSF from brain tissue. Our study reveals a novel facet of the mechanisms by which the brain is protected at a period of high vulnerability, at a time when the astrocytic network is still immature and liver xenobiotic metabolism is limited.
Keywords: blood–brain barriers, brain homeostasis, glutathione S-transferases, neuroprotection, oxidative stress, perinatal
Introduction
The developing brain is sensitive to chemical insults and oxidative stress, and the resulting damage can be devastating, resulting in lifelong neurological impairment (Dammann and Leviton, 1999; Favrais et al., 2011). The blood–brain interfaces, namely the blood–brain barrier located at the endothelium of the cerebral microvessels and the blood–CSF barrier located at the epithelium of the choroid plexuses efficiently protect the adult brain from circulating toxic substances by a set of mechanisms combining tight junctions, efflux transporters, and detoxifying enzymes (Neuwelt et al., 2008; Strazielle and Ghersi-Egea, 2013; Ghersi-Egea et al., 2018). These interfaces have often been considered functionally immature during prenatal and postnatal development, thereby contributing to the vulnerability of the maturing brain. This concept of barrier immaturity is being challenged. The efficacy of tight junctions, which link neighboring barrier cells together, in preventing paracellular diffusion during development has been demonstrated (Johansson et al., 2006). This efficacy results from the early expression and correct membrane localization of the multiple proteins forming these tight junctions in both rat and human brain barrier cells (Kratzer et al., 2012). The functionality of transport and metabolic processes during the perinatal period remains largely unaddressed (Strazielle and Ghersi-Egea, 2015). Glutathione S-transferases (GSTs) form a family of isoenzymes largely involved in detoxification processes (Hayes et al., 2005). Several immunohistochemical analyses have pointed out an apparently robust expression of GSTs in the choroid plexuses and ependyma (for review, see Strazielle et al., 2004). A high enzymatic activity of conjugation to reduced glutathione (GSH) has been measured in homogenates of choroid plexuses isolated from both fetal human and postnatal rat brains (Ghersi-Egea et al., 2006). More recently, a transcriptomic analysis of choroid plexus identified several GST isoenzymes that are expressed during development (Kratzer et al., 2013), revealing perinatal-specific detoxification properties of the blood–CSF barrier. The detoxification properties of this tissue may therefore be especially crucial in protecting the brain during the perinatal period when the CSF, in which the brain develops, represents a large proportion of the cerebral extracellular fluid (Ghersi-Egea et al., 2015).
In this work, we identify the choroid plexuses and the ependyma as major sites for GSH conjugation in the developing brain. We provide the in vivo demonstration that choroidal GSTs support a metabolic barrier function of the blood–CSF interface protecting the maturing brain, by preventing the penetration of blood-borne electrophilic substrates into the CSF. We finally provide the molecular identity and relative level of expression of the different GST isoenzymes responsible for neuroprotection at the blood–CSF barrier and ependyma.
Materials and Methods
Animals, tissue sampling, and processing.
Female OFA rats with their litter, and 35-day-old [postnatal day 35 (P35)] and 200 g adult male OFA rats were obtained from Janvier. Animal care and procedures were conducted according to the guidelines approved by the French Ethical Committee (decree 87-848) and by European Community directive 86-609-EEC. The ethical committee for animal experimentation CEEA-55 has approved the present research project. All animals were kept under identical conditions in standard cages, with free access to food and tap water under a controlled environment (12 h dark/light cycles). Animals were killed at different postnatal ages by decapitation following deep isoflurane gas anesthesia. Following brain extraction in Ca- and Mg-supplemented PBS, lateral ventricle and fourth ventricle choroid plexuses and cerebral cortices devoid of meninges were sampled under a stereomicroscope at 4°C. Tissues were frozen and kept at −80°C for enzymatic analysis or RNA isolation. Alternatively, some brains were either collected in ice-cold Krebs-Ringer (KR) buffer (135 mm NaCl, 4 mm KCl, 2.2 mm CaCl2, 1.2 mm MgCl2, 6 mm NaHCO3, 10 mm HEPES, and 5 mm glucose, pH 7.4) and immediately sliced for imaging studies, or were frozen in −40°C cold isopentane and kept at −80°C until used for laser microdissection. In some animals, the liver was also sampled.
Choroidal glutathione-depleted animals.
Two-day-old rat pups were treated with DL-buthionine-(S,R)-sulfoximine (BSO; catalog B2640, Sigma-Aldrich) to generate developing animals that display a severe (92%) GSH depletion in blood–CSF barrier cells. The pups were treated by three intraperitoneal injections of 2 μmol/g depleting agent in dimethylsulfoxide/NaCl 0.9% (15/85), performed on 2 consecutive days (9:00 A.M. and 4:00 P.M. on P2, and 9:00 A.M. on the following day). Half of the litter was treated with BSO, and the second half was treated with vehicle only. Animals were used for permeability measurement 4 h after the last injection (see Fig. 3).
Figure 3.

Characterization of the choroid plexus glutathione-depleted animal model. A, Levels of GSH in tissue from animals treated with BSO. Choroidal tissue is depleted from GSH by >90%, while 40% of GSH is still present in brain tissue after BSO treatment. Data are calculated as micromoles per milligram of tissue and are expressed as the percentage of values measured in DMSO-treated control animals, and the mean ± SEM (n). *p < 0.05, **p < 0.01, ***p < 0.001, different from control animals. $$p < 0.01, different from Cx or Pons, two-tailed Student's t test for unequal variance. LVCP and 4VCP, choroid plexuses from the lateral and fourth ventricle, respectively; Cx, Cerebral cortex; Hyp, hypothalamus. B, Animal weight from the time of treatment initiation (T0) to the time of killing, 4 h following the third injection (T28). Animals were treated by three injections of BSO or vehicle (control). A typical experiment is shown. Data are expressed as the percentage of weight at T0, mean ± SEM, n = 6. No difference in weight gain was observed between the two groups of animals. C, Enzymatic GST activity toward CDNB in homogenates from tissue sampled from control (black bars) and GSH-depleted (gray bars) animals. BSO treatment did not induce any significant change in GST-specific activity. Data are expressed as the mean ± SEM, n = 3. D, Blood–CSF permeability constant (Kin csf) and apparent blood–cerebral cortex permeability constant (AppKin Cx) for [14C]-sucrose in control (black bars) and BSO-treated (gray bars) animals. No difference in permeability was observed between the two groups of animals, indicating that the integrity of the blood–brain and blood–CSF barriers is preserved following BSO administration. Data are expressed as the mean ± SEM, n = 5.
Glutathione S-transferase activity measurement.
Choroidal tissue, cerebral cortices, and liver were sampled from P2, P7, P35, and adult rats. Tissues were homogenized in a 0.25 m sucrose, 50 mm K-phosphate, 1 mm K-EDTA, and 0.1 mm dithiothreitol buffer, pH 7.4, using a Tenbroeck homogenizer. Glutathione S-transferase activity was determined by a 15 min kinetic analysis in a M200 Pro TECAN plate spectrofluorimeter. Assays were conducted in 250 μl of 0.05 m K-phosphate and 0.02% bovine serum albumin, pH 6.5 at 22°C, using 1 mm GSH (Sigma-Aldrich), and 50 μm monochlorobimane (MCB; Sigma-Aldrich) as a substrate. Excitation and emission wavelengths of 355 and 460 nm, respectively, were used to detect and quantify the rate of appearance of the GSH conjugate GS-MCB. The low rate of nonenzymatic conjugation of MCB to GSH was assessed by omitting the protein homogenate from the complete assay mixture. Production of the conjugate was calibrated in each sample by measuring the increase in fluorescence intensity produced by the addition of 1 nmol of pure GS-MCB at the end of the kinetic analysis. The specific activity was then expressed as nanomoles of conjugate produced per minute and per milligrams of proteins. The total protein content of the samples was determined by the method of Peterson (1977) with bovine serum albumin as the standard. The GS-MCB used as a calibrator was prepared and purified as follows. Known concentrations of MCB were incubated with rat liver homogenate and GSH in excess in a 37°C water bath for 20 min to produce GS-MCB. Final concentration of the standard was calculated after determining the presence of residual unconjugated MCB by HPLC, as previously described for another conjugate (Ghersi-Egea et al., 2006).
For in vitro competition studies, GST activity was measured toward 1-chloro-2,4-dinitrobenzene (CDNB) in the presence of 1 mm GSH and 50 or 200 μm competitor. These competitor concentrations were about twofold and eightfold higher than the apparent affinity constant for CDNB in choroidal tissue. Production of the conjugate was followed at 340 nm by kinetic spectrophotometry, as previously described (Ghersi-Egea et al., 2006). All the potential competitive substrates were tested for their limited absorbance at 340 nm.
Imaging of GSH conjugation in ependyma-containing brain slices and isolated choroid plexuses.
Brains of P6 animals were dissected under a stereomicroscope in ice-cold KR buffer to isolate the midbrain tissue. The latter was sliced in ice-cold KR buffer using a McIllwain tissue chopper to generate 200-μm-thick ependyma-containing brain slices. Freshly cut brain slices were warmed to 22°C in a few microliters of buffer on a glass slide under a Zeiss AxioPlan 2 Microscope, equipped with the Digital Camera F-View II and AnalySISauto software (Olympus Soft Imaging Solutions). Fluorescence images were recorded at 460 nm with a 390 nm excitation beam switched on only intermittently. This protocol prevented photobleaching, the acute cellular toxicity induced by UV waves, and the depletion of the cellular pool of ATP that is necessary for transport processes. An initial image was recorded in the ependymal area (negative control). Buffer was removed and replaced with a 10 μm MCB solution, and fluorescence images were again recorded 0.7 and 4 min after MCB addition. All images were recorded with the same time exposure. On each recorded image, a fluorescence intensity profile was established along a rectangle perpendicular to the ependymal cell layer using ImageJ software. This yielded average fluorescence intensity data in both the ependyma and in the adjacent tissue.
For fluorescence image recording on choroid plexus, the choroidal tissue was rapidly dissected from the brain of P5 animals in ice-cold KR buffer, transferred on a glass slide in a few microliters of buffer, maintained with a 300 μm mesh sieve, and allowed to warm up at 22°C before MCB addition and image recording, as described above.
Intracerebroventricular monochlorobimane injection and imaging of GSH conjugation.
P4 rats were set in a stereotaxic frame under inhaled anesthesia (4–5% isoflurane, 60%/40% O2/air), associated with a local topical application of lidocaine/prilocaine (Anesderm, Pierre Fabre). Two microliters of artificial CSF (124 mm NaCl, 2.5 mm KCl, 2.5 mm CaCl2, 1.3 mm MgCl2, 26 mm NaHCO3, 1.25 mm Na2HPO4, and 10 mm glucose, pH 7.3) containing 0.7% EtOH and 200 μm MCB were injected over 10 s in the lateral ventricle using a 30 gauge needle. Stereotaxic coordinates relative to λ were +4 mm anteroposterior, +1.2 mm lateral, and −2.4 mm dorsoventral. The needle was removed 50 s after the end of the injection, and the head of the anesthetized animal was immediately severed and frozen in −40°C isopentane. The head was then sliced in a cryostat as previously described (Ghersi-Egea et al., 2015), and 20-μm-thick coronal sections were dried immediately on a plate heater set at 37°C. Fluorescence images were instantly recorded using the Zeiss AxioPlan 2 Microscope.
Primary culture of choroidal epithelial cells.
Primary cultures of choroid plexus epithelial cells were prepared from neonate rat choroid plexuses, as previously described (Strazielle and Ghersi-Egea, 1999; Strazielle et al., 2003). The cells were seeded on microporous Transwell polycarbonate filter inserts (either 0.33 or 1.12 cm2 surface area, and 0.4 μm pore size; Corning Life Sciences) and grown in culture medium consisting of Ham's F-12 medium and DMEM (1:1) supplemented with 10% FCS, 2 mm glutamine, 50 mg/ml gentamicin, and several growth factors, as previously reported (Strazielle and Ghersi-Egea, 1999). The medium was renewed every other day until the permeability studies, which were performed 5 d after cells reached confluence.
In vitro cellular permeability and metabolic studies.
Permeability and metabolic studies were performed on a rotating shaker (300 rpm) at 37°C, in Ringer-HEPES buffer (RH; 150 mm NaCl, 5.2 mm KCl, 2.2 mm CaCl2, 0.2 mm MgCl2, 6 mm NaHCO3, 2.8 mm glucose, and 5 mm HEPES, pH 7.4). The volumes used in the two compartments of the inserts (the bottom basolateral one mimicking blood, and the top apical one mimicking CSF) were selected as to be in equilibrium to prevent any hydrostatic pressure effect. Inserts were rinsed once with RH buffer before initiating the transport studies. MCB was added to the basolateral compartment at a concentration of 5 or 50 μm. At regular time intervals, the inserts were transferred into another well containing fresh incubation solution, and four-fifths of the volume in the apical compartment was sampled and replaced with fresh prewarmed RH. Media from the apical and basolateral compartments were analyzed by spectrofluorimetry (excitation/emission at 355/460 nm) for GS-MCB and by HPLC for MCB. The organic anion transporter inhibitor probenecid was used at a concentration of 3 mm in both compartments. It was added 30 min before incubating the cells with MCB, and was kept and renewed in both compartments following each sampling. Monochlorobimane clearance volumes were calculated for each time interval, taking into account the residual volume remaining in the apical compartment from the previous time point, as previously described (Strazielle and Preston, 2003). For quantification of spectrofluorimetric data, the GS-MCB calibrator described above for enzymatic measurement was used. Experiments were run in parallel using cell-covered filters in the absence of MCB, with and without probenecid, to measure the background fluorescence levels associated with the inhibitor or any solute secreted by the epithelial cells during the transfer experiment. Background fluorescence was subtracted from the total fluorescence before calculating the amount of GS-MCB released by the cells in both compartments. The intracellular GS-MCB content was assessed as follows: inserts were rinsed twice for 5 min in RH buffer at 4°C on an orbital shaker; the cell-covered filters were quickly detached from their plastic support and stored at −20°C in 100 μl of water/ethanol (98/2 v/v) containing 250 μm ethacrynic acid. After thawing and homogenization by trituration using a conic plastic pestle, an aliquot of the liquid was analyzed by spectrofluorimetry as described above. Ethacrynic acid was added to the medium to prevent further conjugation of residual MCB to GSH during cell homogenization.
To assess the innocuousness of the different compounds and vehicles added on the epithelial cell layer, [14C]-sucrose permeability was measured as an index of barrier integrity. Data were expressed as permeability coefficients Pe (in centimeters per minute) calculated from the clearance curves generated as previously described in detail (Strazielle and Ghersi-Egea, 1999; Strazielle et al., 2003). Radioactivity was determined by liquid scintillation counting using a β-counter (Tri-Carb 1600 TR, Canberra Packard).
In vivo blood–CSF transport studies.
The permeability of the blood–CSF barrier to CDNB was measured in BSO-treated and untreated animals by perfusing the compound into the vascular system through the heart and sampling CSF. Animals were anesthetized with pentobarbital (5 μl/g), and the heart was exposed. Intracardiac perfusion of a physiological buffer (128 mm NaCl, 2 mm NaHCO3, 4.2 mm KCl, 2.4 mm NaH2PO4, 1.5 mm CaCl2, 0.9 mm MgSO4, and 9 mm d-glucose) was performed at a physiological rate (0.67 ml/min) for 2 min using a dual-perfusion pump (AL-1000, WPI). This was followed by a 5 min perfusion of the same buffer supplemented with CDNB at various concentrations. This cardiac perfusion protocol did not alter the choroid plexus morphology, as assessed by hematoxylin and phloxin histology staining and claudin-1 immunolabeling of sections of brains sampled after perfusion (data not shown). After the perfusion was stopped, the animal was turned over and the skin over the cistern magna was quickly excised. CSF (4–8 μl) was sampled using a glass microcapillary sampler with an outer tip diameter of ∼500 μm. CSF volume was precisely measured. CSF was then mixed with acetonitrile/H2O (50/50) to reach a total volume of 40 μl and analyzed by HPLC for its CDNB content. The blood–CSF barrier integrity in control and BSO-treated animals was assessed by measuring the Kin csf permeability constant for [14C]-sucrose as previously described (Ghersi-Egea et al., 2015). The apparent blood–cerebral cortex permeability constant AppKin Cx for [14C]-sucrose was measured in a similar way, without correcting for the amount of sucrose associated with the tissue for the residual vascular content.
Quantification of monocholorobimane, 1-chloro-2,4-dinitrobenzene, and reduced glutathione.
A Shimadzu LC10 HPLC System was used. Monochlorobimane was quantified using UV detection at 236 nm. CSF, buffers, and media were directly injected on a 150 mm hypersil 5 μm C18 BDS column (Thouzart and Matignon), and the compound was eluted using an isocratic 20/80 acetonitrile/water mobile phase at a 1 ml/min flow rate. The monochlorobimane retention time was 8 min. 1-Chloro-2,4-dinitrobenzene was quantified on an Ultraphere ODS RP-18 Analytical Column (Beckman) using an isocratic 55/10/35 methanol/10 mm phosphate buffer pH 6/water mobile phase at a 1 ml/min flow rate. It was detected at 250 nm with a retention time of 11 min. Reduced glutathione was coupled with 5,5′-dithio-bis(2-nitrobenzoic) acid in 10 mm phosphate buffer, pH 7.4, and analyzed on the Ultrasphere ODS RP-18 column, using a methanol/phosphate buffer elution gradient as previously described (Schmitt et al., 2011). UV detection was set at 324 nm, and the retention time was 6.6 min. For all compounds, the area under the curve of the peak of interest was measured, and quantification was performed using a calibration curve generated from standard solutions.
Total RNA extraction, reverse transcription, and real-time PCR.
Liver, choroid plexuses from lateral and fourth ventricles, and cerebral cortex from P2, P10, and adult Sprague Dawley rats were sampled. When collected from P2 and P10 pups, choroid plexuses were pooled from three to four animals. For comparative purpose, RNA was also extracted from two batches of microvessels prepared from P9 and adult rat brain, as described previously in detail (Gazzin et al., 2008). Total RNA was extracted using RNeasy Mini (liver) or Micro (choroid plexuses, cortex, and capillaries) kits (Qiagen) according to the manufacturer instructions. Before RNA extraction, liver, cerebral cortex, and capillaries were homogenized in the RLT buffer using a FastPrep shaker (QBiogene). Capillary homogenates were treated with Proteinase K (Qiagen) at 55°C for 10 min. Choroid plexus tissues from lateral and fourth ventricles were dissociated by pipetting. All samples were treated on column with DNase I as recommended by the manufacturer. RNA was quantified using a NanoDrop spectrophotometer (Thermo Fisher Scientific), and RNA integrity was assessed using the Agilent 2100 Bioanalyzer.
RNA was reverse transcribed using oligo(dT) primers and the Verso SYBR Green 2-step RT-qPCR Rox Kit (Thermo Fisher Scientific). Total RNA (1 μg) was spiked with 50 pg of AraB RNA (l-ribulokinase, E. coli strain K-12, substrain MG1655, GE Healthcare Bio-Sciences). The bacterial RNA was added in each reaction mixture as an external standard for normalization. Reverse transcription was run at 42°C for 60 min. Quantitative PCR (qPCR) was performed using the FastStart DNA Master SYBR Green I Kit in the LightCycler 1.5 instrument (Roche Applied Science), as previously described (Kratzer et al., 2012). Primer sequences, MgCl2 concentrations, and sizes of the predicted amplicons are presented in Table 1. Results were analyzed using RelQuant 1.01 software (Roche Applied Science), which takes into account the respective amplification efficiencies of the target gene and the external standard (AraB) when calculating the target gene/standard ratio. An approximate ranking of abundance of the different GST gene products was generated first by establishing the expression levels of all genes in a reference sample, arbitrarily chosen as adult liver N°2, and second by calculating for each target gene the expression levels in all individual samples relative to the value in the reference sample, as previously described (Kratzer et al., 2012).
Table 1.
Primers used for RT-qPCR
| Target gene | Forward primer sequence 5′ > 3′ | Reverse primer sequence 5′ > 3′ | Product size (bp) | [MgCl2] |
|---|---|---|---|---|
| GSTM1 | CCA CAT TTT TGA GCC CAA GT | TGA GTG CCA GTG TAG CAA GG | 179 | 3 mm |
| GSTM2 | TTC GCC TGT TCC TGG AGT AT | CCA TAG CCT GGT TCT CCA AA | 267 | 3 mm |
| GSTM3 | CAT CCC GGA GAA GAT GAA AA | CAG GTC TTG GGA GAA AGC TG | 233 | 3 mm |
| GSTM4 | TAC AAG GGT GGC TAC TTG GG | GCC TAG GCT GTA GAG GGC TT | 191 | 3 mm |
| GSTM5 | GGT TTG CAG GAG AAA AGC TG | TGA TTG GCA TCT TGA AGC AG | 186 | 3 mm |
| GSTM6 | ACG GAA GCC TGA GTT CTT GA | CCA CGT GGC CTG TTT TAA GT | 280 | 3 mm |
| GSTA1 | AGT TGG GAG CTG AGT GGA GA | TTT GGT GGC GAT GTA GTT GA | 297 | 3 mm |
| GSTA3 | CAG CCA GAG GAA GCC ATT AG | CCC AGC AAT TCC TCA TCA GT | 228 | 3 mm |
| GSTA4 | GTT CCT GCA GAC AAA ATC CC | GCA TCC ATC CTT TTG CAA CT | 195 | 4 mm |
| GSTP1 | TGT ACT TCC CAG TTC GAG GG | GGG TGA GGT CTC CAT CTT CA | 168 | 3 mm |
| Arab (Escherichia coli) | ATC CCC TGA TCG GTA AAG CA | ACG CCT GAA AGG GGT GAT TA | 126 | 4 mm |
Ependyma laser microdissection and sample processing.
Brains were dissected from P2, P9, and adult Sprague Dawley rats. Twelve-micrometer-thick sections were cut around bregma using a Shandon Cryotome E (Thermo Fisher Scientific) in an RNase-free environment, rapidly air dried, frozen on dry ice, and kept at −80°C until further processed for laser microdissection. Sections were thawed and stained using the Ambion LCM Staining Kit (Thermo Fisher Scientific), according to the manufacturer instructions. Briefly, sections were fixed in 95% ethanol, rehydrated into 75% and 50% ethanol, and stained in cresyl violet. They were dehydrated in 75% to 100% ethanol solutions, then in xylene and kept in a vacuum chamber with dessicant until used to perform microdissection. The PixCell II laser capture microdissection instrument (Arcturus Engineering) was used to isolate the ependymal layer of the lateral ventricle wall and adjacent caudate–putamen tissue (see Fig. 8). The captured tissue was lysed in RLT buffer and RNA was extracted and treated with DNase I using the RNeasy Micro Kit, as described above. RNA integrity was assessed using the Agilent 2100 Bioanalyzer and RNA was quantified using the Quant-iT RiboGreen Reagent (Thermo Fisher Scientific). RNA samples (5 ng) were spiked with 0.125 pg of bacterial AraB RNA. Two amplification rounds were performed using the ExpressArt NANO RNA Linear Amplification Kit (amsbio). The quality of amplified RNA was assessed using the Agilent 2100 Bioanalyzer, and reverse transcription of 1 μg of RNA was performed using the iScript Reverse Transcription Supermix for RT-qPCR (Bio-Rad). Real-time PCR was performed as described above.
Figure 8.
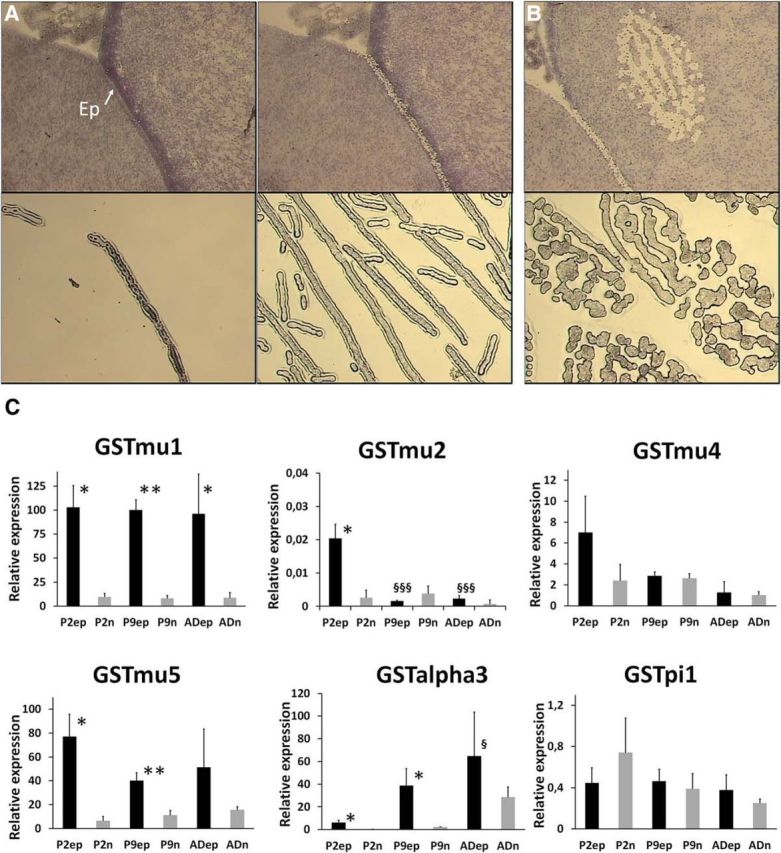
Glutathione S-transferase transcript levels in ependymal cells and adjacent neuropil during postnatal development. A, Top, An example of a tissue section before and after laser microdissection of the ependyma. The lower sections show the ependyma microdissected from the tissue and a preparation of pooled microdissected ependyma. B, The two panels show the area of adjacent neuropil analyzed and the microdissected tissue obtained. C, Transcript levels of selected GST isoenzymes at three developmental stages. The results, expressed relative to the bacterial AraB gene added as an external standard, are shown as the mean ± SD, n = 3. The results are expressed as arbitrary units and are all normalized to the expression level found for GSTμ1 in one sample of ependyma obtained from P2 animals, and set arbitrarily at 100. GSTα1, GSTα4, and GSTμ6 were not detected in microdissected samples. *p < 0.05 and **p < 0.01, statistical significance of the difference in expression levels between the ependyma and the adjacent neuropil, one-tailed paired Student's t test. §p < 0.05 and §§§p < 0.001, statistical significance of the differences in expression levels in P9 and adult animals compared with P2 animals in a given tissue, one-way ANOVA followed by a Tukey's multiple-comparison test. Ep, Ependyma; P2ep and P2n, ependyma and adjacent neuropil from P2 rat; P9ep and P9n, ependyma and adjacent neuropil from P9 rat; ADep and ADn, ependyma and adjacent neuropil from adult rat.
Western blot analysis.
Western blot analysis of GSTs in tissue sampled from P2 or P9 rats was performed using anti-GSTα3 rabbit polyclonal antibody (pAb; MBS713631, MyBioSource), anti-GSTμ1/2 rabbit monoclonal Ab (ab108524, Abcam), anti-GSTμ5 rabbit pAb (MBS712299, MyBioSource), and anti-GSTπ1 rabbit pAb (PAB13020, Abnova). Actin used as a loading control was detected with anti-β-actin mouse monoclonal Ab (A1978, Sigma-Aldrich). Proteins were separated on 10% Bis-Tris Criterion XT Gels (Bio-Rad) run in MOPS [3-(N-morpholino)propanesulfonic acid] buffer (Bio-Rad) at 70 V for 10 min, followed by 120 V for 2 h. The gel was blotted onto 0.2 μm Protran nitrocellulose membrane (GE Healthcare Life Sciences) in Tris-glycine transfer buffer (Euromedex) containing 20% methanol for 50 min at 100 V. The membrane was saturated in 5% skimmed milk, 0.1% Tween 20 in Tris-buffered saline (TBS) at room temperature for 1 h. Primary anti-GST antibodies were diluted in saturation buffer at 0.69 μg/ml for GSTμ1/2, 0.7 μg/ml for GSTμ5, 1 μg/ml for GSTπ1, and 0.31 μg/ml for GSTα3. The anti-β-actin mouse Ab was used at a final dilution of either 0.1 or 0.05 μg/ml, depending on the amount of protein loaded. Membranes were incubated at 4°C overnight, then washed three times for 10 min in TBS containing 0.1% Tween-20 (TBS-T) at room temperature. Horseradish peroxidase (HRP)-conjugated secondary antibodies were from Jackson ImmunoResearch. Donkey anti-rabbit IgG pAb (711-036-152) was used at a final concentration of 0.04 μg/ml for GSTα3, GSTπ1, and GSTμ1/2, and 0.08 μg/ml for GSTμ5, and donkey anti-mouse IgG pAb (catalog #715-036-151, Jackson ImmunoResearch) was used at a final concentration of 0.04 μg/ml. After two washes in TBS-T and a final one in TBS, chemiluminescent HRP substrate (SuperSignal West Pico Chemiluminescent Substrate, Thermo Fisher Scientific) was used and membranes were exposed to x-ray films (Hyperfilm ECL, GE Healthcare Life Sciences). Only one band was observed for GSTμ1/2 (28 kDa), GSTμ5 (26 kDa), GSTα3 (26 kDa), and GSTπ1 (24 kDa) in postnatal choroid plexus. Two bands were observed in liver homogenate for GSTα3 (25 and 26 kDa).
Experimental design and statistical analysis.
Quantitative data related to animal physiology, barrier function, and enzymatic processes are reported as the mean ± SEM. Data from RT-qPCR analysis are expressed as the mean ± SD. Multigroup comparisons were analyzed using one-way ANOVA followed by a Tukey's multiple-comparison test. Statistical analyses involving two groups of data were performed using either a one-tailed or two-tailed Student's t test, as appropriate. Lateral and fourth ventricle choroid plexuses were dissected and analyzed separately in each animal, or in batches of animals. Analyses of differences between these two tissues were therefore performed using a paired t test. For clarity, the statistical tests used are specified in each figure. The number of samples analyzed per group was chosen in reference to similar previously published experimental approaches conducted by our group on choroid plexuses. These numbers are indicated on graphs or in figure legends. All qualitative data presented have been reproduced at least twice on different batches of materials.
Results
Enzymatic glutathione conjugation at the blood–CSF interface efficiently protects the CSF and brain during postnatal development
Substrates like CDNB or MCB are conjugated with GSH by the whole subset of GST isoenzymes involved in xenobiotic detoxification (Eklund et al., 2002; Hayes et al., 2005). Conjugation of either compound therefore reflects the overall GST-dependent detoxifying capacity of a given tissue. While CDNB conjugation produces a metabolite that is readily detected and quantified by HPLC, MCB presents the advantage in generating a fluorescent conjugate that can be detected by both spectrofluorimetry and cellular imaging techniques. We first used MCB to establish the postnatal developmental profile of GST activity in tissue homogenates prepared from lateral and fourth ventricle choroid plexuses, and from cerebral cortex (Fig. 1). Liver, which is the main detoxifying organ in the body, was analyzed in parallel as a reference tissue. Glutathione S-transferase-specific activity increased sharply in liver in P35 animals compared with P2 pups, indicating the relative immaturity of this organ at birth in the rat. In contrast, GST activity was already high in both types of plexuses in newborn animals and was higher than in the brain cortical tissue. From 5 weeks on, the activity toward MCB measured in the choroidal tissue from the fourth ventricle was twice that measured in the choroidal tissue from the lateral ventricle, as also observed when using CDNB as a substrate (Ghersi-Egea et al., 2006). The activity in choroid plexuses from the fourth ventricle remained significantly higher than in the cerebral cortex at all developmental stages.
Figure 1.

Glutathione S-transferase-specific activity in liver and brain tissues in the rat during postnatal development. The specific activity of GST was measured using MCB as a substrate in liver (A) and brain tissue (B) homogenates prepared from P2, P7, P35, and adult (Ad) animals. Data are the mean ± SEM; n = 4–8. ***p < 0.01, statistically different from cerebral cortex activity at matching developmental stage, one-way ANOVA followed by post hoc Tukey-Kramer test. §p < 0.05 and §§§p < 0.001, significantly different from the activity measured in choroid plexus of the fourth ventricle, two-tailed paired Student's t test. CP(VL) and CP(4V), CP of the lateral and fourth ventricle, respectively; Cx, cortex.
To explore the GST-dependent conjugating capacity of live choroidal epithelial cells and investigate the fate of the metabolite, we exposed freshly isolated choroid plexuses to 10 μm MCB and recorded fluorescence images in a plane cutting through both the epithelium and the stroma (Fig. 2A–F). The choroidal epithelium became rapidly and intensely fluorescent (Fig. 2C,E, arrowheads), indicating that MCB is readily conjugated to GSH within epithelial cells. The fluorescence in the epithelial layer decreased thereafter (Fig. 2D,F), likely as a result of the sharp drop in MCB concentration that we measured in the incubation medium (data not shown) and the concomitant cellular export of the fluorescent metabolite, which was observed in the underlying stroma at 4 min (Fig. 2F, arrows). This suggests that the conjugate is exported from the cells at least partially at the basolateral membrane. To get more insight into the conjugation capacity of the choroidal epithelium and to characterize the polarity of conjugate efflux from the choroidal cells, we used an in vitro cellular model of the blood–CSF barrier (Fig. 2G), which closely reproduces the properties of the choroidal epithelium in vivo. In particular, cells in culture maintain the drug metabolism capacity of the native tissue, including its high GST-specific activity (Strazielle and Ghersi-Egea, 1999; Strazielle et al., 2003). Epithelial cells were exposed to 5 μm MCB in the lower compartment of the bichamber culture device. The basolateral to apical (blood to CSF) diffusion of MCB across the epithelial monolayer was completely prevented for up to 30 min, presumably by intracellular MCB conjugation, and remained low thereafter (Fig. 2H). Over the 60 min period, the clearance of MCB did not exceed the clearance of sucrose used as a paracellular marker (data not shown). Adding CDNB as a competitor (data not shown) or raising the MCB concentration from 5 to 50 μm (Fig. 2H) negated the barrier effect. To ascertain the metabolic nature of this barrier mechanism, the appearance of the conjugate GS-MCB in the basolateral and apical compartments and its intracellular concentration were monitored by HPLC analysis after a 30 min exposure to 5 μm MCB. The conjugate was indeed produced and secreted from epithelial cells, and its efflux rate was 10 times higher at the basolateral membrane compared with the CSF-facing membrane (Fig. 2I). The conjugate was below detection level in the cell monolayer. When choroidal epithelial cells were preincubated with probenecid, an inhibitor of large specificity for organic anion transporters, GS-MCB efflux was reduced at both basolateral and apical membranes and its intracellular concentration increased (Fig. 2I). Collectively, this set of data confirms the strong basolateral (blood-facing) polarity of efflux that we observed in freshly isolated choroid plexuses, and demonstrates that cellular export of the GSH conjugate is mediated by organic anions transporters at both membranes.
Figure 2.
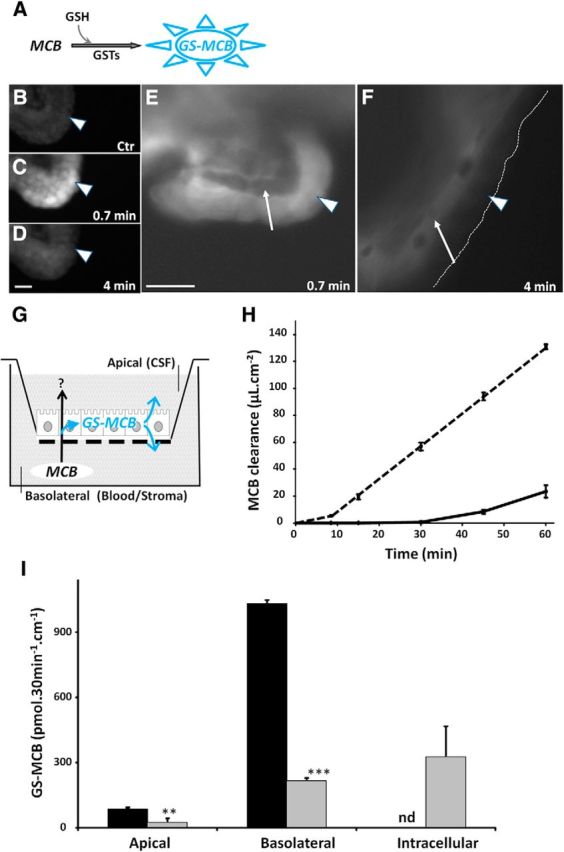
Enzymatic conjugation of MCB to glutathione in the blood–CSF barrier. A, Method: GST-mediated conjugation of the substrate MCB with GSH produces a fluorescent metabolite that can be monitored over time. B–F, Ex vivo exposure of freshly isolated choroid plexuses to MCB led to the rapid formation of the fluorescent GSH-conjugated metabolite within epithelial cells (arrowhead in C and E). After 4 min, the fluorescence signal decreased in the choroidal epithelial cells (arrowheads in D and F), and seemingly concentrated in the underlying vessel containing stroma (arrows in E and F). B is a negative control. B–D and E, F show, respectively, low-magnification and high-magnification images of the choroidal tissue exposed to 10 μm (C–D) and 2.5 μm (E–F) MCB. In F, the dashed line delimits the apical membrane of epithelial cells. Scale bar, 20 μm. G–H, Choroid plexus epithelial cells cultured on a porous filter (G) form an enzymatic barrier preventing the basolateral to apical transfer of MCB. When MCB was added to the basolateral compartment at a concentration of 5 μm, the appearance of the parent compound in the apical compartment was fully impeded for 30 min and remained strongly limited thereafter (H, full line). When the cells were exposed to 50 μm MCB, the enzymatic barrier effect lasted only 10 min, and the MCB clearance rate strongly increased thereafter (H, dashed line). I, Excretion of the conjugate produced within the choroidal epithelium was highly polarized, 90% of the metabolite being found in the basolateral medium. The conjugate was not detected intracellularly (black bars, cells exposed to 5 μm MCB for 30 min). When cells were preincubated with 3 mm probenecid before MCB exposure, GS-MCB efflux was strongly reduced at both basolateral and apical membranes, and the metabolite accumulated in the epithelial cells (gray bars). Data are the mean ± SEM of four filters. **p < 0.01 and ***p < 0.001, statistically different from GS-MCB amount in the absence of probenecid, one-tailed Student's t test for unequal variance. nd, Not detected.
We then assessed in vivo the efficacy of this enzymatic barrier mechanism in preventing the diffusion of GST substrates from blood to CSF during brain development. The blood–CSF barrier permeability to CDNB was measured in rat pups displaying either a normal or a low capacity to conjugate xenobiotics to GSH in the choroid plexus. CDNB was chosen over MCB as it can be unambiguously detected and quantified in biological fluids including CSF by HPLC. It is also more soluble than MCB and could be used at the high concentration required to approach the maximal GSH-dependent conjugation capacity of the blood–CSF barrier. Functional knockdown of GSH conjugation in choroid plexuses of developing animals was achieved by treating P2 rats with three injections of BSO over a 36 h period. Buthionine sulfoximine is a selective inhibitor of γ-glutamyl cysteine ligase, which catalyzes the first and rate-limiting step in GSH synthesis. Four hours after the last injection, the treatment led to an almost complete (92%) depletion of GSH in both the fourth and lateral ventricle choroid plexuses, while the depletion was only partial (60%) in the brain parenchyma (Fig. 3A). This mild BSO treatment did not induce any overt toxicity, as it modified neither the feeding behavior of P2 rats nor their weight gain (Fig. 3B). It did not change the GST intrinsic enzymatic activity in either the brain parenchyma or the choroid plexus (Fig. 3C). Finally, this treatment did not alter the integrity of CNS barriers, as shown by the similar CSF and brain Kin values measured for sucrose in control and treated animals (Fig. 3D).
Four hours following the last BSO injection, animals were underwent intracardiac perfusion of CDNB followed by CSF sampling. In control animals, CDNB was not detected in the CSF when the compound was perfused at concentrations up to 200 μm, but was measured at a low level when the perfusate concentration was raised to 500 μm (Fig. 4). The depletion of choroidal GSH resulted in a substantial increase in CDNB influx in CSF at both concentrations. When the xenobiotic was perfused at 500 μm, its concentration in CSF was 30-fold higher in depleted rats than in control animals. The outflow perfusate collected from the cardiac atrium did not show any significant difference in CDNB concentration between control and BSO-treated animals (Fig. 4, insert), ruling out a possible peripheral effect of the depleting agent on the xenobiotic distribution. The large difference in CDNB levels in CSF observed between control and choroidal GSH-depleted animals demonstrates that the GSH-dependent metabolic activity of the blood–CSF barrier effectively contributes to protect the brain environment from blood-borne chemical substrates during development.
Figure 4.

In vivo efficacy of the glutathione-dependent metabolic activity of the blood–CSF barrier toward CDNB. Levels of CDNB were measured in the CSF sampled from control animals (black) and animals depleted in choroidal GSH (BSO treated, gray) in P3 rats perfused with 200 or 500 μm CDNB. Data are expressed as the mean ± SEM, (n). **p < 0.01, statistically different from control animals, one-tailed Student's t test for unequal variance. Insert shows no changes in CDNB concentration measured in the outflow perfusate collected from the cardiac atrium between control and BSO-treated animals (mean ± SEM, no statistical difference).
The ependyma participates in cerebral glutathione-dependent detoxification
Exposure to toxic molecules for a prolonged period may ultimately overcome this choroidal neuroprotective metabolic mechanism. The ependymal cells lining the ventricular system will become the first targets. To address the possibility that these cells could be a site of subsequent protection, we evaluated their conjugating capacity by infusing MCB in the lateral ventricles of P4 rats. A strong fluorescence reflecting GS-MCB production was observed at the ependymal interface in the ipsilateral ventricle of animals killed within a minute from the beginning of the infusion, indicating that ependymal cells also have a high GSH conjugation capacity (Fig. 5A). The contralateral ependyma, used as a negative control, did not show any fluorescence above background level (Fig. 5B). A similar GSH conjugation activity toward MCB was observed downstream in the ventricular system, namely in the ependyma of the third ventricle (Fig. 5C). Monochlorobimane conjugation was further explored in ependyma-containing brain slices prepared from developing animals (Fig. 5D–F). Six-day-old animals were selected for these studies because at this developmental stage the ependyma appears mostly as a single layer, thus allowing a clear delineation between the ependymocytes and the adjacent tissue, and a more precise quantification of the signal. Following MCB addition to the slice incubation medium, a bright fluorescence reflecting the production of the GSH conjugate rapidly appeared within the ependymal layer, as illustrated at the level of the ventral part of the third ventricle (Fig. 5E). Production of the GSH conjugate in the ependyma was higher than in the adjacent brain parenchyma (Fig. 5E,G). The ependymal fluorescence intensity decreased after a few minutes (Fig. 5F), presumably due to the transporter-mediated efflux of the polar metabolite from ependymocytes. Accordingly, the addition of 2.5 mm probenecid in slice incubation medium prevented the decrease in fluorescence (Fig. 5G). Identical images were recorded for the ependyma bordering the aquaduct (data not shown). Collectively, these data show the ability of the ependyma to act as a second line of defense toward electrophilic compounds that could have bypassed the blood–CSF barrier and reached the CSF.
Figure 5.

Conjugation to glutathione in the ependyma. A–C, Intracerebroventricular injection of MCB induced a rapid and strong fluorescence signal in the ependyma, reflecting GS-MCB production in these cells. In the lateral ventricle close to the site of injection, the signal was seen in both the ependyma and choroid plexus (A). The contralateral ventricle was used as a negative control (B). The ependyma of the third ventricle was also labeled within 1 min following MCB injection (C). D–F, Freshly prepared ependyma-containing brain slices also generated GSH conjugates upon addition of MCB to the medium. D represents the initial image before MCB addition. The fluorescent metabolite GS-MCB was rapidly produced at a higher level in the ependyma than in the adjacent brain tissue (E). The concentration of GS-MCB in ependyma decreased over time (F). G, Relative quantification of fluorescence intensity recorded in the ependyma and adjacent brain tissue in the presence or absence of 2.5 mm probenecid. Data are expressed as percentage values of the signal measured in ependyma in the same slice 0.7 min after MCB addition; data are the mean ± SEM (n = 6). The box in E shows a typical area from which a fluorescent profile was obtained and analyzed (see Materials and Methods). *p < 0.05 and ***p < 0.001, statistically different, paired t test for unequal variance. $p < 0.05, statistically different, one-tailed t test for unequal variance. CP, choroid plexus; Ep, Ependymal layer; VL, lateral ventricle; 3V, third ventricle; PB, probenecid. Scale bars: A–C, 100 μm; D–F, 50 μm.
GSH conjugation at the blood–CSF and CSF–brain interfaces results from the expression of several multispecific isoenzymes
To probe into the diversity of molecules that could be detoxified by the choroid plexus during development, we performed GST activity competitive inhibition studies. The spectrophotometric method using CDNB as the substrate was chosen over the fluorimetric method with MCB to avoid interference of the competitors with the fluorescent signal. Choroidal CDNB conjugation was inhibited by several xenobiotics such as dietary additives, pharmacological drugs, or products of oxidative stress, which are all potentially toxic (Table 2). The inhibitory effect of these compounds on GST activity was similar in choroid plexuses of the lateral ventricle and fourth ventricle. Large differences were observed among molecules in their effective concentration and in the extent of inhibition. As detoxifying GST isoforms display differential substrate specificities toward the competitors tested, while CDNB is conjugated by all these isoenzymes, this suggests that several isoforms are active in choroid plexuses. Extensive multiple-concentration inhibition studies were not performed as they requested an ethically unacceptable number of developing animals to generate the required amount of choroidal tissue.
Table 2.
Inhibition of the choroidal glutathione S-transferase activity by environmental chemicals and therapeutic drugs
| Potential substrate | Potential substrate concentration (μm) | Reference substrate concentration CDNB (μm) | Origin of choroid plexus | Inhibition (%) |
|---|---|---|---|---|
| Ethacrynic acid | 5 | 200 | 4V | 91.7 ± 1.5 |
| LV | 81.2 ± 4.9 | |||
| Bromosulfophtalein | 40 | 200 | 4V | 72.0 ± 3.5 |
| LV | 73.3 ± 3.8 | |||
| Menadione | 80 | 200 | 4V | 41.2 ± 7.4 |
| LV | 50.0 ± 19.5 | |||
| MCB | 200 | 200 | 4V | 77.7 ± 1.5 |
| LV | 82.7 ± 2.1 | |||
| Chlorambucil | 200 | 200 | 4V | 83.5 ± 3.8 |
| LV | 84.6 ± 2.7 | |||
| Benzyl-isothiocyanate | 500 | 200 | 4V | 84.7 ± 3.2 |
| LV | 79.5 ± 6.7 | |||
| Sulforaphane | 200 | 50 | 4V | 26.7 ± 5.5 |
| LV | 19.5 ± 6.2 | |||
| Trans-4-phenyl- | 500 | 50 | 4V | 58.1 ± 7.0 |
| 3-buten-2-one | LV | 53.5 ± 6.9 | ||
| Acrolein | 500 | 50 | 4V | 53.4 ± 10.1 |
| LV | 48.4 ± 10.3 | |||
| Cisplatin | 500 | 50 | 4V | 41.5 ± 9.3 |
| LV | 27.7 ± 10.3 |
The potential substrates include diuretic, anticancer, or diagnostic drugs (ethacrynic acid, bromosulfophthalein, chlorambucil, cisplatin); dietary supplements and flavoring agents (menadione, trans-4-phenyl-3-buten-2-one); phytochemicals (benzyl-isothiocyanate, sulforaphane); or environmental pollutants (acrolein, also a product of lipid peroxidation). The competition was tested using the multispecific GST substrate CDNB as a reference substrate in both lateral ventricle and fourth ventricle choroid plexuses. Data, expressed as percentages of activity inhibition, are mean values ± SD (n = 3–4 from multiple CP pools). Owing to the limited choroidal material available per litter, they were generated for only one efficient competing concentration. The reference substrate concentration was typically 200 μm. When a low inhibition was observed, CDNB was tested at a lower concentration to increase the inhibitory action of the compound. The average specific activities measured in lateral ventricle choroid plexus tissue in the absence of inhibitors were 96 ± 22 and 54 ± 10 nmol/min/mg of proteins toward 200 and 50 μm CDNB, respectively. The same activities measured in fourth ventricle choroid plexus tissue were 87 ± 15 and 48 ± 10 nmol/min/mg of proteins (Data are the mean ± SD of three different batches).
We then determined by RT-qPCR the GST isoenzyme expression profiles in lateral ventricle choroid plexus, fourth ventricle choroid plexus, and cortical tissue at three developmental stages, and compared it to the isoenzyme profile in liver. Two preparations each of brain cortical microvessels isolated from P9 and adult rats were also analyzed (Fig. 6). We focused on isoforms involved in detoxification processes, namely GSTπ1 and the isoforms belonging to the μ and α subfamilies. Differences in y-scale values between graphs provide an approximation of the respective abundance of the various gene products in the samples. Glutathione-S-transferase isoenzymes were expressed at higher levels in the choroidal tissue forming the blood–CSF barrier than in the microvessels forming the blood–brain barrier. Levels of GSTμ isoform mRNAs increased during development in liver, while in choroid plexuses these isoforms were expressed at a higher or similar level in P2 animals compared with adults. GSTμ1 and GSTμ2 RNAs were the most abundant μ-isoform transcripts in the choroid plexuses. At P2, the expression levels of both genes in this tissue exceeded by far those measured in the liver, especially in lateral ventricle choroid plexuses (p < 0.001). The choroidal expression of GSTμ1 and GSTμ2 was also higher than in the cerebral tissue (p < 0.001 and p < 0.05 for lateral and fourth ventricle choroid plexuses, respectively). This was particularly striking for GSTμ2, whose messenger was barely detected in the cerebral parenchyma.
Figure 6.
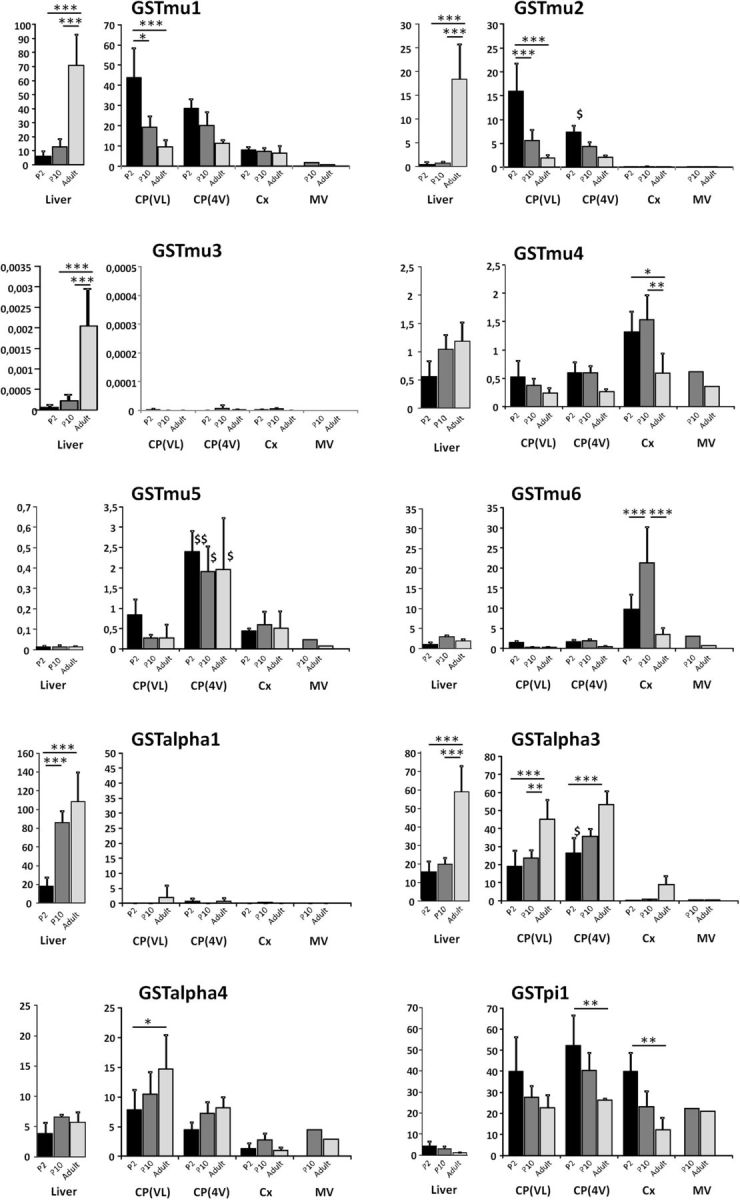
Developmental profiles of glutathione S-transferase transcript levels in choroid plexuses, cerebral cortex, cerebral microvessels, and liver in the rat. RT-qPCR was performed in tissues sampled at three developmental stages. For each isoenzyme gene, the right panel shows data obtained for cerebral structures, and the left panel shows liver data. The results, expressed relative to the bacterial AraB gene added as an external standard, are shown as the mean ± SD, n = 4, except for the microvessels, for which values represent the mean of two preparations. Gene expression levels did not differ by >44% between the two age-matched microvessel preparations or were both low (<0.3). The results are expressed as arbitrary units and are all normalized to the expression level found for GSTμ1 in one sample of liver obtained from an adult animal and set arbitrarily at 100 (see Materials and Methods; for details on calculations, see Kratzer et al., 2013). *p < 0.05, **p < 0.01, ***p < 0.001, statistical significance of differences in expression levels between developmental stages, analyzed by one-way ANOVA followed by a Tukey's multiple-comparison test. $p < 0.05 and $$p < 0.01, statistical significance of differences in expression levels between age-matched choroid plexuses of the lateral and fourth ventricle, two-tailed paired Student's t test. For clarity, other relevant statistical significances, assessed by one-way ANOVA followed by a Tukey's multiple-comparison test are stated in the Results section. CP(LV) and CP(4V), choroid plexuses from the lateral and fourth ventricles, respectively; Cx, Cerebral cortex; MV, cerebral microvessels.
These data were confirmed by Western blot analysis using an antibody recognizing both μ1 and μ2 isoforms (Fig. 7A). The μ5 isoform displayed a differential level of expression between lateral and fourth ventricle choroid plexuses (Fig. 6) that was not reflected at the protein level (Fig. 7B). Both choroidal tissues showed high protein content by Western blot compared with the cerebral cortex and the liver. High levels of GSTα1 expression were detected in the liver, but not in the different brain tissues. In contrast, similar expression levels were observed for GSTα3 in choroid plexuses and liver (Fig. 6; p ≤ 0.05). These levels increased with age and largely exceeded those measured in cortex. This tissue-specific pattern was confirmed by Western blot (Fig. 7C). The expression of GSTα4 did not vary to a great extent among tissues or with age. Finally, the choroid plexuses and the cortex displayed higher amounts of GSTπ1 transcripts than liver at all developmental stages (p ≤ 0.05), the levels in brain tissues being twofold higher in developing animals compared with adults (Fig. 6). The high choroidal expression relative to the liver was also observed by Western blot (Fig. 7D). In summary, the high conjugation activity measured during postnatal development in choroid plexuses compared with the cerebral cortex (Fig. 1) should be mainly catalyzed by the isoforms GSTμ1, GSTμ2, GSTα3, and possibly GSTπ1.
Figure 7.

Western blot analysis of several GST isoforms in choroid plexus isolated from the lateral and fourth ventricles of the developing rat. Tissue was isolated from 2 P2 (A, C, D) and P9 (B) animals. A, The GSTμ1/2 isoenzymes are expressed at a higher level in choroid plexuses than in the cerebral cortex and liver. B, Both types of choroid plexuses display similar levels of the GSTμ5 protein, which are higher than in the cerebral cortex and liver. C, The GSTα3 protein is abundant in both types of choroid plexuses relative to the cortical neuropil. D, The GSTπ1 isoenzyme is highly enriched in the choroid plexuses compared with the liver and is also more abundant than in the cerebral cortex. All analyses were repeated on two different sets of samples. CP(LV) and CP(4V), choroid plexuses from the lateral and fourth ventricles, respectively; Cx, Cerebral cortex; Li, liver; MW, molecular weight markers.
To assess the GST isoenzyme profile in the CSF–brain interface, ependymal cells were isolated by laser microdissection from the lateral ventricle wall at three developmental stages (Fig. 8A), and their mRNA was extracted and analyzed by RT-qPCR. The data were compared with those obtained from laser-microdissected adjacent neuropil (Fig. 8B). In the ependyma, all μ-isoforms were expressed at P2 at levels as high or higher than those measured at later stages (Fig. 8C). The most abundant mRNAs were found for GSTμ1 and GSTμ5. Their expression level was 11 and 12 times higher in the ependyma than in the adjacent neuropil. GSTα3 was also expressed at higher levels in ependyma than in the adjacent neuropil, its expression increasing with age in both tissues. GSTπ1 expression was modest and similar between structures and developmental stages. Thus, the high conjugation capacity of the ependyma, relative to the brain parenchyma that was observed at P6, appears mainly related to the GST isoforms μ1, μ5, and possibly α3.
Discussion
This article describes a GSH-dependent metabolic pathway that is especially active during early postnatal development, and by which the blood–CSF barrier prevents harmful compounds from circulating through the CSF–brain system. The GST activities measured in tissue homogenates at different developmental stages indicate that during the postnatal period the GSH conjugation capacity of the choroid plexuses is especially high compared with that of the cerebral tissue. It is also higher than the activity measured at the same age in the liver, which is the reference organ for detoxification processes (Gundert-Remy et al., 2014). This finding was supported by RT-qPCR and Western blot data showing the elevated expression of multiple GST isoforms in choroid plexus compared with cortex and liver during postnatal development. Live imaging of the metabolite formation in isolated choroid plexus illustrated the early functionality of the GST-mediated detoxifying pathway. Using a functional GSH conjugation knock-down strategy resulting in a selective and extensive GSH depletion in choroid plexuses, we showed that the high conjugation capacity of this interface translates into an efficient metabolic blood–CSF barrier, preventing the penetration and accumulation of GST substrates into the CSF during early postnatal development. Assuming that CDNB plasma concentration increased linearly from 0 to 500 μm over the 5 min perfusion in P3 animals in the present study, the influx permeability constant Kin csf for CDNB calculated as in the study by Ghersi-Egea et al. (2015) would be 0.3 × 10−3/min in control pups. Sucrose, used as a polar marker of the integrity of the blood–CSF barrier, has Kin csf values of 2.9 × 10−3/min in P2 rats and 2.6 × 10−3/min in P9 rats (Ghersi-Egea et al., 2015). The permeability of the blood–CSF barrier to the lipophilic compound CDNB, even when used at a high concentration, was therefore lower than that of cell membrane-nonpermeating molecules in control animals. Choroidal GSH depletion increased CDNB permeability 30 times, without affecting the barrier integrity. These findings demonstrate the great efficacy with which the choroidal enzymatic barrier pathway reduces the blood-to-CSF permeability of lipophilic molecules. The enzymatic mechanism is further coupled to the polarized excretion of the metabolites at the basolateral membrane of the epithelium for their subsequent elimination through the choroidal blood stream. The small fraction of the detoxified polar metabolite that is effluxed at the apical membrane could be further eliminated from the brain by the CSF bulk flow. Glutathione S-transferase activity in choroid plexuses in the developing human brain is as high or higher than the choroidal activity in newborn rats, and is also several fold higher than in the human neuropil (Ghersi-Egea et al., 2006). The potent enzymatic blood–CSF barrier we demonstrated in rat should also be efficient in the human neonate brain.
During the perinatal period, the brain tissue is still poorly vascularized (Caley and Maxwell, 1970; Saunders et al., 2012), and the choroid plexuses represent a major gate of entry into the CNS. The CSF represents 20% of the cranial cavity volume in P2 rats (Ghersi-Egea et al., 2015). It is considered to play a pivotal role in the delivery of multiple guidance molecules and growth factors to brain structures for their development and maturation (Redzic et al., 2005; Lehtinen et al., 2013). Several of these trophic factors are secreted into the CSF by the choroid plexuses, which grow and mature early during the embryonic period (Dziegielewska et al., 2001; Johansson et al., 2013; Lehtinen et al., 2013). By means of their potent enzymatic barrier activity, the plexuses will prevent a similar intracerebral distribution of deleterious electrophilic endobiotics/xenobiotics. They will thereby fulfill a crucial neuroprotective function during development, at a time when the liver is still immature in its xenobiotic metabolism activity (Coughtrie, 2015; Fig. 1).
The GSTs form a multigenic family whose members display different, yet partially overlapping, substrate specificities. Conjugation of CDNB in choroid plexuses was partially or completely inhibited in the presence of various chemicals. Published catalytic efficiency or affinity data showed that some of these molecules are preferred substrates for specific GST isozymes. For instance, bromosulfophthalein and trans-4-phenyl-3-buten-2-one are preferred substrates for rodent GSTμ isoforms (Hayes and Pulford, 1995; Sano et al., 2002). Cisplatin is a preferred substrate for GSTπ1 and ethacrynic acid for both GSTπ1 and α4 (Hayes and Pulford, 1995; Nagar et al., 2015). Both ethacrynic acid and its GSH conjugate also inhibit various other rat GSTs, in particular μ-isoforms, with IC50 values <5 μm (Ploemen et al., 1990). Data generated with human enzymes showed that acrolein is a preferred substrate for GST π while benzyl-isothiocyanate is metabolized by both μ1 and π1 isoforms (Berhane and Mannervik, 1990; Kolm et al., 1995). The pattern of inhibition produced by this large spectrum of molecules toward the multispecific GST substrate CDNB indicates that multiple isoenzymes of GSTs are significantly active in the choroid plexuses during the early postnatal period in rat. This was corroborated by the high levels of expression in the developing choroid plexus of various isoenzymes belonging to different GST classes, namely GSTμ1, GSTμ2, GSTα3, GSTα4, and GSTπ1. This unusually large pattern of isoenzymes is reminiscent of the adult liver GST load. For all compounds tested, differences in the extent of GST inhibition between lateral ad fourth ventricle choroid plexuses were minor, indicating that the forebrain and hindbrain blood–CSF interfaces display a similar broad spectrum of xenobiotic detoxification. The competitive inhibition study also showed that choroid plexuses metabolize a large range of structurally unrelated chemicals, including different drugs; cytotoxic, carcinogenic, or genotoxic electrophilic compounds issued from oxidative insults; environmental pollutants; or food additives (Table 2 legend). The blood–CSF enzymatic barrier should therefore be essential to prevent chemically induced abnormal neural development and reduce the risks of periventricular and meningeal tumors.
Intracerebroventricular injections of MCB in developing animals revealed that, like the choroidal epithelium, the ependyma displays a high conjugation capacity. Ex vivo conjugation experiments performed on brain slices showed that GST-mediated metabolism in the ependyma is markedly more active than in the surrounding neuropil. This indicates that the ependyma could act as a second line of defense for neural cells against electrophilic molecules that would reach the CSF. According to GST mRNA expression analysis, this protection remains active throughout development up to the adult stage. Neural stem cell niches are present in several discrete regions of the ventricular system, the subventricular zone in the lateral ventricle (Chaker et al., 2016), as well as subependymal regions of the third and fourth ventricles (Lin et al., 2015). They contribute to physiological neurogenesis and gliogenesis during brain development, and the lateral subventricular zone remains active later on in the healthy adult brain. All these niches also participate in adult brain repair following injuries such as stroke. The detoxifying functions of the ependyma may be essential to protect these regions throughout life. Astrocytes fulfill neuroprotective functions as well, especially against products of oxidative stress (Baxter and Hardingham, 2016). Because astrocyte growth and maturation occur at a late postnatal stage, it is conceivable that during the perinatal period ependymal GSTs play a major role in conjunction with choroidal enzymes in the detoxification of potentially neurotoxic lipid peroxidation products released by neural cells (Lovell et al., 2000; Hubatsch et al., 2002).
The profile of GST isoenzymes expressed in the ependyma differs from that in the choroid plexuses. Histologically, the choroidal epithelium and the ependyma share a neuroectodermal origin. Yet, the marked differential expression of GSTμ5 and GSTμ2 between these epithelial cell layers implies that they should be considered as independent interfaces with respect to their metabolic properties. The exact functional significance of this difference calls for additional investigations.
To be fully efficient, GSTs need to work in concert with cellular efflux systems that can export the metabolites out of the producing cells. In some instances, the efflux system can govern the cell sensitivity to toxicants (Paumi et al., 2001). We showed that intracellular GSH conjugates are exported by a probenecid-sensitive organic anion transporter at both the ependyma and the choroidal epithelium. The organic anion transporter Slc21a5 and the export pumps Abcc1 and Abcc4, which transport GSH conjugates, are present at the basolateral membrane of the choroidal epithelium (for review, see Ghersi-Egea et al., 2006). They likely participate in the GSH-dependent detoxification pathway in choroid plexus. The identity of the transporters involved in GSH conjugate efflux in the ependyma is more elusive, and the transporter-mediated ependymal efflux process discovered in this work opens a new field of investigation on transport mechanisms in the CSF–brain interface. Of note, the cystine/glutamate antiporter expressed in both the choroid plexus and the ependyma (Sato et al., 2002; Lee et al., 2012) should favor de novo GSH synthesis within these interfaces to replenish the intracellular pool of this essential cofactor.
In conclusion, the GSH-dependent metabolic detoxification capacity of the choroid plexuses and ependyma during the postnatal period identifies these structures as key actors in the protection of the developing brain, at a time when the astrocytic network is still immature and liver xenobiotic metabolism is limited. This reveals a new facet of the means by which the brain can protect itself at a period of high vulnerability. The choroid plexus–CSF pathway is also highly significant for drug delivery into the CNS during the perinatal period (Strazielle and Ghersi-Egea, 2016). Taking into account the metabolic process highlighted in this work will be useful in understanding the mechanisms setting the cerebral bioavailability of therapeutic agents at that developmental stage, and to optimize neonatal and pediatric pharmacological treatments.
Note Added in Proof: The names of the authors were accidentally incorrectly listed backwards in the Early Release version published March 5, 2018. The author names have now been corrected.
Footnotes
This work was funded in part by the European Union Seventh Framework Programme [No. HEALTH-F2-2009-241778 Neurobid, to N.S. (Brain-i) and to J.-F.G.-E. (FLUID)]. In vivo blood–CSF permeability studies and choroidal epithelial cell culture were performed at the BIP facility of Centre de Recherche en Neurosciences de Lyon, supported by ANR-10-IBHU-0003 Cesame grant. We thank Catherine Rey for valuable help and advice on laser microdissection and RT-qPCR, and Ouliana Barnéoud-Rousset and Aïda Roiges-Biel for technical help in enzymatic measurements.
The authors declare no competing financial interests.
References
- Baxter PS, Hardingham GE (2016) Adaptive regulation of the brain's antioxidant defences by neurons and astrocytes. Free Radic Biol Med 100:147–152. 10.1016/j.freeradbiomed.2016.06.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berhane K, Mannervik B (1990) Inactivation of the genotoxic aldehyde acrolein by human glutathione transferases of classes alpha, mu, and pi. Mol Pharmacol 37:251–254. [PubMed] [Google Scholar]
- Caley DW, Maxwell DS (1970) Development of the blood vessels and extracellular spaces during postnatal maturation of rat cerebral cortex. J Comp Neurol 138:31–47. 10.1002/cne.901380104 [DOI] [PubMed] [Google Scholar]
- Chaker Z, Codega P, Doetsch F (2016) A mosaic world: puzzles revealed by adult neural stem cell heterogeneity. Wiley Interdiscip Rev Dev Biol 5:640–658. 10.1002/wdev.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coughtrie MW. (2015) Ontogeny of human conjugating enzymes. Drug Metab Lett 9:99–108. 10.2174/1872312809666150602151213 [DOI] [PubMed] [Google Scholar]
- Dammann O, Leviton A (1999) Brain damage in preterm newborns: might enhancement of developmentally regulated endogenous protection open a door for prevention? Pediatrics 104:541–550. 10.1542/peds.104.3.541 [DOI] [PubMed] [Google Scholar]
- Dziegielewska KM, Ek J, Habgood MD, Saunders NR (2001) Development of the choroid plexus. Microsc Res Tech 52:5–20. [DOI] [PubMed] [Google Scholar]
- Eklund BI, Edalat M, Stenberg G, Mannervik B (2002) Screening for recombinant glutathione transferases active with monochlorobimane. Anal Biochem 309:102–108. 10.1016/S0003-2697(02)00258-0 [DOI] [PubMed] [Google Scholar]
- Favrais G, van de Looij Y, Fleiss B, Ramanantsoa N, Bonnin P, Stoltenburg-Didinger G, Lacaud A, Saliba E, Dammann O, Gallego J, Sizonenko S, Hagberg H, Lelièvre V, Gressens P (2011) Systemic inflammation disrupts the developmental program of white matter. Ann Neurol 70:550–565. 10.1002/ana.22489 [DOI] [PubMed] [Google Scholar]
- Gazzin S, Strazielle N, Schmitt C, Fevre-Montange M, Ostrow JD, Tiribelli C, Ghersi-Egea JF (2008) Differential expression of the multidrug resistance-related proteins ABCb1 and ABCc1 between blood-brain interfaces. J Comp Neurol 510:497–507. 10.1002/cne.21808 [DOI] [PubMed] [Google Scholar]
- Ghersi-Egea JF, Strazielle N, Murat A, Jouvet A, Buénerd A, Belin MF (2006) Brain protection at the blood-cerebrospinal fluid interface involves a glutathione-dependent metabolic barrier mechanism. J Cereb Blood Flow Metab 26:1165–1175. 10.1038/sj.jcbfm.9600267 [DOI] [PubMed] [Google Scholar]
- Ghersi-Egea JF, Babikian A, Blondel S, Strazielle N (2015) Changes in the cerebrospinal fluid circulatory system of the developing rat: quantitative volumetric analysis and effect on blood-CSF permeability interpretation. Fluids Barriers CNS 12:8. 10.1186/s12987-015-0001-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghersi-Egea JF, Strazielle N, Catala M, Silva-Vargas V, Doetsch F, Engelhardt B (2018) Molecular anatomy and functions of the choroidal blood-cerebrospinal fluid barrier in health and disease. Acta Neuropathol 135:337–361. 10.1007/s00401-018-1807-1 [DOI] [PubMed] [Google Scholar]
- Gundert-Remy U, Bernauer U, Blömeke B, Döring B, Fabian E, Goebel C, Hessel S, Jäckh C, Lampen A, Oesch F, Petzinger E, Völkel W, Roos PH (2014) Extrahepatic metabolism at the body's internal-external interfaces. Drug Metab Rev 46:291–324. 10.3109/03602532.2014.900565 [DOI] [PubMed] [Google Scholar]
- Hayes JD, Pulford DJ (1995) The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol 30:445–600. 10.3109/10409239509083491 [DOI] [PubMed] [Google Scholar]
- Hayes JD, Flanagan JU, Jowsey IR (2005) Glutathione transferases. Annu Rev Pharmacol Toxicol 45:51–88. 10.1146/annurev.pharmtox.45.120403.095857 [DOI] [PubMed] [Google Scholar]
- Hubatsch I, Mannervik B, Gao L, Roberts LJ, Chen Y, Morrow JD (2002) The cyclopentenone product of lipid peroxidation, 15-A(2t)-isoprostane (8-isoprostaglandin A(2)), is efficiently conjugated with glutathione by human and rat glutathione transferase A4–4. Chem Res Toxicol 15:1114–1118. 10.1021/tx020027r [DOI] [PubMed] [Google Scholar]
- Johansson PA, Dziegielewska KM, Ek CJ, Habgood MD, Liddelow SA, Potter AM, Stolp HB, Saunders NR (2006) Blood-CSF barrier function in the rat embryo. Eur J Neurosci 24:65–76. 10.1111/j.1460-9568.2006.04904.x [DOI] [PubMed] [Google Scholar]
- Johansson PA, Irmler M, Acampora D, Beckers J, Simeone A, Götz M (2013) The transcription factor Otx2 regulates choroid plexus development and function. Development 140:1055–1066. 10.1242/dev.090860 [DOI] [PubMed] [Google Scholar]
- Kolm RH, Danielson UH, Zhang Y, Talalay P, Mannervik B (1995) Isothiocyanates as substrates for human glutathione transferases: structure-activity studies. Biochem J 311:453–459. 10.1042/bj3110453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer I, Vasiljevic A, Rey C, Fevre-Montange M, Saunders N, Strazielle N, Ghersi-Egea JF (2012) Complexity and developmental changes in the expression pattern of claudins at the blood-CSF barrier. Histochem Cell Biol 138:861–879. 10.1007/s00418-012-1001-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzer I, Liddelow SA, Saunders NR, Dziegielewska KM, Strazielle N, Ghersi-Egea JF (2013) Developmental changes in the transcriptome of the rat choroid plexus in relation to neuroprotection. Fluids Barriers CNS 10:25. 10.1186/2045-8118-10-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A, Anderson AR, Rayfield AJ, Stevens MG, Poronnik P, Meabon JS, Cook DG, Pow DV (2012) Localisation of novel forms of glutamate transporters and the cystine-glutamate antiporter in the choroid plexus: implications for CSF glutamate homeostasis. J Chem Neuroanat 43:64–75. 10.1016/j.jchemneu.2011.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehtinen MK, Bjornsson CS, Dymecki SM, Gilbertson RJ, Holtzman DM, Monuki ES (2013) The choroid plexus and cerebrospinal fluid: emerging roles in development, disease, and therapy. J Neurosci 33:17553–17559. 10.1523/JNEUROSCI.3258-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Cai J, Nathan C, Wei X, Schleidt S, Rosenwasser R, Iacovitti L (2015) Neurogenesis is enhanced by stroke in multiple new stem cell niches along the ventricular system at sites of high BBB permeability. Neurobiol Dis 74:229–239. 10.1016/j.nbd.2014.11.016 [DOI] [PubMed] [Google Scholar]
- Lovell MA, Xie C, Markesbery WR (2000) Acrolein, a product of lipid peroxidation, inhibits glucose and glutamate uptake in primary neuronal cultures. Free Radic Biol Med 29:714–720. 10.1016/S0891-5849(00)00346-4 [DOI] [PubMed] [Google Scholar]
- Nagar R, Khan AR, Poonia A, Mishra PK, Singh S (2015) Metabolism of cisplatin in the organs of rattus norvegicus: role of glutathione S-transferase P1. Eur J Drug Metab Pharmacokinet 40:45–51. 10.1007/s13318-014-0176-y [DOI] [PubMed] [Google Scholar]
- Neuwelt E, Abbott NJ, Abrey L, Banks WA, Blakley B, Davis T, Engelhardt B, Grammas P, Nedergaard M, Nutt J, Pardridge W, Rosenberg GA, Smith Q, Drewes LR (2008) Strategies to advance translational research into brain barriers. Lancet Neurol 7:84–96. 10.1016/S1474-4422(07)70326-5 [DOI] [PubMed] [Google Scholar]
- Paumi CM, Ledford BG, Smitherman PK, Townsend AJ, Morrow CS (2001) Role of multidrug resistance protein 1 (MRP1) and glutathione S-transferase A1–1 in alkylating agent resistance. kinetics of glutathione conjugate formation and efflux govern differential cellular sensitivity to chlorambucil versus melphalan toxicity. J Biol Chem 276:7952–7956. 10.1074/jbc.M009400200 [DOI] [PubMed] [Google Scholar]
- Peterson GL. (1977) A simplification of the protein assay method of Lowry et al. which is more generally applicable. Anal Biochem 83:346–356. 10.1016/0003-2697(77)90043-4 [DOI] [PubMed] [Google Scholar]
- Ploemen JH, van Ommen B, van Bladeren PJ (1990) Inhibition of rat and human glutathione S-transferase isoenzymes by ethacrynic acid and its glutathione conjugate. Biochem Pharmacol 40:1631–1635. 10.1016/0006-2952(90)90465-W [DOI] [PubMed] [Google Scholar]
- Redzic ZB, Preston JE, Duncan JA, Chodobski A, Szmydynger-Chodobska J (2005) The choroid plexus-cerebrospinal fluid system: from development to aging. Curr Top Dev Biol 71:1–52. 10.1016/S0070-2153(05)71001-2 [DOI] [PubMed] [Google Scholar]
- Sano K, Totsuka Y, Ikegami Y, Uesugi T (2002) Metabolism of sulphobromophthalein II. species differences between rats, guinea-pigs and rabbits. J Pharm Pharmacol 54:231–239. 10.1211/0022357021778420 [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Okuno S, Sato K, Keino-Masu K, Masu M, Bannai S (2002) Distribution of cystine/glutamate exchange transporter, system x(c)-, in the mouse brain. J Neurosci 22:8028–8033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders NR, Liddelow SA, Dziegielewska KM (2012) Barrier mechanisms in the developing brain. Front Pharmacol 3:46. 10.3389/fphar.2012.00046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt C, Strazielle N, Richaud P, Bouron A, Ghersi-Egea JF (2011) Active transport at the blood-CSF barrier contributes to manganese influx into the brain. J Neurochem 117:747–756. 10.1111/j.1471-4159.2011.07246.x [DOI] [PubMed] [Google Scholar]
- Strazielle N, Ghersi-Egea JF (1999) Demonstration of a coupled metabolism-efflux process at the choroid plexus as a mechanism of brain protection toward xenobiotics. J Neurosci 19:6275–6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazielle N, Ghersi-Egea JF (2013) Physiology of blood-brain interfaces in relation to brain disposition of small compounds and macromolecules. Mol Pharm 10:1473–1491. 10.1021/mp300518e [DOI] [PubMed] [Google Scholar]
- Strazielle N, Ghersi-Egea JF (2015) Efflux transporters in blood-brain interfaces of the developing brain. Front Neurosci 9:21. 10.3389/fnins.2015.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazielle N, Ghersi-Egea JF (2016) Potential pathways for CNS drug delivery across the blood-cerebrospinal fluid barrier. Curr Pharm Des 22:5463–5476. 10.2174/1381612822666160726112115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strazielle N, Preston JE (2003) Transport across the choroid plexuses in vivo and in vitro. Methods Mol Med 89:291–304. 10.1385/1-59259-419-0:291 [DOI] [PubMed] [Google Scholar]
- Strazielle N, Belin MF, Ghersi-Egea JF (2003) Choroid plexus controls brain availability of anti-HIV nucleoside analogs via pharmacologically inhibitable organic anion transporters. Aids 17:1473–1485. 10.1097/00002030-200307040-00008 [DOI] [PubMed] [Google Scholar]
- Strazielle N, Khuth ST, Ghersi-Egea JF (2004) Detoxification systems, passive and specific transport for drugs at the blood-CSF barrier in normal and pathological situations. Adv Drug Deliv Rev 56:1717–1740. 10.1016/j.addr.2004.07.006 [DOI] [PubMed] [Google Scholar]