

Abstract
N-methyl-d-aspartate receptors (NMDARs) are ionotropic glutamate receptors important for synaptic plasticity, memory, and neuropsychiatric health. NMDAR hypofunction contributes to multiple disorders, including anti-NMDAR encephalitis (NMDARE), an autoimmune disease of the CNS associated with GluN1 antibody-mediated NMDAR internalization. Here we characterize the functional/pharmacological consequences of exposure to CSF from female human NMDARE patients on NMDAR function, and we characterize the effects of intervention with recently described positive allosteric modulators (PAMs) of NMDARs. Incubation (48 h) of rat hippocampal neurons of both sexes in confirmed NMDARE patient CSF, but not control CSF, attenuated NMDA-induced current. Residual NMDAR function was characterized by lack of change in channel open probability, indiscriminate loss of synaptic and extrasynaptic NMDARs, and indiscriminate loss of GluN2B-containing and GluN2B-lacking NMDARs. NMDARs tagged with N-terminal pHluorin fluorescence demonstrated loss of surface receptors. Thus, function of residual NMDARs following CSF exposure was indistinguishable from baseline, and deficits appear wholly accounted for by receptor loss. Coapplication of CSF and PAMs of NMDARs (SGE-301 or SGE-550, oxysterol-mimetic) for 24 h restored NMDAR function following 24 h incubation in patient CSF. Curiously, restoration of NMDAR function was observed despite washout of PAMs before electrophysiological recordings. Subsequent experiments suggested that residual allosteric potentiation of NMDAR function explained the persistent rescue. Further studies of the pathogenesis of NMDARE and intervention with PAMs may inform new treatments for NMDARE and other disorders associated with NMDAR hypofunction.
SIGNIFICANCE STATEMENT Anti-N-methyl-d-aspartate receptor encephalitis (NMDARE) is increasingly recognized as an important cause of sudden-onset psychosis and other neuropsychiatric symptoms. Current treatment leaves unmet medical need. Here we demonstrate cellular evidence that newly identified positive allosteric modulators of NMDAR function may be a viable therapeutic strategy.
Keywords: autoimmune, glutamate, NMDA receptor, schizophrenia
Introduction
Anti-N-methyl-d-aspartate receptor encephalitis (NMDARE) is increasingly recognized as a cause of sudden-onset psychosis and other neuropsychiatric symptoms (Fischer et al., 2016; Dalmau et al., 2017). In fact, NMDARE is now reported as the most common cause of autoimmune encephalitis, with comparable incidence and prevalence to infectious encephalitis (Gable et al., 2012; Dubey et al., 2018). Indicated treatment for NMDARE includes immunosuppressive therapies. With current treatment, ∼80% of patients improve, whereas relapses are reported in 14–25% of cases (Gabilondo et al., 2011; Titulaer et al., 2013). Although reassuring, documented improvement may take place over protracted periods (Titulaer et al., 2013), with persistent psychiatric and cognitive sequelae of disease increasingly recognized (Finke et al., 2016). These findings emphasize the need for complementary approaches to treatment (Panzer and Lynch, 2013), including interventions capable of mitigating symptoms during the acute phase of disease, and strategies that promote receptor recovery and improve longer-term outcomes.
At the cellular level, binding of NMDAR antibodies leads to capping and crosslinking of NMDARs, resulting in receptor dimerization and internalization (Hughes et al., 2010). Other results suggest that human NMDAR antibodies may alter surface NMDAR retention at synapses (Mikasova et al., 2012). The combined effects are thought to be responsible for the unique manifestations of signs and symptoms that characterize patients with NMDARE, a hypothesis reinforced by the observation that several clinical features are reproduced with pharmacological blockade of NMDARs (Krystal et al., 2002; Anticevic et al., 2012; Zorumski et al., 2016). Although loss of surface receptors is key to antibody effects, whether remaining surface receptors exhibit altered function because of antibody binding is unclear.
We examined additional effects of NMDAR antibody exposure by challenging primary cultures of rodent hippocampal neurons through incubation with CSF from NMDARE patients. We found no evidence of altered function of remaining surface NMDARs following 24–48 h of CSF exposure, nor preferential loss of synaptic versus extrasynaptic NMDARS or GluN2A versus GluN2B-containing NMDARs. We provide proof-of-concept, in vitro evidence that a class of oxysterol mimetics, selective positive allosteric modulators (PAMs) of NMDAR function (Paul et al., 2013), rescue the effect of patient CSF when administered midcourse of a 48 h incubation. The results implicate a novel drug class for rapid intervention in NMDARE.
Materials and Methods
Hippocampal cell culture.
Rat hippocampal neuron-astrocyte cocultures were created and maintained as previously described (Mennerick et al., 1995; Moulder et al., 2007; Emnett et al., 2013). Briefly, hippocampi and cortical (astrocyte) tissue were dissected from postnatal day 0–3 rats of either sex using protocols approved by the Institutional Animal Care and Use Committee. Cells were seeded in modified Eagle's medium (Life Technologies) containing 5% horse serum, 5% fetal calf serum, 17 mm d-glucose, 400 mm glutamine, 50 U/ml penicillin, and 50 mg/ml streptomycin. Mass cultures were plated at 650 cells/mm onto 25 mm cover glasses coated with 5 mg/ml collagen or 0.1 mg/ml poly-d-lysine with 1 mg/ml laminin. Micro-island cultures were seeded on microdots of collagen on 0.15% agarose background as described previously (Mennerick et al., 1995; Moulder et al., 2007; Emnett et al., 2013). Experiments were performed at 7–11 DIV.
Patient material.
CSF was collected from patients admitted to Barnes-Jewish Hospital (Washington University in St. Louis; Saint Louis, Missouri) with definite autoimmune encephalitis, including symptoms and signs consistent with NMDARE (Titulaer et al., 2013), and antibodies against the GluN1 subunit of the NMDAR (clinical testing performed by the Mayo Clinic, Rochester, MN). The clinical assay for NMDARE is a cell-based assay using specific anti-IgG immunofluorescence detection methods on transfected cells (EUROIMMUN). We confirmed with anti-human IgG staining of tissue, as described below. Patient A also presented with ovarian teratoma, a classic comorbidity for NMDARE. CSF samples were obtained before induction of appropriate immunotherapy. Antibody titers were 1:4 for Patient A, 1:10 for Patient B, and 1:32 for Patient C. Additional CSF samples were obtained from female patients between 29 and 33 years of age with multiple sclerosis (MS), a chronic neuroinflammatory/autoimmune disease (Polman et al., 2011), as well as from neurologically normal volunteers. Patients were enrolled in ongoing prospective observational research studies at Washington University in St. Louis. Neurologically normal volunteers were recruited from the local community, and CSF was obtained following a brief interview (to establish status as controls) and neurological examination. Study protocols were approved by the Washington University School of Medicine Human Research Protections Office. Written informed consent was obtained from patients (or their delegates) and neurologically normal volunteers, permitting collection, storage, and use of CSF in appropriate clinical research studies. CSF was stored at −70°C until use.
In electrophysiological recording and immunohistochemistry experiments, cells were incubated in 1:12 or 1:24 dilutions of patient CSF, neurologically normal CSF, or artificial CSF (aCSF) containing the following (in mm): 138 NaCl, 4 KCl, 2 CaCl2, 10 glucose, 10 HEPES 2 CaCl2, 1 MgCl2, and 10 glucose. Glutamate receptor antagonists d-APV (10 μm) and NBQX (1 μm) were included to prevent excitotoxicity. Due to limited CSF sample, aliquots were harvested following experiments and reused up to four times; controls and experimental samples were matched for number of uses.
N2a cell transfection.
Murine neuro-2a (N2a; ATCC, CCL-131) cells were cultured in DMEM supplemented with 10% (v/v) FBS, 2 mm glutamine plus 100 U/ml penicillin, and 0.1 mg/ml streptomycin in an atmosphere of 5% CO2 and 95% air. Cells were maintained at subconfluent densities and transiently transfected with GluN1 (0.34 μg) and GluN2B-SEP (2 μg) subunit DNA. GluN1a in pcDNA3 plasmid was a gift kindly provided by Dr. Elias Aizenman (Department of Neurobiology, University of Pittsburgh School of Medicine, Pittsburgh, PA) The pCI-SEP_NR2B subunit was a gift from Roberto Malinow (Addgene plasmid #23998; Kopec et al., 2006). The transfection protocol consisted of 4 h incubation at 37°C in serum-free medium containing plasmids and Lipofectamine 2000 reagent (Invitrogen). Following the incubation, the medium was exchanged for serum-containing medium including 100 μm ketamine to prevent receptor-mediated cell death (Boeckman and Aizenman, 1996).
Slice staining.
Rodent brain tissue was fixed, permeabilized, and incubated in a 1:20 dilution of patient CSF, followed by application of anti-human IgG secondary antibody conjugated to AlexaFluor 555. Images were taken of hippocampus using a 4× objective under epifluorescence illumination.
Electrophysiology.
Whole-cell recording pipettes were pulled from borosilicate glass capillary tubes (World Precision Instruments) and exhibited 2–6 MΩ final open-tip resistances. Unless otherwise stated, neurons were voltage-clamped to −70 mV. Electrophysiological extracellular recording solution typically contained the following: 138 mm NaCl, 4 mm KCl, 2 mm CaCl2, 10 mm glucose, 10 HEPES, 20 μm d-serine, 15 μm gabazine, 1 μm NBQX, and 0.2 μm tetrodotoxin, pH 7.25. Extracellular Mg2+ was excluded during electrophysiological evaluation of NMDAR function. The whole-cell pipette internal solution contained the following (in mm): 140 cesium methanesulfonate, 5 NaCl, 0.5 CaCl2, 5 EGTA, and 10 HEPES, pH 7.25. NMDA receptor function was probed by applying 10 μm NMDA for 5 s.
Evoked EPSCs were obtained from solitary neurons in microcultures with a 1.5 ms depolarization to 0 mV to elicit a breakaway action potential in the cell's axon. For these experiments potassium gluconate replaced cesium methanesulfonate in the pipette solution. Tetrodotoxin was excluded from bath solution, and NBQX was added or excluded as indicated. Access resistance was compensated to 90–95% for evoked autaptic PSC recordings. Some synaptic recordings were obtained in presence of 1 μm NBQX as indicated.
Fluorescence imaging and anti-GFP immunostaining.
To assess surface receptor presence, we performed acid quenching of superecliptic pHluorin (SEP)-tagged receptor subunits in live cells during time-lapse imaging. Imaging was performed 24–48 h post-transfection. Cells were perfused with electrophysiological extracellular saline containing 2 mm CaCl2, 1 mm MgCl2, 10 mm glucose, and 10 μm d-APV. 2-[N-morpholino] ethane sulfonic acid (MES) was then applied by replacing HEPES in the standard saline and setting the pH to 5.5 with HCl. Fluorescence images were acquired with conventional epifluorescence on a Nikon TE2000 inverted microscope and a CCD camera (Roper Scientific) and analyzed using Fiji/ImageJ. For MES-application experiments, fluorescence intensity was measured in a fixed region-of-interest (ROI) near the membrane over the entirety of perfusion (22.5 s). Baseline fluorescence was calculated as the average of pre-MES and post-MES fluorescence to account for time-dependent decreases. This absolute baseline was compared with that at the final image during MES challenge to measure the MES-induced fluorescence change.
For anti-GFP labeling of SEP-tagged surface receptors, transfected N2a cells were incubated in control or patient CSF 24 h post-transfection. After 24 or 48 h of treatment, cultures were fixed for 5 min in 4% paraformaldehyde in PBS. Cells were blocked with 10% normal goat serum and incubated with rabbit anti-GFP antibody (Life Technologies, catalog #A6455) for 3 h at a 1:1000 dilution. Cells were incubated in secondary antibody conjugated to AlexaFluor 568 for 30 min, and mounted onto coverslips for visualization. Cells were imaged on a Nikon TE2000 microscope equipped with a C1 laser scanning confocal module. Single confocal sections through the middle of the cell were analyzed with Fiji/ImageJ. To ensure objectivity, cells were selected for analysis based on SEP fluorescence, without reference to anti-GFP labeling. To assess anti-GFP surface labeling, ROI lines at 30 pixel thickness were drawn through the cell excluding the nucleus. Fluorescence intensity was assessed along the line, using cell exterior as background. Peak fluorescence value within the first 50 pixels was taken as membrane fluorescence, whereas mean center values (middle 50 points) corresponded to intracellular fluorescence.
Experimental design and statistical analysis.
Summary values are mean ± SEM. For all experiments, we used a “yoked” design to guard against ancillary causes of NMDAR alteration. Each set of experimental conditions was performed in parallel on sibling cultures. Each replicate experiment contributed a similar number of cells per experimental condition to overall pooled results.
Electrophysiology data acquisition and analyses were performed using pCLAMP 10 software (Molecular Devices). Image processing and measurement of fluorescence intensity was acquired in Fiji/ImageJ. Data were processed and plotted using Excel 2011 (Microsoft), and Prism 7 (GraphPad). Statistical significance was determined using Student's t tests, one-way ANOVA, or two-way ANOVA as indicated in text and figure legends and as dictated by experimental design. Unless otherwise noted, post hoc two-tailed Student's paired or unpaired t tests with Bonferroni correction were used for multiple comparisons of only those comparisons important for testing a priori hypotheses. Significance was defined as a corrected p value equivalent to <0.05. NMDARE-A CSF-induced depression of NMDA current relative to control was demonstrated in the first figure in two independent replications. In subsequent figures, this statistical comparison was not repeated to reduce the possibility of type II errors in testing of additional hypotheses.
Least-squares minimization algorithms performed in GraphPad Prism or Clampex were used to estimate rate constants using single exponential functions (Emnett et al., 2013).
Results
Characterization of receptor changes with patient CSF exposure
CSF from NMDARE Patient A (32 year-old female, NMDARE-A) yielded staining of rat brain reminiscent of NMDAR expression (Fig. 1A), in contrast with staining using CSF from age- and sex-matched MS Patient A (Fig. 1B; MS-A). These results parallel those of previous studies (Moscato et al., 2014; Dalmau et al., 2017). We next confirmed that 48 h incubation of hippocampal cultures in NMDARE-A CSF (1:12 dilution in conditioned culture medium) depressed NMDAR responsiveness compared with a CSF control (Fig. 1C,D). Strong depression of NMDAR current was observed with dilutions as low as 1:24, and with incubations as brief as 24 h (Fig. 1E). By contrast 1:12 dilution of CSF from neurologically normal volunteers (NN-A, NN-B) or from MS patients (MS-A, MS-B) produced no decrease in NMDAR function relative to aCSF (48 h incubation; Fig. 1F,G). Subsequently, aCSF was used as a comparator for most experiments.
Figure 1.

Depression of NMDA current in hippocampal neurons following patient CSF incubation. A, NMDARE-A CSF (1:20 dilution) labels mouse hippocampus in a pattern typical of NMDAR distribution. Human antibody binding to the tissue section was visualized with anti-human secondary antibody conjugated to AlexaFluor 555. B, CSF from an age- and sex-matched MS patient (1:20 dilution), failed to exhibit an NMDAR-like pattern of labeling. Scale bar: 500 μm. C, NMDA (10 μm) -elicited current from a hippocampal neuron following 48 h incubation with either aCSF (1:12; black trace) or NMDARE-A CSF (1:12; red trace). D, Summary of NMDA current density from cells treated as in C (t(46) = 6.951, *p = 1.08e−8; n = 21 for control and 27 for NMDARE-A). E, Comparison of effects of two dilutions of NMDARE-A CSF, 24 h in either 1:12 dilution (n = 10) or 1:24 dilution (n = 10) in sibling cultures. A one-way ANOVA showed a significant NMDARE-A CSF-mediated current depression (F(2,27) = 14.1, *p = 6.6e−5). Asterisks denote p < 0.05 versus aCSF, determined with Student's unpaired t test, Bonferroni corrected for multiple comparisons. F, Representative traces from an experiment examining NMDA (10 μm) -elicited current from a hippocampal neuron following 48 h incubation of hippocampal cultures in aCSF (1:12; black trace; n = 15) or CSF from MS-A (1:12; gray trace; n = 10). G, Control CSF from age- and sex-matched MS patients (MS-A, MS-B) and neurologically normal controls (NN-A, NN-B). There was no significant difference between any treated group and control; p > 0.05 uncorrected t tests.
Although surface NMDAR internalization is associated with depression of NMDAR current induced by purified patient antibodies or by NMDARE patient CSF (Hughes et al., 2010; Gleichman et al., 2012; Castillo-Gómez et al., 2017), altered function of remaining surface NMDARs could influence clinical expression of disease or response to therapy. To investigate the function of residual receptors, we examined the effect of patient CSF incubation on NMDAR EPSCs, including decay kinetics recorded from solitary cultured neurons. Because of the brief presence of glutamate in the synaptic cleft, EPSC decay renders a good estimate for the elementary lifetime of NMDAR channels when bound by transmitter (Lester et al., 1990). We found that NMDARE-A CSF incubation selectively decreased the NMDAR component of evoked EPSCs (Fig. 2A–C). However, decay kinetics of residual EPSCs were indistinguishable from control (Fig. 2D). Thus, it appears that channel lifetime in response to brief glutamate exposure is not altered by incubation with patient CSF.
Figure 2.

Lack of change in key functional properties of residual NMDARs. A, B, Evoked autaptic EPSCs from a hippocampal neuron incubated with aCSF (A) or NMDARE-A CSF (B). Dual-component AMPAR and NMDAR EPSCs (light traces) and pharmacologically isolated NMDAR EPSCs (dark traces) are shown. C, Summary of ratio of NMDAR to AMPAR peak EPSC (t(14) = 4.2, *p = 1e−9, Student's unpaired t test). D, Summary of weighted time constant values (τw) obtained from a biexponential fit to the decay phase of the NMDAR EPSCs (t(14) = 1.15, p = 0.27, Student's unpaired t test). E, F, The rapid onset/offset open-channel blocker memantine was used to probe changes in NMDAR channel open probability. G, H, Summary of the memantine onset and offset kinetics (obtained from exponential fits) revealed no change following NMDARE-A CSF-mediated depression; (G, t(7) = 1.15, p = 0.29; H, t(13) = 1.33, p = 0.21, respectively, Student's unpaired t tests).
We also evaluated channel open probability of the total NMDAR population by examining onset and offset kinetics of memantine, an uncompetitive open-channel blocker of NMDARs, during application of exogenous NMDA after 24 h incubation in CSF (Fig. 2E,F). In this protocol, decreased Popen of channels results in slower onset/offset kinetics for memantine (Emnett et al., 2015). However, antibody-depressed NMDAR function did not mediate a change in kinetics of memantine block/unblock (Fig. 2G,H). This finding further suggests that residual surface NMDARs exhibit no substantial change in functionality.
Synaptic NMDAR activation is generally considered trophic, while extrasynaptic NMDAR activation is thought to mediate excitotoxic death (Hardingham and Bading, 2010; but see Wroge et al., 2012). Patient-derived NMDAR autoantibodies may differentially affect trafficking of synaptic versus extrasynaptic receptor populations (Mikasova et al., 2012). To determine whether altered balance between synaptic and extrasynaptic receptors might contribute to dysfunction associated with NMDARE, we estimated the contribution of synaptic NMDARs to the total population of NMDARs following incubation with patient CSF. To do so, we used the very slowly dissociating NMDAR open-channel blocker MK-801 to enrich for extrasynaptic receptors (Rosenmund et al., 1995). MK-801 requires channel opening for binding and once bound is nearly irreversible (Huettner and Bean, 1988). Thus, when applied during ongoing synaptic activity, MK-801 preferentially blocks synaptic NMDARs. Upon MK-801 removal, NMDA-elicited current thus arises mainly from the extrasynaptic NMDAR population. Synaptic activity was elevated during MK-801 application (10 μm, 15 min) with coincubation in the GABAA receptor antagonist bicuculline (15 μm), ensuring that subsequent agonist-elicited responses would come nearly exclusively from extrasynaptic receptors (Rosenmund et al., 1995; Hardingham et al., 2002; Wroge et al., 2012). MK-801 treatment was preceded by 24 h incubation with control aCSF or NMDARE Patient A CSF. Following MK-801 removal, our evaluation of NMDA current revealed no difference in the fraction of pharmacologically isolated extrasynaptic receptors in NMDARE-A CSF versus control-incubated cells (Fig. 3A–D). Thus, patient CSF exposure does not induce preferential loss of synaptic receptors or extrasynaptic receptors.
Figure 3.

No evidence of selective effect of NMDARE-A CSF on proportion of extrasynaptic NMDARs or on the proportion of GluN2B-containing receptors. A, B, NMDA-elicited current from neurons without (black) and with (blue) preceding MK-801 challenge to block synaptic receptors. C, Summary of raw NMDA current density of respective ±MK-801 conditions. Analysis of raw current density revealed no interaction between condition (aCSF/NMDARE-A CSF) and treatment (±MK-801; F(1,52) = 0.36, two-way ANOVA, p > 0.55). D, Summary of MK-801 effect, normalized to the mean current density of the respective −MK-801 condition (right). Analysis of normalized data also revealed no change in the proportion of MK-801-insensitive current following NMDARE-A CSF incubation (t(42) = 0.93, p>0.36, Student's unpaired t test). E, F, Representative examples of ifenprodil (10 μm) sensitivity of NMDA-elicited current. G, Summary of raw NMDA current density in baseline and ifenprodil conditions. There was no statistical interaction between condition (aCSF/NMDARE-A CSF) and treatment (±ifenprodil; F(1,48) = 0.7209, p = 0.4, two-way ANOVA, p > 0.05). H, Summary of ifenprodil effect, normalized to the respective baseline current density. Analysis of normalized data also revealed no change in the proportion of ifenprodil-sensitive current following NMDARE-A CSF incubation; (t(24) = 0.74, p=0.47, Student's unpaired t test).
Two major GluN2 subunits, GluN2A and GluN2B, contribute to hippocampal NMDAR responses. The two classes localize and function differently (Chen et al., 1999; Tovar and Westbrook, 1999; Izumi et al., 2005). To determine whether one class is preferentially lost with patient CSF incubation, we used pharmacological isolation with the GluN2B-selective antagonist ifenprodil (10 μm). NMDARE-A CSF incubation caused no change in the ifenprodil sensitivity (Fig. 3E–H), suggesting that the fractional contribution of GluN2A/GluN2B NMDARs does not change as a result of patient CSF incubation. In summary, our evidence supports no preferential loss of NMDARs associated with localization or with particular GluN2 subunit presence.
Our data suggest that residual NMDAR behavior remains unperturbed, and therefore functionally intact following exposure to patient CSF. We would expect from previous work that NMDAR surface expression is diminished to account for the downregulation of NMDAR responses observed (Hughes et al., 2010; Moscato et al., 2014). To test surface expression in live cells, we used a biological assay with N2a cells transfected with heteromeric NMDA receptors composed of GluN1 and SEP-tagged GluN2B subunits. The SEP-tag is a pH-sensitive GFP variant located at the N-terminus. To test functionality of the SEP-tagged subunit, we tested N2a cells in whole-cell, patch-clamp studies. GFP-transfected NMDARs yielded no response to NMDA application, whereas cells transfected with wild-type GluN1 and GluN2B-SEP exhibited robust current in response to NMDA application (219 ± 23 pA, n = 6 control and 6 GluN-transfected cells; t(10) = 8.9, p = < 1e−4, Student's unpaired t test).
In live cells, fluorescence is quenched by the acidity of intracellular organelles but is brighter in surface receptors where the tag is exposed to the neutral pH of the extracellular space (Kopec et al., 2006). Live transfected cells showed robust fluorescence that was quenched by MES, a membrane impermeant acid (Kopec et al., 2006; Fig. 4A). The baseline fluorescence of SEP-transfected cells was weaker in transfected cells incubated in NMDARE-A CSF, and MES quenching was reduced accordingly (Fig. 4B). These results implicate a robust loss of surface receptors with patient CSF treatment.
Figure 4.

NMDARE-A CSF depresses surface NMDAR presence. A, Fluorescence quenching associated with surface NMDARs. N2a cells transfected with GluN1/GluN2B-SEP were used to visualize NMDARs on the plasma membrane. Transient wash with cell-impermeant MES quenched fluorescence in control cells. Scale bar, 10 μm. B, Comparison of MES quenching in control cells and cells incubated in NMDARE-A CSF for 24 h. The absolute MES-induced change in fluorescence between cells treated with MS-A CSF (n = 21 cells) versus NMDARE-A CSF (n = 21 cells from 4 independent replicates) was significantly different (t(40) = 2.4, *p = 0.02, Student's unpaired t test). C–G, Surface receptors detected using anti-GFP labeling of fixed, non-permeabilized transfected N2a cells. Cells were transfected with GluN1/GluN2B-SEP subunits or with cytosolic GFP as a control. Analysis lines (yellow) through labeled cells in various conditions yielded evidence for surface receptors (red; anti-GFP antibody labeling) in GluN2B-SEP-transfected cells labeled with primary antibody but not in other control conditions. Pri, Primary antibody incubated. Significant membrane labeling was detected in SEP-transfected cells labeled with primary antibody, compared with other indicated conditions (Bonferroni corrected Student's unpaired t test, *p < 0.05). G, Numbers on graph bars indicate pooled cell number from four experiments. Scale bar, 10 μm. H–J, Anti-GFP used to label surface NMDARs in transfected N2a cells incubated for 24 h in MS-A CSF or in NMDARE-A CSF, before fixation. NMDARE-A CSF reduced anti-GFP membrane labeling (Bonferroni corrected Student's unpaired t test, *p < 0.05).
As an additional check on surface NMDAR presence in GluN1/GluN2B-SEP transfected N2a cells, we used anti-GFP antibody labeling in fixed, non-permeabilized cells (Fig. 4C–J). Cells transfected with GluN1/GluN2B-SEP exhibited significantly higher membrane-associated anti-GFP labeling than control cells transfected with cytosolic GFP (Fig. 4C–G). In GluN1/GluN2B-SEP-transfected cells incubated in NMDARE-A and MS-A CSF, anti-GFP surface labeling was strongly reduced (Fig. 4H–J). Thus, both live-cell imaging and immunocytochemistry results support loss of surface receptors following incubation with NMDARE patient CSF.
Effects of oxysterol-like PAMs on receptor function following patient CSF exposure.
The above results demonstrate that exposure to NMDARE patient CSF mediates broad loss of surface NMDARs, regardless of synaptic localization or subunit composition. It follows, therefore, that treatment with a broad spectrum NMDAR PAM may be a rational approach to enhancing function of remaining surface NMDARs. The major brain cholesterol metabolite 24S-hydroxycholesterol and several synthetic analogues represent a new class of broad-spectrum PAMs (Sun et al., 2016). Subsequently, we evaluated the ability of SGE-301, a previously characterized synthetic oxysterol analog (Paul et al., 2013), and SGE-550, a similar but previously unreported synthetic PAM, to rescue NMDAR function following exposure to patient CSF.
As expected, the acute (30 s) application of either SGE-301 or SGE-550 (2 μm) potentiated responses to 10 μm NMDA (Fig. 5A–C). We explored effects of PAM incubation as an intervention during patient CSF exposure. Following 24 h incubation of cultured hippocampal cells in NMDARE-A CSF, we intervened with a coapplication of one of the PAMs for an additional 24 h. Following the full 48 h challenge, we removed the culture medium containing CSF and PAM and placed cells in recording saline free of PAM. Despite the absence of PAM in recording solutions, NMDA responses in both aCSF control conditions and NMDARE-A CSF conditions were significantly augmented (Fig. 5D,E). Statistically, there was no interaction between CSF incubation status and PAM treatment status (Fig. 5D,E), indicating that PAMs may not interfere directly with the mechanisms of depressed NMDA current amplitude, including receptor internalization. We followed-up this observation with experimental tests of the mechanism of persistently potentiated NMDA currents following prolonged PAM incubation.
Figure 5.

Intervention by oxysterol-mimetic PAMs. A, B, Acute potentiation of NMDA-elicited current by SGE-301 (2 μm; A) and by SGE-550 (2 μm; B). PAM was pre-applied for 30 s before coapplication with NMDA (colored traces). C, Normalized current showed significant potentiation by both SGE-301(t(27) = 4.3, *p = 2e−4) and SGE-550 (t(63) = 4.3, *p < 1e−4, paired Student's t tests). D, E, Summary of NMDA-elicited current density recorded following 48 h of incubation with either aCSF or NMDARE-A CSF, with or without SGE-301 (D) or SGE-550 (E) treatment at 24 h. Two-way ANOVA with Bonferroni corrected post hoc t tests indicated significant potentiation in aCSF and CSF conditions for both SGE-301 and SGE-550 (*p < 0.05). There was no statistical interaction between CSF condition and either SGE-301 (D; F(1,56) = 0.5132, p = 0.48) or SGE-550 (E; F(1,125) = 2.121, p = 0.15) treatment. Currents were obtained in recording saline, following removal of CSF and PAM in the culture medium.
We have previously shown that oxysterols are resistant to washout, potentially explaining the persistent effects of PAMs (Paul et al., 2013; Linsenbardt et al., 2014). However, we found no correlation between time of recording following PAM removal and the size of NMDA current in our dataset (data not shown). Therefore, we entertained the possibility that oxysterol analogues induce a permanent change in NMDAR function (e.g., by fostering receptor insertion or inhibiting internalization in control and antibody-treated conditions). We used the open-channel blocker memantine to test whether persistently increased NMDA current following PAM incubation is associated with increased channel open probability, as expected of PAM action (Emnett et al., 2015). Indeed, cells incubated 24 h with SGE-301 or with SGE-550, but recorded in the absence of PAM, exhibited faster onset and offset memantine kinetics (Fig. 6A–D). We further evaluated whether the potentiation of NMDA current results from acute drug presence by briefly incubating cells with the steroid scavenger γ-cyclodextrin (γ-CDX; 500 μm, 2 min; Akk et al., 2005; Shu et al., 2007; Paul et al., 2013). Although γ-CDX incubation alone appeared to have a mild potentiating effect, the scavenger effectively reversed the persisting PAM potentiation (Fig. 6E–G). The rapid reduction produced by the scavenger causes us to conclude that persisting potentiation is most readily explained by lingering compound, likely retained within the membrane, and/or sequestered intracellularly.
Figure 6.
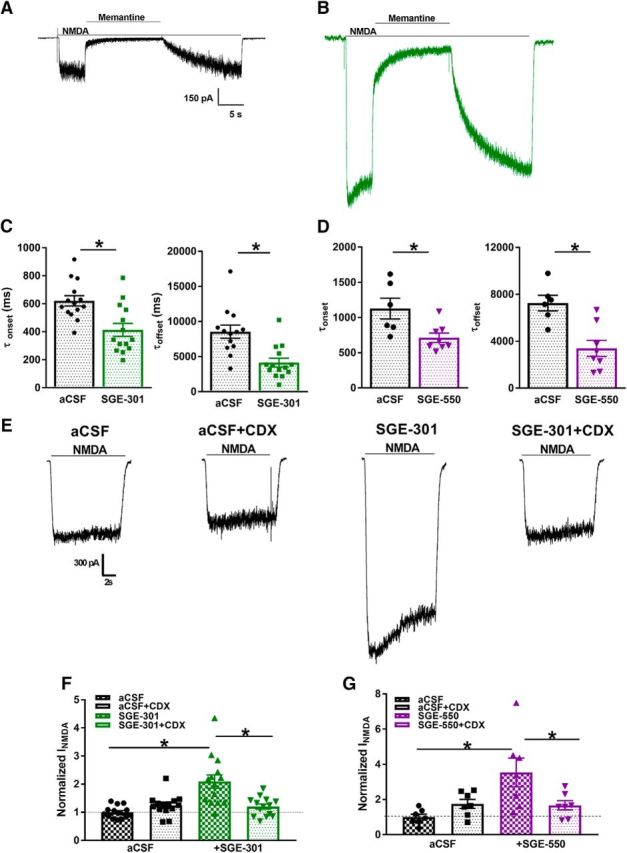
Tests of residual PAM activity to explain persisting potentiation. A, B, Memantine test of NMDAR channel open probability following 24 h aCSF incubation (A) or incubation with SGE-301 (B). C, D, Summary of memantine onset and offset time constant values, obtained from exponential fits; (C) onset: t(13) = 3.1, *p = 1.6e−3; (C) offset: t(25) = 3.9, *p = 6e−4; (D) onset: t(12) = 2.8, *p = 1.6e−2; (D) offset: t(12) = 3.9, *p = 2e−3, Student's unpaired t tests. E, Sample traces from cultures incubated under control or in SGE-301 (2 μm) for 24 h then rinsed with saline or γ-CDX (500 μm) for 2 min before recording. F, G, Summary of effect of brief CDX incubation on residual SGE-301 (F) and SGE-550 (G). A two-way ANOVA showed a significant main effect of SGE-301 (F(13,39) = 11.35, *p < 1e−4). Post hoc testing revealed significant potentiation by SGE-301 and SGE-550 and significant reversal of potentiation by CDX (*p < 0.05, Bonferroni corrected Student's unpaired t tests).
As a final test of whether PAMs interfere directly with the mechanisms of NMDAR removal, we returned to N2a cells transfected with SEP-tagged GluN subunits to visualize surface receptors. We imposed the same CSF challenge and PAM intervention as in Figure 4 on cells to assess surface receptor presence. Cells were incubated for 48 h in NMDARE-A or MS-A CSF with SGE-301 intervention initiated at 24 h. Following the treatment, cells were fixed and labeled with anti-GFP antibody (Fig. 7A–D). We again observed significant depression of surface receptor presence in cells treated with NMDARE Patient A CSF. SGE-301 did not affect membrane or intracellular labeling, nor was there a statistically significant interaction between CSF condition and SGE-301 treatment (Fig. 7E,F). This suggests that PAMs do not alter surface receptor presence, and the observed PAM-associated persisting NMDAR potentiation is most likely a result of SGE-301's direct interaction with NMDARs.
Figure 7.
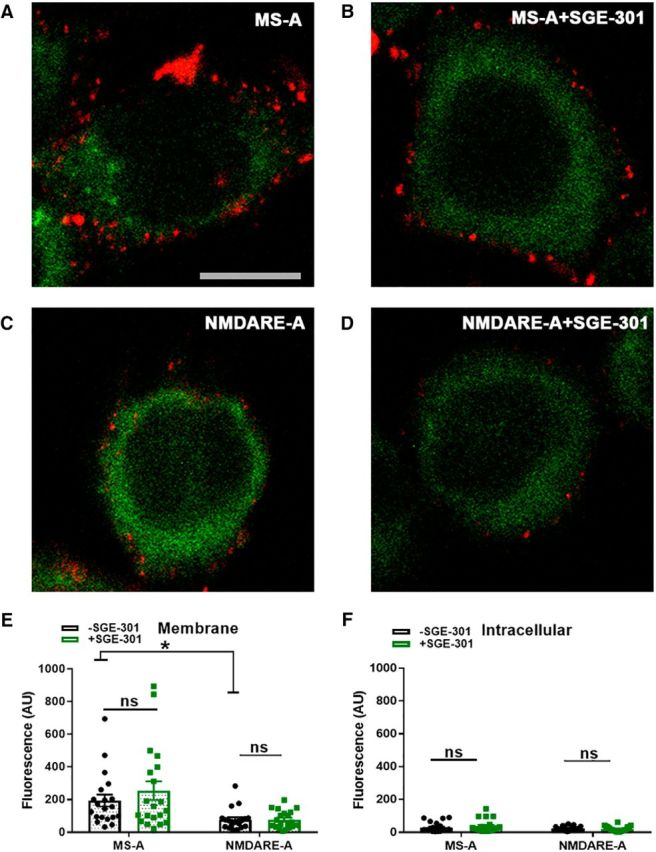
No effect of SGE-301 intervention on surface NMDAR presence. A–D, Examples of GluN1/GluN2B-SEP-transfected N2a cells incubated under the indicated conditions. Cells were incubated in CSF for 48 h and incubation with SGE-301 commenced 24 h following CSF initiation. Anti-GFP labeling (red) in non-permeabilized cells was used to quantify surface NMDARs. Green labeling represents SEP fluorescence following fixation. E, F, Two-way ANOVA with Bonferroni corrected post hoc t tests indicated significant depression of surface labeling after incubation in NMDARE-A CSF (E; t(38) = 3.0, *p = 4.8e−3). There was no statistical interaction between SGE-301 and CSF condition (E; F(1,76) = 0.75, p = 0.39). SGE-301 did not affect membrane (E) or intracellular anti-GFP labeling (F) in post hoc testing (p > 0.05, Bonferroni corrected t tests).
To extend the proof-of-principle for the PAM therapeutic approach, we examined the effect of SGE-301 on CSF incubation from two additional NMDARE patients (NMDARE-B, -C; Fig. 8). Incubation in NMDARE-B and NMDARE-C CSF (23- and 50-year-old females, respectively; 1:12 dilution for 48 h) decreased responses to NMDA, and the depression was circumvented by incubation in SGE-301, similar to results with NMDARE-A CSF (Fig. 8E). These results suggest that the PAM approach may be broadly applicable in patients with NMDARE.
Figure 8.
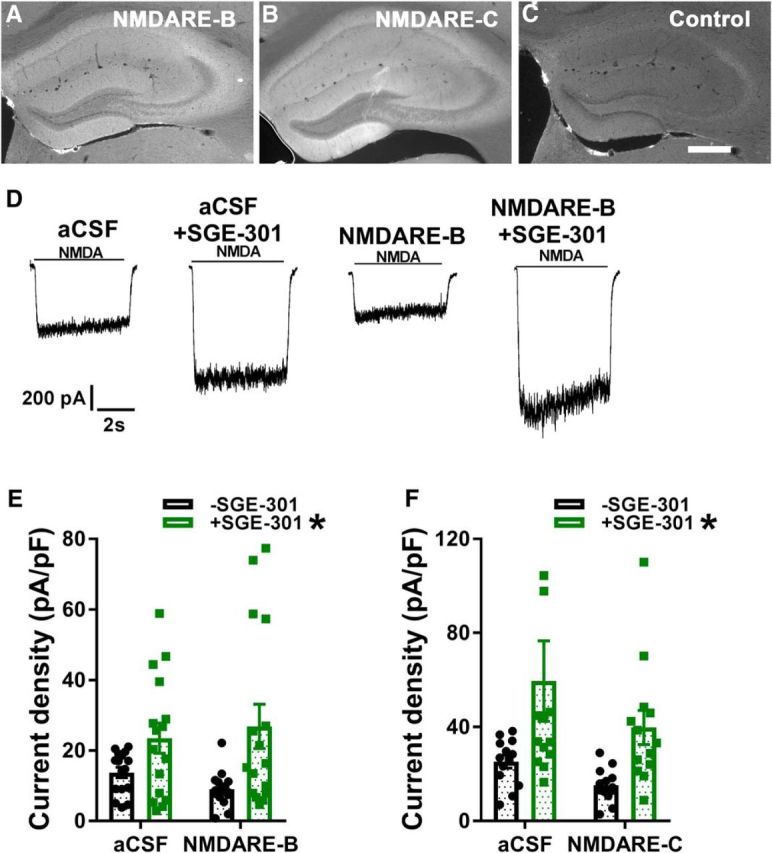
Effect of SGE-301 intervention on depression induced by CSF from a second NMDARE patient. A–C, Staining performed as in Figure 1, A and B, for two additional patient samples and a control sample. Scale bar: 500 μm. D, Sample traces representative of the indicated conditions. NMDARE-B CSF was incubated for 48 h at 1:12 dilution. Intervention with 2 μm SGE-301 commenced at 24 h. E, Summary of effect of incubation with NMDARE-B CSF and intervention with SGE-301. A two-way ANOVA showed a significant main effect of SGE-301 (F(1,59) = 11.9, *p = 1.0e−3; asterisk associated with legend) but no interaction with CSF incubation condition (F(1,59) = 1.02, p = 0.32). Post hoc testing revealed significant depression of NMDA-mediated current induced by NMDARE-B CSF compared with the aCSF condition (t(29) = 2.38, *p = 0.02, Student's unpaired t test). F, Same protocol and statistics as depicted in D and E applied to patient NMDAR-C. A two-way ANOVA showed a significant main effect of SGE-301(F(1,48) = 9.71, *p = 3.1e−3) but no interaction with CSF incubation condition (F(1,48) = 0.26, p = 0.61). Post hoc testing revealed significant depression of NMDA-mediated current induced by NMDARE-B CSF compared with the aCSF condition (t(24) = 2.99, *p = 6.4e−3, Student's unpaired t test). One data point in the aCSF+SGE-301 condition >120 pA/pF is not shown on the graph for clarity but was used in statistics.
Discussion
Our proof of concept studies support the use of NMDAR PAMs as adjunct or complementary treatments for NMDARE and possibly for other disorders associated with NMDAR hypofunction, including schizophrenia (Olney and Farber, 1995; Krystal et al., 2002; Lin et al., 2012; Cioffi, 2013). Using CSF obtained from a patient with a confirmed clinical diagnosis of NMDARE, we functionally characterized NMDARs in cultured rat hippocampal cells following incubation with patient CSF, and we quantified characteristics of rescue with two allosteric modulators of NMDAR function. Persistent effects of compounds restore NMDAR function through allosteric potentiation of residual NMDARs and suggest that signs and symptoms of NMDARE may be ameliorated by this therapeutic approach. These cellular studies set a course for future in vivo tests of the therapeutic approach.
Our study relied primarily on CSF from a single NMDARE patient for which we had sufficient CSF to permit multiple studies. This patient presented with classic features of NMDARE (see Materials and Methods). Nevertheless, we designed several experiments to test that CSF from NMDARE Patient A possesses features characteristic of NMDARE. Features confirmed in our studies included CSF labeling (using IgG-specific secondary antibody) of rodent brain tissue in a pattern expected of NMDAR distribution, functional selective depression of NMDAR current, and loss of surface NMDARs. We replicated key results in two additional patients with IgG against NMDARs, confirmed by testing in commercial clinical laboratories and by our own staining (Fig. 8).
In addition, our experiments characterized several new features of NMDAR suppression studied using NMDARE Patient A sample. In contrast with some recent studies (Mikasova et al., 2012), our results suggest that both synaptic and extrasynaptic receptors are equally targeted for loss. Although the dimensions and molecular complexity of the synaptic cleft might reduce antibody access, direct access of various antibodies to the synapse has been demonstrated in numerous previous studies (Ehlers, 2000; Martens et al., 2008; Mikasova et al., 2012). Thus, it seems likely that both populations of NMDARs are directly targeted by human GluN antibody. Alternatively, interchange between synaptic and extrasynaptic populations (Tovar and Westbrook, 2002) may allow indirect antibody access to synaptic receptors by targeting the extrasynaptic pool. Future time course studies may distinguish these possibilities.
Other previous studies have suggested that antibody may alter the function of remaining surface NMDARs. For instance, within minutes following patient CSF exposure, increased channel open duration was detected (Gleichman et al., 2012). Our studies revealed no evidence for increased or decreased open probability of NMDAR channels following prolonged patient CSF exposure, using an open-channel blocker as a probe of channel open probability in whole-cell recordings (Huettner and Bean, 1988). Discrepancies with previous work may reflect differences in the time at which measurements were made or heterogeneity in the clinical population. These hypotheses can be tested as additional patient material becomes available.
Together, our findings suggest that treatment with PAMs may represent a potential strategy to rescue NMDAR hypofunction, with no detectable direct effect on the mechanisms of NMDAR internalization that characterize the cellular pathophysiology. A more direct approach to maintain or restore NMDAR function would be to target the cell biological mechanisms responsible for receptor endocytosis and insertion; however, at this time, pharmacological interventions targeting these mechanisms are not practical. Therefore, we made use of emerging understanding of a class of positive regulators of NMDAR function that has resulted from study of the endogenous cholesterol metabolite, 24S-hydroxycholesterol (Sun et al., 2016).
SGE-301 and SGE-550 represent synthetic analogues of 24S-hydroxycholesterol with selective NMDAR effects in the sub-to-low micromolar concentration range (Paul et al., 2013; Linsenbardt et al., 2014). These compounds appear to bind to a site independent of other known allosteric modulators of NMDAR function (Paul et al., 2013; Wilding et al., 2016) and increase channel open probability. Results with chimeric GluN-GluK subunits suggest that GluN transmembrane domains are critical for oxysterol modulation (Wilding et al., 2016). A transmembrane interaction site would be consistent with the lipophilic nature of these modulators and with their sensitivity to cyclodextrin extraction (Shu et al., 2007). Interaction of oxysterols with NMDARs results in a slight reduction in the EC50 of agonists and increased agonist efficacy (Linsenbardt et al., 2014). Here we show that several dilutions of NMDARE patient CSF result in similar presumed steady-state NMDAR reductions (Fig. 1). The mechanisms responsible for a fixed steady-state level of functional receptors despite varied antibody concentration are unclear. We found that low micromolar concentrations of allosteric modulators restored the activity of NMDARs from this steady-state to near or above baseline levels (Figs. 5, 6, 8).
A puzzling observation initially was that potentiated NMDAR function persisted beyond the period of direct exposure to compounds in all cells incubated in SGE-301 or SGE-550. We found that the enhanced function was associated with increased channel open probability, was sensitive to a scavenger of lipophilic compounds, and was not associated with increased surface NMDARs. Thus, we attribute the persistence to the strong lipophilicity of the modulators. We have previously shown evidence of modulator persistence for at least tens of seconds (Paul et al., 2013). The present studies confirm that some oxysterol-like compounds can persist for many minutes beyond removal, presumably by adhering to cells or accumulating intracellularly, potentiating NMDAR responses despite constant local perfusion with compound-free saline. This persistence of drug binding becomes relevant when assessing therapeutic viability, recognizing that strong lipophilicity may be advantageous to dosing schedules. However, drug exposure in a clinical setting would presumably last beyond the 24 h incubation period used herein and could result in other changes not appreciated here. Additional in vivo studies are required to clarify the time-dependent effects, and effect of dosing regimen on NMDAR function.
Our work focused on the restorative actions of NMDAR modulators following exposure to CSF from NMDARE patients. The idea that NMDAR hypofunction accounts for the broad spectrum of clinical symptomatology described in NMDARE is supported by two primary lines of evidence. First, pharmacologically-induced NMDAR hypofunction produces psychotomimetic symptoms, not unlike those observed in NMDARE. Second, intrathecal injection of recombinant antibodies can recapitulate key aspects of the disorder in rodents (Malviya et al., 2017). We acknowledge, however, that additional pathogenic processes downstream of NMDAR internalization, or other CSF constituents (e.g., other disease-associated antibodies or inflammatory mediators; Kreye et al., 2016) may affect neuronal function and contribute to symptomatology. Some of these may account for apparent differences in the degree of suppression of NMDAR function (e.g., the stronger suppression observed in NMDARE Patient A vs Patients B and C). Others may be unrelated to NMDAR function ant thus not modulated by NMDAR PAMs. Furthermore, sample quantities limited culture studies to relatively brief periods of exposure, unlike the clinical situation where antibody is constantly renewed, resulting in prolonged exposure. For these reasons, it will be important to adopt robust animal models and follow-up the cellular level studies herein with circuit-level and in vivo tests of the therapeutic potential of NMDAR PAMs in NMDARE (Planagumà et al., 2015; Malviya et al., 2017).
Our work shows that indiscriminate loss of NMDARs occurs following exposure to NMDARE patient CSF, and suggests that PAMs may be developed as a potential therapeutic strategy. Restoration of NMDAR function was achieved with application of oxysterol analogues, which are potent, selective, but broad spectrum PAMs of NMDAR function. The restorative effects were fully accounted for by the acute pharmacological effects of the compounds.
Footnotes
This work was supported by a sponsored project agreement between Sage Pharmaceuticals and Washington University School of Medicine, and the Harry Weaver Neuroscience Scholar of the National Multiple Sclerosis Society (NMSS, JF 2144A2/1) to L.P. We thank Roberto Malinow and Elias Aizenman for making SEP-tagged and WT GluN subunits available and Anne Cross for help in obtaining control patient samples.
Conflict of interest: S.M.P., M.L., J.D., M.Q., and C.F.Z. have financial interests in Sage Therapeutics. The remaining authors declare no competing financial interests.
References
- Akk G, Shu HJ, Wang C, Steinbach JH, Zorumski CF, Covey DF, Mennerick S (2005) Neurosteroid access to the GABAA receptor. J Neurosci 25:11605–11613. 10.1523/JNEUROSCI.4173-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anticevic A, Gancsos M, Murray JD, Repovs G, Driesen NR, Ennis DJ, Niciu MJ, Morgan PT, Surti TS, Bloch MH, Ramani R, Smith MA, Wang XJ, Krystal JH, Corlett PR (2012) NMDA receptor function in large-scale anticorrelated neural systems with implications for cognition and schizophrenia. Proc Natl Acad Sci U S A 109:16720–16725. 10.1073/pnas.1208494109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeckman FA, Aizenman E (1996) Pharmacological properties of acquired excitotoxicity in chinese hamster ovary cells transfected with N-methyl-d-aspartate receptor subunits. J Pharmacol Exp Ther 279:515–523. [PubMed] [Google Scholar]
- Castillo-Gómez E, Oliveira B, Tapken D, Bertrand S, Klein-Schmidt C, Pan H, Zafeiriou P, Steiner J, Jurek B, Trippe R, Prüss H, Zimmermann WH, Bertrand D, Ehrenreich H, Hollmann M (2017) All naturally occurring autoantibodies against the NMDA receptor subunit NR1 have pathogenic potential irrespective of epitope and immunoglobulin class. Mol Psychiatry 22:1776–1784. 10.1038/mp.2016.125 [DOI] [PubMed] [Google Scholar]
- Chen N, Luo T, Raymond LA (1999) Subtype-dependence of NMDA receptor channel open probability. J Neurosci 19:6844–6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cioffi CL. (2013) Modulation of NMDA receptor function as a treatment for schizophrenia. Bioorg Med Chem Lett 23:5034–5044. 10.1016/j.bmcl.2013.07.019 [DOI] [PubMed] [Google Scholar]
- Dalmau J, Geis C, Graus F (2017) Autoantibodies to synaptic receptors and neuronal cell surface proteins in autoimmune diseases of the central nervous system. Physiol Rev 97:839–887. 10.1152/physrev.00010.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon V, Gadoth A, Smith CY, Bryant SC, Klein CJ, Aksamit AJ, Toledano M, Boeve BF, Tilemma JM, Flanagan EP (2018) Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis. Ann Neurol 83:166–177. 10.1002/ana.25131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. (2000) Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28:511–525. 10.1016/S0896-6273(00)00129-X [DOI] [PubMed] [Google Scholar]
- Emnett CM, Eisenman LN, Taylor AM, Izumi Y, Zorumski CF, Mennerick S (2013) Indistinguishable synaptic pharmacodynamics of the N-methyl-D-aspartate receptor channel blockers memantine and ketamine. Mol Pharmacol 84:935–947. 10.1124/mol.113.089334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emnett CM, Eisenman LN, Mohan J, Taylor AA, Doherty JJ, Paul SM, Zorumski CF, Mennerick S (2015) Interaction between positive allosteric modulators and trapping blockers of the NMDA receptor channel. Br J Pharmacol 172:1333–1347. 10.1111/bph.13007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finke C, Kopp UA, Pajkert A, Behrens JR, Leypoldt F, Wuerfel JT, Ploner CJ, Prüss H, Paul F (2016) Structural hippocampal damage following anti-N-methyl-d-aspartate receptor encephalitis. Biol Psychiatry 79:727–734. 10.1016/j.biopsych.2015.02.024 [DOI] [PubMed] [Google Scholar]
- Fischer CE, Golas AC, Schweizer TA, Munoz DG, Ismail Z, Qian W, Tang-Wai DF, Rotstein DL, Day GS (2016) Anti N-methyl-d-aspartate receptor encephalitis: a game-changer? Expert Rev Neurother 16:849–859. 10.1080/14737175.2016.1184088 [DOI] [PubMed] [Google Scholar]
- Gabilondo I, Saiz A, Galán L, González V, Jadraque R, Sabater L, Sans A, Sempere A, Vela A, Villalobos F, Viñals M, Villoslada P, Graus F (2011) Analysis of relapses in anti-NMDAR encephalitis. Neurology 77:996–999. 10.1212/WNL.0b013e31822cfc6b [DOI] [PubMed] [Google Scholar]
- Gable MS, Sheriff H, Dalmau J, Tilley DH, Glaser CA (2012) The frequency of autoimmune N-methyl-d-aspartate receptor encephalitis surpasses that of individual viral etiologies in young individuals enrolled in the California encephalitis project. Clin Infect Dis 54:899–904. 10.1093/cid/cir1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleichman AJ, Spruce LA, Dalmau J, Seeholzer SH, Lynch DR (2012) Anti-NMDA receptor encephalitis antibody binding is dependent on amino acid identity of a small region within the GluN1 amino terminal domain. J Neurosci 32:11082–11094. 10.1523/JNEUROSCI.0064-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Bading H (2010) Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11:682–696. 10.1038/nrn2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardingham GE, Fukunaga Y, Bading H (2002) Extrasynaptic NMDARs oppose synaptic NMDARs by triggering CREB shut-off and cell death pathways. Nat Neurosci 5:405–414. 10.1038/nn835 [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP (1988) Block of N-methyl-d-aspartate- activated current by the anticonvulsant MK-801: selective binding to open channels. Proc Natl Acad Sci U S A 85:1307–1311. 10.1073/pnas.85.4.1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes EG, Peng X, Gleichman AJ, Lai M, Zhou L, Tsou R, Parsons TD, Lynch DR, Dalmau J, Balice-Gordon RJ (2010) Cellular and synaptic mechanisms of anti-NMDA receptor encephalitis. J Neurosci 30:5866–5875. 10.1523/JNEUROSCI.0167-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi Y, Nagashima K, Murayama K, Zorumski CF (2005) Acute effects of ethanol on hippocampal long-term potentiation and long-term depression are mediated by different mechanisms. Neuroscience 136:509–517. 10.1016/j.neuroscience.2005.08.002 [DOI] [PubMed] [Google Scholar]
- Kopec CD, Li B, Wei W, Boehm J, Malinow R (2006) Glutamate receptor exocytosis and spine enlargement during chemically induced long-term potentiation. J Neurosci 26:2000–2009. 10.1523/JNEUROSCI.3918-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreye J, Wenke NK, Chayka M, Leubner J, Murugan R, Maier N, Jurek B, Ly LT, Brandl D, Rost BR, Stumpf A, Schulz P, Radbruch H, Hauser AE, Pache F, Meisel A, Harms L, Paul F, Dirnagl U, Garner C, et al. (2016) Human cerebrospinal fluid monoclonal N-methyl-d-aspartate receptor autoantibodies are sufficient for encephalitis pathogenesis. Brain 139:2641–2652. 10.1093/brain/aww208 [DOI] [PubMed] [Google Scholar]
- Krystal JH, Anand A, Moghaddam B (2002) Effects of NMDA receptor antagonists: implications for the pathophysiology of schizophrenia. Arch Gen Psychiatry 59:663–664. 10.1001/archpsyc.59.7.663 [DOI] [PubMed] [Google Scholar]
- Lester RA, Clements JD, Westbrook GL, Jahr CE (1990) Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature 346:565–567. 10.1038/346565a0 [DOI] [PubMed] [Google Scholar]
- Lin CH, Lane HY, Tsai GE (2012) Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharmacol Biochem Behav 100:665–677. 10.1016/j.pbb.2011.03.023 [DOI] [PubMed] [Google Scholar]
- Linsenbardt AJ, Taylor A, Emnett CM, Doherty JJ, Krishnan K, Covey DF, Paul SM, Zorumski CF, Mennerick S (2014) Different oxysterols have opposing actions at N-methyl-d-aspartate receptors. Neuropharmacology 85:232–242. 10.1016/j.neuropharm.2014.05.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malviya M, Barman S, Golombeck KS, Planagumà J, Mannara F, Strutz-Seebohm N, Wrzos C, Demir F, Baksmeier C, Steckel J, Falk KK, Gross CC, Kovac S, Bönte K, Johnen A, Wandinger KP, Martín-García E, Becker AJ, Elger CE, Klöcker N, et al. (2017) NMDAR encephalitis: passive transfer from man to mouse by a recombinant antibody. Ann Clin Transl Neurol 4:768–783. 10.1002/acn3.444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens H, Weston MC, Boulland JL, Grønborg M, Grosche J, Kacza J, Hoffmann A, Matteoli M, Takamori S, Harkany T, Chaudhry FA, Rosenmund C, Erck C, Jahn R, Härtig W (2008) Unique luminal localization of VGAT-C terminus allows for selective labeling of active cortical GABAergic synapses. J Neurosci 28:13125–13131. 10.1523/JNEUROSCI.3887-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Que J, Benz A, Zorumski CF (1995) Passive and synaptic properties of hippocampal neurons grown in microcultures and in mass cultures. J Neurophysiol 73:320–332. 10.1152/jn.1995.73.1.320 [DOI] [PubMed] [Google Scholar]
- Mikasova L, De Rossi P, Bouchet D, Georges F, Rogemond V, Didelot A, Meissirel C, Honnorat J, Groc L (2012) Disrupted surface cross-talk between NMDA and ephrin-B2 receptors in anti-NMDA encephalitis. Brain 135:1606–1621. 10.1093/brain/aws092 [DOI] [PubMed] [Google Scholar]
- Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ (2014) Acute mechanisms underlying antibody effects in anti-N-methyl-d-aspartate receptor encephalitis. Ann Neurol 76:108–119. 10.1002/ana.24195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Shin W, Gillis KD, Mennerick S (2007) Vesicle pool heterogeneity at hippocampal glutamate and GABA synapses. J Neurosci 27:9846–9854. 10.1523/jneurosci.2803-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Farber NB (1995) Glutamate receptor dysfunction and schizophrenia. Arch Gen Psychiatry 52:998–1007. 10.1001/archpsyc.1995.03950240016004 [DOI] [PubMed] [Google Scholar]
- Panzer JA, Lynch DR (2013) Neuroimmunology: treatment of anti-NMDA receptor encephalitis-time to be bold? Nat Rev Neurol 9:187–189. 10.1038/nrneurol.2013.31 [DOI] [PubMed] [Google Scholar]
- Paul SM, Doherty JJ, Robichaud AJ, Belfort GM, Chow BY, Hammond RS, Crawford DC, Linsenbardt AJ, Shu HJ, Izumi Y, Mennerick SJ, Zorumski CF (2013) The major brain cholesterol metabolite 24(S)-hydroxycholesterol is a potent allosteric modulator of N-methyl-d-aspartate receptors. J Neurosci 33:17290–17300. 10.1523/JNEUROSCI.2619-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planagumà J, Leypoldt F, Mannara F, Gutiérrez-Cuesta J, Martín-Garcia E, Aguilar E, Titulaer MJ, Petit-Pedrol M, Jain A, Balice-Gordon R, Lakadamyali M, Graus F, Maldonado R, Dalmau J (2015) Human N-methyl D-aspartate receptor antibodies alter memory and behaviour in mice. Brain 138:94–109. 10.1093/brain/awu310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O'Connor P, Sandberg-Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS (2011) Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol 69:292–302. 10.1002/ana.22366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Feltz A, Westbrook GL (1995) Synaptic NMDA receptor channels have a low open probability. J Neurosci 15:2788–2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu HJ, Zeng CM, Wang C, Covey DF, Zorumski CF, Mennerick S (2007) Cyclodextrins sequester neuroactive steroids and differentiate mechanisms that rate limit steroid actions. Br J Pharmacol 150:164–175. 10.1038/sj.bjp.0706973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun MY, Linsenbardt AJ, Emnett CM, Eisenman LN, Izumi Y, Zorumski CF, Mennerick S (2016) 24(S)-Hydroxycholesterol as a modulator of neuronal signaling and survival. Neuroscientist 22:132–144. 10.1177/1073858414568122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titulaer MJ, McCracken L, Gabilondo I, Armangué T, Glaser C, Iizuka T, Honig LS, Benseler SM, Kawachi I, Martinez-Hernandez E, Aguilar E, Gresa-Arribas N, Ryan-Florance N, Torrents A, Saiz A, Rosenfeld MR, Balice-Gordon R, Graus F, Dalmau J (2013) Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol 12:157–165. 10.1016/S1474-4422(12)70310-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (1999) The incorporation of NMDA receptors with a distinct subunit composition at nascent hippocampal synapses in vitro. J Neurosci 19:4180–4188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar KR, Westbrook GL (2002) Mobile NMDA receptors at hippocampal synapses. Neuron 34:255–264. 10.1016/S0896-6273(02)00658-X [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Lopez MN, Huettner JE (2016) Chimeric glutamate receptor subunits reveal the transmembrane domain is sufficient for NMDA receptor pore properties but some positive allosteric modulators require additional domains. J Neurosci 36:8815–8825. 10.1523/JNEUROSCI.0345-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroge CM, Hogins J, Eisenman L, Mennerick S (2012) Synaptic NMDA receptors mediate hypoxic excitotoxic death. J Neurosci 32:6732–6742. 10.1523/JNEUROSCI.6371-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorumski CF, Izumi Y, Mennerick S (2016) Ketamine: NMDA receptors and beyond. J Neurosci 36:11158–11164. 10.1523/JNEUROSCI.1547-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]