

Abstract
Acetaminophen (paracetamol) is a widely used analgesic and antipyretic drug with only incompletely understood mechanisms of action. Previous work, using models of acute nociceptive pain, indicated that analgesia by acetaminophen involves an indirect activation of CB1 receptors by the acetaminophen metabolite and endocannabinoid reuptake inhibitor AM 404. However, the contribution of the cannabinoid system to antihyperalgesia against inflammatory pain, the main indication of acetaminophen, and the precise site of the relevant CB1 receptors have remained elusive. Here, we analyzed acetaminophen analgesia in mice of either sex with inflammatory pain and found that acetaminophen exerted a dose-dependent antihyperalgesic action, which was mimicked by intrathecally injected AM 404. Both compounds lost their antihyperalgesic activity in CB1−/− mice, confirming the involvement of the cannabinoid system. Consistent with a mechanism downstream of proinflammatory prostaglandin formation, acetaminophen also reversed hyperalgesia induced by intrathecal prostaglandin E2. To distinguish between a peripheral/spinal and a supraspinal action, we administered acetaminophen and AM 404 to hoxB8-CB1−/− mice, which lack CB1 receptors from the peripheral nervous system and the spinal cord. These mice exhibited unchanged antihyperalgesia indicating a supraspinal site of action. Accordingly, local injection of the CB1 receptor antagonist rimonabant into the rostral ventromedial medulla blocked acetaminophen-induced antihyperalgesia, while local rostral ventromedial medulla injection of AM 404 reduced hyperalgesia in wild-type mice but not in CB1−/− mice. Our results indicate that the cannabinoid system contributes not only to acetaminophen analgesia against acute pain but also against inflammatory pain, and suggest that the relevant CB1 receptors reside in the rostral ventromedial medulla.
SIGNIFICANCE STATEMENT Acetaminophen is a widely used analgesic drug with multiple but only incompletely understood mechanisms of action, including a facilitation of endogenous cannabinoid signaling via one of its metabolites. Our present data indicate that enhanced cannabinoid signaling is also responsible for the analgesic effects of acetaminophen against inflammatory pain. Local injections of the acetaminophen metabolite AM 404 and of cannabinoid receptor antagonists as well as data from tissue-specific CB1 receptor-deficient mice suggest the rostral ventromedial medulla as an important site of the cannabinoid-mediated analgesia by acetaminophen.
Keywords: acetaminophen, AM 404, analgesia, inflammation, N-arachidonoylphenolamin, paracetamol
Introduction
In the past decades, several potential molecular mechanisms have been proposed that may explain how acetaminophen exerts its analgesic action. These include the inhibition of cyclooxygenases (COXs) (Flower and Vane, 1972; Hanel and Lands, 1982; Graham and Scott, 2005), the activation of spinal serotonergic descending projections (Tjølsen et al., 1991; Pini et al., 1996), an involvement of the brain opioid system (Tjølsen et al., 1991; Herrero and Headley, 1996; Pini et al., 1996; Sandrini et al., 2001), inhibition of nitric oxide generation (Björkman et al., 1994; Bujalska, 2004), and activation of spinal TRPA1 channels by the acetaminophen metabolites N-acetyl-p-benzoquinoneimine (NAPQI) and p-benzoquinone (Andersson et al., 2011). In addition, the generation of N-arachidonoylphenolamin (AM 404) from acetaminophen through deacetylation to p-aminophenol and the subsequent conjugation with arachidonic acid by CNS fatty amide hydrolase (FAAH) (Högestatt et al., 2005) has drawn the attention to a possible involvement of the endocannabinoid system. AM 404 increases tissue concentrations of the endocannabinoid arachidonoyl ethanolamide, also known as anandamide, through an inhibition of anandamide reuptake into neurons and astrocytes (Beltramo et al., 1997; Fegley et al., 2004). After spinal or systemic application, AM 404 exerts analgesic activity against acute pain, evoked by noxious chemical stimuli, as well as against inflammatory and neuropathic pain (Gühring et al., 2002; La Rana et al., 2006). In line with an important contribution of the endocannabinoid system, acetaminophen-mediated antinociception was lost in CB1 receptor-deficient (CB1−/−) mice (Mallet et al., 2008) as well as in mice lacking FAAH (FAAH−/− mice) (Mallet et al., 2010). Accordingly, acetaminophen-induced analgesia was also reduced by the FAAH inhibitor URB 597 (Mallet et al., 2008) and by the CB1 receptor antagonists AM 251 and rimonabant (Ottani et al., 2006; Dani et al., 2007; Mallet et al., 2008).
The studies discussed above support a contribution of the endocannabinoid system to acetaminophen-mediated analgesia. However, most of these studies (Ottani et al., 2006; Mallet et al., 2008, 2010) tested acetaminophen in models of acute nociceptive pain (i.e., pain evoked by acute noxious thermal, mechanical, or chemical stimuli applied to naive animals in the absence of nociceptive sensitization by inflammation or neuropathy). These acute pain models only poorly reflect the clinical indications for acetaminophen, which is primarily used to treat mild inflammatory pain (Bradley et al., 1991). Indeed, acute antinociceptive effects of acetaminophen in humans are rather vague or do not exist at all (Olesen et al., 2012; Tiippana et al., 2013). In the present study, we have analyzed the antihyperalgesic properties of acetaminophen in mice with inflammatory hyperalgesia and demonstrate a critical contribution of CB1 receptors to the effects of acetaminophen against inflammatory hyperalgesia. Additional experiments with tissue-specific CB1−/− mice and local injections of AM 404 or the CB1 receptor antagonist rimonabant suggest that the CB1 receptors relevant for inflammatory antihyperalgesia reside in the rostral ventromedial medulla (RVM), which is a well-known site for endogenous pain control.
Materials and Methods
Mice.
Experiments were performed in wild-type mice (C57BL/6J; www.jax.org/strain/000664), CB1−/− mice (genetic background C57BL/6N; www.informatics.jax.org/allele/MGI:2182924) (Marsicano et al., 2002), and hoxb8-CB1−/− mice (genetic background C57BL/6; http://www.informatics.jax.org/allele/MGI:4881836) (Witschi et al., 2010). hoxb8-CB1−/− mice were obtained by crossing mice carrying floxed CB1 receptor alleles (CB1fl/fl mice; www.informatics.jax.org/allele/MGI:3045419) (Marsicano et al., 2003) with mice expressing in addition the cre recombinase in spinal cord neurons and glial cells as well as in neurons of the DRGs (hoxb8-cre mice) (Witschi et al., 2010). Behavioral experiments on hoxb8-CB1−/− mice were performed with hoxb8-cre-negative CB1fl/fl littermates as “wild-type” controls. Animals were housed under controlled environmental conditions (22°C, 12/12 light/dark cycle) and were allowed to take food and water ad libitum.
Behavioral testing.
Experiments were performed in adult (7 to 9-week-old) female and male mice. Mice were randomly assigned to treatment groups. On the first day of the experiments, each mouse was tested several times to obtain baseline paw withdrawal thresholds (PWTs). Animals were placed in Plexiglas boxes on a metal grid and allowed to accommodate to the test confinement for at least 1 h before starting behavioral experiments. Mechanical sensitivity was measured using electronically controlled von Frey filaments (IITC). At least three measurements were made for each time point. The experimenter was blind to the genotype or to the type of treatment (vehicle or drug) in all experiments. Permission for animal experiments was obtained from the Veterinäramt des Kantons Zürich (licenses 92/2007, 126/2012, and 031/2016).
Inflammatory hyperalgesia was induced using the yeast extract zymosan A (Meller and Gebhart, 1997). Zymosan A (Fluka) was suspended in 0.9% NaCl and injected subcutaneously (0.06 mg/20 μl) into the plantar side of the left hindpaw 24 h before the administration of acetaminophen or AM 404. Spinal PGE2-induced hyperalgesia was evoked through intrathecal injection of prostaglandin E2 (PGE2; Sigma; 0.4 nmol/4 μl, dissolved in 1% ethanol and 99% aCSF). Intrathecal injections were made 1 h before application of acetaminophen (Reinold et al., 2005).
Drug administration, intrathecal and intra-RVM injections.
Acetaminophen (Sigma) was dissolved in 0.9% NaCl. The acetaminophen-containing solution or vehicle (0.9% NaCl, 400 μl) was given orally through stainless-steel tubes (Delvo). Rimonabant (SR141716A; Tocris Bioscience) (Rinaldi-Carmona et al., 1994) was dissolved in a mixture of 43% (v/v) DMSO, 43% aCSF, and 14% ethanol. Injection volumes were 5 and 4 μl for AM 404 (Tocris Bioscience) and PGE2, respectively. AM 404 (Tocris Bioscience) was dissolved in 40% DMSO and 60% 0.9% NaCl. Intrathecal injections were performed under isoflurane anesthesia at the level of the lumbar spine using a Hamilton syringe (Ahmadi et al., 2001). A small amount of black ink (1% v/v) was added to permit post hoc verification of proper intrathecal injections. Injections into the RVM were performed with stainless-steel cannulas. Fully anesthetized mice were placed in a Kopf stereotaxic frame and implanted with a cannula using the following coordinates, which were calibrated to the cranial bregma points: x = −5.7; y = 0; zcranium = 4.2. The cannula was fixed with dental cement, and the cement was secured at the skull with 2 or 3 screws. The fixed cannula was used to insert a 30 G needle attached to a Hamilton syringe 5.8 mm deep. A volume of 300 nl was injected. For post hoc verification of correct targeting of the RVM, 1% v/v Evans blue was included in the injection solution.
Hepatotoxicity assays.
Mice were treated orally with vehicle (0.9% NaCl), 200, 300, or 400 mg/kg acetaminophen. Twenty-four hours later, blood was collected after decapitation, and the liver was dissected. To quantify liver damage, we determined the blood levels of three enzymes, alanine aminotransferase (ALT), aspartic aminotransferase (AST), and lactate dehydrogenase (LDH), which are released upon acute liver damage from hepatocytes into the bloodstream using the UniCel DxC 800 Synchron Clinical Systems (Beckman Coulter). Livers were put in 4% formalin overnight and subsequently embedded in paraffin. Tissue sections (3 μm) were cut and stained with hematoxylin-eosin following standard procedures (Fischer et al., 2008). Liver degeneration was defined by the presence of vacuolar degeneration and pink-red tissue discoloration due to sinusoidal congestion and apoptotic cell body formation, as described previously (Zhao et al., 2016). For quantification of liver degeneration, the ratio of venules surrounded by healthy or discolored tissue was calculated.
Immunohistochemistry and in situ hybridization.
For immunohistochemistry, 3 mice of each genotype were deeply anesthetized with a mixture of 25 mg/ml ketamine, 5 mg/ml xylazine, and 0.1 w/w% promethazine in H2O (1 ml/100 g, i.p.) and subsequently perfused transcardially through the ascending aorta with 0.9% NaCl for 2 min, followed by 100 ml of a fixative containing 4% PFA in 0.1 m phosphate buffer (PB; pH 7.4) for another 20 min. After perfusion, spinal cords and brains were immediately isolated and postfixed in 4% PFA for 2 h and washed in 0.1 m PB. Transverse sections of the spinal cord at a lumbar level as well as coronal sections of the cerebral hemispheres and the cerebellum (all 50 μm thick) were cut using a vibratome (Leica, VTS-1000). Free-floating sections were collected in 0.1 m PB. For immunoperoxidase staining, the sections were first extensively washed in 0.1 m PB. To block endogenous peroxidase activity, sections were afterward incubated in 1% H2O2 in 0.1 m PB for 10 min and again washed in 0.1 m PB. Following washing in 0.05 m TBS, pH 7.4, conditioning TBST, the sections were blocked in 10% normal donkey serum (Vector Laboratories) for 45 min. Sections were then incubated with polyclonal affinity-purified guinea pig anti-CB1 antibodies (1:250; ∼1 μg/ml) (Fukudome et al., 2004) at 4°C for 48 h. The antibodies were dissolved in 0.05 m TBS. After multiple washings, the sections were treated in TBS with biotinylated goat anti-guinea pig IgG (1: 300; Vector Laboratories) for 2 h and after further washing in TBS incubated with avidin-biotinylated HRP complex (1: 500; Elite-ABC, Vector Laboratories) for 1.5 h. Development of the immunoperoxidase reaction was done with DAB as chromogen and 0.01% H2O2 dissolved in TB, pH 7.6. Sections were briefly submerged in chrome gelatin (0.05% chromium potassium sulfate dodecahydrate, 0.5% gelatin, and 0.05% NaN3 in DW), dried, soaked in xylene (2 × 15 min), and covered in DePeX (Serva). Sections containing the RVM were treated with 0.5% OsO4 in PB for 20 min at 4°C, dehydrated in an ascending series of ethanol and propylene oxide, and embedded in Durcupan (ACM, Fluka) following DAB development. During dehydration, sections were treated with 1% uranyl acetate in 70% ethanol for 15 min at 4°C. Light microscopic analysis of immunostaining was performed with a Nikon Eclipse 80i upright microscope. Micrographs were taken with a Nikon DS-Fi1 digital camera.
Statistical analyses.
Data are presented as mean ± SEM; n indicates the number of animals tested. For dose–response curves, PWTs were transformed into percent maximum possible effects, with 0% and 100% being the inflamed predrug value and the full return to preinflammation value, respectively. Data from the dose–response relationship of acetaminophen and AM 404 were fitted to the Hill equation y = ymax/(1 + (ED50/D)nH)]; with ymax, maximum percent maximum possible effects reached with saturating doses; D, actual dose; ED50, half-maximum effective dose; and nH, Hill coefficient. To compare the magnitude of antihyperalgesic effects of acetaminophen or AM 404 in wild-type and CB1−/− mice or in the presence or absence of antagonists, areas under the curve (AUC) were calculated for the changes of PWTs from predrug baseline over 150 min or 80 min, following application of acetaminophen or AM 404, respectively. When more than two groups were compared, statistical analyses were done by one-way ANOVA followed by Bonferroni or Dunnett's post hoc tests or two-way ANOVA, when two factors were analyzed. In all other experiments, statistical analyses were performed using the unpaired Student's t test (two-tailed). Statistical significance was accepted for p ≤ 0.05.
Results
Antihyperalgesic actions of acetaminophen and AM 404 in inflammatory pain
Because acetaminophen is an antipyretic analgesic whose main indication is mild inflammatory pain, we analyzed its analgesic effects in the zymosan A model of inflammatory pain (Meller and Gebhart, 1997; Reinold et al., 2005). Subcutaneous zymosan A injection (0.06 mg in 20 μl 0.9% NaCl) into one hindpaw decreased mechanical PWT from 4.11 ± 0.06 g (mean ± SEM, n = 30 mice) to 1.10 ± 0.06 g within 24 h after injection. For the first experiments, we chose a dose of 200 mg/kg (p.o.) because this dose has successfully been used in studies by others (e.g., Högestätt et al., 2005; Mallet et al., 2010; Dalmann et al., 2015; Gentry et al., 2015). Acetaminophen caused a time-dependent partial reversal of zymosan A-induced decreases in PWT. Acetaminophen reached a maximum effect at 60–80 min after administration (Fig. 1A). PWTs in the contralateral noninflamed paws were not affected. Accordingly, acetaminophen had no effects on PWT in naive mice (Fig. 1B). Testing the effects of different doses of acetaminophen revealed significant antihyperalgesic effects at doses ≥30 mg/kg. Dose–response curves (Fig. 1D) display percentage maximum possible analgesia determined for the time interval between 60 and 80 min after drug application. Data were fitted to the Hill equation revealing an ED50 of 30.1 ± 4.9 mg/kg and a maximal effect of 44.3 ± 3.4%.
Figure 1.
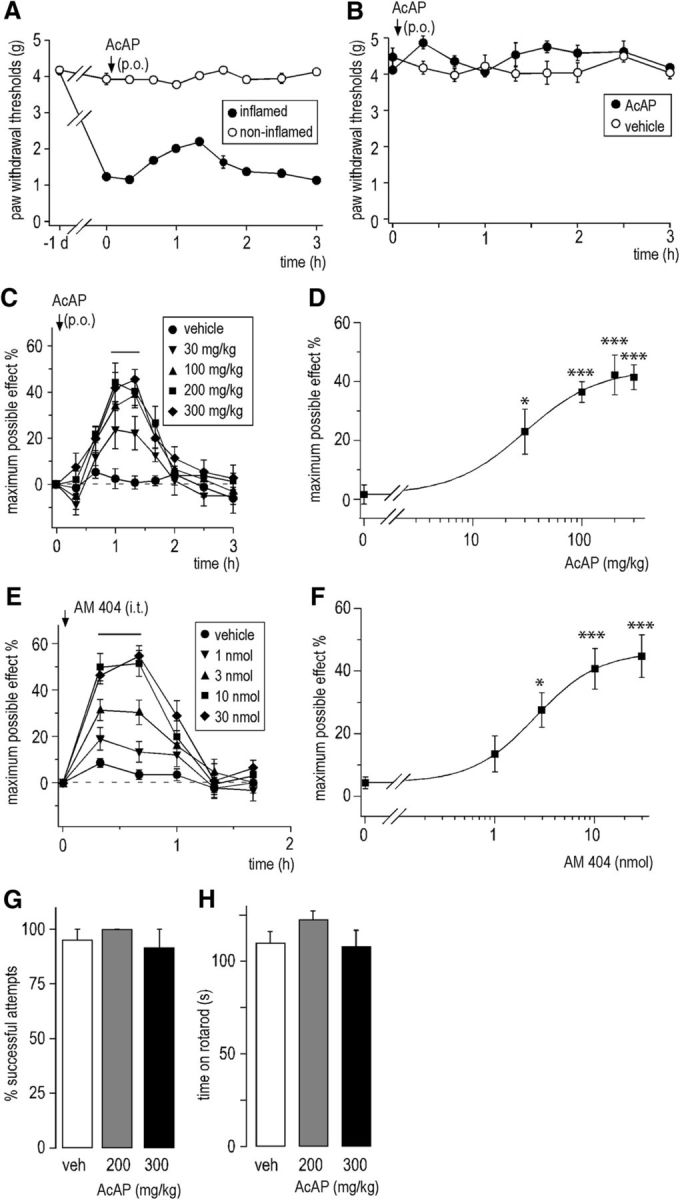
Antihyperalgesic actions of acetaminophen (p.o.) and AM 404 (intrathecal) in the zymosan A model of inflammatory hyperalgesia. A, Partial reversal of reduction in PWT (g) by acetaminophen 200 mg/kg. n = 6 mice. B, The same dose of acetaminophen had no significant effect on PWT in naive mice. p = 0.66 (unpaired Student's t test). n = 5 for acetaminophen; n = 7 for vehicle. Horizontal line indicates the time interval used to determine the maximal effects. C, Effects of different doses of systemic acetaminophen administered 24 h following subcutaneous injection of zymosan A (n = 6 mice per dose) on mechanical PWTs quantified as percent maximal possible effect (mean ± SEM). D, Dose–response curve. Average percentage maximum possible analgesia determined for the intervals 60 and 80 min after drug administration was calculated for each group and fitted to the Hill equation. *p ≤ 0.05 (ANOVA followed by Dunnett's post hoc test). ***p < 0.001 (ANOVA followed by Dunnett's post hoc test). F(4,25) = 10.11 with Fcrit = 2.76. E, F, Same as C, D, but with intrathecal AM 404 (n = 6 mice per group). Average percentage maximum possible analgesia was determined for the time interval between 20 and 40 min after drug injection. *p ≤ 0.05 (ANOVA followed by Dunnett's post hoc test). ***p < 0.001 (ANOVA followed by Dunnett's post hoc test). F(4,25) = 25.15. G,H, Impact of systemic acetaminophen on muscle strength (percent successful attempts in the horizontal wire test) (G) and on motor coordination (time on rotarod) (H) at 60 or 90 min after oral acetaminophen administration. No statistically significant effects were found in the two tests. G, ANOVA followed by Dunnett's post hoc test. F(2,22) = 1.46. p = 0.33 and p = 0.92, for 200 and 300 mg/kg, respectively. n = 7 or 8 mice. H, F(2,22) = 1.43. p = 0.33 and p = 0.97, for 200 and 300 mg/kg, respectively. n = 7 or 8 mice.
We next tested whether this antihyperalgesia would be mimicked by CNS injection of the acetaminophen metabolite AM 404. Different doses of AM 404 were injected directly into the mouse spinal canal 24 h after zymosan A injection (Fig. 1E,F). Mechanical sensitivities were measured for 100 min at 20 min intervals. Similar to acetaminophen, AM 404 caused a significant dose-dependent increase in PWTs (Fig. 1E). Dose–response curves (Fig. 1F) revealed an ED50 of 2.55 ± 0.04 nmol and maximal effect of 46.2 ± 0.2%. These experiments demonstrate that acetaminophen and its metabolite AM 404 exert potent dose-dependent antihyperalgesic actions against inflammatory pain.
We also examined whether acetaminophen exerted behavioral effects that might interfere with the read-outs of pain tests (Fig. 1G,H). To this end, we assessed effects of acetaminophen on motor coordination and sedation in the rotarod test and on muscle strength in the horizontal wire test. At doses of 200 and 300 mg/kg (p.o.), acetaminophen did not impair performance in these two tests (for statistics, see figure legends).
Liver toxicity of acute treatment with acetaminophen
Compared with clinically used doses in humans (1 g in a 70 kg person is equivalent to 15 mg/kg), the acetaminophen doses required in the present study to achieve at least 40% reduction in hyperalgesia (≥200 mg/kg) appear rather high. In humans, doses >150–250 mg/kg may induce hepatotoxicity (Brunton et al., 2011). On the other hand, a 10- to 15-fold difference between effective doses in humans and rodents is not unusual given the much higher metabolic rate of mice (Sharma and McNeill, 2009). However, because this ratio provides only an estimate and may differ between drugs, we tested whether the doses used here would cause acute liver toxicity in mice (Fig. 2). We measured blood levels of ALT, AST, and LDH 24 h after administration of different doses of acetaminophen (Fig. 2A–C). For all three enzymes, increases in enzyme activities were minor at a dose of 200 mg/kg and did not reach significance (ALT 63 ± 10, 214 ± 104, and 3624 ± 2010 IU/L, for vehicle, 200 mg/kg and 300 mg/kg, respectively; AST 281 ± 42, 457 ± 48.6, and 1349 ± 730 IU/L; LDH 1072 ± 170, 1674 ± 147, and 7498 ± 4663 IU/L; for statistics, see Fig. 2). At a dose of 300 mg/kg, blood levels of all three enzymes increased severalfold and increases became statistically significant for ALT. We also investigated potential changes in liver histology caused by acetaminophen (Fig. 2D). Tissue damage was quantified by counting the number of venules surrounded by healthy or discolored liver tissue per field of view. No detectable liver degeneration was observed after 200 mg/kg. At 300 mg/kg, the number of venules in degenerating tissue was increased, but this increase did not reach statistical significance. Statistically significant tissue damage was, however, found after 400 mg/kg. Based on these results, we decided to perform all subsequent experiments with an acetaminophen dose of 200 mg/kg.
Figure 2.
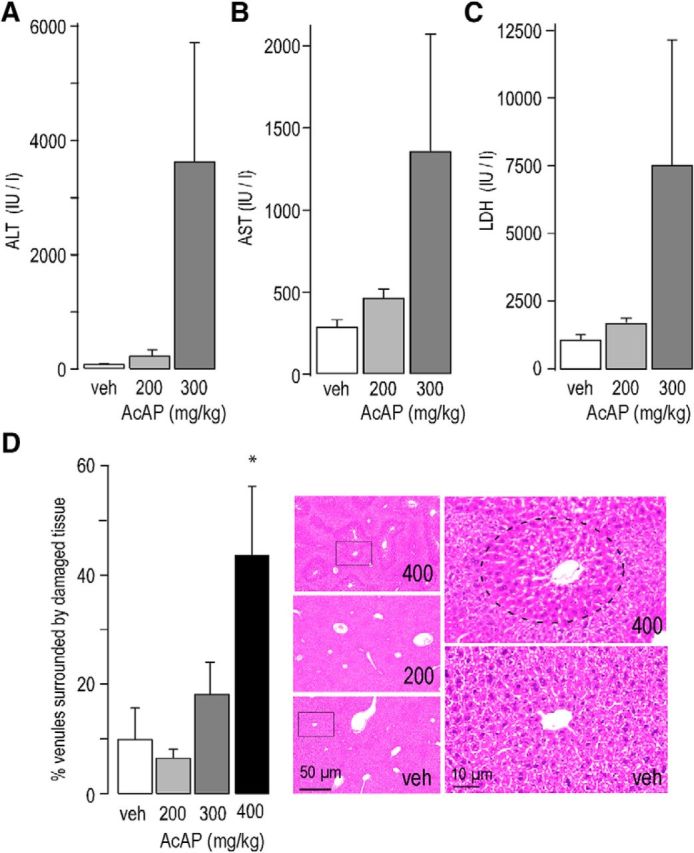
Acute liver toxicity of acetaminophen. A–C, Plasma levels of enzymatic markers of liver damage were quantified in mice 24 h after oral treatment with vehicle, 200 mg/kg or 300 mg/kg acetaminophen. Statistical comparisons were made with ANOVA followed by Dunnett's post hoc test. A, ALT: F(2,21) = 2.55, p = 0.99 and p = 0.02, for 200 and 300 mg/kg, respectively. n = 6–8 mice. B, AST: F(2,21) = 2.67, p = 0.91 and p = 0.08, for 200 and 300 mg/kg, respectively. n = 7 or 8 mice. C, LDH: F(2,20) = 5.28, p = 0.97 and p = 0.09, for 200 and 300 mg/kg, respectively. n = 7 or 8 mice. D, Histological changes caused by acetaminophen treatment were assessed 24 h after drug administration. The percent venules surrounded by discolored tissue was calculated. No significant changes were observed after 200 and 300 mg/kg; however, 400 mg/kg caused statistically significant liver damage. F(3,20) = 6.05, p = 0.69, p = 0.78, and *p = 0.014, for 200, 300, and 400 mg/kg, respectively. n = 6 mice for all four groups. Right micrographs represent magnifications of the indicated areas with healthy tissue surrounding a venule in the section taken from a vehicle-treated mouse (veh) and damaged tissue around a venue in the section prepared from a mouse treated with 400 mg/kg. Top left, Dotted line indicates the damage area around the venule in the center.
Contribution of CB1 receptors to antihyperalgesia by acetaminophen
To test for a possible contribution of the cannabinoid systems to acetaminophen and AM 404-mediated analgesia in inflammatory pain conditions, we tested the effects of acetaminophen and AM 404 in global CB1 receptor-deficient (CB1−/−) mice with an inflamed hindpaw. Wild-type and CB1−/− mice did not differ in their baseline mechanical sensitivities: PWTs were 3.9 ± 0.1 g, n = 15 and 4.0 ± 0.09 g, n = 13, for naive wild-type and CB1−/−, respectively, and developed similar inflammatory hyperalgesia. PWTs were 0.93 ± 0.10 g, n = 15, and 1.00 ± 0.05 g, n = 13, for zymosan A-injected wild-type and CB1−/− mice, respectively. Antihyperalgesic effects of acetaminophen were virtually absent in the CB1−/− mice. For statistical analyses, we calculated the AUC over time for the difference between postdrug PWTs and the predrug PWT baseline. AUC were 0.30 ± 0.34 g·h, n = 6, versus 1.23 ± 0.16 g·h, n = 8, in wild-type mice (p = 0.012, unpaired Student's t test) (Fig. 3A). We next assessed whether the antihyperalgesic action of the acetaminophen metabolite AM 404 would also be lost in CB1−/− mice (Fig. 3B). To this end, we injected 10 nmol of AM 404 intrathecally. AM 404 again reversed mechanical hyperalgesia in wild-type mice (AUC: 1.07 ± 0.14 g·h; n = 7) but completely failed to reduce hyperalgesia in CB1−/− mice (AUC: −0.22 ± 0.03 g·h, n = 6, p < 0.001, unpaired Student's t test). The lack of a pain-relieving action of acetaminophen and AM 404 in CB1−/− mice corresponds well with the reversal of acetaminophen- and AM 404-mediated analgesia by the CB1 receptor antagonists (inverse agonists) AM 251 and rimonabant described previously by others in different pain models (La Rana et al., 2006; Ottani et al., 2006; Dani et al., 2007; Mallet et al., 2008). It strongly suggests that antihyperalgesia by systemic acetaminophen requires activation of CB1 receptors. A lack of CB1 receptors during development may cause changes in neuronal circuits (Berghuis et al., 2007) that could potentially interfere with the actions of acetaminophen. To exclude this possibility, we tested whether systemic antagonism of CB1 receptors with rimonabant would recapitulate the effect of genetic ablation of CB1 receptors. Rimonabant (5 mg/kg, i.p.) administered immediately before acetaminophen indeed completely prevented the antihyperalgesic action of acetaminophen (Fig. 3C).
Figure 3.

Effect of CB1 receptor ablation on the antihyperalgesic actions of by acetaminophen and AM 404. A, Acetaminophen (200 mg/kg, p.o.). Time course of changes in PWT. Acetaminophen was given 24 h after injection of zymosan A to wild-type mice (n = 6) and to CB1−/− mice (n = 8). Bar chart represents AUC (g·h, mean ± SEM). *p ≤ 0.05 (unpaired Student's t test). B, AM 404 (10 nmol, intrathecal) was administered 24 h after injection of zymosan A in wild-type and CB1−/− mice (n = 7 each). ***p < 0.001 (unpaired Student's t test). C, Systemic pretreatment with rimonabant (rim, 5 mg/kg, i.p.) completely blocked antihyperalgesia by acetaminophen. Two-way ANOVA: F(1,22) = 9.08, p = 0.007 for pretreatment × treatment interaction. n = 4–8 per group. **p < 0.01. n = 6 and n = 8 mice for vehicle- and rimonabant-pretreated mice (unpaired Student's t test).
Analgesic effect of acetaminophen in PGE2-induced inflammatory pain
It has previously been suggested that acetaminophen might act through an inhibition of COX-dependent prostaglandin formation in the CNS (Flower and Vane, 1972; Hanel and Lands, 1982; Chandrasekharan et al., 2002; Graham and Scott, 2005). To test whether acetaminophen reduces inflammatory hyperalgesia through a mechanism downstream of central prostaglandin production, we induced hyperalgesia through intrathecal PGE2 injection (Taiwo and Levine, 1986; Uda et al., 1990; Reinold et al., 2005). One hour after PGE2 injection (0.4 nmol), PWTs decreased from a baseline value of 3.50 ± 0.08 g to 0.90 ± 0.06 g (n = 13) (Fig. 4A). Acetaminophen (200 mg/kg, p.o.), but not vehicle (0.9% NaCl, p.o.), administered 1 h after PGE2 injection partially reversed PGE2-induced hyperalgesia. The AUC (g·h) were calculated between the postdrug PWTs and a straight line between the PWT at 1.5 and 4.0 h after PGE2 injection. In wild-type mice, the average AUC (antihyperalgesia) in acetaminophen-treated mice (AUC: 1.51 ± 0.14 g·h, n = 7) was significantly higher than that of the vehicle-treated group (AUC. 0.073 ± 0.073 g·h, n = 6 mice, p < 0.001, unpaired Student's t test) (Fig. 4B). We also assessed the hyperalgesic effect of intrathecal PGE2 in CB1−/− mice and the potential reversal of PGE2-induced hyperalgesia by acetaminophen in these mice. PGE2 induced the same level of hyperalgesia, but acetaminophen was again completely devoid of antihyperalgesic effects in CB1−/− mice. Average AUC in acetaminophen-treated CB1−/− mice (AUC: 0.20 ± 0.58 g·h, n = 6) were virtually identical to those in vehicle-treated CB1−/− mice (AUC: 0.064 ± 0.46 g·h, n = 6, p = 0.95, unpaired Student's t test). Two-way ANOVA yielded a significant genotype × treatment interaction (F(1,25) = 5.46, p = 0.03). These results suggest that acetaminophen alleviates inflammatory hyperalgesia through a mechanism independent of prostaglandin formation.
Figure 4.

Effect of acetaminophen (200 mg/kg, p.o.) on mechanical hyperalgesia evoked by intrathecal PGE2 (0.4 nmol) in wild-type and CB1−/− mice. A, Change in PWTs (mean ± SEM). PGE2 was injected intrathecally at time 0. Acetaminophen or vehicle was given orally (1 h after PGE2 injection; n = 7 for acetaminophen; n = 6 for vehicle). B, AUC (mean ± SEM). Two-way ANOVA yielded a significant genotype × treatment interaction (F(1,25) = 5.46, p = 0.03). n = 6 or 7 mice per group.
Ablation of CB1 receptors from the periphery and the spinal cord does not block antihyperalgesia by systemic acetaminophen
We next aimed at identifying the anatomical origin of acetaminophen-induced antihyperalgesia. Our first analyses concentrated on CB1 receptors in the spinal cord for two reasons: (1) intrathecal injection of AM 404 mimicked the antihyperalgesia induced by systemic treatment with acetaminophen in several respects; and (2) activation of spinal CB1 receptors inhibits transmission for nociceptive signals between primary nociceptors and second-order dorsal horn neurons in vitro (Liang et al., 2004; Kato et al., 2012). The latter action might be considered a prime candidate mechanism for acetaminophen-induced antihyperalgesia. To distinguish a peripheral/spinal from a supraspinal site of action, we made use of hoxb8-CB1−/− mice, which were generated by crossing hoxb8-cre mice with CB1fl/fl mice. During development, hoxb8-cre is expressed in all DRG neurons and in all neurons and astrocytes of the spinal cord up to level C4. hoxb8-cre is, however, virtually absent from the brain (Witschi et al., 2010). We verified the specific ablation of CB1 receptors from the spinal cord by comparing CB1 receptor expression in the spinal dorsal horn and in the periaqueductal gray (PAG), a midbrain area rich in CB1 receptors (Fig. 5). In wild-type (CB1fl/fl) mice, intense CB1 receptor staining was observed in the gray matter of the superficial dorsal horn and in the dorsolateral funiculus as well as around the cerebral aqueduct in the PAG (Fig. 5A,D,D′,G). This staining was completely absent in spinal cord and PAG sections obtained from global CB1−/− mice (Fig. 5B,E,E′,H), indicating the specificity of the CB1 receptor antibody (see also Nyilas et al., 2009). As expected, hoxb8-CB1−/− mice exhibited a drastic reduction in CB1 receptor expression in the spinal dorsal horn (Fig. 5C,F,F′), but not in the PAG (Fig. 5I). A side-by-side comparison of global CB1−/− and conditional hoxb8-CB1−/− mice showed some remaining CB1 immunoreactivity in the dorsal horn of the hoxb8-CB1−/− mice, especially in the most superficial layers of the dorsal horn, which might result from terminals of axons descending from supraspinal sites to the dorsal horn.
Figure 5.
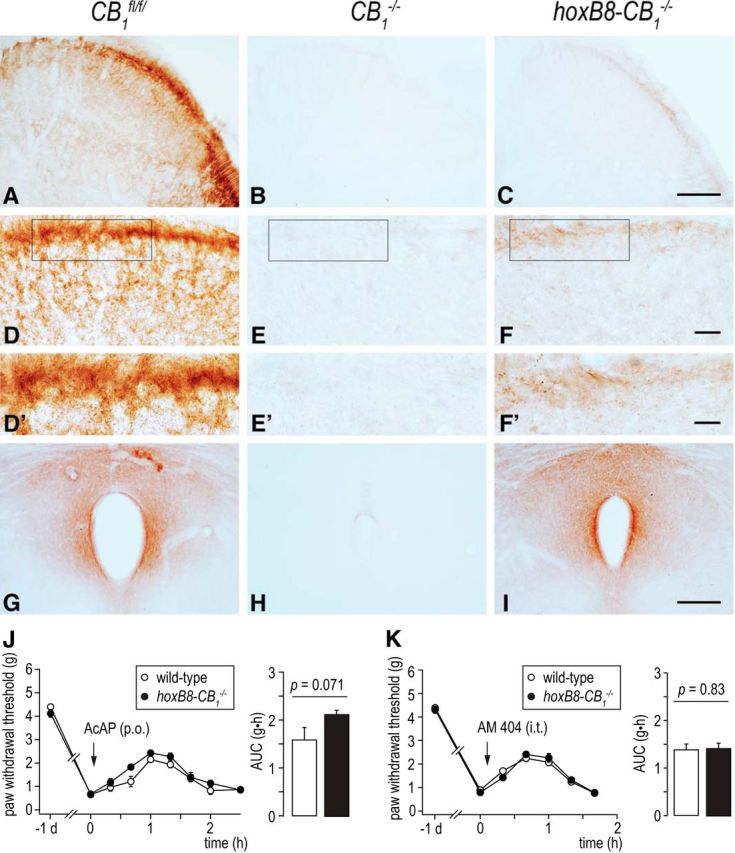
Morphological and behavioral analysis of hoxb8-CB1−/− mice.A–I, CB1 receptor expression in the spinal dorsal horn and PAG of wild-type, CB1−/−, and hoxb8-CB1−/− mice.A, High density of CB1 receptor immunostaining is found in the superficial layers in the dorsal horn of wild-type (CB1fl/fl) mouse spinal cord. D, D′, At higher magnification, an abundant punctate staining pattern corresponding mostly to axon terminals is observed. B, E, E′, The specificity of this staining pattern is validated by the complete lack of immunostaining on spinal cord sections derived from global CB1−/− animals. C, F, F′, Deletion of CB1 receptors from DRG and spinal neurons as well as from astrocytes in hoxb8-CB1−/− animals did not fully eliminate CB1 receptor immunostaining. A remaining weak staining pattern was found in lamina I and II, where most descending monoaminergic fibers terminate. G, Immunostaining for CB1 receptors in the midbrain PAG is concentrated around the dorsal and central part of the PAG. H, This staining pattern is completely eliminated in the global CB1−/− animals but remains fully intact in hoxB8-CB1−/− mice. Similar results were obtained in 3 mice of both genotypes. Scale bars: A–C, 100 μm; D–F, 20 μm; D′, E′, F′, 10 μm; G–I, 200 μm. J, K, Behavioral analysis. Changes in PWTs induced by the acetaminophen (200 mg/kg, p.o.; J) in hoxb8-CB1−/− (n = 6) and wild-type (CB1fl/fl) mice (n = 6), and by AM 404 (10 nmol, intrathecal; K) in hoxb8-CB1−/− (n = 6) and wild-type (CB1fl/fl) mice (n = 9). Acetaminophen and AM 404 were administered 24 h after zymosan A injection. Differences in AUC were statistically insignificant (unpaired Student's t test).
In behavioral experiments, hoxb8-CB1−/− mice and wild-type (hoxB8-cre negative CB1fl/fl) littermates did not differ in their baseline sensitivity to mechanical stimulation: PWTs were 4.21 ± 0.10 g (n = 15) and 4.39 ± 0.07 g (n = 12) in naive hoxb8-CB1−/− mice and CB1fl/fl littermates and developed virtually identical inflammatory hyperalgesia with PWTs of 0.79 ± 0.07 g and 0.73 ± 0.08 g in hoxb8-CB1−/− mice and CB1fl/fl littermates. Both genotypes also exhibited virtually identical antihyperalgesic responses to systemic acetaminophen treatment. AUC were 2.15 ± 0.08 g·h (n = 6) and 1.59 ± 0.27 g·h (n = 6) for hoxB8-CB1−/− and cre-negative wild-type (CB1fl/fl) mice, respectively (Fig. 5J). Very similar results were obtained with AM 404. AUC were 1.41 ± 0.12 g·h (n = 9) and 1.38 ± 0.11 g·h (n = 6), for hoxB8-CB1−/− and cre-negative littermates (Fig. 5K). Together with the complete lack of antihyperalgesia by acetaminophen and AM 404 in CB1−/− mice, these results suggest that acetaminophen acted through CB1 expressed at supraspinal sites. Alternatively, acetaminophen might act via CB1 receptors expressed in the spinal cord on the terminals of neurons descending from supraspinal sites, which are not targeted by the hoxB8-cre (compare Fig. 5C,F,F′). To distinguish between these two possibilities, we continued with local injections of AM 404 and of the CB1 receptor antagonist rimonabant.
Local injection of rimonabant and AM 404 suggests a critical role of the RVM in antihyperalgesia by systemic acetaminophen
The RVM serves well-established roles in endogenous pain control (Heinricher and Fields, 2013) and as a site of action of centrally acting analgesic drugs, including cannabinoid ligands (Meng et al., 1998; Suplita et al., 2005). We therefore tested whether the RVM was also involved in the antihyperalgesic actions of acetaminophen. To this end, we analyzed whether local injection into the RVM of the CB1 receptor antagonist rimonabant would interfere with antihyperalgesia by systemic acetaminophen (Fig. 6). Rimonabant (and vehicle) injections were made via chronic cannulas that had been preimplanted into the RVM 1 week before the experiment. Proper RVM injections were verified by addition of a small amount of Evans Blue to the injection solution and post hoc anatomical analysis of mouse brain sections (Fig. 6A,B). Injection of rimonabant (0.67 μg in 300 nl) completely prevented the antihyperalgesic action of systemic acetaminophen (200 mg/kg) (Fig. 6C,D). The AUC were 4.89 ± 1.35 g·h (n = 5) versus 0.67 ± 0.54 g·h (n = 6), in aCSF and rimonabant pretreated mice, respectively (p = 0.013, unpaired Student's t test). RVM injection of rimonabant per se did not affect inflammatory hyperalgesia, and RVM injection of vehicle neither affected the inflammatory hyperalgesia nor changed the antihyperalgesic response of acetaminophen. Injection of rimonabant or vehicle or cannula implantation into the RVM of naive mice was tested in 5–7 mice per group. These interventions had no effect on mechanical pain response threshold (data not shown). We next tested whether the effect of acetaminophen would be mimicked by local RVM injection of AM 404. As expected, AM 404 (1 μg, equivalent to 2.5 nmol) significantly alleviated inflammatory hyperalgesia in wild-type mice but not in CB1−/− mice (Fig. 6E,F). In naive mice, RVM injection of AM 404 did not significantly change PWTs (4.65 ± 0.56 g vs 4.23 ± 0.36 g, for AM 404 and vehicle, p = 0.54, n = 4 mice per group). In this series of experiments, we finally tested whether injection of acetaminophen into the RVM would reduce hyperalgesia (Fig. 6G). Consistent with an only very low conversion of acetaminophen in AM 404 in the brain (Högestätt et al., 2005), acetaminophen (1 μg in 300 nl) failed to significantly change PWTs (n = 6).
Figure 6.

Local RVM injection of rimonabant blocks and local RVM injection of AM 404 mimic the antihyperalgesic action of systemic acetaminophen. A, Sagittal brain section taken from a mouse after RVM injection verifies proper local RVM injection procedures. Red represents Evans Blue. Blue represents DAPI. B, Respective brain regions (sagittal section at −0.04 mm) redrawn and simplified from Paxinos and Franklin (2001) for comparison. C, D, Local injection of rimonabant (0.67 μg in 300 nl) prevented antihyperalgesia by systemic acetaminophen. Cannulation of the RVM and injection of vehicle or rimonabant were per se without effect on mechanical pain thresholds. C, Time course. D, Two-way ANOVA revealed a significant pretreatment × treatment interaction (F(1,23) = 10.8, p < 0.004). n = 5–7 mice per group. *p < 0.05, acetaminophen in aCSF (n = 5) or rimonabant (n = 6) pretreated mice (unpaired Student's t test). E, F, Local injection of AM 404 (1 μg in 300 nl) into the RVM mimicked acetaminophen-induced antihyperalgesia. E, Time course. F, Statistics. ANOVA followed by Bonferroni post hoc test (F(2,17) = 13.4). ***p ≤ 0.001. n = 6 mice per group. G, Local injection of acetaminophen (1 μg in 300 nl) into the RVM had no effect on paw withdrawal threshold.
Distribution of CB1 receptor mRNA and protein in the RVM
In many parts of the CNS, cannabinoid receptors are located on the presynaptic axon terminal where they control neuronal activity through the inhibition of neurotransmitter release. The experiments described above suggest that acetaminophen exerts its antihyperalgesia action through a perhaps indirect activation of antinociceptive fiber tracts descending from the RVM. To gain insights into the distribution of CB1 receptors at this site, we performed immunohistochemistry and in situ hybridization experiments in wild-type and global CB1−/− mice (Fig. 7). The immunohistochemical experiments revealed that CB1 receptors at the protein level were abundantly distributed throughout the RVM (Fig. 7A–D), which is consistent with a central role of the RVM in the CB1-mediated antihyperalgesic action of acetaminophen. In contrast, CB1 receptor mRNA was only detected in a few selected cells in the RVM close to the midline (Fig. 7E). No such cells were detected in tissue from CB1−/− mice (Fig. 7F). The low-density CB1 immunolabeling found in the dorsal horn of the spinal cord of hoxB8-CB1−/− mice (Fig. 5F,F′) likely reflects those descending fibers, which originate from the few RVM CB1 mRNA-expressing cells.
Figure 7.

CB1 receptor immunoreactivity and in situ hybridization in the RVM. A, B, CB1 receptor immunoreactivity in coronal sections through the brainstem of wild-type (A) and CB1−/− mice (B). The lack of brownish color of the DAB precipitate in the CB1−/− tissue (B) confirms the specificity of CB1 immunolabeling. Squares represent the area of the RVM shown at high magnification in C–F. C, CB1 protein is present in high density within the RVM of wild-type mice. There are dense DAB puncta around the cell bodies, which are always devoid of labeling. D, No CB1 immunostaining can be found in control sections from CB1−/− mice, which were processed together with the wild-type sections throughout the whole immunostaining procedure. The dark yellow color of the white matter bundles is due to an osmification step of tissue dehydration. E, CB1 in situ hybridization signal in the RVM. Only a few scattered neurons (blue) express CB1 receptor mRNA at rather low levels. F, No labeled cells are present in control sections prepared from CB1 receptor-deficient mice. Scale bars: A, B, 250 μm; C–F, 50 μm.
Local ablation of CB1 receptors in the RVM does not prevent the antihyperalgesic actions of acetaminophen
The results obtained with local injection into the RVM of rimonabant and AM 404 suggest a critical role of the RVM in the antihyperalgesic actions of acetaminophen. The relevant CB1 receptors in the RVM may either reside on RVM neurons themselves or may be located on axon terminals of neurons innervating the RVM. To distinguish between these possibilities, we selectively ablated receptors on intrinsic RVM neurons by local injection of CB1fl/fl mice with adeno-associated virus (AAV) carrying a cre recombinase expression cassette. AAV-cre virus injections were performed 1 week before acetaminophen treatment. Successful cre-mediated ablation of the CB1 receptor gene was verified with real-time RT-PCR. The number of CB1 receptor transcripts in the RVM was reduced to ∼25% (Fig. 8A). However, despite this significant downregulation of CB1 receptors, acetaminophen-induced antihyperalgesia remained largely unaffected (Fig. 8B,C). These results suggest that the relevant CB1 receptors reside on axon terminals of neurons projecting to the RVM rather than on intrinsic RVM neurons.
Figure 8.

Local knockdown of CB1 receptor expression in intrinsic RVM neurons fails to prevent acetaminophen-induced antihyperalgesia. A, Changes in CB1 receptor mRNA levels 7 d after AAV-cre injection in CB1fl/fl mice. mRNA levels have been normalized to β-actin mRNA copy numbers. **p < 0.01 (unpaired Student's t test). n = 19 for AAV-Cre; n = 14 for AAV-GFP. B, Antihyperalgesia by acetaminophen (200 mg/kg). RVM cannula implantation and AAV-cre injections were made 7 d before acetaminophen treatment. Zymosan A was injected 1 d before acetaminophen treatment. Mechanical PWTs were determined before AAV-cre injection, after zymosan A injection, and after acetaminophen or vehicle administration. C, Statistical analyses. Comparisons of acetaminophen effects in the three treatment groups (AAV-cre, AAV-eGFP, sham-operated mice) revealed significant acetaminophen versus vehicle effects (*p < 0.05, n = 6–8/group) but no significant treatment × pretreatment interaction (two-way ANOVA, F(2,39) = 0.41, p = 0.67).
Figure 9 illustrates a possible scenario: AM 404 in the RVM would increase the concentration of endocannabinoids (anandamide and 2-AG) and thereby indirectly activate CB1 receptors on inhibitory neurons that project to the RVM to tonically inhibit antinociceptive fiber tracts descending to the spinal cord. Increased activation of CB1 receptors on these neurons will reduce GABA release and disinhibit endogenous descending pain control units. Because many of the descending fibers release serotonin (Heinricher and Fields, 2013), this scenario is consistent with previous reports proposing not only a central site of action of acetaminophen but also a contribution of spinal serotonin receptors (Pelissier et al., 1995; Bonnefont et al., 2005).
Figure 9.
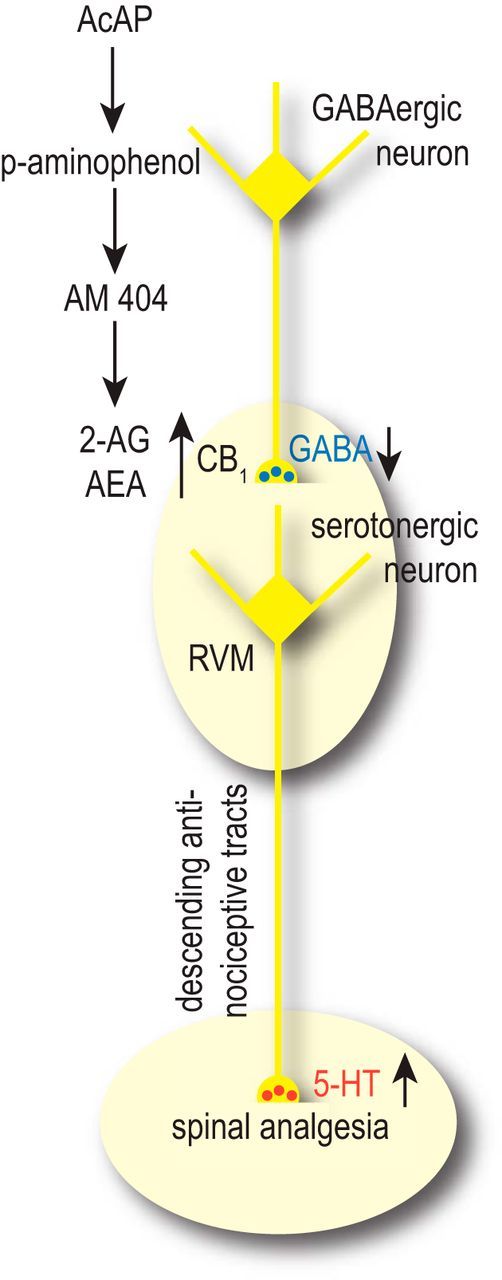
Hypothetical scheme of the central site of action of acetaminophen in inflammatory pain conditions. AM 404 produced from systemically administered acetaminophen increases the concentration of endocannabinoids (endocannabinoid arachidonoyl ethanolamide and 2-AG) in the RVM by inhibiting their uptake or degradation. This increase activates CB1 receptors on axon terminals of neurons projecting to the RVM from upstream brain regions, such as the PAG. These terminals normally release GABA to tonically inhibit serotonergic antinociceptive fiber tracts, which descend from the RVM to the spinal cord. Increased activation of CB1 receptors in the RVM would then reduce GABA release in the RVM and disinhibit descending pain control units. For a detailed discussion on the role of serotonergic neurons in the RVM, see Heinricher and Fields (2013).
Discussion
Our study demonstrates that acetaminophen exerts antihyperalgesic actions in a mouse model of inflammatory pain consistent with previous experimental (Vinegar et al., 1976; McQueen et al., 1991; Abbadie and Besson, 1994) and clinical studies (Skjelbred et al., 1977; Bradley et al., 1991; Bjørnsson et al., 2003; Brandt et al., 2006). These previous data have shown analgesia in adjuvant-induced monoarthritis or postoperative swelling and against secondary pain in oral surgery or osteoarthritic knee pain. Activity against inflammatory hyperalgesia and the well-known antipyretic effect of acetaminophen have led researchers to speculate about an inhibitory action of acetaminophen on prostaglandin formation (e.g., through COX inhibition). However, acetaminophen is largely devoid of anti-inflammatory activity (Clissold, 1986; Bertolini et al., 2006; Brunton et al., 2011), which is a hallmark effect of classical COX inhibitors. Significant activity against inflammatory hyperalgesia in the absence of general anti-inflammatory efficacy could be due to a specific inhibition of prostaglandin production in the CNS or to an analgesic mechanism independent of the inhibition of prostaglandin formation. Several studies have supported a contribution of the endocannabinoid system. However, most of these studies used models of acute nociceptive pain, which do not necessarily permit conclusions about the mechanisms of antihyperalgesic actions.
As shown in a previous study from our group, zymosan A-induced hyperalgesia strongly depends on spinally produced PGE2 (Reinold et al., 2005). This model is therefore well suited to investigate mechanisms of drugs with antihyperalgesic actions in inflammatory conditions and should permit a straightforward detection of prostaglandin-dependent drug actions. The reversal of inflammatory hyperalgesia by acetaminophen observed in our study would hence be consistent with a block of PGE2 production by acetaminophen. However, acetaminophen was still active when hyperalgesia was induced by local spinal injection of PGE2 favoring a mechanism different from inhibition of prostaglandin formation. Several results of the present study support instead the involvement of central CB1 receptors: the reversal of PGE2-induced hyperalgesia by acetaminophen was absent in CB1−/− mice, and both AM 404 and acetaminophen failed to reverse zymosan A-induced hyperalgesia in CB1−/− mice. Furthermore, the congruent pattern of efficacy of acetaminophen and of AM 404 in different (global and spinal cord-specific) CB1 receptor-deficient mouse lines supports the contribution of AM 404 to the antihyperalgesic actions of acetaminophen. These results also correspond well with previous findings demonstrating that acetaminophen-induced analgesia was lost in FAAH−/− mice, which do not convert acetaminophen into AM 404 (Högestätt et al., 2005; Dalmann et al., 2015). However, neither the present nor previously published results (Ottani et al., 2006; Dani et al., 2007; Mallet et al., 2008) exclude an involvement of COX-1 or COX-2 (Flower and Vane, 1972; Hanel and Lands, 1982; Muth-Selbach et al., 1999; Boutaud et al., 2002; Graham and Scott, 2005). An ex vivo study performed in human volunteers demonstrated inhibition of COX-1 and COX-2 following the oral administration of acetaminophen (Hinz et al., 2008), and AM 404 has also been shown to block COX-1 and COX-2 in lipopolysaccharide-stimulated macrophages (Högestätt et al., 2005). In this context, it is important to note that COX-2 contributes to the metabolism of endocannabinoids (Yu et al., 1997; Kozak et al., 2000). The extent to which inhibition of COX-dependent endocannabinoid degradation or blockade of endocannabinoid transporters contribute to acetaminophen-induced analgesia remains to be determined.
Our results can also be reconciled with a report by Mallet et al. (2010), who have proposed a role of supraspinal TRPV1 receptors as additional targets in acetaminophen and AM 404-induced analgesia. AM 404 is not only an inhibitor of anandamide reuptake but also an agonist at TRPV1 receptors (De Petrocellis et al., 2000). The observation that AM 404-induced analgesia was absent in TRPV1−/− mice and abolished by intracerebroventricular injection of the TRPV1 receptor antagonist capsazepine may suggest functional interactions of CB1 and TRPV1 receptors in the CNS (Fioravanti et al., 2008). More difficult to reconcile with our findings is the report by Andersson et al. (2011). These authors ascribe the analgesic action of acetaminophen to the activation of TRPA1 channels on the spinal terminals of nociceptive fibers by the acetaminophen metabolites NPQI and p-benzoquinone, and a subsequent inhibition of transmitter release via primary afferent depolarization. Because antihyperalgesia by acetaminophen was retained in hoxb8-CB1−/− mice, an interaction of TRPA1 channels with CB1 receptors cannot explain these findings. It is likely that distinct mechanisms underlie the acute analgesic and the antihyperalgesic actions of acetaminophen.
Comparing the effects of classical cannabinoids with those of acetaminophen reveals similarities and differences. Classical cannabinoids exert a tetrad of actions in rodents, which includes analgesia, hypothermia, sedation (reduced locomotor activity), and catalepsy (Little et al., 1988). Analgesia, sedation, and hypothermia do also occur in mice in response to acetaminophen (Mallet et al., 2010). Although our data provide strong support for the involvement of cannabinoid signaling in acetaminophen-induced antihyperalgesia, cannabinoid-independent actions are likely more relevant for the hypothermic and antipyretic effects of acetaminophen (Gentry et al., 2015). Such CB1 receptor-independent mechanisms include the inhibition of hypothalamic COX by AM 404 (Högestätt et al., 2005) and the activation of TRPA1 via the acetaminophen metabolite NAPQI (Gentry et al., 2015). The mechanisms of acetaminophen-induced sedation in mice have not been identified so far, and catalepsy is not seen in mice. Furthermore, the psychotropic actions seen with classical CB1 receptor agonists in humans do not occur with acetaminophen. Local differences in the conversion of the acetaminophen metabolite p-aminophenol into pharmacologically active AM 404, caused, for example, by varying FAAH activity in different CNS regions, or differences in the local activity of endocannabinoid system, may explain these discrepancies. Such differences may also account for another discrepancy. Whereas a previous report has suggested that CB1 receptor agonists exert most of their analgesic action through CB1 receptors on peripheral nociceptors (Agarwal et al., 2007), our experiments in hoxB8-CB1−/−, which lack CB1 receptors also from these cells, suggest that this is not the case for acetaminophen (see also Dalmann et al., 2015).
In our experiments, we also aimed at a better definition of the site of acetaminophen's action. To this end, we used hoxb8-CB1−/− mice, which lack CB1 receptors specifically from the spinal cord and peripheral sensory neurons. Because CB1 receptors are densely expressed on different types of intrinsic spinal dorsal horn neurons and on sensory fiber terminals (Tsou et al., 1998; Farquhar-Smith et al., 2000; Bridges et al., 2003; Hegyi et al., 2009; Nyilas et al., 2009), experiments first focused on a possible spinal site of action. However, the antihyperalgesia by acetaminophen was completely preserved in hoxb8-CB1−/− mice.
At least two explanations may account for these findings. The CB1 receptors responsible for acetaminophen analgesia might reside on the spinal terminals of fibers descending from supraspinal sites, which are spared from hoxb8-cre mediated gene deletion. This scenario is consistent with the presence of CB1 receptors in the termination area of descending fiber tracts in spinal cords of hoxb8-CB1−/− mice, and with the efficacy of AM 404 after intrathecal injection. However, AM 404 might have diffused to supraspinal sites after lumbar intrathecal injection. Such diffusion has been demonstrated earlier for radioactively labeled morphine (Gustafsson et al., 1985). Alternatively, acetaminophen might act via CB1 receptors at supraspinal sites located, for example, in the brainstem, where the somata of descending antinociceptive fiber tracts are located. Our experiments with local injection of rimonabant and AM 404 into the RVM provide strong support for this scenario (see also Högestätt et al., 2005; Mallet et al., 2008, 2010; Dalmann et al., 2015). According to these previous studies, acetaminophen acts via a CB1 receptor-mediated reinforcement of descending serotonergic bulbospinal pathways originating from the RVM (Mallet et al., 2008) with subsequent activation of pain-suppressing serotonin receptors in the spinal cord (Tjølsen et al., 1991; Pelissier et al., 1995; Pini et al., 1996; Bonnefont et al., 2005). Our results are thus in line with the important role of supraspinal CB1 receptors in stress-induced analgesia (Hohmann et al., 2005; Suplita et al., 2006).
Strong CB1 receptor immune reactivity but weak in situ hybridization signals in the RVM suggest that the relevant CB1 receptors reside on processes of neurons that project to the RVM from other brain areas. In this scenario, it is likely that the acetaminophen metabolite AM 404 promotes the activation of CB1 receptors on GABAergic axon terminals that tonically inhibit serotonergic antinociceptive fiber tracts descending from the RVM to the spinal cord. Because the PAG controls RVM activity via descending axons (Heinricher and Fields, 2013), it is conceivable that the CB1 receptors relevant for the analgesic action of acetaminophen reside on the terminals of fibers reaching the RVM from the PAG. Acetaminophen would thus indirectly reduce GABA release from these projections and disinhibit descending serotonergic fibers to facilitate endogenous pain control.
In conclusion, our results shed new light on the mechanisms and sites of action of the antihyperalgesic action of the widely used analgesic acetaminophen. They support the involvement of the endocannabinoid system in the analgesic action of acetaminophen against inflammatory pain and identify the RVM and descending antinociceptive fiber tracts as a likely site and mechanism of action.
Footnotes
This work was supported in part by Federal Government of Switzerland, Swiss Contribution Grant SH7/2/18 to I.K. and H.U.Z., and Hungarian Academy of Sciences Momentum Program LP-54/2013 to I.K., E.N. was supported by a Deutsche Forschungsgemeinschaft scholarship. We thank Drs. Beat Lutz and Giovanni Marsicano for providing CB1fl/fl mice; Dr. Masahiko Watanabe for the CB1 receptor antibody; Sébastien Druart, Andreas Pospischil, and Roseline Weilenmann for the analyses of biomarkers of liver damage; and Isabelle Kellenberger, Balázs Pintér, Erika Tischler, and Louis Scheurer for technical assistance.
The authors declare no competing financial interests.
References
- Abbadie C, Besson JM (1994) Chronic treatments with aspirin or acetaminophen reduce both the development of polyarthritis and Fos-like immunoreactivity in rat lumbar spinal cord. Pain 57:45–54. 10.1016/0304-3959(94)90106-6 [DOI] [PubMed] [Google Scholar]
- Agarwal N, Pacher P, Tegeder I, Amaya F, Constantin CE, Brenner GJ, Rubino T, Michalski CW, Marsicano G, Monory K, Mackie K, Marian C, Batkai S, Parolaro D, Fischer MJ, Reeh P, Kunos G, Kress M, Lutz B, Woolf CJ, et al. (2007) Cannabinoids mediate analgesia largely via peripheral type 1 cannabinoid receptors in nociceptors. Nat Neurosci 10:870–879. 10.1038/nn1916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmadi S, Kotalla C, Gühring H, Takeshima H, Pahl A, Zeilhofer HU (2001) Modulation of synaptic transmission by nociceptin/orphanin FQ and nocistatin in the spinal cord dorsal horn of mutant mice lacking the nociceptin/orphanin FQ receptor. Mol Pharmacol 59:612–618 [DOI] [PubMed] [Google Scholar]
- Andersson DA, Gentry C, Alenmyr L, Killander D, Lewis SE, Andersson A, Bucher B, Galzi JL, Sterner O, Bevan S, Högestätt ED, Zygmunt PM (2011) TRPA1 mediates spinal antinociception induced by acetaminophen and the cannabinoid Δ9-tetrahydrocannabiorcol. Nat Commun 2:551. 10.1038/ncomms1559 [DOI] [PubMed] [Google Scholar]
- Beltramo M, Stella N, Calignano A, Lin SY, Makriyannis A, Piomelli D (1997) Functional role of high-affinity anandamide transport, as revealed by selective inhibition. Science 277:1094–1097. 10.1126/science.277.5329.1094 [DOI] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T (2007) Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science 316:1212–1216. 10.1126/science.1137406 [DOI] [PubMed] [Google Scholar]
- Bertolini A, Ferrari A, Ottani A, Guerzoni S, Tacchi R, Leone S (2006) Paracetamol: new vistas of an old drug. CNS Drug Rev 12:250–275. 10.1111/j.1527-3458.2006.00250.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkman R, Hallman KM, Hedner J, Hedner T, Henning M (1994) Acetaminophen blocks spinal hyperalgesia induced by NMDA and substance P. Pain 57:259–264. 10.1016/0304-3959(94)90001-9 [DOI] [PubMed] [Google Scholar]
- Bjørnsson GA, Haanaes HR, Skoglund LA (2003) A randomized, double-blind crossover trial of paracetamol 1000 mg four times daily vs ibuprofen 600 mg: effect on swelling and other postoperative events after third molar surgery. Br J Clin Pharmacol 55:405–412. 10.1046/j.1365-2125.2003.01779.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnefont J, Chapuy E, Clottes E, Alloui A, Eschalier A (2005) Spinal 5-HT1A receptors differentially influence nociceptive processing according to the nature of the noxious stimulus in rats: effect of WAY-100635 on the antinociceptive activities of paracetamol, venlafaxine and 5-HT. Pain 114:482–490. 10.1016/j.pain.2005.01.019 [DOI] [PubMed] [Google Scholar]
- Boutaud O, Aronoff DM, Richardson JH, Marnett LJ, Oates JA (2002) Determinants of the cellular specificity of acetaminophen as an inhibitor of prostaglandin H2 synthases. Proc Natl Acad Sci U S A 99:7130–7135. 10.1073/pnas.102588199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley JD, Brandt KD, Katz BP, Kalasinski LA, Ryan SI (1991) Comparison of an antiinflammatory dose of ibuprofen, an analgesic dose of ibuprofen, and acetaminophen in the treatment of patients with osteoarthritis of the knee. N Engl J Med 325:87–91. 10.1056/NEJM199107113250203 [DOI] [PubMed] [Google Scholar]
- Brandt KD, Mazzuca SA, Buckwalter KA (2006) Acetaminophen, like conventional NSAIDs, may reduce synovitis in osteoarthritic knees. Rheumatology (Oxford) 45:1389–1394. 10.1093/rheumatology/kel100 [DOI] [PubMed] [Google Scholar]
- Bridges D, Rice AS, Egertová M, Elphick MR, Winter J, Michael GJ (2003) Localisation of cannabinoid receptor 1 in rat dorsal root ganglion using in situ hybridisation and immunohistochemistry. Neuroscience 119:803–812. 10.1016/S0306-4522(03)00200-8 [DOI] [PubMed] [Google Scholar]
- Brunton L, Chabner B, Knollmann B (2011) Goodman and Gilman's the pharmacological basis of therapeutics, Ed 12 New York: McGraw-Hill. [Google Scholar]
- Bujalska M. (2004) Effect of nitric oxide synthase inhibition on antinociceptive action of different doses of acetaminophen. Pol J Pharmacol 56:605–610. 10.1211/0022357023367 [DOI] [PubMed] [Google Scholar]
- Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS, Simmons DL (2002) COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A 99:13926–13931. 10.1073/pnas.162468699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clissold SP. (1986) Paracetamol and phenacetin. Drugs 32 [suppl 4]:46–59. [DOI] [PubMed] [Google Scholar]
- Dalmann R, Daulhac L, Antri M, Eschalier A, Mallet C (2015) Supra-spinal FAAH is required for the analgesic action of paracetamol in an inflammatory context. Neuropharmacology 91:63–70. 10.1016/j.neuropharm.2014.11.006 [DOI] [PubMed] [Google Scholar]
- Dani M, Guindon J, Lambert C, Beaulieu P (2007) The local antinociceptive effects of paracetamol in neuropathic pain are mediated by cannabinoid receptors. Eur J Pharmacol 573:214–215. 10.1016/j.ejphar.2007.07.012 [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Bisogno T, Davis JB, Pertwee RG, Di Marzo V (2000) Overlap between the ligand recognition properties of the anandamide transporter and the VR1 vanilloid receptor: inhibitors of anandamide uptake with negligible capsaicin-like activity. FEBS Lett 483:52–56. 10.1016/S0014-5793(00)02082-2 [DOI] [PubMed] [Google Scholar]
- Farquhar-Smith WP, Egertová M, Bradbury EJ, McMahon SB, Rice AS, Elphick MR (2000) Cannabinoid CB(1) receptor expression in rat spinal cord. Mol Cell Neurosci 15:510–521. 10.1006/mcne.2000.0844 [DOI] [PubMed] [Google Scholar]
- Fegley D, Kathuria S, Mercier R, Li C, Goutopoulos A, Makriyannis A, Piomelli D (2004) Anandamide transport is independent of fatty-acid amide hydrolase activity and is blocked by the hydrolysis-resistant inhibitor AM1172. Proc Natl Acad Sci U S A 101:8756–8761. 10.1073/pnas.0400997101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fioravanti B, De Felice M, Stucky CL, Medler KA, Luo MC, Gardell LR, Ibrahim M, Malan TP Jr, Yamamura HI, Ossipov MH, King T, Lai J, Porreca F, Vanderah TW (2008) Constitutive activity at the cannabinoid CB1 receptor is required for behavioral response to noxious chemical stimulation of TRPV1: antinociceptive actions of CB1 inverse agonists. J Neurosci 28:11593–11602. 10.1523/JNEUROSCI.3322-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer AH, Jacobson KA, Rose J, Zeller R (2008) Hematoxylin and eosin staining of tissue and cell sections. CSH Protoc 2008:pdb prot4986. 10.1101/pdb.prot4986 [DOI] [PubMed] [Google Scholar]
- Flower RJ, Vane JR (1972) Inhibition of prostaglandin synthetase in brain explains the anti-pyretic activity of paracetamol (4-acetamidophenol). Nature 240:410–411. 10.1038/240410a0 [DOI] [PubMed] [Google Scholar]
- Fukudome Y, Ohno-Shosaku T, Matsui M, Omori Y, Fukaya M, Tsubokawa H, Taketo MM, Watanabe M, Manabe T, Kano M (2004) Two distinct classes of muscarinic action on hippocampal inhibitory synapses: M2-mediated direct suppression and M1/M3-mediated indirect suppression through endocannabinoid signalling. Eur J Neurosci 19:2682–2692. 10.1111/j.0953-816X.2004.03384.x [DOI] [PubMed] [Google Scholar]
- Gentry C, Andersson DA, Bevan S (2015) TRPA1 mediates the hypothermic action of acetaminophen. Sci Rep 5:12771. 10.1038/srep12771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham GG, Scott KF (2005) Mechanism of action of paracetamol. Am J Ther 12:46–55. 10.1097/00045391-200501000-00008 [DOI] [PubMed] [Google Scholar]
- Gühring H, Hamza M, Sergejeva M, Ates M, Kotalla CE, Ledent C, Brune K (2002) A role for endocannabinoids in indomethacin-induced spinal antinociception. Eur J Pharmacol 454:153–163. 10.1016/S0014-2999(02)02485-8 [DOI] [PubMed] [Google Scholar]
- Gustafsson LL, Post C, Edvardsen B, Ramsay CH (1985) Distribution of morphine and meperidine after intrathecal administration in rat and mouse. Anesthesiology 63:483–489. 10.1097/00000542-198511000-00003 [DOI] [PubMed] [Google Scholar]
- Hanel AM, Lands WE (1982) Modification of anti-inflammatory drug effectiveness by ambient lipid peroxides. Biochem Pharmacol 31:3307–3311. 10.1016/0006-2952(82)90565-2 [DOI] [PubMed] [Google Scholar]
- Hegyi Z, Kis G, Holló K, Ledent C, Antal M (2009) Neuronal and glial localization of the cannabinoid-1 receptor in the superficial spinal dorsal horn of the rodent spinal cord. Eur J Neurosci 30:251–262. 10.1111/j.1460-9568.2009.06816.x [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Fields HL (2013) Central nervous system mechanisms of pain modulation. In: Wall and Melzack's textbook of pain, Ed 6 (McMahon SB, Koltzenburg M, Tracey I, Turk D, eds), pp 129–142. Philadelphia: Saunders. [Google Scholar]
- Herrero JF, Headley PM (1996) Reversal by naloxone of the spinal antinociceptive actions of a systemically-administered NSAID. Br J Pharmacol 118:968–972. 10.1111/j.1476-5381.1996.tb15494.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinz B, Cheremina O, Brune K (2008) Acetaminophen (paracetamol) is a selective cyclooxygenase-2 inhibitor in man. FASEB J 22:383–390. 10.1096/fj.07-8506com [DOI] [PubMed] [Google Scholar]
- Högestätt ED, Jönsson BA, Ermund A, Andersson DA, Björk H, Alexander JP, Cravatt BF, Basbaum AI, Zygmunt PM (2005) Conversion of acetaminophen to the bioactive N-acylphenolamine AM404 via fatty acid amide hydrolase-dependent arachidonic acid conjugation in the nervous system. J Biol Chem 280:31405–31412. 10.1074/jbc.M501489200 [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D (2005) An endocannabinoid mechanism for stress-induced analgesia. Nature 435:1108–1112. 10.1038/nature03658 [DOI] [PubMed] [Google Scholar]
- Kato A, Punnakkal P, Pernía-Andrade AJ, von Schoultz C, Sharopov S, Nyilas R, Katona I, Zeilhofer HU (2012) Endocannabinoid-dependent plasticity at spinal nociceptor synapses. J Physiol 590:4717–4733. 10.1113/jphysiol.2012.234229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozak KR, Rowlinson SW, Marnett LJ (2000) Oxygenation of the endocannabinoid, 2-arachidonylglycerol, to glyceryl prostaglandins by cyclooxygenase-2. J Biol Chem 275:33744–33749. 10.1074/jbc.M007088200 [DOI] [PubMed] [Google Scholar]
- La Rana G, Russo R, Campolongo P, Bortolato M, Mangieri RA, Cuomo V, Iacono A, Raso GM, Meli R, Piomelli D, Calignano A (2006) Modulation of neuropathic and inflammatory pain by the endocannabinoid transport inhibitor AM404 [N-(4-hydroxyphenyl)-eicosa-5,8,11,14-tetraenamide]. J Pharmacol Exp Ther 317:1365–1371. 10.1124/jpet.105.100792 [DOI] [PubMed] [Google Scholar]
- Liang YC, Huang CC, Hsu KS, Takahashi T (2004) Cannabinoid-induced presynaptic inhibition at the primary afferent trigeminal synapse of juvenile rat brainstem slices. J Physiol 555:85–96. 10.1113/jphysiol.2003.056986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little PJ, Compton DR, Johnson MR, Melvin LS, Martin BR (1988) Pharmacology and stereoselectivity of structurally novel cannabinoids in mice. J Pharmacol Exp Ther 247:1046–1051. [PubMed] [Google Scholar]
- Mallet C, Daulhac L, Bonnefont J, Ledent C, Etienne M, Chapuy E, Libert F, Eschalier A (2008) Endocannabinoid and serotonergic systems are needed for acetaminophen-induced analgesia. Pain 139:190–200. 10.1016/j.pain.2008.03.030 [DOI] [PubMed] [Google Scholar]
- Mallet C, Barrière DA, Ermund A, Jönsson BA, Eschalier A, Zygmunt PM, Högestätt ED (2010) TRPV1 in brain is involved in acetaminophen-induced antinociception. PLoS One 5:e12748. 10.1371/journal.pone.0012748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsicano G, Wotjak CT, Azad SC, Bisogno T, Rammes G, Cascio MG, Hermann H, Tang J, Hofmann C, Zieglgänsberger W, Di Marzo V, Lutz B (2002) The endogenous cannabinoid system controls extinction of aversive memories. Nature 418:530–534. 10.1038/nature00839 [DOI] [PubMed] [Google Scholar]
- Marsicano G, Goodenough S, Monory K, Hermann H, Eder M, Cannich A, Azad SC, Cascio MG, Gutiérrez SO, van der Stelt M, López-Rodriguez ML, Casanova E, Schütz G, Zieglgänsberger W, Di Marzo V, Behl C, Lutz B (2003) CB1 cannabinoid receptors and on-demand defense against excitotoxicity. Science 302:84–88. 10.1126/science.1088208 [DOI] [PubMed] [Google Scholar]
- McQueen DS, Iggo A, Birrell GJ, Grubb BD (1991) Effects of paracetamol and aspirin on neural activity of joint mechanonociceptors in adjuvant arthritis. Br J Pharmacol 104:178–182. 10.1111/j.1476-5381.1991.tb12404.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller ST, Gebhart GF (1997) Intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat. Eur J Pain 1:43–52. 10.1016/S1090-3801(97)90052-5 [DOI] [PubMed] [Google Scholar]
- Meng ID, Manning BH, Martin WJ, Fields HL (1998) An analgesia circuit activated by cannabinoids. Nature 395:381–383. 10.1038/26481 [DOI] [PubMed] [Google Scholar]
- Muth-Selbach US, Tegeder I, Brune K, Geisslinger G (1999) Acetaminophen inhibits spinal prostaglandin E2 release after peripheral noxious stimulation. Anesthesiology 91:231–239. 10.1097/00000542-199907000-00032 [DOI] [PubMed] [Google Scholar]
- Nyilas R, Gregg LC, Mackie K, Watanabe M, Zimmer A, Hohmann AG, Katona I (2009) Molecular architecture of endocannabinoid signaling at nociceptive synapses mediating analgesia. Eur J Neurosci 29:1964–1978. 10.1111/j.1460-9568.2009.06751.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen AE, Andresen T, Staahl C, Drewes AM (2012) Human experimental pain models for assessing the therapeutic efficacy of analgesic drugs. Pharmacol Rev 64:722–779. 10.1124/pr.111.005447 [DOI] [PubMed] [Google Scholar]
- Ottani A, Leone S, Sandrini M, Ferrari A, Bertolini A (2006) The analgesic activity of paracetamol is prevented by the blockade of cannabinoid CB1 receptors. Eur J Pharmacol 531:280–281. 10.1016/j.ejphar.2005.12.015 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin K (2001) Paxinos and Franklin's the mouse brain in stereotaxic coordinates, Ed 2 San Diego: Academic. [Google Scholar]
- Pelissier T, Alloui A, Paeile C, Eschalier A (1995) Evidence of a central antinociceptive effect of paracetamol involving spinal 5HT3 receptors. Neuroreport 6:1546–1548. 10.1097/00001756-199507310-00020 [DOI] [PubMed] [Google Scholar]
- Pini LA, Sandrini M, Vitale G (1996) The antinociceptive action of paracetamol is associated with changes in the serotonergic system in the rat brain. Eur J Pharmacol 308:31–40. 10.1016/0014-2999(96)00261-0 [DOI] [PubMed] [Google Scholar]
- Reinold H, Ahmadi S, Depner UB, Layh B, Heindl C, Hamza M, Pahl A, Brune K, Narumiya S, Müller U, Zeilhofer HU (2005) Spinal inflammatory hyperalgesia is mediated by prostaglandin E receptors of the EP2 subtype. J Clin Invest 115:673–679. 10.1172/JCI23618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi-Carmona M, Barth F, Héaulme M, Shire D, Calandra B, Congy C, Martinez S, Maruani J, Néliat G, Caput D, et al. (1994) SR141716A, a potent and selective antagonist of the brain cannabinoid receptor. FEBS Lett 350:240–244. 10.1016/0014-5793(94)00773-X [DOI] [PubMed] [Google Scholar]
- Sandrini M, Romualdi P, Vitale G, Morelli G, Capobianco A, Pini LA, Candeletti S (2001) The effect of a paracetamol and morphine combination on dynorphin A levels in the rat brain. Biochem Pharmacol 61:1409–1416. 10.1016/S0006-2952(01)00623-2 [DOI] [PubMed] [Google Scholar]
- Sharma V, McNeill JH (2009) To scale or not to scale: the principles of dose extrapolation. Br J Pharmacol 157:907–921. 10.1111/j.1476-5381.2009.00267.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skjelbred P, Album B, Lokken P (1977) Acetylsalicylic acid vs paracetamol: effects on post-operative course. Eur J Clin Pharmacol 12:257–264. 10.1007/BF00607424 [DOI] [PubMed] [Google Scholar]
- Suplita RL 2nd, Farthing JN, Gutierrez T, Hohmann AG (2005) Inhibition of fatty-acid amide hydrolase enhances cannabinoid stress-induced analgesia: sites of action in the dorsolateral periaqueductal gray and rostral ventromedial medulla. Neuropharmacology 49:1201–1209. 10.1016/j.neuropharm.2005.07.007 [DOI] [PubMed] [Google Scholar]
- Suplita RL 2nd, Gutierrez T, Fegley D, Piomelli D, Hohmann AG (2006) Endocannabinoids at the spinal level regulate, but do not mediate, nonopioid stress-induced analgesia. Neuropharmacology 50:372–379. 10.1016/j.neuropharm.2005.10.007 [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD (1986) Indomethacin blocks central nociceptive effects of PGF2α. Brain Res 373:81–84. 10.1016/0006-8993(86)90317-3 [DOI] [PubMed] [Google Scholar]
- Tiippana E, Hamunen K, Kontinen V, Kalso E (2013) The effect of paracetamol and tropisetron on pain: experimental studies and a review of published data. Basic Clin Pharmacol Toxicol 112:124–131. 10.1111/j.1742-7843.2012.00935.x [DOI] [PubMed] [Google Scholar]
- Tjølsen A, Lund A, Hole K (1991) Antinociceptive effect of paracetamol in rats is partly dependent on spinal serotonergic systems. Eur J Pharmacol 193:193–201. 10.1016/0014-2999(91)90036-P [DOI] [PubMed] [Google Scholar]
- Tsou K, Brown S, Sañudo-Peña MC, Mackie K, Walker JM (1998) Immunohistochemical distribution of cannabinoid CB1 receptors in the rat central nervous system. Neuroscience 83:393–411. 10.1016/S0306-4522(97)00436-3 [DOI] [PubMed] [Google Scholar]
- Uda R, Horiguchi S, Ito S, Hyodo M, Hayaishi O (1990) Nociceptive effects induced by intrathecal administration of prostaglandin D2, E2, or F2α to conscious mice. Brain Res 510:26–32. 10.1016/0006-8993(90)90723-O [DOI] [PubMed] [Google Scholar]
- Vinegar R, Truax JF, Selph JL (1976) Quantitative comparison of the analgesic and anti-inflammatory activities of aspirin, phenacetin and acetaminophen in rodents. Eur J Pharmacol 37:23–30. 10.1016/0014-2999(76)90004-2 [DOI] [PubMed] [Google Scholar]
- Witschi R, Johansson T, Morscher G, Scheurer L, Deschamps J, Zeilhofer HU (2010) Hoxb8-Cre mice: a tool for brain-sparing conditional gene deletion. Genesis 48:596–602. 10.1002/dvg.20656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Ives D, Ramesha CS (1997) Synthesis of prostaglandin E2 ethanolamide from anandamide by cyclooxygenase-2. J Biol Chem 272:21181–21186. 10.1074/jbc.272.34.21181 [DOI] [PubMed] [Google Scholar]
- Zhao H, Si ZH, Li MH, Jiang L, Fu YH, Hong W, Ruan LY, Wang JS (2016) Pyrazinamide-induced hepatotoxicity and gender differences in rats as revealed by a 1H NMR based metabolomics approach. Toxicol Res 6:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]