

Abstract
Chronic cerebral hypoperfusion is a characteristic seen in widespread CNS diseases, including neurodegenerative and mental disorders, and is commonly accompanied by cognitive impairment. Recently, several studies demonstrated that chronic cerebral hypoperfusion can induce the excessive inflammatory responses that precede neuronal dysfunction; however, the precise mechanism of cognitive impairment due to chronic cerebral hypoperfusion remains unknown. Transient receptor potential melastatin 2 (TRPM2) is a Ca2+-permeable channel that is abundantly expressed in immune cells and is involved in aggravation of inflammatory responses. Therefore, we investigated the pathophysiological role of TRPM2 in a mouse chronic cerebral hypoperfusion model with bilateral common carotid artery stenosis (BCAS). When male mice were subjected to BCAS, cognitive dysfunction and white matter injury at day 28 were significantly improved in TRPM2 knock-out (TRPM2-KO) mice compared with wild-type (WT) mice, whereas hippocampal damage was not observed. There were no differences in blood–brain barrier breakdown and H2O2 production between the two genotypes at 14 and 28 d after BCAS. Cytokine production was significantly suppressed in BCAS-operated TRPM2-KO mice compared with WT mice at day 28. In addition, the number of Iba1-positive cells gradually decreased from day 14. Moreover, daily treatment with minocycline significantly improved cognitive perturbation. Surgical techniques using bone marrow chimeric mice revealed that activated Iba1-positive cells in white matter could be brain-resident microglia, not peripheral macrophages. Together, these findings suggest that microglia contribute to the aggravation of cognitive impairment by chronic cerebral hypoperfusion, and that TRPM2 may be a potential target for chronic cerebral hypoperfusion-related disorders.
SIGNIFICANCE STATEMENT Chronic cerebral hypoperfusion is manifested in a wide variety of CNS diseases, including neurodegenerative and mental disorders that are accompanied by cognitive impairment; however, the underlying mechanisms require clarification. Here, we used a chronic cerebral hypoperfusion mouse model to investigate whether TRPM2, a Ca2+-permeable cation channel highly expressed in immune cells, plays a destructive role in the development of chronic cerebral hypoperfusion-induced cognitive impairment, and propose a new hypothesis in which TRPM2-mediated activation of microglia, not macrophages, specifically contributes to the pathology through the aggravation of inflammatory responses. These findings shed light on the understanding of the mechanisms of chronic cerebral hypoperfusion-related inflammation, and are expected to provide a novel therapeutic molecule for cognitive impairment in CNS diseases.
Keywords: cerebral hypoperfusion, cognitive impairment, cytokine, microglia, TRPM2, white matter injury
Introduction
Chronic cerebral hypoperfusion, resulting in an inadequate supply of blood to the brain, is elicited by aging (Aanerud et al., 2012) and a variety of risk factors such as atherosclerosis, hypertension, and obesity (Daulatzai, 2017). It has been previously suggested that chronic cerebral hypoperfusion is highly related to dementia diseases such as Alzheimer's disease (Zlokovic, 2005) and vascular dementia (Gorelick et al., 2011), and other CNS diseases including multiple sclerosis (D'haeseleer et al., 2013), major depression (Nagafusa et al., 2012), and epilepsy (Sone et al., 2017). Recently, emerging evidence has suggested that cognitive impairment, which causes difficulties in memory functioning and information processing, is associated not only with dementia (Gorelick et al., 2011), but also a number of CNS diseases, including multiple sclerosis (Chiaravalloti and DeLuca, 2008), Parkinson's disease (Kehagia et al., 2010), major depression (Marazziti et al., 2010), schizophrenia (Aquila and Citrome, 2015), and epilepsy (Holmes and Lenck-Santini, 2006). These facts could imply that chronic cerebral hypoperfusion is an upstream change in a wide variety of CNS diseases presenting with cognitive impairment. In line with this hypothesis, several studies have reported that chronic cerebral hypoperfusion leads to a variety of CNS pathologies associated with the above-mentioned neurodegenerative and mental disorders (Daulatzai, 2017), with the precise molecular mechanism of chronic cerebral hypoperfusion-induced cognitive impairment still needing to be identified.
Current studies indicate that chronic cerebral hypoperfusion contributes to the progression of inflammatory responses (Akiguchi et al., 1997). It was previously reported that chronic cerebral hypoperfusion-induced cognitive impairment is highly associated with inflammation in mice (Saggu et al., 2016) and humans (Kawamoto et al., 2006). Moreover, the application of anti-inflammatory drugs reduces chronic cerebral hypoperfusion-induced cognitive impairment (Fu et al., 2014), suggesting that regulation of the inflammatory response is a potential target for cognitive decline in chronic cerebral hypoperfusion.
Transient receptor potential melastatin 2 (TRPM2) is a member of the TRPM superfamily, and is a Ca2+-permeable channel with a unique C-terminal adenosine diphosphate ribose (ADPR) pyrophosphatase domain (Perraud et al., 2001). TRPM2 is functionally expressed in the brain, including neurons (Kaneko et al., 2006) and microglia (Kraft et al., 2004; Miyake et al., 2014), and peripheral inflammatory cells including monocytes/macrophages (Yamamoto et al., 2008) and neutrophils (Hiroi et al., 2013). In addition, TRPM2 is involved in the production of proinflammatory cytokines/chemokines in monocytes/macrophages, and aggravation of inflammation-related disorders such as ulcerative colitis (Yamamoto et al., 2008) and neuropathic pain (Haraguchi et al., 2012), implying that TRPM2 could be involved in chronic inflammation during chronic cerebral hypoperfusion.
In this study, we used genetically engineered mice to investigate the possible involvement of TRPM2 in chronic cerebral hypoperfusion, and evaluated the effects of TRPM2 deficiency on cognitive impairment using a mouse bilateral common carotid artery stenosis (BCAS) model. In this model, mild chronic cerebral hypoperfusion, but not a stroke-like condition, is observed (Temma et al., 2017); the extent of this may be more like that seen in non-stroke-related CNS diseases accompanied by cognitive impairment.
Materials and Methods
Animals.
The experiments were conducted in accordance with the ethical guidelines of the Kyoto University animal experimentation committee and the guidelines of the Japanese Pharmacological Society. Male C57BL/6J mice (RRID:IMSR_JAX:000664) and TRPM2 knock-out (TRPM2-KO) mice (9–12 weeks old, 20–30 g) used in the study were maintained in our laboratory. TRPM2-KO mice were generated as reported previously (Yamamoto et al., 2008), and backcrossed with C57BL/6J mice for 10 generations to eliminate any background effects on the phenotype. C57BL/6J mice and C57BL/6-Tg (CAG-EGFP) transgenic mice (GFP-transgenic mice) were purchased from Japan SLC. They were kept at a constant ambient temperature of 22 ± 2°C under a 12 h light/dark cycle and fed water and chow ad libitum.
Bilateral common carotid artery stenosis.
Mice were subjected to BCAS using microcoils with an internal diameter of 0.18 mm (Sawane Spring), as previously described (Temma et al., 2017). First, mice were anesthetized with 3% isoflurane in 30% O2 and 70% N2O, and maintained on 1.5% isoflurane in 30% O2 and 70% N2O using a face mask. After a midline skin incision, the bilateral common carotid artery was isolated and a microcoil was applied to it. Regional cerebral blood flow (rCBF) in the middle cerebral artery territory was monitored by laser Doppler flowmetry (Omegaflow, Omegawave). A flexible probe was fixed to the skull (2 mm posterior and 6 mm lateral to the bregma) before BCAS. A sham operation was performed in the same fashion as the BCAS operation, but without using a microcoil: the common carotid artery was isolated and the Doppler probe was fixed for monitoring rCBF while the sham control mice were subjected to anesthesia for the same amount of time as the BCAS-operated mice.
Y-maze test.
The Y-maze test was conducted at days 14 and 28 after the operation. The Y-maze had three arms (40 cm long, 12 cm high, and 3 cm wide) at equal angles. The arm where the mice were initially placed was labeled A, with the other two arms being labeled B and C. Experiments were performed over an 8 min period, and the total number and sequence of arm entries were recorded with a video camera. Alternation behavior was defined and counted as when mice entered all three of the arms consecutively (ABC, CAB, or BCA, but not BAB). The alternation count was divided by the maximum alternation (the total number of entries minus 2) and multiplied by 100 to create the percentage of alternation behavior. The total number of arm entries was recorded as spontaneous activity. Different cohorts of mice were assessed in experiments 14 and 28 d after BCAS. Mice that made <15 arm entries were excluded.
Novel object recognition test.
Cognitive assessment by novel object recognition test was performed on days 14 and 28 after surgery. Experiments were conducted in a dimly illuminated situation (30 lux), with mice being habituated to the black box (30 × 30 × 30 cm) for 3 d (10 min a day) before the training. In the training session, two different objects (a yellow triangular prism and a blue quadrangular pyramid) were placed in the box and mice were allowed to freely interact with the objects for 10 min. Six hours after the training session, the test session was performed, and the blue quadrangular object was replaced by a wooden ball to serve as a novel object. Exploratory time was defined as the time spent exploring the blue quadrangular object in the training session and the wooden ball in the test session, and was considered an indicator of locomotor activity. Exploratory preference was defined as the ratio of the exploratory time versus the total time spent exploring both of the objects, and was considered to be an indicator of recognition memory.
Fluoromyelin staining.
Mice were intraperitoneally injected with 50 mg/kg pentobarbital and perfused transcardially with 0.1 m PBS followed by 4% paraformaldehyde in 0.1 m PBS. Brains were stored in the fixative for 3 h and then transferred into 15% sucrose in 0.1 m phosphate buffer for 24 h. Coronal sections of 20 μm thickness were cut using a cryomicrotome (Leica). For fluoromyelin staining, brain sections were soaked in 0.2% Triton X-100 for at least 1 h. The sections were incubated with Fluoromyelin Green fluorescent myelin stain (1:300; Invitrogen) for 1 h at room temperature. Fluorescence was visualized with an Olympus Fluoview microscope equipped with a laser scanning confocal imaging system. The mean intensity of fluoromyelin staining in the corpus callosum was measured in a 200 × 200 μm field at approximately the bregma + 0.7 mm.
Immunofluorescence.
The coronal sections were then incubated with the primary antibodies for GSTpi (rabbit anti-GSTpi antibody, 1:200; MBL Life Science), Iba1 (rabbit anti-Iba1 antibody, 1:500; Wako Pure Chemical Industries), GFAP (rabbit anti-GFAP antibody, 1:1000; Dako), or CD3 (rat anti-CD3 antibody, 1:100; BD Biosciences), at 4°C overnight. Sections were then labeled with fluorescence-labeled secondary antibodies (AlexaFluor 594-labeled donkey anti-rabbit or anti-rat IgG, 1:300; Invitrogen) at room temperature for 1.5 h in the dark. Images were captured with a confocal fluorescence microscope. GSTpi-, Iba1-, and GFAP-positive cells in a 0.125 mm2 field of the corpus callosum at the bregma + 0.7 mm, and in the hippocampus at the bregma −2.8 mm, were counted.
NeuN and IgG staining.
For NeuN staining, the coronal sections were then incubated with primary antibody for NeuN (mouse anti-NeuN antibody, 1:500) at 4°C overnight. Sections were then labeled with biotinylated horse anti-mouse IgG (1:200; Vector Laboratories) for 1.5 h, followed by ABC Elite reagent (1:200; Vector Laboratories) for 1 h. Immunoreactivity was visualized using diaminobenzidine (Dojindo) as a chromogen. The numbers of NeuN-positive cells in the cortex at the bregma + 0.7 mm and the hippocampus at the bregma −2.8 mm were evaluated. For IgG staining, IgG leakage was immunostained with biotinylated horse anti-mouse IgG (1:200; Vector Laboratories) for 1.5 h, and then visualized by an avidin-biotin complex, as described above. The mean intensity of the leakage area in the corpus callosum was calculated per image.
Measurement of H2O2 content.
The corpus callosum and hippocampus were removed from the brains, frozen in liquid nitrogen, and stored at −80°C until use. They were homogenized with a Polytron homogenizer in ice-cold 50 mm phosphate buffer, pH 7.4, with 0.5% hexadecyltrimethyl ammonium bromide. The homogenates were centrifuged at 22,000 × g at 4°C for 10 min to remove debris. The supernatants were heated at 95°C for 5 min followed by centrifugation at 22,000 × g at 4°C for 10 min. The H2O2 contents in the supernatants were determined using an Amplex Red Assay Kit (Invitrogen) in accordance with the manufacturer's protocol.
ELISA.
The brain tissues were homogenized with a Polytron homogenizer in ice-cold homogenizing buffer (PBS containing 0.1% Triton X-100 and 1% protease-inhibitor mixture). The homogenates were centrifuged to remove debris, and protein concentrations in the supernatants were measured using DC protein assay reagents (Bio-Rad). The tissue homogenates and culture media suspected to have higher cytokine/chemokine levels than the linear range of the standard curve were diluted with homogenizing buffer or fresh culture media respectively, as appropriate. The IL1β, TNFα, and IL6 levels in the tissue homogenates and culture media were determined using mouse IL1β, TNFα, and IL6 ELISA kits (R&D Systems), in accordance with the manufacturer's protocols. Every sample was assayed in duplicate in the same plate.
Real-time PCR.
The removed brains were flash-frozen in liquid nitrogen and stored at −80°C until use. Total RNA from brain tissues was isolated with ISOGEN reagent (Nippon Gene) in accordance with the manufacturer's suggested protocols, total RNA from the cells was isolated with the FastPure RNA Kit (Takara Bio), and cDNA was synthesized with ReverTra Ace (Toyobo) with 1 μg of total RNA, as described previously (Shirakawa et al., 2017). Real-time quantitative PCRs were performed using the StepOne real-time PCR system (Life Technologies) in a final volume of 20 μl containing 1 μg of cDNA with THUNDERBIRD SYBR qPCR Mix (Toyobo). Each PCR amplification consisted of heat activation for 10 min at 95°C followed by 40 cycles of 95°C for 10 s and 60°C for 1 min. The quantitative measurements of mRNA expression levels were performed using the oligonucleotide primers (TRPM2: 5′-TGG ATC ATG AGT GTG CAG GT-3′ and 5′-ACA GAC AAT GCC TGG ATC G-3′; CX3CR1: 5′-TCA CCG TCA TCA GCA TCG A-3′ and 5′-CTG CAC TGT CCG GTT GTT CA-3′; CX3CL1: 5′-GCG ACA AGA TGA CCT CAC GA-3′ and 5′-TGT CGT CTC CAG GAC AAT GG-3′; MHCII (H2-Aa): 5′-GAC GCT CAA CTT GTC CCA AAA C-3′ and 5′-GCA GCC GTG AAC TTG TTG AAC-3′; CD68: 5′-TTC TGC TGT GGA AAT GCA AG-3′ and 5′-AGA GGG GCT GGT AGG TTG AT-3′; TLR4: 5′-TCC AGC CAC TGA AGT TCT GA-3′ and 5′-ACC AGG AAG CTT GAA TCC CT-3′; CD14: 5′-AGG GTA CAG CTG CAA GGA C-3′ and 5′-CTT CAG CCC AGT GAA AGA CA-3′; TREM2: 5′-GCA CCT CCA GGA ATC AAG AG-3′ and 5′-GGG TCC AGT GAG GAT CTG AA-3′; IL1β: 5′-TGA GCA CCT TCT TTT CCT TCA-3′ and 5′-TTG TCT AAT GGG AAC GTC ACA C-3′; TNFα: 5′-TGC CTA TGT CTC AGC CTC TTC-3′ and 5′-GAG GCC ATT TGG GAA CTT CT-3′; IL6: 5′-GTG GCT AAG GAC CAA GAC CA-3′ and 5′-TAA CGC ACT AGG TTT GCC GA-3′; 18S: 5′-GCA ATT ATT CCC CAT GAA CG-3′ and 5′-GGC CTC ACT AAA CCA TCC A-3′). Measurement of the 18S rRNA level of the samples was used to normalize mRNA contents, with the mRNA level being expressed relative to the corresponding control.
Drug treatment.
Thirty minutes after BCAS, mice were intraperitoneally injected with 50 mg/kg−1 minocycline hydrochloride (Sigma-Aldrich). Thereafter, the animals were injected once a day after BCAS, at a dose of 50 mg/kg−1. Control groups were treated with PBS 30 min after stroke and once a day thereafter.
Generation of bone marrow chimeric mice.
Homozygous TRPM2−/− male mice were crossed with GFP-transgenic female mice to produce GFP+ TRPM2+/− mice. GFP+ TRPM2+/+ (WT) mice were obtained by the hetero-mating of GFP+ TRPM2+/− female and male mice to obtain suitable bone marrow (BM) donor mice. BM transplantation was performed as previously reported (Isami et al., 2013), with slight modifications. BM recipients were male 6-week-old C57BL/6J WT or TRPM2-KO mice. Recipient mice were irradiated with 8 Gy total body irradiation to kill the BM cells. GFP+ WT donor mice were killed by decapitation, their femurs were isolated, and both ends were cut and placed into a microtube. The femurs were centrifuged at 400 × g for 10 min, and the pellet of GFP+ BM cells was suspended in sterile PBS. Between 3 and 5 h after the irradiation, the WT or TRPM2-KO recipient mice were transplanted with 4.0 × 106 BM cells by an intravenous injection into the tail vein. WT recipient mice transplanted with WT donor mouse-derived GFP+ BM cells (WTWT-BM), and TRPM2-KO recipient mice transplanted with WT donor mouse-derived GFP+ BM cells (TRPM2-KOWT-BM), were housed in an environment of specific pathogen-free conditions with ad libitum access to autoclaved pellets and autoclaved water. After 6 weeks, all chimeric animals were housed in a conventional environment, and male BM chimeric mice were used for BCAS surgery at the age of 12 weeks.
Flow cytometry.
Flow cytometry was used to identify the purity of GFP+ cells in the blood after the BM transplantation. Peripheral blood (100 μl) was collected from the tail vein of each chimeric mouse at 6 weeks after BM transplantation. Collected blood was dissolved in 300 μl of saline diluted three times for ∼10 s to hemolyze the erythrocytes. After adding 1000 μl of saline to restore the osmotic pressure, the solution was centrifuged for 5 min at 2000 × g. After pipetting off the supernatant, the cells were washed by adding 1000 μl of saline and were again centrifuged for 5 min at 2000 g. After again pipetting off the supernatant, 500 μl of fluorescence-activated cell sorting (FACS) buffer (0.02 m ethylene diaminetetraacetic acid and 0.01% bovine serum albumin in PBS) was added. The purity of the GFP+ cells was assessed by FACS (BD FACSAriaII, Becton Dickinson).
Experimental design and statistical analysis.
Statistical analysis was performed using Prism 6 software (GraphPad Software; RRID:SCR_002798). Details on statistical analyses and experimental design, including tests performed, exact p values, and sample sizes, are provided with the results describing each figure, or within the legend of each figure. Briefly, for comparisons between a single experimental group and a control group Student's t test or a Mann–Whitney test was used. For comparisons between multiple experimental groups, one-way ANOVA followed by Dunnett's post hoc comparison test or Kruskal–Wallis test followed by Dunn's post hoc comparison test was used. In addition, two-way ANOVA with Bonferroni's post hoc test was used as appropriate. In all cases, differences of p < 0.05 were considered statistically significant. Data are given as mean ± SEM.
Each data point represents one sample (section or extracts from specific brain regions, such as corpus callosum or hippocampus) of one individual mouse. The numbers of animals used in each experiment are indicated in the figure legends. The assessor was blinded to treatment condition assignment.
Results
Assessment of BCAS-induced cognitive impairment
When mice were subjected to BCAS, the rCBF 60 min after the operation was successfully decreased to ∼60% compared with the sham operation, with the change not differing between the WT and TRPM2-KO mice (WT BCAS: 60.72 ± 8.18%, TRPM2-KO BCAS: 58.08 ± 6.09%; sham vs BCAS: F(1,11) = 34.35, p = 0.0001, WT vs TRPM2-KO: F(1,11) = 0.04, p = 0.8508, two-way ANOVA; WT sham vs WT BCAS: t = 4.099, p = 0.0035, TRPM2-KO sham vs TRPM2-KO BCAS: t = 4.189, p = 0.0030, Bonferroni post hoc test). The Y-maze test was conducted to assess the involvement of TRPM2 in cognitive impairment induced by chronic cerebral hypoperfusion (Fig. 1A–D). At day 14 after BCAS, alternation behavior had not decreased in either of the BCAS-operated mouse groups in comparison with the sham groups (Fig. 1A; sham vs BCAS: F(1,48) = 0.18, p = 0.6761, WT vs TRPM2-KO: F(1,48) = 0.55, p = 0.4609, two-way ANOVA). At day 28, however, there were significant decreases in BCAS-operated WT mice, whereas the reduction was significantly suppressed in BCAS-operated TRPM2-KO mice (Fig. 1B; sham vs BCAS: F(1,71) = 7.05, p = 0.0098, WT vs TRPM2-KO: F(1,71) = 6.04, p = 0.0164, two-way ANOVA; WT sham vs WT BCAS: t = 2.525, p = 0.0270, WT BCAS vs TRPM2-KO BCAS: t = 2.735, p = 0.0157, Bonferroni post hoc test). The number of arm entries as an index of locomotor activity did not differ between the genotypes on days 14 and 28 [(Fig. 1C) sham vs BCAS: F(1,48) = 3.55, p = 0.0655, WT vs TRPM2-KO: F(1,48) = 0.37, p = 0.5443, two-way ANOVA; (D) sham vs BCAS: F(1,71) = 0.77, p = 0.3840, WT vs TRPM2-KO: F(1,71) = 1.30, p = 0.2584, two-way ANOVA]. The novel object recognition test was performed to further evaluate cognitive performance (Fig. 1E,F). Exploratory preferences to the two different objects were ∼50%, with no significant difference being observed in the training sessions on days 14 and 28 (day 14; sham vs BCAS: F(1,40) = 0.05, p = 0.8284, WT vs TRPM2-KO: F(1,40) = 2.64, p = 0.1118, two-way ANOVA, data not shown; day 28; sham vs BCAS: F(1,44) = 0.06, p = 0.8024, WT vs TRPM2-KO: F(1,44) = 1.97, p = 0.1673, two-way ANOVA, data not shown). In the test session, exploratory preferences to the novel object after the training session and interval did not differ between the groups at day 14 after BCAS, whereas at day 28 there were significant decreases in BCAS-operated WT mice, with the reduction being significantly suppressed in BCAS-operated TRPM2-KO mice [(Fig. 1E) sham vs BCAS: F(1,40) = 0.69, p = 0.4123, WT vs TRPM2-KO: F(1,40) = 0.04, p = 0.8484, two-way ANOVA; (F) sham vs BCAS: F(1,44) = 20.13, p < 0.0001, WT vs TRPM2-KO: F(1,44) = 7.38, p = 0.0094, two-way ANOVA; WT sham vs WT BCAS: t = 5.777, p < 0.0001, WT BCAS vs TRPM2-KO BCAS: t = 4.498, p < 0.0001, Bonferroni post hoc test]. These results suggest that TRPM2 could be involved in the development of cognitive impairment induced by chronic cerebral hypoperfusion.
Figure 1.
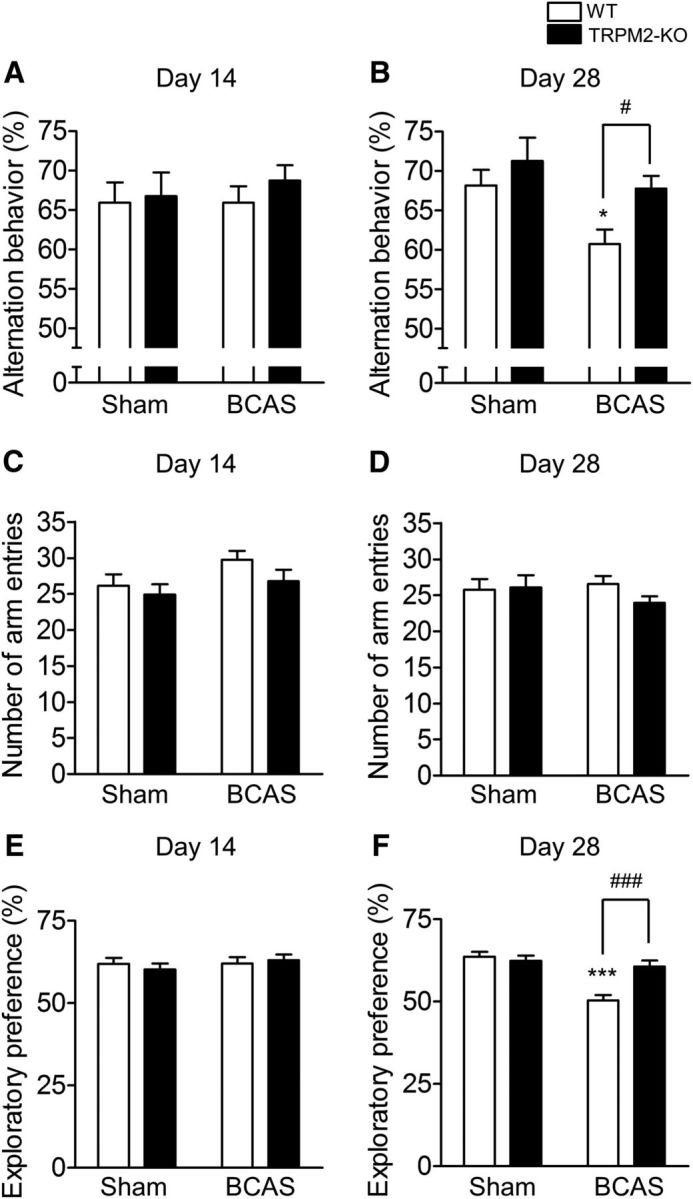
BCAS-induced cognitive decline at day 28 was not observed in TRPM2-KO mice. A–D, Alternation behavior at day 14 (A) and day 28 (B) and the number of arm entries at day 14 (C) and day 28 (D) after BCAS in the Y-maze test. E, F, Exploratory preference to the novel object in the novel object recognition test session at days 14 (E) and 28 (F) after BCAS. *p < 0.05 and ***p < 0.001 vs WT sham; #p < 0.05 and ###p < 0.001 vs WT BCAS. Values are mean ± SEM. A, C, n = 12–14; B, D, n = 14–25; E, n = 10–13; F, n = 11–13.
Evaluation of BCAS-induced white matter injury
White matter damage is a characteristic of a variety of CNS diseases, and is associated with cognitive impairment in dementia cases (Douaud et al., 2013) and other psychological disorders (de Groot et al., 2000). To evaluate the involvement of TRPM2 in white matter injury due to chronic cerebral hypoperfusion, myelin staining was performed in the corpus callosum at days 14 and 28 after BCAS (Fig. 2A–C). In BCAS-operated WT mice, there was a gradual decrease in myelin density compared with the sham mice (Fig. 2A,B; sham vs BCAS: F(2,28) = 3.163, p = 0.0578, one-way ANOVA; sham vs D28: q = 2.513, p = 0.0333, Dunnett's multiple-comparison test). By contrast, such a decrease was not observed in BCAS-operated TRPM2-KO mice (Fig. 2A,C; sham vs BCAS: F(2,25) = 0.6848, p = 0.5134, one-way ANOVA). For a further assessment of white matter damage, immunostaining of GSTpi, a marker of oligodendrocytes, was performed. In BCAS-operated WT mice, the number of GSTpi-positive cells in the corpus callosum decreased in a time-dependent manner, similar to myelin density, whereas in TRPM2-KO mice, such a decrease was not observed [(Fig. 2D,E) sham vs BCAS: F(2,22) = 2.914, p = 0.0754, one-way ANOVA; sham vs D28: q = 2.411, p = 0.0452, Dunnett's multiple-comparison test; (D,F) sham vs BCAS: F(2,18) = 0.1525, p = 0.8596, one-way ANOVA]. These results imply that TRPM2 plays a critical part in the aggravation of white matter injury.
Figure 2.

BCAS-induced white matter injury at day 28 was not observed in TRPM2-KO mice. A–C, Representative images of white matter in the corpus callosum by fluoromyelin staining (A) and the relative myelin density in WT (B) and TRPM2-KO mice (C). D–F, Representative images of immunostaining with GSTpi antibody in the corpus callosum (D) and the number of positive cells counted in WT (E) and TRPM2-KO mice (F). A, D, Bottom, Magnified images from the location marked by the boxed area of the above panels. *p < 0.05 vs WT sham. Values are mean ± SEM. B, C, n = 8–12; E, F, n = 6–9.
Next, we investigated the condition of gray matter when mice were subjected to chronic cerebral hypoperfusion for 28 d. Immunostaining with the neuron marker NeuN revealed that, in comparison with the sham groups, there was no clear pathological change in the number of NeuN-positive cells after BCAS surgery in the hippocampus, including CA1 (Fig. 3A,E), CA3 (Fig. 3B,F), dentate gyrus (DG; Fig. 3C,G), and the cerebral cortex (Fig. 3D,H), which is a finding consistent with those of previous studies [Nishio et al., 2010; Yamada et al., 2011; (Fig. 3A,E) sham vs BCAS: F(1,15) = 0.01, p = 0.9146, two-way ANOVA; (B,F) sham vs BCAS: F(1,15) = 0.13, p = 0.7207, two-way ANOVA; (C,G) sham vs BCAS: F(1,15) = 0.20, p = 0.6579, two-way ANOVA; (D,H) sham vs BCAS: F(1,15) = 0.03, p = 0.8710, two-way ANOVA].
Figure 3.

Neuronal death was not observed in either WT or TRPM2-KO mice at day 28 after BCAS. A–D, Representative images of NeuN-positive neurons in CA1 (A), CA3 (B), DG (C), and cerebral cortex (D) at 28 d after BCAS. E–H, The number of NeuN-positive cells measured in each area. Values are mean ± SEM. n = 3–6.
Inflammatory changes in the corpus callosum after BCAS
We attempted further investigations into the pathological changes in the white matter area at days 14 and 28 after BCAS. As blood–brain barrier (BBB) breakdown appears to be one of the exacerbating factors in this chronic cerebral hypoperfusion model (Seo et al., 2013), IgG staining in the corpus callosum was performed to explore the leakage of serum. At days 14 and 28 after BCAS, IgG leakage tended to increase in both the WT and TRPM2-KO mice in comparison with the sham operation mice, although there were no significant differences between the WT and TRPM2-KO groups (Fig. 4A,B; sham vs BCAS: F(1,26) = 1.11, p = 0.3441, WT vs TRPM2-KO: F(1,26) = 0.01, p = 0.9363, two-way ANOVA). Next, we measured the tissue content of H2O2, as it is an endogenous stimulator for TRPM2 (Hara et al., 2002). The H2O2 levels were significantly increased from day 14 after BCAS surgery in comparison with the sham operation, although there was no significant difference between WT and TRPM2-KO mice (Fig. 4C; sham vs BCAS: F(2,28) = 7.22, p = 0.0030, WT vs TRPM2-KO: F(1,28) = 0.06, p = 0.8089, two-way ANOVA; WT sham vs WT D14: t = 3.209, p = 0.0100, TRPM2-KO sham vs TRPM2-KO D14: t = 2.109, p = 0.1322, Bonferroni post hoc test).
Figure 4.

No differences were observed in BBB permeability and ROS production in the corpus callosum between WT and TRPM2-KO mice at days 14 and 28 after BCAS. A, B, Representative images (A) and relative leakage (B) evaluated by IgG staining. C, The level of measured H2O2 content. **p < 0.01 vs WT sham. Values are mean ± SEM. B, n = 4–8; C, n = 5–6. n.s., Not significant.
We next determined inflammatory signals at days 14 and 28 after BCAS. TRPM2 mRNA expression in the corpus callosum was increased at day 28 after BCAS surgery (Fig. 5A; p = 0.00064, Kruskal–Wallis test; sham vs D28: p = 0.0033, Dunn's multiple-comparison test). In addition, proinflammatory cytokines including IL1β (Fig. 5B), TNFα (C), and IL6 (D) were detected in the corpus callosum after chronic cerebral hypoperfusion, and the protein levels of IL1β [(Fig. 5B) sham vs BCAS: F(2,59) = 10.45, p = 0.0001, WT vs TRPM2-KO: F(1,59) = 16.30, p = 0.0002, two-way ANOVA; WT sham vs WT D28: t = 5.148, p < 0.0001, WT D28 vs TRPM2-KO D28: t = 4.888, p < 0.0001, Bonferroni post hoc test) and TNFα (C) sham vs BCAS: F(2,59) = 1.83, p = 0.1695, WT vs TRPM2-KO: F(1,59) = 4.46, p = 0.0389, two-way ANOVA; WT sham vs WT D28: t = 2.583, p = 0.0369, WT D28 vs TRPM2-KO D28: t = 2.775, p = 0.0222, Bonferroni post hoc test] at day 28 were significantly decreased in TRPM2-KO mice compared with WT mice, when the change in the IL6 level between genotypes was slight [(Fig. 5D) sham vs BCAS: F(2,58) = 5.48, p = 0.0066, WT vs TRPM2-KO: F(1,58) = 2.00, p = 0.1628, two-way ANOVA; WT sham vs WT D28: t = 3.402, p = 0.0037, Bonferroni post hoc test]. These findings indicate that TRPM2 is needed for inflammatory responses after chronic cerebral hypoperfusion.
Figure 5.

TRPM2 mRNA upregulation and proinflammatory cytokine production were detected in the corpus callosum at days 14 and 28 after BCAS. A, The mRNA expression level of TRPM2 by real-time PCR. B–D, The protein levels of IL1β (B), TNFα (C), and IL6 (D) measured by ELISA. *p < 0.05, **p < 0.01, and ***p < 0.001 vs WT sham; #p < 0.05 and ###p < 0.001 vs WT BCAS. Values are mean ± SEM. A, n = 10–13; B–D, n = 9–14. n.s., Not significant.
Involvement of Iba1-positive cells in the white matter after BCAS
To identify the type of cells responsible for the exacerbation of chronic cerebral hypoperfusion-induced inflammatory events, we performed immunostaining with the astrocyte marker GFAP (Fig. 6A,B), and Iba1 (C,D) as a marker for microglia and macrophages in the corpus callosum. The number of GFAP-positive cells was increased in both WT and TRPM2-KO mice from day 14 after the BCAS surgery, although there were no differences between these two groups (Fig. 6B; sham vs BCAS: F(2,31) = 12.33, p = 0.0001, WT vs TRPM2-KO: F(1,31) = 0.38, p = 0.5408, two-way ANOVA; WT sham vs WT D14: t = 2.904, p = 0.0202, TRPM2-KO sham vs TRPM2-KO D14: t = 2.598, p = 0.0427, WT sham vs WT D28: t = 3.065, p = 0.0134, TRPM2-KO sham vs TRPM2-KO D28: t = 3.477, p = 0.0046, Bonferroni post hoc test). By contrast, an observed increase in Iba1-positive cells gradually decreased to day 28 after BCAS in TRPM2-KO mice compared with the WT mice, when a significant change was observed (Fig. 6D; sham vs BCAS: F(2,41) = 8.37, p = 0.0009, WT vs TRPM2-KO: F(1,41) = 9.43, p = 0.0038, two-way ANOVA; WT sham vs WT D14: t = 3.827, p = 0.0013, WT sham vs WT D28: t = 3.086, p = 0.0109, WT D14 vs TRPM2-KO D14: t = 2.702, p = 0.0299, WT D28 vs TRPM2-KO D28: t = 2.927, p = 0.0167, Bonferroni post hoc test). Gr1-immunopositive neutrophils were not detected in brain parenchyma at day 28 after the BCAS surgery (data not shown). We then investigated the possible involvement of microglia/macrophage-related genes. In brain tissue, CX3CR1 is exclusively expressed on microglia (Y. Liu et al., 2015), whereas in the circulating blood, CX3CR1 is primarily expressed on circulating monocytes and tissue macrophages, and also on a subset of T cells and natural killer cells (Böttcher et al., 2015). The mRNA level of this receptor was significantly increased in WT mice after BCAS and TRPM2 deletion significantly suppressed an increase (Fig. 6E; sham vs BCAS: F(2,64) = 4.37, p = 0.0166, WT vs TRPM2-KO: F(2,64) = 8.20, p = 0.0057, two-way ANOVA; WT sham vs WT D14: t = 3.421, p = 0.0033, WT sham vs WT D28: t = 3.054, p = 0.0099, WT D14 vs TRPM2-KO D14: t = 2.522, p = 0.0425, WT D28 vs TRPM2-KO D28: t = 2.487, p = 0.0465, Bonferroni post hoc test), whereas the mRNA level of CX3CL1, which is a specific ligand for CX3CR1 and expressed primarily in neurons (Y. Liu et al., 2015), showed no changes between sham and BCAS groups (Fig. 6F; sham vs BCAS: F(1,51) = 0.23, p = 0.6301, WT vs TRPM2-KO: F(2,51) = 0.09, p = 0.9119, two-way ANOVA). In addition, the expression levels of the activated microglia/macrophage markers, such as MHCII and CD68 (Perry and Holmes, 2014), were significantly increased in WT mice at day 14 after BCAS, whereas no change was observed in TRPM2-KO mice [Fig. 6G; sham vs BCAS: F(2,50) = 5.27, p = 0.0084, WT vs TRPM2-KO: F(1,50) = 3.61, p = 0.0630, two-way ANOVA; WT sham vs WT D14: t = 3.748, p = 0.0014, WT D14 vs TRPM2-KO D14: t = 2.972, p = 0.0136, Bonferroni post hoc test; (H) sham vs BCAS: F(2,47) = 4.68, p = 0.0140, WT vs TRPM2-KO: F(1,47) = 7.18, p = 0.0101, two-way ANOVA; WT sham vs WT D14: t = 3.777, p = 0.0013, WT D14 vs TRPM2-KO D14: t = 3.550, p = 0.0027, Bonferroni post hoc test]. We also examined the key innate immune receptor toll-like-receptor 4 (TLR4) and its coreceptor CD14, and found that the mRNA levels of TLR4 and CD14 were significantly increased in WT mice after the BCAS surgery; however, there was no difference between WT and TRPM2-KO mice, implying that significant changes in TLR4 in microglia/macrophages might be masked by the upregulated TLR4 expression in neurons (Vindiš et al., 2014) and astrocytes [Shen et al., 2016; (Fig. 6I) sham vs BCAS: F(2,41) = 6.39, p = 0.0038, WT vs TRPM2-KO: F(1,41) = 0.23, p = 0.6305, two-way ANOVA; (J) sham vs BCAS: F(2,49) = 3.43, p = 0.0402, WT vs TRPM2-KO: F(1,49) = 0.20, p = 0.6542, two-way ANOVA]. Moreover, we checked the expression of triggering receptor by myeloid cells 2 (TREM2), which has anti-inflammatory properties and is linked to phagocytosis on microglia/macrophages (Jay et al., 2017). However, throughout the hypoperfusion period there was no difference between WT and TRPM2-KO mice, and between sham and BCAS groups (Fig. 6K; sham vs BCAS: F(2,55) = 0.76, p = 0.4717, WT vs TRPM2-KO: F(1,55) = 0.00, p = 0.9908, two-way ANOVA).
Figure 6.

The number of Iba1-positive cells in the corpus callosum and microglia-related genes were decreased in TRPM2-KO mice at days 14 and 28, with the pharmacological inhibition ameliorating the cognitive outcomes in BCAS-operated mice at day 28. A, B, Representative images (A) and the number (B) of GFAP-positive cells in the corpus callosum. C, D, Representative images (C) and the number (D) of Iba1-positive cells in the corpus callosum. A, C, Bottom, Magnified images from the location marked by the boxed area of the above panels. *p < 0.05 and **p < 0.01 vs sham groups; #p < 0.05 vs WT BCAS. E–K, The mRNA expression levels of CX3CR1 (E), CX3CL1 (F), MHCII (H2-Aa, G), CD68 (H), TLR4 (I), CD14 (J), and TREM2 (K) by real-time PCR. **p < 0.01 vs WT sham; #p < 0.05 and ##p < 0.01 vs WT BCAS. L–N, Representative images (L) and the number (M) of Iba1-positive cells in the corpus callosum, and exploratory preference to the novel object in the test session (N), at day 28 after daily treatment with minocycline or vehicle. L, Bottom, Magnified images from the location marked by the boxed area of the above panels. *p < 0.05 and ***p < 0.001 vs vehicle sham; ##p < 0.01, and ###p < 0.001 vs vehicle BCAS. Values are mean ± SEM. B, D, n = 5–12; E–K, n = 6–16; M, n = 4–5; N, n = 12–13.
To investigate whether suppression of Iba1-positive cells affects chronic cerebral hypoperfusion-induced cognitive impairment, we systemically administered minocycline, an inhibitor for microglia and macrophages, into BCAS-operated mice. Daily treatment with minocycline significantly diminished the number of Iba1-positive cells in comparison with vehicle-treated mice at day 28 after BCAS, as expected (Fig. 6L,M; sham vs BCAS: F(1,14) = 3.132, p = 0.0986, vehicle vs minocycline: F(1,14) = 6.63, p = 0.0221, two-way ANOVA; vehicle sham vs vehicle BCAS: t = 2.716, p = 0.0335, vehicle BCAS vs minocycline BCAS: t = 3.484, p = 0.0073, Bonferroni post hoc test). Furthermore, in the test session of the novel object recognition test, minocycline-treated mice showed significant reductions in cognitive disturbance compared with vehicle-treated mice (Fig. 6N; sham vs BCAS: F(1,45) = 11.53, p = 0.0014, vehicle vs minocycline: F(1,45) = 9.23, p = 0.0040, two-way ANOVA; vehicle sham vs vehicle BCAS: t = 4.020, p = 0.0004, vehicle BCAS vs minocycline BCAS: t = 3.764, p = 0.0010; Bonferroni post hoc test). These results suggest that Iba1-positive cells are involved in the aggravation of cognitive dysfunction induced by chronic cerebral hypoperfusion.
Involvement of Iba1-positive cells in the hippocampus after BCAS
As with the corpus callosum, we further examined the possible involvement of astrocytes, microglia/macrophages, and neutrophils in the hippocampus, which is important for cognitive function. Immunostaining revealed that the numbers of GFAP-positive astrocytes in CA1, CA3, and DG areas of the hippocampus were either significantly increased or showed a tendency to increase in both WT and TRPM2-KO mice at day 28 after the BCAS surgery; however, there were no differences between these genetic groups [(Fig. 7A,B) sham vs BCAS: F(1,15) = 15.19, p = 0.0014, WT vs TRPM2-KO: F(1,15) = 0.00, p = 0.9612, two-way ANOVA; WT sham vs WT D28: t = 3.079, p = 0.0153, TRPM2-KO sham vs TRPM2-KO D28: t = 2.421, p = 0.0572, Bonferroni post hoc test; (E,F) sham vs BCAS: F(1,15) = 11.89, p = 0.0036, WT vs TRPM2-KO: F(1,15) = 0.07, p = 0.7983, two-way ANOVA; WT sham vs WT D28: t = 2.357, p = 0.0649, TRPM2-KO sham vs TRPM2-KO D28: t = 2.523, p = 0.0468, Bonferroni post hoc test; (I,J) sham vs BCAS: F(1,15) = 16.71, p = 0.0010, WT vs TRPM2-KO: F(1,15) = 0.22, p = 0.6424, two-way ANOVA; WT sham vs WT D28: t = 3.038, p = 0.0166, TRPM2-KO sham vs TRPM2-KO D28: t = 2.738, p = 0.0305, Bonferroni post hoc test]. By contrast, significant increases in the numbers of Iba1-positive microglia/macrophages in CA1, CA3, and DG areas of the hippocampus were observed in WT mice, whereas they were significantly decreased or showed a tendency to decrease in TRPM2-KO mice at day 28 after the BCAS operation [(Fig. 7C,D) sham vs BCAS: F(1,22) = 11.93, p = 0.0023, WT vs TRPM2-KO: F(1,22) = 5.02, p = 0.0355, two-way ANOVA; WT sham vs WT D28: t = 2.984, p = 0.0137, TRPM2-KO sham vs TRPM2-KO D28: t = 1.888, p = 0.1446, WT D28 vs TRPM2-KO D28: t = 2.353, p = 0.0559, Bonferroni post hoc test; (G,H) sham vs BCAS: F(1,22) = 11.57, p = 0.0026, WT vs TRPM2-KO: F(1,22) = 1.49, p = 0.2357, two-way ANOVA; WT sham vs WT D28: t = 3.443, p = 0.0046, TRPM2-KO sham vs TRPM2-KO D28: t = 1.343, p = 0.3862, WT D28 vs TRPM2-KO D28: t = 2.113, p = 0.0924, Bonferroni post hoc test; (K,L) sham vs BCAS: F(1,22) = 9.96, p = 0.0046, WT vs TRPM2-KO: F(1,22) = 2.57, p = 0.1229, two-way ANOVA; WT sham vs WT D28: t = 3.324, p = 0.0062, TRPM2-KO sham vs TRPM2-KO D28: t = 1.112, p = 0.5562, WT D28 vs TRPM2-KO D28: t = 2.467, p = 0.0437, Bonferroni post hoc test]. Gr1-positive neutrophils were not detected in any of the regions of the hippocampus (data not shown). We then investigated the expression of TRPM2 and microglia/macrophage-related genes in the hippocampus at day 28 after BCAS operation. TRPM2 mRNA expression in the hippocampus showed a tendency to increase in BCAS-operated WT mice (Fig. 7M). CX3CR1 mRNA expression was significantly increased at day 28 after BCAS, although this increase was not observed in TRPM2-KO mice (Fig. 7N; sham vs BCAS: F(1,31) = 4.70, p = 0.0379, WT vs TRPM2-KO: F(1,31) = 2.61, p = 0.1162, two-way ANOVA; WT sham vs WT D28: t = 3.210, p = 0.0062, WT D28 vs TRPM2-KO D28: t = 2.429, p = 0.0422, Bonferroni post hoc test.). CX3CL1 mRNA expression was not changed (Fig. 7O; sham vs BCAS: F(1,19) = 0.16, p = 0.6970, WT vs TRPM2-KO: F(1,19) = 0.39, p = 0.5398, two-way ANOVA.). Moreover, in comparison with the sham operation, there was only a slight increase in the production of cytokines in the hippocampus after BCAS operation, including IL1β, TNFα, and IL6 [(Fig. 7P) sham vs BCAS: F(1,32) = 3.35, p = 0.0767, WT vs TRPM2-KO: F(1,32) = 4.33, p = 0.0456, two-way ANOVA; (Q) sham vs BCAS: F(1,31) = 6.75, p = 0.0142, WT vs TRPM2-KO: F(1,31) = 0.10, p = 0.7540, two-way ANOVA; (R) sham vs BCAS: F(1,33) = 3.84, p = 0.0587, WT vs TRPM2-KO: F(1,33) = 2.33, p = 0.1361, two-way ANOVA]. H2O2 production in the hippocampus was not changed at day 28 after BCAS (Fig. 7S; sham vs BCAS: F(1,19) = 0.05, p = 0.8185, WT vs TRPM2-KO: F(1,19) = 0.07, p = 0.7917, two-way ANOVA). Together with the observation in Figure 3, these findings indicate that hippocampal damage did not occur at day 28 after BCAS operation.
Figure 7.

The numbers of GFAP- and Iba1-positive cells were increased but there was a slight change of cytokines in the hippocampus of WT mice at 28 d after BCAS. A, B, E, F, I, J, Representative images (A, E, I) and the number (B, F, J) of GFAP-positive cells in CA1 (A, B), CA3 (E, F), and DG (I, J). C, D, G, H, K, L, Representative images (C, G, K) and the number (D, H, L) of Iba1-positive cells in CA1 (C, D), CA3 (G, H), and DG (K, L). *p < 0.05 and **p < 0.01 vs WT sham; #p < 0.05 vs WT BCAS. M–R, The mRNA expression level of TRPM2 (M), CX3CR1 (N), CX3CL1 (O), IL1β (P), TNFα (Q), and IL6 (R) by real-time PCR. **p < 0.01 vs WT sham; #p < 0.05 vs WT BCAS. S, The level of measured H2O2 content. Values are mean ± SEM. B, D, F, H, J, L, n = 4–6; M–R, n = 5–13; S, n = 5–6.
Identification of Iba1-positive cells after BCAS
Finally, we addressed the remaining question as to whether these accumulated Iba1-positive cells are peripheral macrophages or brain-resident microglia. To identify these cells, we generated BM chimeric mice, whose BM-derived cells were replaced with GFP-labeled BM-derived cells harvested from GFP-positive WT mice. These chimeric mice were divided into two groups: WT chimeric mice transplanted with GFP-positive WT BM (WTWT-BM mice), and TRPM2-KO mice transplanted with GFP-positive WT BM (TRPM2-KOWT-BM mice). BM-derived cells in these chimeric mice were successfully replaced with >90% GFP-positive BM (data not shown). There were no changes in the number of Iba1-positive cells in sham-operated chimeric mice (Fig. 8A,C; t(4) = 0.4392, p = 0.6832, unpaired Student's t test), which showed a similarly small number as the sham-operated groups shown in Figure 6. Almost no GFP-positive cells were detected in these groups (Fig. 8B; t(4) = 0.5000, p = 0.6433, unpaired Student's t test), demonstrating that the irradiation did not affect the general morphology and proliferation of microglia. At day 28 after performing BCAS on the chimeric mice, infiltration of GFP-positive cells was observed in the corpus callosum, in contrast to the situation in the sham-operated groups, whereas the numbers were not different between BCAS-operated WTWT-BM mice and TRPM2-KOWT-BM mice (Fig. 8D,E; t(16) = 0.4793, p = 0.6382, unpaired Student's t test). Importantly, immunostaining with Iba1 revealed that Iba1-positive cells did not emerge with GFP fluorescence (Fig. 8D). This observation suggests that peripheral Iba1-positive cells no longer infiltrate into the brain in this chronic cerebral hypoperfusion model. Furthermore, BCAS-operated TRPM2-KOWT-BM mice showed a significant decrease in Iba1-positive cells, similar to that shown in Figure 6 (Fig. 8D,F; t(16) = 3.449, p = 0.0036, unpaired Student's t test), although their BM was WT. In addition, the number of GSTpi-positive cells after BCAS was significantly higher in TRPM2-KOWT-BM mice than in WTWT-BM mice (Fig. 8G,H; t(13) = 3.266, p = 0.0061, unpaired Student's t test). Approximately 50% of GFP-positive cells merged with CD3, a marker of T cells (Fig. 8I,J; t(23) = 0.1065, p = 0.9161, unpaired Student's t test). To assess the cognitive performance of these chimeric mice, Y-maze tests (Fig. 8K) and novel object recognition tests (Fig. 8L) were conducted. BCAS-operated TRPM2-KOWT-BM mice showed significantly higher alternation behavior (Fig. 8K; t(28) = 2.241, p = 0.0331, unpaired Student's t test) and exploratory preference to the novel object than the WTWT-BM mice (Fig. 8L; t(13) = 3.704, p = 0.0026, unpaired Student's t test). Together, these findings indicate that the Iba1-positive cells that accumulated in the brain were not macrophages, but resident microglia, and central TRPM2 activation is crucial for microglial activation.
Figure 8.
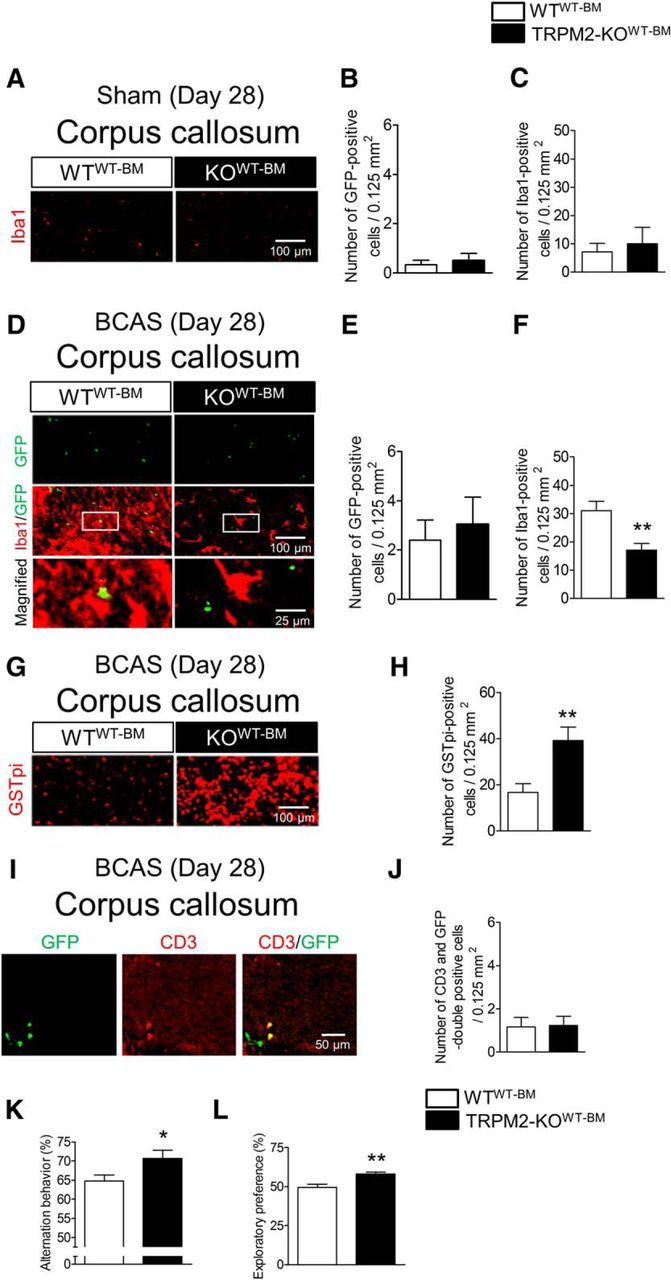
In chimeric mice, Iba1-positive cells activated 28 d after BCAS were microglia, not macrophages. A–C, Representative images (A) and the number of GFP-positive cells (B) and Iba1-positive cells (C) in the corpus callosum of sham-operated chimeric mice. D–F, Representative images (D) and the number of GFP-positive cells (E) and Iba1-positive cells (F) in the corpus callosum of BCAS-operated chimeric mice. D, Bottom, Magnified images from the location marked by the boxed area of the above panels. G, H, Representative images (G) and the number of GSTpi-positive cells (H) in the corpus callosum. I, J, Representative image (I) and the number of GFP and CD3 double-positive cells (J) in the corpus callosum. K, Alternation behavior in the Y-maze test at day 28 after BCAS. L, Exploratory preferences to the novel object in the novel object recognition test session at day 28 after BCAS. *p < 0.05 and **p < 0.01 vs WTWT-BM. Values are mean ± SEM. B, C, n = 3; E, F, H, J–L, n = 9–15.
Discussion
In the present study, we provide the first evidence that TRPM2 in resident microglia plays critical roles in the pathogenesis of chronic cerebral hypoperfusion through the aggravation of inflammatory responses, as supported by the following findings. (1) TRPM2 deficiency suppressed cognitive function and white matter injury after BCAS. (2) Releases of proinflammatory cytokines, such as TNFα and IL6, were reduced in TRPM2-KO mice. (3) The expression of TRPM2 mRNA was increased at the lesion site with the production of reactive oxygen species (ROS). (4) TRPM2 deficiency significantly decreased the accumulation of Iba1-positive cells in the corpus callosum, without a change in the number of GFAP-positive cells between genotypes. (5) Minocycline, which is an inhibitor of microglia/macrophage activation, prevented cognitive decline in WT mice. (6) BM chimeric TRPM2-KOWT-BM mice showed a significant decrease in the number of Iba1-positive cells, as well as attenuation of cognitive impairment, in comparison with WTWT-BM mice, the extent of which was similar to that observed in TRPM2-global knock-out mice. Figure 9 shows the schematics for the role of TRPM2 under chronic cerebral hypoperfusion.
Figure 9.

Schematics for the role of TRPM2 under chronic cerebral hypoperfusion.
It would be of interest to know the precise mechanisms of TRPM2-mediated cognitive impairment induced by chronic cerebral hypoperfusion. A number of studies on BCAS models have indicated that one of the initial pathophysiological changes induced by chronic cerebral hypoperfusion is BBB breakdown. Seo et al. (2013) reported that IgG leakages (namely, BBB breakdown) in the corpus callosum are detected by immunohistochemistry at days 3 and 7 after BCAS; these are times when activation of glial cells, including astrocytes and microglia, is not observed (Duan et al., 2009). In patients with neuromyelitis optica, the severity of BBB breakdown was shown to correlate with the levels of proinflammatory markers (Uchida et al., 2017). Given that peripheral bloodborne substances such as thrombin (Yang et al., 2015) and CCL11 (Villeda et al., 2011) aggravate CNS inflammation, BBB integrity could play an important role as a barrier contributing to prevention of extravasation of these toxic factors from the circulation into the brain parenchyma. In fact, BBB permeability is associated with white matter hyperintensity in patients with manifestations of vascular dementia and Alzheimer's disease (Farrall and Wardlaw, 2009). In the present study, there were no differences between WT and TRPM2-KO mice in the extent of BBB breakdown, implying that TRPM2 is not involved in BBB disruption, and may therefore be involved in more delayed phases of pathogenesis after chronic cerebral hypoperfusion.
ROS have detrimental roles for oligodendrocyte vulnerability (Volpe et al., 2011) and BBB permeability (Toyama et al., 2014), which are involved in chronic cerebral hypoperfusion-induced cognitive impairment (H. Liu and Zhang, 2012). More importantly, ROS are well known as endogenous agonists for TRPM2 (Hara et al., 2002). Several studies using scavengers or markers for ROS suggest that they are produced and released in chronic cerebral hypoperfusion (Dong et al., 2011; Miyamoto et al., 2013; Yang et al., 2017). In this study, we directly detected the levels of ROS in the corpus callosum at day 14 after BCAS and found them to be similar between WT and TRPM2-KO mice, indicating that TRPM2 is not involved in ROS production itself after the induction of cerebral hypoperfusion, and that the protective effect in TRPM2-KO mice is not attributed to the suppression of direct oxidative damage to white matter.
Next, we investigated changes in proinflammatory cytokines and white matter injury during chronic cerebral hypoperfusion, and found that knocking out TRPM2 markedly suppressed the production of proinflammatory cytokines such as TNFα and IL1β, and white matter injury. It was previously reported that IL1β levels are upregulated in serum from patients with vascular dementia (Zuliani et al., 2007) and monocytes derived from patients with schizophrenia (Uranova et al., 2017). In the mouse chronic cerebral hypoperfusion model, IL1R deletion and neutralization improved white matter damage and facilitated the migration of oligodendrocyte precursor cells (OPCs) from the subventricular zone (Zhou et al., 2017). TNFα, a proinflammatory cytokine, as well as IL1β, were expressed abundantly in patients with subcortical vascular dementia (Tarkowski et al., 2003) and schizophrenia (Pandey et al., 2018). TNFα levels have also been found to be associated with white matter hyperintensities in patients with bipolar disorder (Benedetti et al., 2016) and premature newborns (Ellison et al., 2005), indicating that excessive production and release of proinflammatory cytokines is highly involved in white matter injury. In the context of these studies, it has been reported that TRPM2 is involved in NLRP3 inflammasome-mediated IL1β secretion (Zhong et al., 2013), and that TRPM2 is required for the LPS-induced production of TNFα and IL6 in human monocytes (Wehrhahn et al., 2010) and Aβ42-induced generation of TNFα in mouse cultured microglia (Alawieyah Syed Mortadza et al., 2018), findings consistent with our own in vivo observations. Therefore, it is conceivable that TRPM2 contributes to chronic cerebral hypoperfusion-related cognitive impairment through the enhancement of cytokine production and subsequent white matter injury.
One could raise a question as to which types of cells are responsible for the TRPM2-mediated signaling that can result in the upregulation of proinflammatory cytokines in the corpus callosum. TRPM2 is expressed broadly in immune cells, including microglia (Kraft et al., 2004), monocytes/macrophages (Yamamoto et al., 2008), and neutrophils (Hiroi et al., 2013). We therefore investigated the activation of glial cells and the infiltration of neutrophils during chronic cerebral hypoperfusion, and found that knocking out TRPM2 suppressed the activation of Iba1-positive microglia/macrophages, but not GFAP-positive astrocytes and Gr1-positive neutrophils. In addition, the expression of CX3CR1, MHCII, and CD68, which were predominantly expressed in activated microglia/macrophages (Perry and Holmes, 2014; Y. Liu et al., 2015), was significantly upregulated in WT mice after BCAS, whereas the changes in these markers were not observed in TRPM2-KO mice. Consistent with these observations, previous studies reported that immunosuppressants ameliorate white matter injury in a rat chronic cerebral hypoperfusion model (Wakita et al., 1998). Moreover, TRPM2 plays an important role in microglial activation (Haraguchi et al., 2012; Miyake et al., 2014; Jeong et al., 2017) and the development of inflammatory processes in in vivo mouse models (Yamamoto et al., 2008; Haraguchi et al., 2012; Huang et al., 2017). Together, our data suggest that TRPM2 expressed in microglia/macrophages is responsible for TRPM2-mediated upregulation of proinflammatory cytokines in the white matter.
In the present study, we performed an experiment using BM chimeric mice to identify whether Iba1-positive cells are brain-resident microglia or peripheral macrophages, as the number of Iba1-positive cells was markedly decreased in TRPM2-KO mice compared with WT mice. We concluded that the decreased number of Iba1-positive cells were brain-resident microglia, but not peripheral macrophages, because these Iba1-positive cells were almost all GFP-negative, defining them as brain-resident microglia. Intriguingly, TRPM2-KOWT-BM mice showed a significant decrease in the number of Iba1-positive cells and a higher cognitive performance, the extents of which were similar to those of TRPM2-global knock-out mice, which clearly indicates the exclusive role of resident microglia in a mouse chronic cerebral hypoperfusion model for the first time. It was recently reported that microglial CX3CR1 silencing reduces cytokine release, white matter injury, and cognitive impairment (Y. Liu et al., 2015), which is consistent with our findings.
It is well known that an overwhelming production of proinflammatory cytokines results in cumulative damage to tissue (Smith et al., 2012). In this context, in the present study, increased production of proinflammatory cytokines, such as IL1β, TNFα, and IL6, and tissue damage were markedly observed in the white matter, but only slightly in the hippocampus, although the numbers of Iba1-positive microglia and GFAP-positive astrocytes in both regions increased to the same extent. The reason why there was a significant difference between these brain regions remains unclear. However, proinflammatory cytokines released by activated microglia are known to play an important role in oligodendrocyte toxicity (Peferoen et al., 2014), which is maturation-dependent (i.e., greater toxicity to actively proliferating OPCs and immature oligodendrocytes than to mature oligodendrocytes; Bradl and Lassmann, 2010; Volpe et al., 2011). Taking into consideration that oligodendrocytes and their precursor OPCs are more abundant in the white matter than in the hippocampus, it is conceivable that a wide variety of damage-associated molecular patterns (DAMPs), which are passively released from dying, dead, or injured cells (Seong and Matzinger, 2004), could be more dominant in the white matter than the hippocampus. We believe that enhanced activation of microglia with DAMPs results in increased release of proinflammatory cytokines (Perry and Holmes, 2014), which subsequently leads to more severe tissue injury in the white matter, unlike in the hippocampus.
There remains controversy over whether astrocytes are harmful or protective for chronic cerebral hypoperfusion. NFκB suppression in GFAP-positive cells reduced white matter injury and cognitive impairment (Saggu et al., 2016), whereas BDNF deletion in GFAP-positive cells worsened cognitive outcomes (Miyamoto et al., 2015). Although the present study revealed that the number of GFAP cells was increased by the BCAS operation, there were no differences between WT and TRPM2-KO mice, suggesting that TRPM2 is not involved in astrocyte activation by the BCAS operation. However, global knock-out of TRPM2 diminishes the number of both Iba1- and GFAP-positive cells in neonatal hypoxic-ischemic (HI) brain injury (Huang et al., 2017), suggesting that TRPM2 plays a role in reactive astrogliosis following HI injury. Future investigations will help in understanding the precise roles of astrocytic TRPM2 in chronic cerebral hypoperfusion.
TRPM2 is also expressed in CNS neurons including cortical neurons (Kaneko et al., 2006) and hippocampal neurons (Alim et al., 2013). In the present study, there was no obvious change in hippocampal and cortical neurons, although microglia and astrocytes were clearly activated after BCAS operation. A high concentration of H2O2, when present at 0.1–1 mm, can open TRPM2 (Hara et al., 2002) and induce neuronal death (Kaneko et al., 2006), whereas a relatively low level of ROS can induce TRPM2-mediated chemokine production in monocytes/macrophages (Yamamoto et al., 2008) and augment nitric oxide production in microglia (Miyake et al., 2014), implying that the ROS produced after the BCAS operation could be insufficient to cause TRPM2-mediated neuronal injury in the CNS. Future investigations are required to explore the different sensitivities of TRPM2-expressing cells to ROS in chronic cerebral hypoperfusion.
Yata et al. (2014) reported that two-photon imaging in the small cerebral arteries showed rolling of leukocytes in BCAS-operated mice; however, the mechanism of leukocyte infiltration was not fully described. In the present study using BM chimeric mice, the numbers of GFP-positive BM-derived cells were similar between the different BCAS-chimeric groups. Moreover, a few GFP-positive BM-derived cells were observed in BCAS-operated chimeric mice, and among them ∼50% population of these cells merged with CD3, suggesting that a few of the cells infiltrating into the brain parenchyma are T cells and the other BM-derived cells, but not macrophages or neutrophils.
In conclusion, TRPM2 plays a critical role in the development of inflammation by chronic cerebral hypoperfusion, and this may be mediated through the activation of microglia. TRPM2 in microglia is a potential therapeutic target for hypoperfusion-induced CNS diseases.
Footnotes
This work was supported by a Grant-in-aid for Scientific Research from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; from the Japan Society for the Promotion of Science; the Naito Foundation, Takeda Science Foundation, and Kanzawa Medical Research Foundation; and irradiation experiments were supported by the Radiation Biology Center Cooperative Research Program (Kyoto University). We thank S. Teramukai, Department of Biostatistics, Graduate School of Medical Science, Kyoto Prefectural University of Medicine for helpful advice in the statistical analysis.
The authors declare no competing financial interests.
References
- Aanerud J, Borghammer P, Chakravarty MM, Vang K, Rodell AB, Jónsdottir KY, Møller A, Ashkanian M, Vafaee MS, Iversen P, Johannsen P, Gjedde A (2012) Brain energy metabolism and blood flow differences in healthy aging. J Cereb Blood Flow Metab 32:1177–1187. 10.1038/jcbfm.2012.18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiguchi I, Tomimoto H, Suenaga T, Wakita H, Budka H (1997) Alterations in glia and axons in the brains of Binswanger's disease patients. Stroke 28:1423–1429. 10.1161/01.STR.28.7.1423 [DOI] [PubMed] [Google Scholar]
- Alawieyah Syed Mortadza S, Sim JA, Neubrand VE, Jiang LH (2018) A critical role of TRPM2 channel in Aβ42-induced microglial activation and generation of tumor necrosis factor-α. Glia 66:562–575. 10.1002/glia.23265 [DOI] [PubMed] [Google Scholar]
- Alim I, Teves L, Li R, Mori Y, Tymianski M (2013) Modulation of NMDAR subunit expression by TRPM2 channels regulates neuronal vulnerability to ischemic cell death. J Neurosci 33:17264–17277. 10.1523/JNEUROSCI.1729-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquila R, Citrome L (2015) Cognitive impairment in schizophrenia: the great unmet need. CNS Spectr 20:35–39; quiz 40. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Poletti S, Hoogenboezem TA, Mazza E, Ambrée O, de Wit H, Wijkhuijs AJ, Locatelli C, Bollettini I, Colombo C, Arolt V, Drexhage HA (2016) Inflammatory cytokines influence measures of white matter integrity in bipolar disorder. J Affect Disord 202:1–9. 10.1016/j.jad.2016.05.047 [DOI] [PubMed] [Google Scholar]
- Böttcher JP, Beyer M, Meissner F, Abdullah Z, Sander J, Höchst B, Eickhoff S, Rieckmann JC, Russo C, Bauer T, Flecken T, Giesen D, Engel D, Jung S, Busch DH, Protzer U, Thimme R, Mann M, Kurts C, Schultze JL, et al. (2015) Functional classification of memory CD8(+) T cells by CX3CR1 expression. Nat Commun 6:8306. 10.1038/ncomms9306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradl M, Lassmann H (2010) Oligodendrocytes: biology and pathology. Acta Neuropathol 119:37–53. 10.1007/s00401-009-0601-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiaravalloti ND, DeLuca J (2008) Cognitive impairment in multiple sclerosis. Lancet Neurol 7:1139–1151. 10.1016/S1474-4422(08)70259-X [DOI] [PubMed] [Google Scholar]
- Daulatzai MA. (2017) Cerebral hypoperfusion and glucose hypometabolism: key pathophysiological modulators promote neurodegeneration, cognitive impairment, and Alzheimer's disease. J Neurosci Res 95:943–972. 10.1002/jnr.23777 [DOI] [PubMed] [Google Scholar]
- de Groot JC, de Leeuw FE, Oudkerk M, Hofman A, Jolles J, Breteler MM (2000) Cerebral white matter lesions and depressive symptoms in elderly adults. Arch Gen Psychiatry 57:1071–1076. 10.1001/archpsyc.57.11.1071 [DOI] [PubMed] [Google Scholar]
- D'haeseleer M, Beelen R, Fierens Y, Cambron M, Vanbinst AM, Verborgh C, Demey J, De Keyser J (2013) Cerebral hypoperfusion in multiple sclerosis is reversible and mediated by endothelin-1. Proc Natl Acad Sci U S A 110:5654–5658. 10.1073/pnas.1222560110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong YF, Kataoka K, Toyama K, Sueta D, Koibuchi N, Yamamoto E, Yata K, Tomimoto H, Ogawa H, Kim-Mitsuyama S (2011) Attenuation of brain damage and cognitive impairment by direct renin inhibition in mice with chronic cerebral hypoperfusion. Hypertension 58:635–642. 10.1161/HYPERTENSIONAHA.111.173534 [DOI] [PubMed] [Google Scholar]
- Douaud G, Menke RA, Gass A, Monsch AU, Rao A, Whitcher B, Zamboni G, Matthews PM, Sollberger M, Smith S (2013) Brain microstructure reveals early abnormalities more than two years prior to clinical progression from mild cognitive impairment to Alzheimer's disease. J Neurosci 33:2147–2155. 10.1523/JNEUROSCI.4437-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Gui L, Zhou Z, Liu Y, Tian H, Chen JF, Zheng J (2009) Adenosine A2A receptor deficiency exacerbates white matter lesions and cognitive deficits induced by chronic cerebral hypoperfusion in mice. J Neurol Sci 285:39–45. 10.1016/j.jns.2009.05.010 [DOI] [PubMed] [Google Scholar]
- Ellison VJ, Mocatta TJ, Winterbourn CC, Darlow BA, Volpe JJ, Inder TE (2005) The relationship of CSF and plasma cytokine levels to cerebral white matter injury in the premature newborn. Pediatr Res 57:282–286. 10.1203/01.PDR.0000148286.53572.95 [DOI] [PubMed] [Google Scholar]
- Farrall AJ, Wardlaw JM (2009) Blood–brain barrier: ageing and microvascular disease—systematic review and meta-analysis. Neurobiol Aging 30:337–352. 10.1016/j.neurobiolaging.2007.07.015 [DOI] [PubMed] [Google Scholar]
- Fu X, Zhang J, Guo L, Xu Y, Sun L, Wang S, Feng Y, Gou L, Zhang L, Liu Y (2014) Protective role of luteolin against cognitive dysfunction induced by chronic cerebral hypoperfusion in rats. Pharmacol Biochem Behav 126:122–130. 10.1016/j.pbb.2014.09.005 [DOI] [PubMed] [Google Scholar]
- Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, Launer LJ, Laurent S, Lopez OL, Nyenhuis D, Petersen RC, Schneider JA, Tzourio C, Arnett DK, Bennett DA, Chui HC, Higashida RT, Lindquist R, Nilsson PM, Roman GC, et al. (2011) Vascular contributions to cognitive impairment and dementia. Stroke 42:2672–2713. 10.1161/STR.0b013e3182299496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara Y, Wakamori M, Ishii M, Maeno E, Nishida M, Yoshida T, Yamada H, Shimizu S, Mori E, Kudoh J, Shimizu N, Kurose H, Okada Y, Imoto K, Mori Y (2002) LTRPC2 Ca2+-permeable channel activated by changes in redox status confers susceptibility to cell death. Mol Cell 9:163–173. 10.1016/S1097-2765(01)00438-5 [DOI] [PubMed] [Google Scholar]
- Haraguchi K, Kawamoto A, Isami K, Maeda S, Kusano A, Asakura K, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2012) TRPM2 contributes to inflammatory and neuropathic pain through the aggravation of pronociceptive inflammatory responses in mice. J Neurosci 32:3931–3941. 10.1523/JNEUROSCI.4703-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiroi T, Wajima T, Negoro T, Ishii M, Nakano Y, Kiuchi Y, Mori Y, Shimizu S (2013) Neutrophil TRPM2 channels are implicated in the exacerbation of myocardial ischaemia/reperfusion injury. Cardiovasc Res 97:271–281. 10.1093/cvr/cvs332 [DOI] [PubMed] [Google Scholar]
- Holmes GL, Lenck-Santini PP (2006) Role of interictal epileptiform abnormalities in cognitive impairment. Epilepsy Behav 8:504–515. 10.1016/j.yebeh.2005.11.014 [DOI] [PubMed] [Google Scholar]
- Huang S, Turlova E, Li F, Bao MH, Szeto V, Wong R, Abussaud A, Wang H, Zhu S, Gao X, Mori Y, Feng ZP, Sun HS (2017) Transient receptor potential melastatin channels (TRPM2) mediate neonatal hypoxic-ischemic brain injury in mice. Exp Neurol 296:32–40. 10.1016/j.expneurol.2017.06.023 [DOI] [PubMed] [Google Scholar]
- Isami K, Haraguchi K, So K, Asakura K, Shirakawa H, Mori Y, Nakagawa T, Kaneko S (2013) Involvement of TRPM2 in peripheral nerve injury-induced infiltration of peripheral immune cells into the spinal cord in mouse neuropathic pain model. PLoS One 8:e66410. 10.1371/journal.pone.0066410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay TR, von Saucken VE, Landreth GE (2017) TREM2 in neurodegenerative diseases. Mol Neurodegener 12:56. 10.1186/s13024-017-0197-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong H, Kim YH, Lee Y, Jung SJ, Oh SB (2017) TRPM2 contributes to LPC-induced intracellular Ca2+ influx and microglial activation. Biochem Biophys Res Commun 485:301–306. 10.1016/j.bbrc.2017.02.087 [DOI] [PubMed] [Google Scholar]
- Kaneko S, Kawakami S, Hara Y, Wakamori M, Itoh E, Minami T, Takada Y, Kume T, Katsuki H, Mori Y, Akaike A (2006) A critical role of TRPM2 in neuronal cell death by hydrogen peroxide. J Pharmacol Sci 101:66–76. 10.1254/jphs.FP0060128 [DOI] [PubMed] [Google Scholar]
- Kawamoto Y, Akiguchi I, Tomimoto H, Shirakashi Y, Honjo Y, Budka H (2006) Upregulated expression of 14-3-3 proteins in astrocytes from human cerebrovascular ischemic lesions. Stroke 37:830–835. 10.1161/01.STR.0000202587.63936.37 [DOI] [PubMed] [Google Scholar]
- Kehagia AA, Barker RA, Robbins TW (2010) Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson's disease. Lancet Neurol 9:1200–1213. 10.1016/S1474-4422(10)70212-X [DOI] [PubMed] [Google Scholar]
- Kraft R, Grimm C, Grosse K, Hoffmann A, Sauerbruch S, Kettenmann H, Schultz G, Harteneck C (2004) Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am J Physiol Cell Physiol 286:C129–C137. 10.1152/ajpcell.00331.2003 [DOI] [PubMed] [Google Scholar]
- Liu H, Zhang J (2012) Cerebral hypoperfusion and cognitive impairment: the pathogenic role of vascular oxidative stress. Int J Neurosci 122:494–499. 10.3109/00207454.2012.686543 [DOI] [PubMed] [Google Scholar]
- Liu Y, Wu XM, Luo QQ, Huang S, Yang QW, Wang FX, Ke Y, Qian ZM (2015) CX3CL1/CX3CR1-mediated microglia activation plays a detrimental role in ischemic mice brain via p38MAPK/PKC pathway. J Cereb Blood Flow Metab 35:1623–1631. 10.1038/jcbfm.2015.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marazziti D, Consoli G, Picchetti M, Carlini M, Faravelli L (2010) Cognitive impairment in major depression. Eur J Pharmacol 626:83–86. 10.1016/j.ejphar.2009.08.046 [DOI] [PubMed] [Google Scholar]
- Miyake T, Shirakawa H, Kusano A, Sakimoto S, Konno M, Nakagawa T, Mori Y, Kaneko S (2014) TRPM2 contributes to LPS/IFNγ-induced production of nitric oxide via the p38/JNK pathway in microglia. Biochem Biophys Res Commun 444:212–217. 10.1016/j.bbrc.2014.01.022 [DOI] [PubMed] [Google Scholar]
- Miyamoto N, Maki T, Pham LD, Hayakawa K, Seo JH, Mandeville ET, Mandeville JB, Kim KW, Lo EH, Arai K (2013) Oxidative stress interferes with white matter renewal after prolonged cerebral hypoperfusion in mice. Stroke 44:3516–3521. 10.1161/STROKEAHA.113.002813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto N, Maki T, Shindo A, Liang AC, Maeda M, Egawa N, Itoh K, Lo EK, Lok J, Ihara M, Arai K (2015) Astrocytes promote oligodendrogenesis after white matter damage via brain-derived neurotrophic factor. J Neurosci 35:14002–14008. 10.1523/JNEUROSCI.1592-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagafusa Y, Okamoto N, Sakamoto K, Yamashita F, Kawaguchi A, Higuchi T, Matsuda H (2012) Assessment of cerebral blood flow findings using 99mTc-ECD single-photon emission computed tomography in patients diagnosed with major depressive disorder. J Affect Disord 140:296–299. 10.1016/j.jad.2012.03.026 [DOI] [PubMed] [Google Scholar]
- Nishio K, Ihara M, Yamasaki N, Kalaria RN, Maki T, Fujita Y, Ito H, Oishi N, Fukuyama H, Miyakawa T, Takahashi R, Tomimoto H (2010) A mouse model characterizing features of vascular dementia with hippocampal atrophy. Stroke 41:1278–1284. 10.1161/STROKEAHA.110.581686 [DOI] [PubMed] [Google Scholar]
- Pandey GN, Rizavi HS, Zhang H, Ren X (2018) Abnormal gene and protein expression of inflammatory cytokines in the postmortem brain of schizophrenia patients. Schizophr Res 192:247–254. 10.1016/j.schres.2017.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peferoen L, Kipp M, van der Valk P, van Noort JM, Amor S (2014) Oligodendrocyte-microglia cross-talk in the central nervous system. Immunology 141:302–313. 10.1111/imm.12163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perraud AL, Fleig A, Dunn CA, Bagley LA, Launay P, Schmitz C, Stokes AJ, Zhu Q, Bessman MJ, Penner R, Kinet JP, Scharenberg AM (2001) ADP-ribose gating of the calcium permeable LTRPC2 channel revealed by nudix motif homology. Nature 411:595–599. 10.1038/35079100 [DOI] [PubMed] [Google Scholar]
- Perry VH, Holmes C (2014) Microglial priming in neurodegenerative disease. Nat Rev Neurol 10:217–224. 10.1038/nrneurol.2014.38 [DOI] [PubMed] [Google Scholar]
- Saggu R, Schumacher T, Gerich F, Rakers C, Tai K, Delekate A, Petzold GC (2016) Astroglial NF-κB contributes to white matter damage and cognitive impairment in a mouse model of vascular dementia. Acta Neuropathol Commun 4:76. 10.1186/s40478-016-0350-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo JH, Miyamoto N, Hayakawa K, Pham LD, Maki T, Ayata C, Kim KW, Lo EH, Arai K (2013) Oligodendrocyte precursors induce early blood–brain barrier opening after white matter injury. J Clin Invest 123:782–786. 10.1172/JCI65863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seong SY, Matzinger P (2004) Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol 4:469–478. 10.1038/nri1372 [DOI] [PubMed] [Google Scholar]
- Shen Y, Qin H, Chen J, Mou L, He Y, Yan Y, Zhou H, Lv Y, Chen Z, Wang J, Zhou YD (2016) Postnatal activation of TLR4 in astrocytes promotes excitatory synaptogenesis in hippocampal neurons. J Cell Biol 215:719–734. 10.1083/jcb.201605046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirakawa H, Katsumoto R, Iida S, Miyake T, Higuchi T, Nagashima T, Nagayasu K, Nakagawa T, Kaneko S (2017) Sphingosine-1-phosphate induces Ca2+ signaling and CXCL1 release via TRPC6 channel in astrocytes. Glia 65:1005–1016. 10.1002/glia.23141 [DOI] [PubMed] [Google Scholar]
- Smith JA, Das A, Ray SK, Banik NL (2012) Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull 87:10–20. 10.1016/j.brainresbull.2011.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sone D, Watanabe M, Ota M, Kimura Y, Sugiyama A, Maekawa T, Okura M, Enokizono M, Imabayashi E, Sato N, Matsuda H (2017) Thalamic hypoperfusion and disrupted cerebral blood flow networks in idiopathic generalized epilepsy: arterial spin labeling and graph theoretical analysis. Epilepsy Res 129:95–100. 10.1016/j.eplepsyres.2016.12.009 [DOI] [PubMed] [Google Scholar]
- Tarkowski E, Tullberg M, Fredman P, Wikkelsö C (2003) Correlation between intrathecal sulfatide and TNF-alpha levels in patients with vascular dementia. Dement Geriatr Cogn Disord 15:207–211. 10.1159/000068780 [DOI] [PubMed] [Google Scholar]
- Temma T, Yamazaki M, Miyanohara J, Shirakawa H, Kondo N, Koshino K, Kaneko S, Iida H (2017) Sequential PET estimation of cerebral oxygen metabolism with spontaneous respiration of 15O-gas in mice with bilateral common carotid artery stenosis. J Cereb Blood Flow Metab 37:3334–3343. 10.1177/0271678X17692815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyama K, Koibuchi N, Uekawa K, Hasegawa Y, Kataoka K, Katayama T, Sueta D, Ma MJ, Nakagawa T, Yasuda O, Tomimoto H, Ichijo H, Ogawa H, Kim-Mitsuyama S (2014) Apoptosis signal-regulating kinase 1 is a novel target molecule for cognitive impairment induced by chronic cerebral hypoperfusion. Arterioscler Thromb Vasc Biol 34:616–625. 10.1161/ATVBAHA.113.302440 [DOI] [PubMed] [Google Scholar]
- Uchida T, Mori M, Uzawa A, Masuda H, Muto M, Ohtani R, Kuwabara S (2017) Increased cerebrospinal fluid metalloproteinase-2 and interleukin-6 are associated with albumin quotient in neuromyelitis optica: their possible role on blood–brain barrier disruption. Mult Scler 23:1072–1084. 10.1177/1352458516672015 [DOI] [PubMed] [Google Scholar]
- Uranova NA, Bonartsev PD, Androsova LV, Rakhmanova VI, Kaleda VG (2017) Impaired monocyte activation in schizophrenia: ultrastructural abnormalities and increased IL-1β production. Eur Arch Psychiatry Clin Neurosci 267:417–426. 10.1007/s00406-017-0782-1 [DOI] [PubMed] [Google Scholar]
- Villeda SA, Luo J, Mosher KI, Zou B, Britschgi M, Bieri G, Stan TM, Fainberg N, Ding Z, Eggel A, Lucin KM, Czirr E, Park JS, Couillard-Després S, Aigner L, Li G, Peskind ER, Kaye JA, Quinn JF, Galasko DR, et al. (2011) The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 477:90–94. 10.1038/nature10357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindiš B, Gašperšič R, Skalerič U, Kovačič U (2014) Toll-like receptor 4 expression in trigeminal neurons is increased during ligature-induced periodontitis in rats. J Periodontol 85:170–177. 10.1902/jop.2013.130039 [DOI] [PubMed] [Google Scholar]
- Volpe JJ, Kinney HC, Jensen FE, Rosenberg PA (2011) The developing oligodendrocyte: key cellular target in brain injury in the premature infant. Int J Dev Neurosci 29:423–440. 10.1016/j.ijdevneu.2011.02.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakita H, Tomimoto H, Akiguchi I, Kimura J (1998) Dose dependent, protective effect of FK506 against white matter changes in the rat brain after chronic cerebral ischemia. Brain Res 792:105–113. 10.1016/S0006-8993(98)00126-7 [DOI] [PubMed] [Google Scholar]
- Wehrhahn J, Kraft R, Harteneck C, Hauschildt S (2010) Transient receptor potential melastatin 2 is required for lipopolysaccharide-induced cytokine production in human monocytes. J Immunol 184:2386–2393. 10.4049/jimmunol.0902474 [DOI] [PubMed] [Google Scholar]
- Yamada M, Ihara M, Okamoto Y, Maki T, Washida K, Kitamura A, Hase Y, Ito H, Takao K, Miyakawa T, Kalaria RN, Tomimoto H, Takahashi R (2011) The influence of chronic cerebral hypoperfusion on cognitive function and amyloid β metabolism in APP overexpressing mice. PLoS One 6:e16567. 10.1371/journal.pone.0016567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, Kaneko S, Lange I, Fleig A, Penner R, Nishi M, Takeshima H, Mori Y (2008) TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nat Med 14:738–747. 10.1038/nm1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Zhang M, Kang X, Jiang C, Zhang H, Wang P, Li J (2015) Thrombin-induced microglial activation impairs hippocampal neurogenesis and spatial memory ability in mice. Behav Brain Funct 11:30. 10.1186/s12993-015-0075-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Ju J, Deng M, Wang J, Liu H, Xiong L, Zhang J (2017) Hypoxia inducible factor 1α promotes endogenous adaptive response in rat model of chronic cerebral hypoperfusion. Int J Mol Sci 18:e3. 10.3390/ijms18010003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yata K, Nishimura Y, Unekawa M, Tomita Y, Suzuki N, Tanaka T, Mizoguchi A, Tomimoto H (2014) In vivo imaging of the mouse neurovascular unit under chronic cerebral hypoperfusion. Stroke 45:3698–3703. 10.1161/STROKEAHA.114.005891 [DOI] [PubMed] [Google Scholar]
- Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, Qiao L (2013) TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun 4:1611. 10.1038/ncomms2608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Zhang J, Wang L, Chen Y, Wan Y, He Y, Jiang L, Ma J, Liao R, Zhang X, Shi L, Qin Z, Zhou Y, Chen Z, Hu W (2017) Interleukin-1β impedes oligodendrocyte progenitor cell recruitment and white matter repair following chronic cerebral hypoperfusion. Brain Behav Immun 60:93–105. 10.1016/j.bbi.2016.09.024 [DOI] [PubMed] [Google Scholar]
- Zlokovic BV. (2005) Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci 28:202–208. 10.1016/j.tins.2005.02.001 [DOI] [PubMed] [Google Scholar]
- Zuliani G, Ranzini M, Guerra G, Rossi L, Munari MR, Zurlo A, Volpato S, Atti AR, Blè A, Fellin R (2007) Plasma cytokines profile in older subjects with late onset Alzheimer's disease or vascular dementia. J Psychiatr Res 41:686–693. 10.1016/j.jpsychires.2006.02.008 [DOI] [PubMed] [Google Scholar]