

Abstract
Methods to promote myelin regeneration in response to central myelin loss are essential to prevent the progression of clinical disability in demyelinating diseases. The neurotrophin brain-derived neurotrophic factor (BDNF) is known to promote myelination during development via oligodendrocyte expressed TrkB receptors. Here, we use a structural mimetic of BDNF to promote myelin regeneration in a preclinical mouse model of central demyelination. In female mice, we show that selective targeting of TrkB with the BDNF-mimetic enhances remyelination, increasing oligodendrocyte differentiation, the frequency of myelinated axons, and myelin sheath thickness after a demyelinating insult. Treatment with exogenous BDNF exerted an attenuated effect, increasing myelin sheath thickness only. Further, following conditional deletion of TrkB from premyelinating oligodendrocytes, we show the effects of the BDNF-mimetic on oligodendrocyte differentiation and remyelination are lost, indicating these are dependent on oligodendrocyte expression of TrkB. Overall, these studies demonstrate that targeting oligodendrocyte TrkB promotes in vivo remyelination in the brain.
SIGNIFICANCE STATEMENT Novel strategies to promote myelin regeneration are required to prevent progressive neurodegeneration and clinical disability in patients with central demyelinating disease. Here, we test whether selectively targeting the TrkB receptor on the myelin-producing oligodendrocytes, can promote remyelination in the brain. Using a structural mimetic of its native ligand, BDNF, we show that stimulation of TrkB enhances remyelination, increasing oligodendrocyte differentiation, the frequency of myelinated axons and thickness of the myelin sheath following a demyelinating insult. Further, we show that these effects are dependent on the phosphorylation of oligodendrocyte expressed TrkB receptors in vivo. Overall, we demonstrate that selective targeting of TrkB has therapeutic potential to promote remyelination in the brain.
Keywords: BDNF, CNS, neurotrophins, oligodendrocytes, remyelination, TrkB
Introduction
Innate myelin regeneration is often incomplete in CNS demyelinating diseases such as multiple sclerosis (MS). This leaves axons exposed, resulting in conduction deficits, loss of metabolic and trophic support to axons, and contributes to secondary irreversible axonal damage that ultimately drives the progressive clinical disability in patients (Lucchinetti et al., 1999; Chang et al., 2002; Albert et al., 2007). Current commercially available immunomodulatory therapies for MS effectively decrease the frequency of relapses, but do not directly stimulate remyelination (Stangel et al., 2017). As such there is a compelling need to complement these therapies with new strategies to promote myelin regeneration. The neurotrophin brain-derived neurotrophic factor (BDNF) enhances CNS myelination, acting through oligodendrocyte-expressed TrkB receptors (Du et al., 2003; Xiao et al., 2010). Despite the relative potency with which BDNF enhances myelination, its promiscuity to both p75NTR and TrkB receptors, brief half-life (Poduslo and Curran, 1996), large molecular size, and relatively poor ability to cross the blood–brain barrier make it a poor therapeutic candidate, likely contributing to its failure in clinical trials for neurodegenerative disease (The BDNF Study Group, 1999).
To overcome these limitations, a range of small molecular weight neurotrophin receptor agonists have been developed (Longo and Massa, 2013). This includes tricyclic-dimeric peptide-6 (TDP6), a peptide mimetic structurally based on the loop-2 region of the BDNF homodimer that interacts with TrkB (O'Leary and Hughes, 2003). When BDNF binds and activates TrkB, it triggers autophosphorylation of the intracellular domain, recruitment of cytosolic adaptor proteins, and activation of many intracellular signaling cascades, including PI3K/Akt and MAPK/Erk (Chao, 2003). We have previously shown that TDP6, like BDNF, enhances oligodendrocyte myelination in vitro, and does this through phosphorylation of oligodendrocyte-expressed TrkB receptors (Wong et al., 2014).
There is a clinical need to develop remyelinating therapies capable of complementing existing immunomodulatory treatments for MS. Here we test whether oligodendrocyte expressed TrkB receptors are a rational target to achieve this, by assessing whether TDP6 promotes myelin repair in vivo. We show infusion of TDP6, but not BDNF, into the CNS increased oligodendrocyte differentiation, the frequency of myelinated axons and myelin sheath thickness in the cuprizone model of toxic demyelination. Importantly, we demonstrate that this effect is driven by phosphorylation of oligodendrocyte expressed TrkB receptors in vivo. These data suggest selective targeting of TrkB is a rational approach to promote myelin repair in vivo.
Materials and Methods
Experimental animals and cuprizone-induced demyelination.
Female C57BL/6 mice, aged 8–10 weeks were fed 0.2% cuprizone in normal chow (Teklad Custom Research Diets) for 6 weeks to induce demyelination. Cuprizone feed was removed and mice were killed or received intracerebroventricular osmotic pumps for 7 d.
For experiments in conditional knock-out mice, 8- to 10-week-old CNPaseCre+/− female progeny of CNPaseCre+/− mice (Lappe-Siefke et al., 2003) crossed to TrkBfl/fl mice (Lulkart et al., 2005) bred to a C57BL/6 background for five generations, underwent procedures described above. Additional Cre−/− and Cre+/− female mice aged 14–16 weeks were used as healthy controls.
All mice were housed in specific pathogen-free conditions at the Melbourne Brain Centre Animal Facility. All animal procedures were approved by The Florey Institute for Neuroscience and Mental Health Animal Ethics Committee and followed the Australian Code of Practice for the Care and Use of Animals for Scientific Purposes.
Intracerebroventricular delivery of BDNF and TDP6.
Following cuprizone feeding, mice received either 4 μm BDNF carried in 0.1% BSA in artificial CSF (aCSF; n = 8), 40 μm TDP6 in aCSF (n = 9) or the aCSF vehicle (n = 7) via intracerebroventricular osmotic pumps with a flow rate of 0.5 μl/h (ALZET). Infusion concentrations were based on effective concentrations of BDNF and TDP6 in previously published in vitro myelination assays (Wong et al., 2014). Cannulae were implanted at coordinates +0.5 mm rostral and −0.7 mm lateral of bregma to administer into the right lateral ventricle. Anesthesia was induced with 4–5% isoflurane and 0.5% oxygen, and maintained at 2.5–1% isoflurane and 0.5% oxygen through a nose cone during stereotaxic manipulation. All mice were placed in a recovery chamber maintained at 32°C and monitored immediately following surgery for adverse reactions, and then daily. After 7 d of continuous infusion, mice were killed and the brain removed for immunostaining and electron microscopic (EM) analysis.
Tissue processing and immunofluorescence.
Mice were anesthetized and transcardially perfused with 0.1 m sterile mouse isotonic PBS followed by 4% paraformaldehyde (PFA). Brains were collected and postfixed in 4% PFA overnight. The first millimeter of the right hemisphere from the sagittal midline (Fig. 1B) was selected using a sagittal mouse brain matrix and placed in Kanovsky's buffer overnight and washed in 0.1 m sodium cacodylate before embedding in epoxy resin at the Peter MacCallum Centre for Advanced Histology and Microscopy for EM analysis. The left hemisphere (Fig. 1B) was cryoprotected in 30% sucrose and frozen in OCT in isopentane over dry ice.
Figure 1.
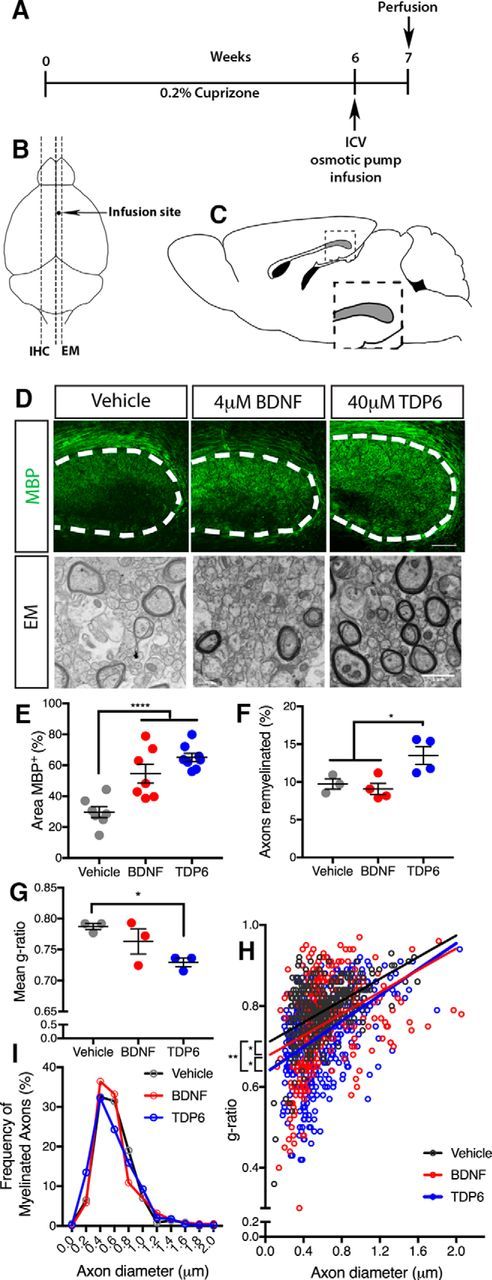
BDNF and TDP6 treatment enhance remyelination following cuprizone-induced demyelination. A, Schematic of experimental procedures. B, Schematic of tissue selection; contralateral hemisphere to infusion site was processed for immunohistochemistry, whereas ipsilateral hemisphere was taken for EM. C, Schematic of sagittal brain section 0.36 mm lateral from midline adapted from Paxinos and Franklin (2001); caudal region of the corpus callosum used for analysis is shaded in gray (inset). D, Representative micrographs of MBP immunostaining, and electron micrographs of myelinated and unmyelinated axons in the caudal corpus callosum of vehicle-, BDNF-, and TDP6-treated mice respectively. Dotted line indicates the boundary of caudal corpus callosum in sagittal plane. MBP micrographs: Scale bar, 20 μm. EM: Scale bar, 1 μm. E, Percentage area positive for MBP immunostaining was significantly enhanced in the caudal corpus callosum of mice treated with 4 μm BDNF and 40 μm TDP6 for 7 d after 6 weeks cuprizone demyelination, compared with the vehicle-treated mice (****p < 0.0001, n = 7–9/group). F, Percentage of myelinated axons was significantly increased with TDP6, but not BDNF, compared with vehicle-treated controls (*p = 0.018, n = 3–4/group). G, Mean g-ratio was significantly decreased with TDP6 treatment compared with vehicle controls, indicative of thicker myelin sheaths (*p = 0.019, n = 3–4/group). E–G, One-way ANOVA with Tukey's post hoc multiple comparisons. Mean ± SEM plotted. H, Scatter-plot of g-ratio against axon diameter. Linear regression analysis revealed a significant decrease in the y-intercepts between BDNF and TDP6-treated mice compared with vehicle-treated controls, and between TDP6 compared with BDNF treatment (*p = 0.0042), but no significant change in slope (p = 0.45). I, Frequency distribution plot of myelinated axon diameters indicating no change with treatment in the frequency of myelinated axons based on size (p = 0.43, χ2 distribution test). F–I, Minimum 150 axons/animal, n = 3/group.
Sagittal sections were cut at 10 μm using a cryostat maintained between −20 to −17°C and collected on Superfrost+ slides, air-dried and stored at −80°C until use. Approximately 70–100 μm separated adjacent sections on each slide. Sections cut beyond ±2.64 mm lateral from the midline were excluded.
For immunofluorescence, slides were washed in PBS before overnight incubation at room temperature with primary antibodies diluted in 10% normal donkey serum (NDS) with 0.3% Triton X-100. Slides were washed in PBS before 2 h incubation with the appropriate fluorophore-conjugated secondary antibody in the dark. After washing with PBS, slides were counterstained with nuclear marker Hoechst 33442 and coverslipped using aqueous mounting media (Dako). For all stains, immunohistochemistry was performed in batches.
Antibodies used were as follows: rat anti-myelin basic protein (MBP; 1:200; MAB386, Millipore) as a marker for remyelination, rabbit anti-Olig2 (1:200; AB9610, Millipore), mouse anti-CC1 (1:200; APC, OP80, CalBioChem), goat anti-platelet derived growth factor receptor-α (PDGFRα; 1:200; AF1062, R&D Systems) to identify stages of the oligodendrocyte lineage, goat anti-Iba1 (1:200; ab5076, Abcam) for microglia and mouse anti-glial fibrillary acidic protein (GFAP; 1:100; MA360, Millipore) for astrocytes. Rabbit anti-Ki67 (1:200; RM-9160, Thermo Scientific) was used to assess cell proliferation following heat-induced antigen retrieval with citrate buffer (10 mm, pH 6). To identify the level of TrkB expression rabbit anti-TrkB (1:500; R-149-100, Biosensis) was used, and TrkB activation was detected with antibodies against phosphorylated TrkB (pTrkBS478; 1:200; R-1718-50, Biosensis).
For MBP immunostaining sections were postfixed with ice-cold 100% methanol for 10 min before the first wash. For pTrkBS478, tris-based saline with 0.3% Triton X-100 was used for all washes and the antibody diluent contained 1% bovine serum albumin in addition to 10% NDS and primary antibody incubation was performed overnight at 4°C.
Electron microscopy and analysis.
Semithin (0.5–1 μm) sections of the caudal corpus callosum in a sagittal plane were collected on glass slides and stained with 1% toluidine blue. Ultrathin (0.1 μm) sections were subsequently collected on 3 × 3 mm copper grids. Specimens were examined using JEOL 1011 transmission electron microscope, and images were collected using MegaView III CCD cooled camera operated with iTEM AnalySIS software (Olympus Soft Imaging Systems). Six distinct fields-of-view were imaged at 10,000× magnification per animal. Images were used to count myelinated axons, measure axon diameters, myelin thickness, and g-ratios in FIJI/ImageJ (ImageJ 1.51K, NIH). A minimum of three fields-of-view (142 μm2) were examined per animal with three to four mice per treatment group. For g-ratios, at least 100 axons from three mice per group were measured. Resin embedding, sectioning, poststaining, and EM imaging were performed at the Peter MacCallum Centre for Advanced Histology and Microscopy.
Fluorescence imaging and analysis.
All imaging was performed blinded to treatment group and restricted to the caudal region of the corpus callosum, ∼−1.1 to −3.0 mm from bregma (Fig. 1C). Analysis was restricted to the splenium of the corpus callosum; tracts contributing to the dorsal hippocampal commissure were excluded. For each analysis, a minimum of three sections per animal were imaged.
To quantify the level of remyelination images of MBP-stained sections were collected with an AxioVision Hr camera attached to a Zeiss Axioplan2 epifluorescence microscope under a 20× objective. Uniform exposure times were used.
Remaining analyses were performed using images acquired with a Zeiss LSM780 or LSM880 confocal microscope with 405, 488, 561, and 633 nm laser lines. For each fluorescent stain, uniform settings were used.
MBP and GFAP staining were quantified as described by Fletcher et al. (2014) using the threshold function in FIJI/ImageJ and limited to a standard region-of-interest (ROI) of 625,000 μm2 for each section. Data were expressed as the percentage area of positive staining in a single ROI.
Cell counts.
All cell counts were performed using maximum intensity projection images generated from z-stacks. A standard ROI was set (625,000 μm2) and nuclei masks identified by Hoechst or positive Olig2 staining were segmented and counted in FIJI/ImageJ using the “Analyze Particles …” function to create masks. The threshold function was then applied to identified positive CC1, PDGFRα, Iba1-positive staining and to generate a binary image. Positive nuclei were identified using the Binary Reconstruction plugin (Legland et al., 2016), and counted with the Analyze Particles function. Automated counts were verified by manually counting a subsample of images. For pTrkBS478 and Ki67 cell counts were performed manually. Data were expressed as the number of cells per square millimeter or the proportion out of the total number of nuclei.
HPLC and mass spectroscopy analysis of TDP6 after 7 d minipump incubation in vivo.
TDP6 prepared the day before pump administration (day 0) and retrieved from the reservoir of osmotic mini-pumps implanted in conditional knock-out mice (day 7) were analyzed by reverse-phase HPLC (RP-HPLC), using an Agilent 1200 series unit, fitted with a Phenomenex Luna C8 column (5u; 50×4.6 mm), running 0–60% acetonitrile over a 14 min window. UV spectra were measured for each sample using a 214 nm wavelength, showing a single TDP6 peptide peak at 7.3 min (Fig. 6). A sample of aCSF was run as a control.
Figure 6.

TDP6 was retrieved and detected after 7 d incubation within osmotic minipumps implanted in conditional knock-out mice. A, Reverse-phase HPLC UV trace of aCSF (“blank/control” sample). B, HPLC UV trace of TDP6 at day before animal administration. C, HPLC UV trace of TDP6 at day 0. D, HPLC UV trace of TDP6 at day 7. All traces measured using 214 nm wavelength. Note: Additional peak in D was determined not to be a peptide degradation product. Area under the peptide peak was quantified in B–D and no significant change was observed.
Liquid-chromatography mass spectrometry, using an Agilent 6100 series single quadrupole system, was used to confirm the molecular weight of the peptide, TDP6 showing the predicted m/z of 1207.5 [M + 2H]2+.
Experimental design and statistical analyses.
All data collection was performed blind to sample identity. To compare between treatment groups, one-way or two-way ANOVA or unpaired Student's t tests were performed with post hoc multiple-comparison tests as appropriate. For each ANOVA the effect size was estimated using ηp2, whereas for t tests Cohen's d and SD were calculated. For g-ratios, linear regressions were performed, and axon diameter frequency distribution was assessed using χ2 distribution tests. All statistical tests were performed in GraphPad Prism 7 with p < 0.05 considered significant.
Results
BDNF, and its structural mimetic TDP6, enhance myelin repair following cuprizone-induced demyelination
Having previously shown that BDNF and TDP6 enhance oligodendrocyte myelination in vitro (Wong et al., 2014), here we tested the capacity of BDNF and TDP6 to promote remyelination in vivo. To do this, demyelination was induced in 8- to 10-week-old female C57BL/6 mice with 0.2% cuprizone in normal chow for 6 weeks (Fig. 1A). Efficacy of cuprizone-induced demyelination was confirmed through EM and immunostaining for MBP in the brains of mice collected at the end of the 6-week cuprizone challenge (minimum n = 2/cohort). These mice demonstrated demyelination, with loss of myelin from axons identified by EM and severe reduction in the level of MBP expression within the corpus callosum (data not shown). Remaining mice received 4 μm BDNF, 40 μm TDP6, or aCSF vehicle delivered at a rate of 0.5 μl/h by intracerebroventricular osmotic pump for 7 d to the right lateral ventricle. Dose was chosen based on our previous in vitro data (Wong et al., 2014). After 7 d infusion, immunostaining (Fig. 1D) revealed that BDNF and TDP6 significantly increased the area of the contralateral caudal corpus callosum positive for MBP (Fig. 1E; ****p < 0.001, ηp2 = 0.66), indicative of greater remyelination. EM analysis of myelin ultrastructure (Fig. 1D) revealed that in the caudal corpus callosum of mice treated with TDP6, the proportion of myelinated axons was significantly increased compared with vehicle controls (Fig. 1F; *p < 0.018, ηp2 = 0.63). In contrast, in those treated with BDNF, the proportion of myelinated axons was similar to those that only received the vehicle (Fig. 1F). Assessment of myelin sheath thickness by g-ratio revealed that TDP6 treatment significantly reduced the mean g-ratio compared with the vehicle (Fig. 1G; *p = 0.019, ηp2 = 0.63) indicating thicker myelin sheaths. Linear regression indicated that BDNF also significantly increased myelin sheath thickness (Fig. 1H; *p = 0.04) compared with the vehicle, but that TDP6 had a more pronounced effect with significantly thicker myelin than both BDNF (Fig. 1H; *p = 0.02) and vehicle treatment (Fig. 1H; **p = 0.014). There was no change in the frequency distribution of myelinated axons based on axon diameter (Fig. 1I). Overall, these findings indicate TDP6 significantly enhances remyelination in vivo.
Treatment with TDP6, but not BDNF, increases the density of postmitotic oligodendrocytes
To identify whether BDNF or TDP6 treatment exerted an effect on oligodendroglial subpopulations, we next examined the density of total Olig2+ oligodendroglia, Olig2+PDGFRα+ oligodendrocyte progenitor cells (OPCs) and postmitotic Olig2+CC1+ oligodendrocytes in the caudal corpus callosum using immunohistochemistry (Fig. 2A). The density of Olig2+ oligodendroglia was significantly increased with TDP6 treatment, above that seen with BDNF or vehicle treatment (Fig. 2B; *p = 0.024, ηp2 = 0.30). Examination of the contribution of OPCs to this increase in oligodendroglia, revealed there was no significant change in the density of Olig2+PDGFRα+ OPCs in mice treated with TDP6 compared with those treated with BDNF or the vehicle (Fig. 2C; p = 0.06, ηp2 = 0.22). In contrast, the density of Olig2+CC1+ oligodendrocytes in TDP6-treated mice was significantly increased compared with treatment with BDNF and the vehicle (Fig. 2D; **p = 0.0051, ηp2 = 0.38). To examine whether this increase in postmitotic oligodendrocytes was due to TDP6 exerting an effect on OPC proliferation post-cuprizone, we assessed the proportion of PDGFRα+ cells colabeled with proliferative cell marker, Ki67. This revealed that TDP6 infusion did not alter the proportion of OPCs proliferating at 7 d post-cuprizone removal (6 weeks cuprizone: 0.17 ± 0.06; Vehicle: 0.14 ± 0.08; TDP6: 0.09 ± 0.03, p = 0.67, ηp2 = 0.12). This increased density of oligodendrocytes, but not OPCs, suggests TDP6 may increase overall oligodendrocyte differentiation and/or their survival during remyelination. Together, the increase in oligodendrocyte density is consistent with the increased proportion of remyelinated axons in mice treated with TDP6, but not BDNF.
Figure 2.
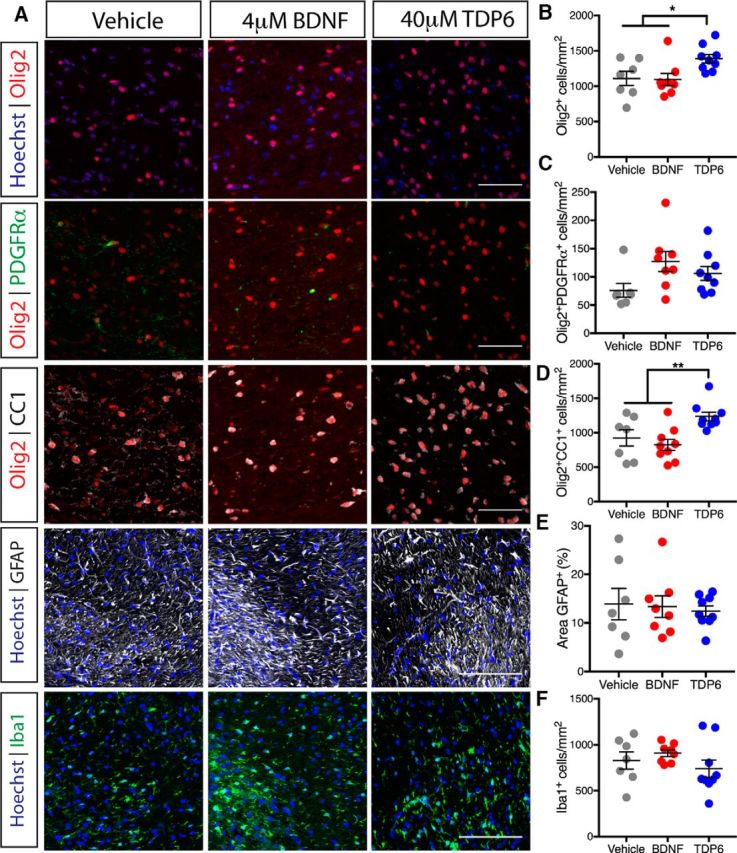
Treatment with TDP6, but not BDNF, enhances oligodendrocyte density and differentiation during remyelination. A, Representative micrographs of immunostaining in the caudal region of the corpus callosum for total Olig2+ oligodendrocytes, Olig2+PDGFRα+ OPCs, and Olig2+CC1+ oligodendrocytes (Scale bar, 50 μm), as well as GFAP+ astrocytosis and Iba1+ microglia (Scale bar, 100 μm). B, Density of total Olig2+ oligodendrocytes was significantly increased in TDP6-treated mice compared with treatment with BDNF or vehicle (*p = 0.024). C, Density of Olig2+PDGFRα+ OPCs was unchanged across treatments (p = 0.06). D, Density of Olig2+CC1+ oligodendrocytes significantly increased with TDP6 treatment compared with BDNF and vehicle treatments (*p = 0.0051). E, Percentage area of GFAP+ astrocytes was unchanged across treatments (p = 0.89). F, Density of Iba1+ microglia was unchanged across treatments (p = 0.34). B–F, n = 7–9/group, one-way ANOVA with Tukey's post hoc multiple comparisons. Mean ± SEM plotted.
To determine whether BDNF or TDP6 treatment altered gliosis, the contralateral caudal corpus callosum was immunolabeled with GFAP for astrocytosis and Iba1 for microgliosis (Fig. 2A). Astrocytosis persisted with both BDNF and TDP6 treatments, and the level of GFAP staining was unchanged across the three groups (Fig. 2E; p = 0.34, ηp2 = 0.011). Similarly, the density of Iba1+ microglia was unchanged across treatments (Fig. 2F; p = 0.34, ηp2 = 0.099). This suggests treatment with either TDP6 or BDNF exerts no reductive effects on neuroinflammatory cell populations, indicating their effect on myelin repair is not secondary to an anti-inflammatory effect.
TDP6 and BDNF increase TrkB phosphorylation in oligodendrocytes during remyelination
As a structural-mimetic of the loop-2 region of the BDNF homodimer, TDP6 is designed to selectively interact with and initiate phosphorylation of TrkB receptors (O'Leary and Hughes, 2003). Indeed, we have previously shown that TDP6 phosphorylates oligodendrocyte-expressed TrkB receptors and promotes myelination in vitro (Wong et al., 2014). To examine whether BDNF and TDP6 treatment successfully stimulated TrkB phosphorylation in oligodendrocytes in vivo, the contralateral caudal corpus callosum was coimmunolabeled with antibodies directed against the phosphorylated serine 478 of TrkB (pTrkBS478), and the oligodendroglial markers CC1 and PDGFRα (Fig. 3A). This revealed that the proportion of total pTrkBS478+ cells in the corpus callosum was significantly increased following TDP6 infusion, but not BDNF, compared with the vehicle (Fig. 3B; **p = 0.0047, ηp2 = 0.49). Assessing the proportion of pTrkBS478+ cells that were PDGFRα+ OPCs, indicated a trend toward increased pTrkBS478+ OPCs in mice treated with TDP6 (Fig. 3C; p = 0.12, ηp2 = 0.22). In PDGFRα+ cells, pTrkBS478 was observed to be colocalized either with PDGFRα or intracellularly (Fig. 3A) in all groups. In contrast, pTrkBS478 was only seen intracellularly in CC1+ cells. The proportion of CC1-pTrkBS478 double-positive cells in the corpus callosum was significantly increased in mice treated with BDNF and TDP6 (Fig. 3D; ****p < 0.0001, ηp2 = 0.25). These results demonstrate that both BDNF and TDP6 successfully reached their cellular targets following intracerebroventricular delivery, and were capable of phosphorylating TrkB on oligodendrocytes.
Figure 3.

Treatment with TDP6 increased TrkB phosphorylation in the remyelinating corpus callosum. A, Representative micrographs of pTrkBS478 immunostaining in the caudal region of the corpus callosum. Scale bar, 20 μm. Asterisk, PDGFRα+ cell featured in inset; arrowhead, CC1+ cell featured in inset. B, Proportion of pTrkBS478+ cells in the caudal corpus callosum was increased with TDP6 treatment compared with BDNF and vehicle controls (**p = 0.0047). C, Proportion of pTrkBS478+ cells that were PDGFRα+ OPCs tended toward an increase with TDP6 treatment (p = 0.12). D, Proportion of pTrkBS478+ cells that were CC1+ oligodendrocytes significantly increased with both BDNF and TDP6 treatment. B–D, n = 7–9/group, one-way ANOVA with Tukey's post hoc multiple comparisons. Mean ± SEM plotted.
Adult myelination and demyelination is unaltered by deletion of oligodendroglial TrkB
To test whether the pro-remyelinating effect of TDP6 required TrkB expression on oligodendrocytes, we generated mice with an oligodendrocyte-specific deletion of TrkB (Lulkart et al., 2005) driven by the CNPase promoter (Lappe-Siefke et al., 2003; CNPaseCre+/− TrkBfl/fl mice). These mice had ∼3-fold reduction in Olig2+TrkB+ cells in the caudal region of the corpus callosum at 14–16 weeks of age (Fig. 4A, quantified in B).
Figure 4.
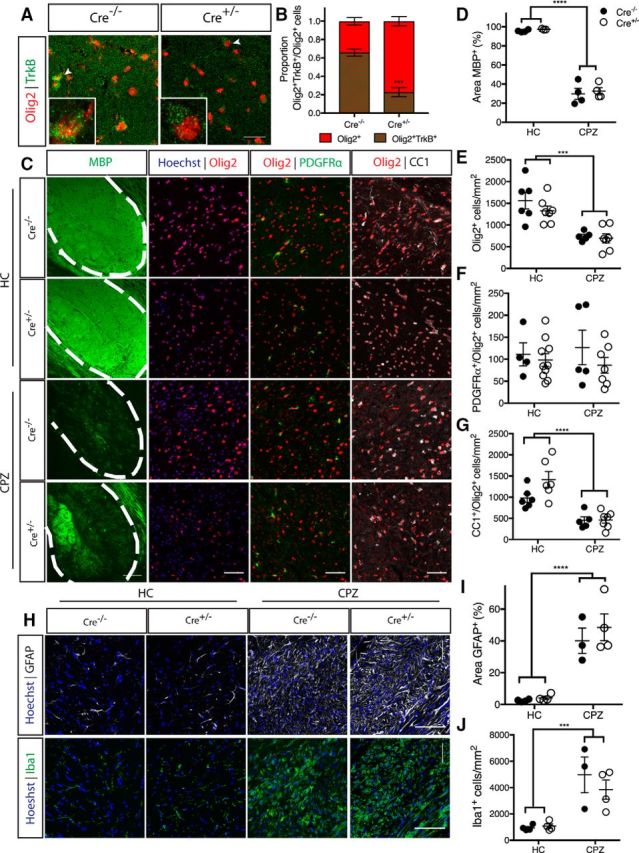
CNPaseCre+/− × TrkBfl/fl mice exhibit normal adult myelination and response to cuprizone-demyelination. A, Representative micrographs of TrkB and Olig2 immunostaining in the caudal corpus callosum of CNPaseCre−/− and CNPaseCre+/− TrkBfl/fl healthy controls aged 16 weeks. Scale bar, 20 μm. B, Proportion of Olig2+ cells positive for TrkB was significantly decreased in CNPaseCre+/− TrkBfl/fl mice compared with Cre−/− controls. Student's t test, n = 4–6/group. ***p = 0.0003. Mean ± SEM plotted. C, Representative micrographs of immunostaining for MBP and oligodendrocyte lineage markers Olig2, PDGFRα, and CC1 in the caudal corpus callosum of CNPaseCre−/− (closed circles) and CNPaseCre+/− (open circles) TrkBfl/fl healthy controls (HC), and cuprizone-fed (CPZ) mice. Scale bar, 50 μm. D, Area of MBP immunostaining and (E) Olig2+ cell density were unchanged due to genotype in the healthy controls, but were significantly decreased in both genotypes following cuprizone treatment (****p < 0.0001 and ***p < 0.001, respectively). F, Density of Olig2+PDGFRα+ OPCs was unchanged with cuprizone treatment or genotype (p = 0.94). G, Olig2+CC1+ oligodendrocytes were unchanged between genotypes in healthy controls, while cuprizone feeding significantly reduced the density of these cells in the caudal corpus callosum of both genotypes (****p < 0.0001). H, Representative micrographs of GFAP+ astrocytosis and Iba1+ microglia in the caudal corpus callosum of Cre−/− and Cre+/− HC and CPZ mice. Scale bar, 50 μm. I, Percentage area GFAP+ and (J) density of Iba1+ microglia were similar in both genotypes in HCs, and were increased in both genotypes following CPZ (****p < 0.0001, ***p = 0.0003). D–G, I, J, n = 3–7/group, two-way ANOVA.
To assess the effect of TrkB deletion from CNPase-expressing cells on oligodendrocyte density and myelin, we first compared unchallenged conditional knock-out and wild-type littermate mice, then compared the effect of cuprizone-induced demyelination on the two genotypes. Female CNPaseCre−/− TrkBfl/fl and CNPaseCre+/− TrkBfl/fl mice were unchallenged, or fed 0.2% cuprizone in normal chow for 6 weeks from 8 to 10 weeks of age, killed and the caudal corpus callosum immunohistochemically analyzed (Fig. 4C). In unchallenged mice, similar levels of MBP staining were observed regardless of genotype, and both genotypes demyelinated to a similar extent following cuprizone (Fig. 4C, quantitated in D; pgenotype = 0.55, ηp2 = 0.033). Similarly, the density of Olig2+ oligodendroglia (Fig. 4E), Olig2+PDGFRα+ OPCs (Fig. 4F), and Olig2+CC1+ oligodendrocytes (Fig. 4G) was similar in unchallenged mice, with cuprizone exerting no differences between genotypes (Olig2+ cells: pgenotype = 0.28, ηp2 = 0.055; OPCs: pgenotype = 0.27, ηp2 = 0.054; Olig2+CC1+: pgenotype = 0.074, ηp2 = 0.15). Assessment of astrogliosis (GFAP staining; Fig. 4H quantitated in I; pgenotype = 0.38, ηp2 = 0.070) and microgliosis (Iba1 staining; Fig. 4H quantitated in J; pgenotype = 0.47, ηp2 = 0.048) also exhibited no differences between genotype in either unchallenged or cuprizone challenged conditions. These data indicate that oligodendroglial TrkB does not exert an essential role in oligodendroglial or myelin maintenance in the adult, or following cuprizone-induced demyelination
TDP6-enhanced oligodendrocyte differentiation is dependent on oligodendroglial TrkB
To determine whether the pro-remyelinating effects of TDP6 required oligodendrocyte expression of TrkB, we repeated the cuprizone experiments and 7 d osmotic pump infusions with either TDP6 (n = 4) or vehicle (n = 3) in CNPaseCre+/− × TrkBfl/fl conditional knock-out mice. Immunohistochemistry for MBP (Fig. 5A) revealed the pro-remyelinating effect exerted by TDP6 in wild-type mice was lost, with both vehicle and TDP6-treated conditional knock-out mice exhibiting the same level of MBP staining (Fig. 5B; p = 0.45, d = 0.63 ± 0.78). Further, the density of total Olig2+ oligodendroglia was also unchanged following TDP6 treatment (Fig. 5C; p = 0.55, d = 0.71 ± 0.79) and there was no change in the density of Olig2+PDGFRα+ OPCs or Olig2+CC1+ oligodendrocytes between treatment groups in the conditional knock-out mice (Fig. 5D,E; p = 0.80, d = 0.26 ± 0.77 and p = 0.17, d = 1.59 ± 0.87, respectively). These data suggest the effect TDP6 exerts to promote remyelination and increase oligodendrocyte density is dependent on oligodendrocyte expressed TrkB.
Figure 5.
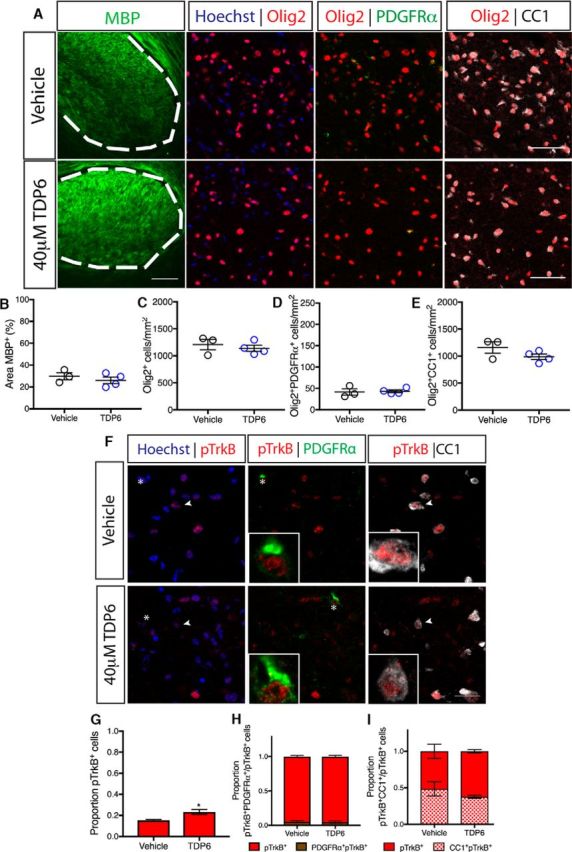
Enhanced oligodendrocyte differentiation by TDP6 during remyelination requires oligodendrocyte TrkB. A, Representative micrographs of MBP immunostaining and oligodendrocyte lineage markers in the caudal corpus callosum of CNPasCre+/− TrkBfl/fl cuprizone-demyelinated mice treated with vehicle or TDP6. Scale bar, 20 μm. B, Percentage area of MBP immunostaining (p = 0.45), density of (C) Olig2+ cells (p = 0.55), (D) Olig2+PDGFRα+ OPCs (p = 0.80), and (E) Olig2+CC1+ oligodendrocytes (p = 0.17) were unchanged between vehicle and TDP6-treated TrkB conditional knock-out mice. Unpaired Student's t test, n = 3–4/group. F, Representative micrographs of pTrkBS478 immunostaining in the caudal corpus callosum of CNPaseCre+/− TrkBfl/fl cuprizone-demyelinated mice treated with vehicle or TDP6. Scale bar, 20 μm. G, Proportion of pTrkBS478+ cells were significantly increased in TDP6-treated conditional knock-out mice (p = 0.037), but (H) the proportion of pTrkBS478+ PDGFRα+ OPCs were unchanged (p = 0.80) as were (I) the proportion of pTrkBS478+ CC1+ oligodendrocytes (p = 0.25). Unpaired Student's t test, n = 3–4/group.
Successful delivery of TDP6 was confirmed by immunohistochemistry for pTrkBS478 (Fig. 5F) which revealed that the proportion of total pTrkBS478+ cells in the corpus callosum significantly increased in conditional knock-out mice that received TDP6, compared with those receiving the vehicle (Fig. 5G; *p = 0.037, d = 0.10 ± 0.76). Consistent with reduced levels of TrkB expression in the CNPaseCre+/− TrkBfl/fl mice, there was a low proportion of pTrkBS478+ PDGFRα+ OPCs (Fig. 5H) and pTrkBS478+ CC1+ oligodendrocytes (Fig. 5I), and these were unchanged by treatment with TDP6 (p = 0.80, d = 0.0065 ± 0.76 and p = 0.25, d = 0.14 ± 0.76, respectively). Delivery of TDP6 was further verified by pooling residual TDP6-solution remaining in the osmotic pump reservoir at the end of the treatment period from the conditional knock-out mice, and run through reverse-phase HPLC (Fig. 6). This demonstrated that TDP6 was stable within the reservoir throughout the treatment period (Fig. 6A–D).
Despite no change in MBP staining, EM analysis (Fig. 7A) revealed that the proportion of axons remyelinated in the caudal corpus callosum was significantly increased with TDP6 treatment in conditional knock-out mice (Fig. 7D; *p = 0.031, d = 4.50 ± 1.53). However, the mean g-ratio was unchanged (Vehicle: 0.74 ± 0.02; TDP6: 0.75 ± 0.02, n = 3/group, p = 0.76, d = 0.015 ± 0.82), indicating that whereas TDP6 increased the proportion of myelinated axons, it exerted no effect on myelin sheath thickness in the conditional knock-out mice. Linear regression analysis revealed that compared with vehicle treatment, TDP6 in the conditional knock-out mice resulted in a significant increase in slope (Fig. 7B; p < 0.0001), suggestive of an altered relationship between myelin profiles and axonal diameter. To dissect the driver of this effect, g-ratios were categorized based on axon diameter. This revealed that there was no change in myelin sheath thickness across axons of different diameter (Fig. 7E), suggesting the change in slope was likely driven by an increase in the number of axons myelinated in each size category between treatments. Consistent with this, analysis of the frequency distribution of myelinated axons based on their diameter demonstrated that there were significantly more axons between 0.3 and 0.6 μm range that were myelinated in TDP6-treated conditional knock-out mice (Fig. 7F; *p = 0.0032). Examination of the motor cortex (M2) in available sections (n = 2/group) indicated that there was a trend toward more pTrkBS478-positive neurons in TDP6-treated conditional knock-out mice (Fig. 7C). Collectively, these data suggest that TDP6 may exert a small oligodendroglial-TrkB independent effect to initiate myelin ensheathment for a selective subset of axons, but not increase myelin sheath thickness.
Figure 7.

Increases in myelin sheath thickness by TDP6 requires oligodendrocyte TrkB. A, Electron micrographs of the caudal corpus callosum of CNPaseCre+/− TrkBfl/fl cuprizone-demyelinated mice treated with vehicle or TDP6. Scale bar, 1 μm. B, Scatter plot of g-ratio against axon diameter. Linear regression analysis revealed a significant increase in slope between CNPaseCre+/− TrkBfl/fl mice treated with TDP6 compared with vehicle. C, Representative micrographs of pTrkBS478 in the M2 cortex (top) and colabeled with neuronal marker NeuN (bottom). D, Proportion of myelinated axons was significantly increased in TrkB conditional knock-out mice that received TDP6 compared with vehicle controls (*p = 0.0314). E, Mean g-ratio categorized by axonal diameter revealed no significant change between treatment groups, indicative of no change in myelin sheath thickness with TDP6 treatment in TrkB conditional knock-out mice (p = 0.959), n = 3/group, two-way ANOVA. F, Frequency plot of myelinated axon diameters revealing a significant increase in the frequency of axons myelinated ranging from 0.3 to 0.6 μm diameter in CNPaseCre+/− TrkBfl/fl mice treated with TDP6 (p = 0.0032, χ2 distribution test). B, D–F, Minimum 100 axons/animal, n = 3/group.
Discussion
Selective targeting of TrkB with the BDNF mimetic, TDP6 in the cuprizone-demyelinated brain enhanced remyelination. TDP6 increased the density of Olig2+CC1+ oligodendrocytes, proportion of remyelinated axons, and myelin sheath thickness following 7 d recovery. Infusion with BDNF, the neurotrophin from which TDP6 is derived, demonstrated an attenuated response, only increasing myelin sheath thickness. Importantly, the effects of TDP6 on myelin sheath thickness and oligodendrocyte differentiation required oligodendroglial TrkB. Intriguingly, following deletion of TrkB from oligodendroglia, TDP6 retained some capacity to increase the proportion of remyelinated axons. This may be due to the action of TDP6 on non-recombined oligodendroglial cells. However, TDP6 is not targeted toward specific cell types, and there is also the possibility that TDP6 may activate alternate sources of TrkB, potentially neuronal, to initiate myelin ensheathment, effectively increasing frequency of remyelinated axons.
The therapeutic potential of BDNF has been tested in numerous intervention studies and clinical trials for neurologic conditions over the past several decades (McTigue et al., 1998; The BDNF Study Group, 1999; Fulmer et al., 2014; Ramos-Cejudo et al., 2015). In demyelinating diseases, direct infusion (Ramos-Cejudo et al., 2015), cell-based gene therapy to overexpress BDNF (McTigue et al., 1998), and indirect modulation of endogenous BDNF secretion (Fulmer et al., 2014) in animal models have shown promise. However, by and large, these studies used indirect measures of remyelination, such as OPC proliferation and increased expression of myelin proteins, MBP, MOG, and PLP (McTigue et al., 1998; Fulmer et al., 2014; Ramos-Cejudo et al., 2015). Here, direct BDNF infusion to the demyelinated CNS increased MBP expression, and tended to increase OPC density, consistent with previous reports (McTigue et al., 1998; Fulmer et al., 2014; Ramos-Cejudo et al., 2015), but did not increase the proportion of myelinated axons, or oligodendrocyte differentiation. These abilities are essential to a remyelinating strategy, particularly for MS where OPC differentiation appears arrested (Chang et al., 2002). In contrast, by targeting TrkB, the direct molecular mechanism used by BDNF to promote myelination in development (Xiao et al., 2010; Wong et al., 2013), we increased the density of Olig2+CC1+ oligodendrocytes and the proportion of myelinated axons.
It is well appreciated that BDNF promotes CNS myelination through activation of oligodendroglial TrkB (Xiao et al., 2010; Wong et al., 2013). We found selective targeting of TrkB with TDP6, led to greater TrkB phosphorylation in the corpus callosum, more mature oligodendrocytes, and higher frequency of remyelinated axons than exogenous BDNF. Why TDP6 elicited this significantly greater remyelinating effect remains unclear. Deductive reasoning suggests potential differences in specificity, stability, dosage, or a combination of all three, may contribute to this discrepancy. The concentrations used were based on previous in vitro myelinating cocultures where 25-fold more TDP6 was required to increase the number of MBP+ myelinated axonal segments to the same level as BDNF, and induce TrkB and Erk1/2 phosphorylation (Wong et al., 2014). From this, BDNF is substantially more potent than TDP6, suggesting the lower concentration of BDNF unlikely caused the attenuated response. The notoriously labile and highly charged nature of BDNF may contribute to its reduced efficacy in vivo. BDNF has a very short half-life (Poduslo and Curran, 1996), although we have shown that TDP6 can be retrieved from osmotic pump reservoirs after 7 d in vivo. Finally, the receptor specificity may also play a role. TDP6 selectively interacts with TrkB and has not shown any ability to interact with p75NTR (O'Leary and Hughes, 2003; Xiao et al., 2010; Wong et al., 2014). It is possible that BDNF-p75NTR interactions attenuate the BDNF response. Regardless, the attenuated remyelinating response to BDNF is consistent with reduced TrkB phosphorylation observed compared with TDP6. Importantly, compared with other BDNF-mimetics (Jiang et al., 2013; Simmons et al., 2013), we have shown using conditional knock-out mice, the effects of TDP6 are predominantly dependent on TrkB expression by the target cell in vivo. This is the first time the effect of a neurotrophin mimetic has been identified to require its targeted receptor on a specific cell type. Overall, our results demonstrate the poor therapeutic potential of BDNF, and support the strategy to develop functional BDNF mimics as therapies for neurologic disease.
A feature consistent with BDNF and TDP6 treatment was increased myelin sheath thickness, indicated by reduced g-ratios. This is the first time exogenous BDNF has been demonstrated to affect the myelin sheath in vivo. A role for BDNF in modulating myelin sheath thickness is not unexpected. BDNF−/−, BDNF+/− and MBPCre+/− × TrkBfl/fl mice all demonstrate developmental hypomyelination with thinner myelin compared with wild-type mice (Cellerino et al., 1997; Xiao et al., 2010; Wong et al., 2013). In addition, BDNF-TrkB signaling activates the MAPK/Erk pathway (Du et al., 2006; Xiao et al., 2012), which in oligodendrocytes increases myelin sheath thickness (Ishii et al., 2012, 2016; Fyffe-Maricich et al., 2013). The effect BDNF and TDP6 exert on myelin sheath thickness requires oligodendroglial TrkB expression, as there was no change in g-ratio when TDP6 was delivered in the conditional knock-out mice.
Similarly, the effect TDP6 exerted to increase oligodendrocyte density required oligodendroglial TrkB. It is unclear whether this effect is due to TrkB stimulation of OPC proliferation, or enhanced survival. A role for BDNF in OPC proliferation has been suggested, with reduced OPC proliferation exhibited in BDNF+/− mice following cuprizone demyelination (VonDran et al., 2011; Fulmer et al., 2014). Despite reduced OPC proliferation, the density of mature oligodendrocytes was unchanged between BDNF+/− and wild-type mice following recovery (VonDran et al., 2011). Deletion of TrkB from maturing oligodendrocytes in development causes OPC hyperproliferation, but this too, exerts no effect on the density of mature oligodendrocytes in adulthood (Wong et al., 2013). Collectively, these data suggest BDNF-TrkB signaling is not essential for oligodendrocyte differentiation, but rather TrkB activation regulates OPC proliferation in vivo. Labeling proliferating cells at the time of cuprizone withdrawal and TDP6 delivery in wild-type and conditional knock-out mice would determine whether TrkB signaling exerts a controlling influence upon OPC proliferation following myelin injury. Given the increase in Olig2+CC1+ oligodendrocytes in response to TDP6 depended on oligodendroglial expression of TrkB, sustained levels TrkB activation may also override apoptotic signals. This would enhance OPC survival, increasing the density of differentiated oligodendrocytes. Regardless, the ultimate fate and persistence of all Olig2+CC1+ oligodendrocytes produced by TDP6 treatment in the long-term remyelinating CNS, as well as the incorporation and persistence of the thicker myelin sheath generated by these cells is a pertinent question to be answered in the context of chronic demyelination in MS.
Intriguingly, although deletion of TrkB from oligodendrocytes abrogated the effects of TDP6 on myelin sheath thickness and oligodendrocyte differentiation, the proportion of myelinated axons still increased. This suggests TDP6 may have effects independent of oligodendroglial TrkB. No significant effect of TDP6 on astrogliosis and microgliosis suggests TDP6 does not directly modulate these cell populations, although it may alter the composition of their secreted factors (Djalali et al., 2005). It is also possible increased myelination in TDP6-treated conditional knock-out mice was mediated by a small subset of oligodendrocytes that escape recombination and continue to express TrkB. This view would be consistent with previous work by us (Xiao et al., 2011; Wong et al., 2013) and others (Minichiello et al., 1999; Medina et al., 2004) indicating the role of TrkB in CNS myelination is specific to oligodendrocytes. However, what is striking is that the increase in myelinated axons in TDP6-treated conditional knock-out mice was driven by an increase in the frequency of small diameter axons (0.3–0.6 μm) becoming myelinated. This is suggestive of a selective axonal response driving initiation of myelination on this axonal subpopulation. The recent focus on activity-dependent myelination has indicated efficient remyelination, on small diameter axons in particular, may require neuronal signals (Gautier et al., 2015; Bechler et al., 2018). Optogenetic stimulation of cortical neurons increased neuronal secretion of BDNF (Venkatesh et al., 2015), and this manipulation in healthy cortex increased callosal myelination (Gibson et al., 2014). This suggests a possible role for neuronally derived autocrine BDNF-TrkB signaling in adaptive myelination, and importantly remyelination (Lundgaard et al., 2013). This potential role for neuronally-derived TrkB in promoting remyelination may be fortuitous. TrkB expression is found in neurons adjacent to demyelinated lesions in MS (Stadelmann et al., 2002) and could be directly targeted with TDP6, or by other indirect strategies to sustain TrkB phosphorylation and remyelination. The mechanism mediating the TDP6-dependent increase in the frequency of myelinated axons in conditional knock-out mice is unclear. Adoption of neuronal TrkB knock-out strategies will ultimately confirm whether neuronal TrkB directly influences remyelination, or whether this effect is due to an indirect response stimulated by TDP6.
We demonstrated that TDP6, a structural mimetic of the TrkB binding region of BDNF, enhances remyelination in a preclinical animal model of MS. By targeting TrkB after demyelination, oligodendrocyte differentiation, the proportion of myelinated axons, and myelin sheath thickness all increased. These are key outcomes for a potential remyelinating strategy. Deletion of TrkB from oligodendrocytes abrogated the effect of TDP6 on myelin sheath thickness and oligodendrocyte differentiation, but an increase in myelinated axons persisted, revealing a potential for targeting additional non-oligodendrocyte-specific effects of TrkB in remyelination. Overall, our results support the development of therapeutic strategies to sustain elevations of TrkB signaling to promote myelin repair in demyelinating disease.
Footnotes
This work was supported by Australian National Health and Medical Research Council (NHMRC) Project Grants (APP1058647 to J.X. and APP1105108 to S.S.M.), a Multiple Sclerosis Research Australia (MSRA) Project Grant (13-039; S.S.M. and J.X.), a MSRA Postdoctoral Fellowship (14-056 to J.L.F.), and a MSRA/NHMRC Early Career Fellowship (APP111041 to D.G.G.). We thank Cameron Nowell (Monash University) and Dr. Verena Wimmer (The Florey Institute) for advice on image analysis procedures and automation. Confocal imaging was performed at the Biological Optical Microscope Platform, The University of Melbourne, and the Florey Advanced Microscopy and Immunohistochemistry Service, The Florey Institute of Mental Health and Neuroscience, and Peter MacCallum Centre for Advanced Histology and Microscopy f assistance with EM processing and imaging.
The authors declare no competing financial interests.
References
- Albert M, Antel J, Brück W, Stadelmann C (2007) Extensive cortical remyelination in patients with chronic multiple sclerosis. Brain Pathol 17:129–138. 10.1111/j.1750-3639.2006.00043.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechler ME, Swire M, Ffrench-Constant C (2018) Intrinsic and adaptive myelincation—a sequential mechanism for smart wiring in the brain. Dev Neurobiol 78:68–79. 10.1002/dneu.22518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, Carroll P, Thoenen H, Barde YA (1997) Reduced size of retinal ganglion cell axons and hypomyelination in mice lacking brain-derived neurotrophic factor. Mol Cell Neurosci 9:397–408. 10.1006/mcne.1997.0641 [DOI] [PubMed] [Google Scholar]
- Chang A, Tourtellotte WW, Rudick R, Trapp BD (2002) Premyelinating oligodendrocytes in chronic lesions of multiple sclerosis. N Engl J Med 346:165–173. 10.1056/NEJMoa010994 [DOI] [PubMed] [Google Scholar]
- Chao MV. (2003) Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci 4:299–309. 10.1038/nrn1078 [DOI] [PubMed] [Google Scholar]
- Djalali S, Höltje M, Groβe G, Rothe T, Stroh T, Groβe J, Deng DR, Hellweg R, Grantyn R, Hörtnagl H, Ahnert-Hilger G (2005) Effects of brain-derived neurotrophic factor (BDNF) on glial cells and serotonergic neurones during development. J Neurochem 92:616–627. 10.1111/j.1471-4159.2004.02911.x [DOI] [PubMed] [Google Scholar]
- Du Y, Fischer TZ, Lee LN, Lercher LD, Dreyfus CF (2003) Regionally specific effects of BDNF on oligodendrocytes. Dev Neurosci 25:116–126. 10.1159/000072261 [DOI] [PubMed] [Google Scholar]
- Du Y, Lercher LD, Zhou R, Dreyfus CF (2006) Mitogen-activated protein kinase pathway mediates effects of brain derived neurotrophic factor on basal forebrain oligodendrocytes. J Neurosci Res 84:1692–1702. 10.1002/jnr.21080 [DOI] [PubMed] [Google Scholar]
- Fletcher JL, Kondagari GS, Vite CH, Williamson P, Taylor RM (2014) Oligodendrocyte loss during the disease course in a canine model of the lysosomal storage disease fucosidosis. J Neuropathol Exp Neurol 73:536–547. 10.1097/NEN.0000000000000075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulmer CG, VonDran MW, Stillman AA, Huang Y, Hempstead BL, Dreyfus CF (2014) Astrocyte-derived BDNF supports myelin protein synthesis after cuprizone-induced demyelination. J Neurosci 34:8186–8196. 10.1523/JNEUROSCI.4267-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyffe-Maricich SL, Schott A, Karl M, Krasno J, Miller RH (2013) Signaling through ERK1/2 controls myelin thickness during myelin repair in the adult central nervous system. J Neurosci 33:18402–18408. 10.1523/JNEUROSCI.2381-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier HO, Evans KA, Volbracht K, James R, Sitnikov S, Lundgaard I, James F, Lao-Peregrin C, Reynolds R, Franklin RJ, Káradóttir RT (2015) Neuronal activity regulates remyelination via glutamate signalling to oligodendrocyte progenitors. Nat Commun 6:8518. 10.1038/ncomms9518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, Barres BA, Woo PJ, Vogel H, Monje M (2014) Neuronal activity promotes oligodendrogenesis and adaptive myelinaton in the mammalian brain. Science 344:1252304. 10.1126/science.1252304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Fyffe-Maricich SL, Furusho M, Miller RH, Bansal R (2012) ERK1/ERK2 MAPK signaling is required to increase myelin thickness independent of oligodendrocyte differentiation and initiation of myelination. J Neurosci 32:8855–8864. 10.1523/JNEUROSCI.0137-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii A, Furusho M, Dupree JL, Bansal R (2016) Strength of ERK1/2 MAPK activation determines its effect on myelin and axonal integrity in the adult CNS. J Neurosci 36:6471–6487. 10.1523/JNEUROSCI.0299-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang M, Peng Q, Liu X, Jin J, Hou Z, Zhang J, Mori S, Ross CA, Ye K, Duan W (2013) Small-molecule TrkB receptor agonists improve motor function and extend survival in a mouse model of huntington's disease. Hum Mol Genet 22:2462–2470. 10.1093/hmg/ddt098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappe-Siefke C, Goebbels S, Gravel M, Nicksch E, Lee J, Braun PE, Griffiths IR, Nave KA (2003) Disruption of Cnp1 uncouples oligodendroglial functions in axonal support and myelination. Nat Genet 33:366–374. 10.1038/ng1095 [DOI] [PubMed] [Google Scholar]
- Legland D, Arganda-Carreras I, Andrey P (2016) MorphoLibJ: integrated library and plugins for mathematical morphology with ImageJ. Bioinformatics 32:3532–3534. 10.1093/bioinformatics/btw413 [DOI] [PubMed] [Google Scholar]
- Longo FM, Massa SM (2013) Small-molecule modulation of neurotrophin receptors: a strategy for the treatment of neurological disease. Nat Rev Drug Discov 12:507–525. 10.1038/nrd4024 [DOI] [PubMed] [Google Scholar]
- Lucchinetti C, Brück W, Parisi J, Scheithauer B, Rodriguez M, Lassmann H, Bruck W, Parisi JE, Scheithauer B, Rodriguez M, Lassmann H (1999) A quantitative analysis of oligodendrocytes in multiple sclerosis lesions. Brain 122:2279–2295. 10.1093/brain/122.12.2279 [DOI] [PubMed] [Google Scholar]
- Lulkart BW, Net S, Virmani T, Lush ME, Liu Y, Kavalali ET, Parada LF (2005) TrkB has a cell-autonomous role in the establishment of hippocampal Schaffer collateral synapses. J Neurosci 25:3774–3786. 10.1523/JNEUROSCI.0041-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgaard I, Luzhynskaya A, Stockley JH, Wang Z, Evans KA, Swire M, Volbracht K, Gautier HO, Franklin RJ, ffrench-Constant C, Attwell D, Káradóttir RT (2013) Neuregulin and BDNF induce a switch to NMDA receptor-dependent myelination by oligodendrocytes. PLoS Biol 11:e1001743. 10.1371/journal.pbio.1001743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTigue DM, Horner PJ, Stokes BT, Gage FH (1998) Neurotrophin-3 and brain-derived neurotrophic factor induce oligodendrocyte proliferation and myelination of regenerating axons in the contused adult rat spinal cord. J Neurosci 18:5354–5365. 10.1523/JNEUROSCI.18-14-05354.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina DL, Sciarretta C, Calella AM, Von Bohlen Und Halbach O, Unsicker K, Minichiello L (2004) TrkB regulates neocortex formation through the Shc/PLCγ-mediated control of neuronal migration. EMBO J 23:3803–3814. 10.1038/sj.emboj.7600399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minichiello L, Korte M, Wolfer D, Kühn R, Unsicker K, Cestari V, Rossi-Arnaud C, Lipp HP, Bonhoeffer T, Klein R (1999) Essential tole for TrkB receptors in hippocampus-mediated learning. Neuron 24:401–414. 10.1016/S0896-6273(00)80853-3 [DOI] [PubMed] [Google Scholar]
- O'Leary PD, Hughes RA (2003) Design of potent peptide mimetics of brain-derived neurotrophic factor. J Biol Chem 278:25738–25744. 10.1074/jbc.M303209200 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, Ed 2 London: Academic. [Google Scholar]
- Poduslo JF, Curran GL (1996) Permeability at the blood–brain and blood–nerve barriers of the neurotrophic factors: NGF, CNTF, NT-3, BDNF. Brain Res Mol Brain Res 36:280–286. 10.1016/0169-328X(95)00250-V [DOI] [PubMed] [Google Scholar]
- Ramos-Cejudo J, Gutiérrez-Fernández M, Otero-Ortega L, Rodríguez-Frutos B, Fuentes B, Vallejo-Cremades MT, Hernanz TN, Cerdán S, Díez-Tejedor E (2015) Brain-derived neurotrophic factor administration mediated oligodendrocyte differentiation and myelin formation in subcortical ischemic stroke. Stroke 46:221–228. 10.1161/STROKEAHA.114.006692 [DOI] [PubMed] [Google Scholar]
- Simmons DA, Belichenko NP, Yang T, Condon C, Monbureau M, Shamloo M, Jing D, Massa SM, Longo FM (2013) A small molecule TrkB ligand reduces motor impairment and neuropathology in R6/2 and BACHD mouse models of Huntington's disease. J Neurosci 33:18712–18727. 10.1523/JNEUROSCI.1310-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmann C, Kerschensteiner M, Misgeld T, Brück W, Hohlfeld R, Lassmann H (2002) BDNF and gp145trkB in multiple sclerosis brain lesions: neuroprotective interactions between immune and neuronal cells? Brain 125:75–85. 10.1093/brain/awf015 [DOI] [PubMed] [Google Scholar]
- Stangel M, Kuhlmann T, Matthews PM, Kilpatrick TJ (2017) Achievements and obstacles of remyelinating therapies in multiple sclerosis. Nat Rev Neurol 13:742–754. 10.1038/nrneurol.2017.139 [DOI] [PubMed] [Google Scholar]
- The BDNF Study Group (Phase III) (1999) A controlled trial of recombinant methionyl human BDNF in ALS. Neurology 52:1427–1433. 10.1212/WNL.52.7.1427 [DOI] [PubMed] [Google Scholar]
- Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, Woo PJ, Malenka RC, Vogel H, Bredel M, Mallick P, Monje M (2015) Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell 161:803–816. 10.1016/j.cell.2015.04.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonDran MW, Singh H, Honeywell JZ, Dreyfus CF (2011) Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J Neurosci 31:14182–14190. 10.1523/JNEUROSCI.6595-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AW, Xiao J, Kemper D, Kilpatrick TJ, Murray SS (2013) Oligodendroglial expression of TrkB independently regulates myelination and progenitor cell proliferation. J Neurosci 33:4947–4957. 10.1523/JNEUROSCI.3990-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AW, Giuffrida L, Wood R, Peckham H, Gonsalvez D, Murray SS, Hughes RA, Xiao J (2014) TDP6, a brain-derived neurotrophic factor-based trkB peptide mimetic, promotes oligodendrocyte myelination. Mol Cell Neurosci 63:132–140. 10.1016/j.mcn.2014.10.002 [DOI] [PubMed] [Google Scholar]
- Xiao J, Wong AW, Willingham MM, van den Buuse M, Kilpatrick TJ, Murray SS (2010) Brain-derived neurotrophic factor promotes central nervous system myelination via a direct effect upon oligodendrocytes. NeuroSignals 18:186–202. 10.1159/000323170 [DOI] [PubMed] [Google Scholar]
- Xiao J, Ferner AH, Wong AW, Denham M, Kilpatrick TJ, Murray SS (2012) Extracellular signal-regulated kinase 1/2 signaling promotes oligodendrocyte myelination in vitro. J Neurochem 122:1167–1180. 10.1111/j.1471-4159.2012.07871.x [DOI] [PubMed] [Google Scholar]