

Abstract
Pain associated with skeletal pathology or disease is a significant clinical problem, but the mechanisms that generate and/or maintain it remain poorly understood. In this study, we explored roles for GDNF, neurturin, and artemin signaling in bone pain using male Sprague Dawley rats. We have shown that inflammatory bone pain involves activation and sensitization of peptidergic, NGF-sensitive neurons via artemin/GDNF family receptor α-3 (GFRα3) signaling pathways, and that sequestering artemin might be useful to prevent inflammatory bone pain derived from activation of NGF-sensitive bone afferent neurons. In addition, we have shown that inflammatory bone pain also involves activation and sensitization of nonpeptidergic neurons via GDNF/GFRα1 and neurturin/GFRα2 signaling pathways, and that sequestration of neurturin, but not GDNF, might be useful to treat inflammatory bone pain derived from activation of nonpeptidergic bone afferent neurons. Our findings suggest that GDNF family ligand signaling pathways are involved in the pathogenesis of bone pain and could be targets for pharmacological manipulations to treat it.
SIGNIFICANCE STATEMENT Pain associated with skeletal pathology, including bone cancer, bone marrow edema syndromes, osteomyelitis, osteoarthritis, and fractures causes a major burden (both in terms of quality of life and cost) on individuals and health care systems worldwide. We have shown the first evidence of a role for GDNF, neurturin, and artemin in the activation and sensitization of bone afferent neurons, and that sequestering these ligands reduces pain behavior in a model of inflammatory bone pain. Thus, GDNF family ligand signaling pathways are involved in the pathogenesis of bone pain and could be targets for pharmacological manipulations to treat it.
Keywords: artemin, bone pain, GDNF, neurturin, pain, skeletal pain
Introduction
Pain associated with skeletal pathology, including bone cancer, bone marrow edema syndromes, osteomyelitis, osteoarthritis, and fractures causes a major burden (both in terms of quality of life and cost) on individuals and health care systems worldwide. This burden is expected to increase with advances in modern medicine that prolong life expectancy, because many of the conditions that cause bone pain develop late in life. A dominant feature common to almost all conditions that produce bone pain is the release of inflammatory mediators by cells associated with the disease (Bennett, 1988; Goldring, 2000; Haegerstam, 2001; Mantyh, 2004; Urch, 2004; Starr et al., 2008; Berenbaum, 2013). Opioids and nonsteroidal anti-inflammatory drugs (NSAIDs) are generally used to treat mild to severe inflammatory pain, but therapeutic use for bone pain is limited by side effects associated with prolonged use, and because they may interfere with bone remodeling/healing (Bove et al., 2009; Pountos et al., 2012; Chrastil et al., 2013). There is a clear need to find alternative strategies to treat bone pain that do not involve the use of NSAIDs or opioids, and are targeted more specifically at mechanisms that generate and/or maintain inflammatory bone pain.
Nociceptors can be classified into two groups based on their response to nerve growth factor (NGF) or glial cell line-derived neurotrophic factor (GDNF; Molliver et al., 1995; Snider and McMahon, 1998). Much is known of the role of NGF in inflammatory pain, and this has led to the development of NGF sequestration to treat inflammatory pain, including that associated with bone pathologies. Sequestering NGF by systemic administration of anti-NGF antibodies reduces pain-like behaviors in animal models of bone cancer and fracture-induced pain (Sevcik et al., 2005; Jimenez-Andrade et al., 2007; Koewler et al., 2007), and also inflammatory pain of other tissue systems (Woolf et al., 1994, 1997; Ma and Woolf, 1997). Furthermore, clinical trials reveal that they resolve pain in at least some human inflammatory pain conditions (Kumar and Mahal, 2012). However, many patients with osteoarthritis receiving anti-NGF antibodies in a clinical trial developed rapidly progressive osteoarthritis, particularly when used in conjunction with NSAIDs (Seidel et al., 2013; Hochberg, 2015). We propose that manipulation of signaling through the GDNF family of ligands (GFLs) might constitute an alternative target for therapeutic treatment of inflammatory bone pain.
The GFLs (GDNF, neurturin, artemin, and persephin) act through the receptor tyrosine kinase RET and one of four accessory subunits [GDNF family receptor α-1(GFRα1) to GFRα4], which confer ligand specificity for the receptor complexes (GDNF/GFRα1, neurturin/GFRα2, artemin/GFRα3, persephin/GFRα4; Lindsay and Yancopoulos, 1996; Sah et al., 2005). Further specificity of ligand actions is conferred by the following restricted expression patterns: GFRα1 and GFRα2 are confined mostly to nonpeptidergic small diameter GDNF-sensitive sensory neurons (Bennett et al., 1998), whereas GFRα3 is found principally in a subpopulation of peptidergic small-diameter NGF-sensitive sensory neurons (Orozco et al., 2001) that are likely to be artemin sensitive. GFRα4 is not found in peripheral sensory neurons. There is documented heterogeneity of GFL receptor expression in sensory neurons projecting to different target tissues (Dolatshad and Saffrey, 2007; Malin et al., 2011), but whether this is true of bone afferent neurons is yet to be determined. There is accumulating evidence for a role of GFL signaling in pain (Lindsay and Yancopoulos, 1996; Sah et al., 2005; Bespalov and Saarma, 2007), and there is evidence that the GFLs and GDNF-sensitive neurons contribute to inflammatory pain (Amaya et al., 2004; Lindfors et al., 2006; Jeon et al., 2008; Malin and Davis, 2008). Of key relevance is the finding that GFL levels are increased by inflammation of peripheral tissues in both humans (Okragly et al., 1999; von Boyen et al., 2006) and animals (Amaya et al., 2004; Hashimoto et al., 2005; Malin et al., 2006; Ikeda-Miyagawa et al., 2015). In this study, we show evidence of interactions for each of the GFLs with bone afferent neurons, and that blocking some of these interactions prevents inflammatory bone pain.
Materials and Methods
Male Sprague Dawley rats weighing between 200 and 350 g were used in this study. Animals were housed in pairs or groups of four, in a 12 h light/dark cycle, and were provided with food and water ad libitum. All experiments conformed to the Australian National Health and Medical Research Council code of practice for the use of animals in research, and were approved by the University of Melbourne Animal Experimentation Ethics Committee.
Sensory neurons that innervate the bone marrow (bone afferent neurons) are described as small-diameter myelinated or unmyelinated fibers (Furusawa, 1970; Seike, 1976; Mach et al., 2002; Ivanusic et al., 2006; Ivanusic, 2009); contain neuropeptide markers for nociceptors, such as substance P and calcitonin gene-related peptide (CGRP; Bjurholm et al., 1988; Mach et al., 2002; Ivanusic, 2009); and/or are responsive to algesic substances and noxious mechanical stimuli (Furusawa, 1970; Seike, 1976; Nencini and Ivanusic, 2017; Nencini et al., 2017). These characteristics are consistent with a role in nociception. We used chemical and mechanical stimuli to activate and sensitize bone afferent neurons, and weight-bearing assays to explore pain behavior, because bone pain is predominantly associated with the release of algesic substances and mechanical, but not thermal, stimuli.
Retrograde tracing.
Retrograde labeling of bone afferent neurons was performed in the same way as we have previously described (Ivanusic, 2009; Nencini and Ivanusic, 2017; Nencini et al., 2017). Animals were anesthetized with isoflurane (4% induction; 2.5% maintenance). A skin incision was made over the medial aspect of the tibia, and a small hole was made in the cortical bone on the medial aspect of the tibial diaphysis using a sterile 26 gauge needle. A Hamilton syringe was used to inject the retrograde tracer Fast Blue (FB; 2 μl; 10% in dH2O) through the hole and directly into the medullary cavity. The hole was sealed with bone wax to prevent leakage into surrounding tissues. The entire area was washed extensively with 0.1 m PBS (pH 7.4) and inspected for tracer leakage using a hand-held UV illumination device. Animals that showed evidence of tracer leakage to surrounding tissues were excluded from this study. Skin incisions were closed with stainless steel autoclips. For studies of constitutive expression of GFL signaling molecules in bone afferent neurons, animals were left for a 7 d survival period to allow for transport of the tracer to neuronal cell bodies in the dorsal root ganglion (DRG). For studies of changes in the expression of GFL signaling molecules in inflammatory bone pain, an inflammatory agent [complete Freund's adjuvant (CFA); see below) or saline was coinjected with the retrograde tracer, and the animals were left for a 4 d survival period chosen to coincide with the peak pain time-point in the animal model.
Tissue preparation and immunohistochemistry.
Each animal was given an overdose of sodium pentobarbitone (Lethobarb; 80 mg/kg, i.p.) and was perfused via the ascending aorta with 500 ml of heparinized PBS followed by 500 ml of 4% paraformaldehyde in PBS. Lumbar DRGs L3 and L4 were dissected and left overnight in a solution containing the above fixative and 20% sucrose, and were sectioned at 14 μm using a cryostat the next day. Multiple series of sections were collected on gelatinized glass slides (0.1% chrome alum and 0.5% gelatin) and processed for immunolabeling. Sections were immunolabeled to determine whether retrograde labeled bone afferent neurons expressed GFRα1 to GFRα3, TrkA, CGRP, and/or transient receptor potential vanilloid 1 (TRPV1). Details of the primary and secondary antisera, including references to work that cite the specificity of the antibodies, are given in Table 1. All antisera were diluted in PBS containing 0.3% Triton X-100 and 0.1% sodium azide. Sections were washed three times in PBS and incubated overnight in the primary antisera at room temperature. Following three further washes in PBS, the sections were incubated in secondary antibody for 2 h and washed again three times in PBS. The slides were coverslipped using DAKO fluorescence mounting medium.
Table 1.
Source and concentrations of the primary and secondary antisera
| Immunogen | Manufacturer information/product code | Dilution used | References/characterization | |
|---|---|---|---|---|
| Primary antibody antigen | ||||
| GFRα1 | Mouse myeloma cell line NS0-derived recombinant rat GFRα1 (25–445) | Goat polyclonal; catalog #AF560, R&D Systems/RRID:AB_2110307 | 1:300 | Rakowicz et al. (2002); Pierchala et al. (2006)/manufacturer information |
| GFRα2 | Mouse myeloma cell line NS0-derived recombinant mouse GFRα2 (22–441) | Goat polyclonal; catalog #AF429, R&D Systems/RRID:AB_2294621 | 1:300 | Võikar et al. (2004); Wanigasekara et al. (2004)/manufacturer information |
| GFRα3 | Spodoptera frugiperda insect ovarian cell line Sf 21-derived recombinant mouse GFRα3 (34–379) | Goat polyclonal; catalog #AF2645, R&D Systems/RRID:AB_2110295 | 1:300 | Malin et al. (2006); Keast et al. (2010)/manufacturer information |
| CGRP | Synthetic rat CGRP conjugated to KLH | Rabbit polyclonal; catalog #C8198; Sigma-Aldrich/RRID:AB_259091 | 1:500 | Zhang et al. (2001); Peleshok and Ribeiro-da-Silva (2011)/manufacturer information |
| TRPV1 | Intracellular C terminus of rat TRPV1 (824–838) | Rabbit polyclonal; catalog #ACC-030, Alomone Labs/RRID:AB_2313819 | 1:500 | Everaerts et al. (2009); Alamri et al. (2015)/manufacturer information |
| Rabbit anti-TrkA 1:2000 | Extracellular domain of embryonic chick TrkA isolated from e8 DRG | Rabbit polyclonal; gift from Louis Reichardt, University of California, San Francisco (San Francisco, United States)/RRID:AB_2315490 | 1:2000 | Schröpel et al. (1995); Lefcort et al. (1996); George et al. (2010) |
| Secondary antibody | ||||
| Donkey anti-Rabbit 594 | Catalog #A21207, Molecular Probes, Invitrogen/RRID:AB_141637 | 1:200 | ||
| Donkey anti-Goat 488 | catalog #A11055, Molecular Probes, Invitrogen/RRID:AB_2534102 | 1:200 |
Image acquisition and data analysis.
Sections of DRGs were examined and photographed with a 20× objective using a Zeiss Axioskop Fluorescence Microscope fitted with an AxioCam MRm camera. FITC, Texas Red, and UV filter sets were used to discriminate labeling with the Alexa Fluor 488 and 594 fluorophores, and Fast Blue, respectively. Immunolabeling for expression of GFRα1 to GFRα3 was examined in a separate series of DRG sections for each marker. Counts, soma size measurements (cross-sectional area of soma), and intensity measurements were made directly from the images using Zen Lite Software (Zen 2011, Carl Zeiss; RRID:SCR_013672). Only cells with a nucleus visible under the microscope were examined. The counts presented are estimates of the total number of bone afferent neurons and may overestimate afferents with large cell bodies. For constitutive expression analysis, we determined the proportion of retrograde labeled neurons that expressed each antibody marker for each animal. For analysis of changes associated with inflammatory bone pain, we tested whether there was a difference in the proportion of bone afferent neurons expressing each marker and/or in the intensity of labeling in these same neurons, in inflamed versus saline-treated control rats. Changes in the proportion of neurons that express each molecule likely reflect de novo expression and/or retrograde transport from the periphery to the soma. If there is no change in the proportion of neurons that express a marker, then changes in the intensity of immunolabeling likely reflect increased expression in neurons that already contain the protein, not de novo expression. Intensity of immunolabeling was determined for each cell by calculating the mean pixel intensity in the soma and was expressed relative to the average of the mean pixel intensities of five surrounding cells, in the same section, that were clearly not immunolabeled. Figures were prepared using CorelDraw software (CorelDraw Graphics Suite, Corel; RRID:SCR_014235). Individual images were contrast and brightness adjusted. No other manipulations were made to the images.
Electrophysiological recordings using an in vivo bone–nerve preparation.
Electrophysiological recordings were made using our recently developed in vivo bone-nerve preparation (Nencini and Ivanusic, 2017; Nencini et al., 2017) in rats anesthetized with urethane (50% w/v; 1.5 g/kg, i.p). In this preparation, a fine branch of the tibial nerve that innervates the marrow cavity of the rat tibia is identified and carefully teased away from its associated blood vessels and surrounding tissues over ∼1 cm, and placed over a platinum hook electrode for recording (Fig. 1A). Whole-nerve electrical activity was amplified (1000×) and filtered (high pass, 100 Hz; low pass, 3 kHz) using a differential amplifier (DP-311, Warner Instruments), sampled at 20 kHz (Powerlab, ADInstruments) and stored to a PC using the recording software LabChart (ADInstruments; RRID:SCR_001620). Mechanical stimulation was delivered by injecting heparinized physiological saline (0.9% sodium chloride) into the marrow cavity through a needle, which was implanted in a small hole in the proximal tibia (size, 0.8 mm), that was connected to a feedback-controlled syringe pump (PHD ULTRA Pump, Harvard Apparatus) via polyethylene tubing. Changes in the intraosseous pressure were measured using a bridge-amplified (TAM-D Amplifier, Harvard Apparatus) signal derived from a pressure transducer (APT300 Transducer, Harvard Apparatus) placed to measure the input pressure to the bone (Fig. 1A). The pump uses this as feedback to adjust flow through the system to control and maintain constant input pressures. We used this feature to apply a ramp-and-hold stimulus with a constant 300 mmHg of pressure delivered for 20 s duration during the hold phase (Fig. 1B). These data were stored to a PC in parallel with the nerve recordings. GDNF, neurturin, or artemin (10 μl of 20 μg/ml; #450-10, #450-11, #450-17, PeproTech) was delivered to the marrow cavity through a second hole using a Hamilton syringe attached with polyethylene tubing. The doses chosen for these experiments reflect those known to produce behavioral sensitivity on injection into the hindpaw footpad of mice (Malin et al., 2006) and in the tibial marrow cavity of naive rats (see below). However, we do not know the actual concentration of the ligands at the nerve terminal endings in the bone marrow because there will be dilution in the bone marrow compartment.
Figure 1.

Experimental setup of the in vivo bone–nerve preparation and spike discrimination. A, Schematic of the electrophysiological setup. B, Whole-nerve recording and rasters of single-unit activity in response to a ramp-and-hold pressure stimulus applied to the marrow cavity. Action potentials from single mechanically activated units were discriminated by their amplitude and duration using Spike Histogram software. An example of a single spike for each unit is at the left of each raster. C, Relationship between conduction velocity and amplitude of action potentials. Action potential amplitude (in μV; peak-to-peak) was plotted against conduction velocity (in m/s) for 51 single units activated by high-intensity intraosseous pressure stimuli (≥300 mmHg) during recordings made from seven animals. Units that also responded to stimulation of surrounding tissues (white circles) had very large action potential amplitudes (>120 μV) and conducted in the Aβ range (≥14.3 m/s). Units with conduction velocities in the C-fiber range (<2.5 m/s) had the smallest action potential amplitudes (<40 μV peak-to-peak; gray circles). All other units, with action potential amplitudes of >40 μV peak-to-peak that did not respond to the stimulation of surrounding tissues, had Aδ conduction velocities (black circles). A/D, Analog to digital.
Single units were discriminated in the whole-nerve recordings according to their amplitude and duration using Spike Histogram software (LabChart 8, ADInstruments; RRID:SCR_001620; Fig. 1B). Only action potentials with positive and/or negative peaks clearly above noise were sampled. We have previously documented a linear relationship between action potential amplitude and conduction velocity for single units in response to a 300 mmHg pressure stimulus applied to the marrow cavity (Nencini and Ivanusic, 2017), and we have updated this dataset in the present study (Fig. 1C; an extra 21 units have been included). Conduction velocities were determined using two recording electrodes with a distance of 7–10 mm between electrodes. Action potentials recorded at the second electrode, which were time locked to those recorded at the first electrode, were considered as originating in the same axon. For these units, conduction velocities were estimated by dividing conduction time between the electrodes by the distance between electrodes. Units with Aδ conduction velocities (2.5–12.5 m/s) had action potential amplitudes between 47 and 145 μV (peak-to-peak; n = 31; Fig. 1C) and units with C conduction velocities (<2.5 m/s) had small-amplitude action potentials (<40 μV, n = 6; Fig. 1C). Thus, in the present study we define spikes with peak-to-peak amplitudes of between 40 and 145 μV as being derived from Aδ fibers, and spikes with peak-to-peak amplitudes of <40 μV as derived from C fibers. Some very fast conducting units were observed, but these had very large-amplitude spikes (>145 μV) and had receptive fields outside of the marrow cavity (they responded to gentle probing of the periosteum or muscle adjacent to the bone). It is possible that these units represent low-threshold mechanoreceptors, in surrounding periosteum or muscle, that were mechanically activated by the leakage of saline from the marrow cavity, through Volkmann's canals and into surrounding tissue. They were relatively rare and were excluded from further analysis.
The first series of experiments was conducted to determine whether each of the GFLs could rapidly activate bone afferent neurons in whole-nerve recordings (GDNF, neurturin, or artemin, 10 μl of 20 μg/ml; n = 5 animals each; saline, n = 5 animals). In these experiments, each of the GFLs was applied to the marrow cavity while whole-nerve recordings were being made, and ongoing activity was assessed continuously for up to 1 h. Analysis was performed on data derived from all spikes in the whole-nerve recordings, regardless of whether they belonged to Aδ or C fiber units, and on data derived from spikes with amplitudes consistent with Aδ or C conduction velocities (see above). The number of spikes was reported as frequency histograms with 30 s bin widths. We defined the onset of GFL-induced activity as the time at which the mean frequency of discharge in two consecutive 30 s bins exceeded that of the maximum mean frequency of discharge in all bins preceding injection of the GFL. A second series of experiments was conducted to determine whether single mechanically activated bone afferent neurons could be sensitized by each of the GFLs (GDNF, neurturin, or artemin, 10 μl of 20 μg/ml; N = 12 units each; saline, N = 10 units). We report data here only for single Aδ units because we could not unequivocally isolate single C-fiber units using our recording configuration (Nencini and Ivanusic, 2017). In these experiments, responses of the single Aδ units to pressure (300 mmHg) were assessed before, and 15, 30, 45, and 60 min after application of the GFLs or saline. Thresholds for activation and overall discharge frequency during the pressure stimulus were determined following injection of each GFL and were expressed relative to preinjection baseline levels. Decreases in the mechanical threshold assessed on the rising phase of the pressure ramp and/or increases in overall discharge frequency during the pressure stimulus were used as indicators of sensitization. There was some variability in the discharge frequency of mechanically activated units before and after saline injection, but none had increases that were >15% above the baseline frequency of discharge. Thus, single units were classified as being sensitized by each GFL if they increased their discharge frequency by ≥20% relative to the baseline level. Animals were killed by decapitation at the end of the recordings.
Pain behavior in response to application of GFLs to the marrow cavity.
Behavioral testing was used to test whether GFLs produce pain behaviors when applied directly to bone marrow. Animals were anesthetized and prepared as above for retrograde tracing. A Hamilton syringe was used to inject GDNF, neurturin, or artemin (10 μl of 20 μg/ml; n = 6 animals each; PeproTech) or saline (10 μl of volume control; n = 7 animals) directly into the marrow cavity. Recovery from isoflurane anesthesia occurred within a few minutes of the end of the surgery, allowing behavioral testing to be performed within 15 min of surgery. Changes in weight bearing, assessed with an incapacitance meter (IITC Life Science), were used to assay pain behavior in response to injection of each of the GFLs (at 0, 15, and 30 min, 1, 2, and 5 h, and 1 and 2 d after injection). The incapacitance meter independently measures weight bearing on the injected and uninjected limb. Testing was always performed at the same times each day. Animals were first habituated with exposure to the equipment for 15 min/d over the 3 d before the surgery. At each testing time-point, five weight-bearing readings of 3 s duration were collected. The weight bearing on the injected hindlimb of each animal was then calculated as a percentage of the total weight bearing, using the following equation:
 |
Animals were killed by CO2 asphyxiation at the end of the behavioral testing.
Sequestration of GFLs in CFA-induced inflammatory bone pain.
To test whether the sequestration of each of the GFLs could be used to reduce or prevent inflammatory bone pain, we used an animal model of CFA-induced inflammation of the rat tibial marrow cavity. We have used this approach previously, and have confirmed that pain-like behavior peaks 4 d after CFA injection and that inflammation is present in the marrow cavity of CFA-injected animals at this peak pain time-point (Nencini et al., 2017). For the experiments outlined in the present study, animals were anesthetized and prepared as above for retrograde tracing. A Hamilton syringe was used to coinject polyclonal goat anti-GDNF (10 μl of 20 μg/ml; n = 12 animals; AB212-NA, R&D Systems; RRID:AB_354287), rabbit anti-neurturin (10 μl of 20 μg/ml; n = 10 animals; 500-P102, PeproTech; RRID:AB_147717), or goat anti-artemin antibodies (10 μl of 20 μg/ml; n = 12 animals; AF1085, R&D Systems; RRID:AB_354587) with CFA (50 μl; Mycobacterium tuberculosis, suspended in a 1:1 oil/saline mixture; Sigma-Aldrich) into the marrow cavity. Isotype control antibodies (10 μl of 20 μg/ml; n = 14 animals; goat IgG AB108C, R&D Systems, RRID:AB_354267; n = 14 animals; rabbit IgG 500-P00, PeproTech; RRID:AB_2722620) were also coinjected with CFA to confirm the effects observed were specific to the sequestering antibodies. Changes in weight bearing, assessed with an incapacitance meter (as above), were used to assay pain-like behavior at days 0, 4, 7, 10, 14, 18, 22, and 26. The habituation and testing protocols for this experiment are the same as described above. Animals were killed by CO2 asphyxiation at the end of the behavioral testing.
Changes in the expression of GFL signaling molecules in inflammatory bone pain.
The same model of CFA-induced inflammatory bone pain was used to test whether inflammatory bone pain, which peaks at 4 d, is correlated with changes in the expression of GFL signaling molecules. Animals were anesthetized and prepared as above for retrograde tracing. A Hamilton syringe was used to inject CFA (50 μl; n = 4 animals; Mycobacterium tuberculosis, suspended in a 1:1 oil/saline mixture; Sigma-Aldrich) or saline (volume control; n = 4 animals) through the hole directly into the medullary cavity. To identify bone afferent neurons, the retrograde tracer FB (2 μl; 10% in dH2O) was coinjected with CFA (or saline). Behavioral testing was performed (as above) to confirm pain-like behaviors in each animal injected with CFA, and a lack of pain-like behaviors in those injected with saline. Testing for this was performed before (baseline) and at day 4 (peak pain time-point determined from timeline studies above). Weight bearing of <45% on the injected hindlimb was used to confirm pain behavior in CFA-injected animals because most CFA-injected animals bore less weight than this on their injected limb at the 4 d time-point, while all saline-injected animals had an even distribution of weight between the hindlimbs at this same time-point (Nencini et al., 2017). The protocols for tissue preparation, immunohistochemistry, imaging, and data analysis were the same as those described above.
Experimental design and statistical analysis.
Statistical analyses were performed using GraphPad Prism (GraphPad Software; RRID:SCR_002798). Comparisons of treatment effects for each of the GFLs on the ongoing activity and sensitization of bone afferent neurons, and pain behavior, were analyzed using an ANOVA or a two-way ANOVA with repeated measures, as required. ANOVAs were followed by Bonferroni's post hoc testing only if the ANOVA indicated a significant difference. A Mann–Whitney test was used to test for inflammation-induced differences in the proportion or the intensity of immunolabeling of bone afferent neurons that expressed each of the GFLs. In all cases, p < 0.05 was used to define statistical significance.
Results
Receptors for each of the GFLs are constitutively expressed in bone afferent neurons
We used a combination of retrograde tracing and immunohistochemistry to determine the proportion of bone afferent neurons that express GFRα1 to GFRα3 receptors, and/or other pain-signaling molecules (TRPV1, TrkA, and CGRP; Fig. 2; Table 2). Antibodies were directed against GFRα1, GFRα2, and GFRα3 receptors; TRPV1 (to identify polymodal nociceptors); TrkA (to identify NGF-sensitive neurons); and CGRP (to identify peptidergic nociceptors). A total of 1578 retrograde-labeled bone afferent neurons were counted in L3 DRGs taken from three animals. Almost all bone afferent neurons were small or medium sized (>95% of those counted were <1800 μm2). A substantial proportion of bone afferent neurons were GFRα3 immunoreactive (IR; 40 ± 2.9%; n = 3 animals). Most GFRα3-IR bone afferent neurons were TrkA-IR (80 ± 4.9%; n = 3 animals) and CGRP-IR (80 ± 7.3%; n = 3 animals), and many were TRPV1-IR (39 ± 4.4%; n = 3 animals), suggesting that they are predominantly NGF-sensitive peptidergic nociceptors. Approximately 20% of the bone afferent neurons were GFRα1-IR (19 ± 1.9; n = 3 animals) or GFRα2-IR (20 ± 3.2; n = 3 animals). None of the GFRα1-IR bone afferent neurons were TRPV1-IR (0%; n = 3 animals), but some were TrkA-IR (27 ± 10.6%; n = 3 animals) or CGRP-IR (13 ± 12.5%; n = 3 animals). Only some of the GFRα2-IR bone afferent neurons were TRPV1-IR (14 ± 9.8%; n = 3 animals), TrkA-IR (19 ± 2.3%; n = 3 animals), or CGRP-IR (37 ± 7.2%; n = 3 animals). Thus, most bone afferent neurons that expressed GFRα1 and/or GFRα2 are likely to be NGF-insensitive nonpeptidergic nociceptors. These findings indicate that bone afferent neurons express the receptors required for GFL signaling. Artemin is likely to act predominantly on NGF-sensitive peptidergic bone afferent neurons, whereas neurturin and GDNF are likely to act on GDNF-sensitive nonpeptidergic bone afferent neurons.
Figure 2.

Constitutive expression of GFRα1 to GFRα3 in bone afferent neurons. A, Schematic representation of the retrograde tracing approach used in this study. FB was injected into the marrow cavity (MC) of the rat tibia. The tracer was taken up by nerve terminals and transported back to their soma in the DRG, permitting the identification of sensory neurons that innervate the rat tibia. B, Size/frequency distribution of all retrograde-labeled bone afferent neurons analyzed in this study. Greater than 95% of these were small- or medium-sized sensory neurons (<1800 μm2). C, Images of retrograde-labeled and immunolabeled bone afferent neurons in sections through the DRG. Each horizontal set of panels shows the same field of a single section. Left, Incorporation of the retrograde tracer FB. Middle, GFRα1–3 immunolabeling. Right, A merged image. Arrowheads identify retrograde-labeled bone afferent neurons throughout. Asterisks (*) indicate bone afferent neurons that express GFRα1, GFRα2, or GFRα3. Scale bars, 50 μm. D, Size/frequency distributions of retrograde labeled bone afferent neurons (black) and those that that also express GFRα1 (blue), GFRα2 (green), or GFRα3 (red). SC, Spinal cord.
Table 2.
Constitutive expression of GFL-signaling molecules
| Number of animals | Number of retrograde-labeled bone afferent neurons | % | |
|---|---|---|---|
| Percentage of bone afferent neurons that are | |||
| GFRα1+ | 3 | 99 | 19 ± 1.9 |
| GFRα2+ | 3 | 90 | 20 ± 3.2 |
| GFRα3+ | 3 | 142 | 40 ± 2.9 |
| Proportion of GFRα1 + bone afferent neurons that express CGRP | 3 | 99 | 13 ± 12.5 |
| Proportion of GFRα2 + bone afferent neurons that express CGRP | 3 | 101 | 37 ± 7.2 |
| Proportion of GFRα3 + bone afferent neurons that express CGRP | 3 | 118 | 80 ± 7.3 |
| Proportion of GFRα1 + bone afferent neurons that express TRPV1 | 3 | 73 | 0 |
| Proportion of GFRα2 + bone afferent neurons that express TRPV1 | 3 | 90 | 14 ± 9.8 |
| Proportion of GFRα3 + bone afferent neurons that express TRPV1 | 3 | 142 | 39 ± 4.4 |
| Proportion of GFRα1 + bone afferent neurons that express TrkA | 3 | 246 | 27 ± 10.6 |
| Proportion of GFRα2 + bone afferent neurons that express TrkA | 3 | 236 | 19 ± 2.3 |
| Proportion of GFRα3 + bone afferent neurons that express TrkA | 3 | 142 | 80 ± 4.9 |
Values are given as the mean ± SEM, unless otherwise indicated.
Exogenous application of GFLs to the marrow cavity activates bone afferent neurons
We used an in vivo electrophysiological bone–nerve preparation to determine whether artemin, neurturin, and GDNF can activate bone afferent neurons. The application of artemin, neurturin, or GDNF to the marrow cavity increased ongoing activity in whole-nerve recordings (Fig. 3). The time course of the changes in ongoing whole-nerve activity differed for each of the GFLs. GDNF and neurturin produced an increase in whole-nerve activity that began between 8 and 13 min following application to the marrow cavity (mean ± SEM, 9 min and 54 ± 51 s; n = 5 animals) or 15 and 29 min (18 min and 30 s ± 2 min and 36 s; n = 5 animals) respectively. This increase in activity was maintained for the entire length of the recordings (up to 60 min; Fig. 3A). In contrast, the application of artemin to the marrow cavity resulted in an earlier increase in activity that began between 3 and 6 min (mean ± SEM, 3 min and 54 ± 33 s; n = 5 animals), peaked within 10 min of application, and then slowly decreased to preapplication levels by ∼30 min (Fig. 3A). Saline injection resulted in no change in activity (Fig. 3A; n = 5 animals). Figure 3B shows group data for the 5, 15, 30, 45, and 60 min time-points. Two-way ANOVA with repeated measures showed significant treatment effects for each of the GFLs relative to saline (GDNF: F(1,8) = 2006, p < 0.0001, n = 5 animals; neurturin: F(1,8) = 34.78, p = 0.0004, n = 5 animals; artemin: F(1,8) = 11.95, p = 0.0086, n = 5 animals). GDNF and neurturin significantly increased activity relative to saline control from the 15 and 30 min time-points, respectively (Fig. 3B). The peak level of activity induced by GDNF and neurturin was ∼10 times that of the baseline. In contrast, artemin significantly increased activity relative to saline at 5 and 15 min, but not at the later time-points (Fig. 3B). The magnitude of the peak artemin-induced activity was six times that of the baseline. When spikes derived from Aδ- or C-fiber bone afferent neurons were isolated from the whole-nerve recordings, it was clear that C-fiber units contributed most to the prolonged changes in activity after the application of each of the three GFLs, and that Aδ-fiber units produced most of the early activity induced by artemin (Fig. 3C).
Figure 3.

GDNF, neurturin, and artemin applied directly to the marrow cavity activates bone afferent neurons. A, Frequency histograms of the total number of spikes isolated from whole-nerve recordings before and after the application of GDNF, neurturin, or artemin (n = 5 animals each). Bin width, 30 s. Error bars indicate the SEM. B, Group data showing the number of spikes recorded (mean ± SEM) before and 5, 15, 30, 45, and 60 min after the application of GDNF, neurturin, artemin, or saline (n = 5 animals each). At each time-point, the number of spikes has been determined over five consecutive minutes and is represented as the mean ± SEM. Application of GDNF and neurturin resulted in an increase in whole-nerve activity, relative to saline, from the 15 and 30 min time-points, respectively (Bonferroni's post hoc test, *p < 0.05). The application of artemin resulted in a significant increase in whole-nerve activity, relative to saline, at the 5 and 15 min time-points, but not at the later time-points (Bonferroni's post hoc test, *p < 0.05). C, Frequency histograms showing the number of spikes with amplitudes consistent with Aδ- or C-fiber conduction velocities isolated from the same whole-nerve recordings. C-fiber spikes contributed more than Aδ-fiber spikes to the prolonged changes in activity after the application of GDNF, neurturin, and artemin, whereas Aδ-fiber spikes contributed to early activity induced by artemin.
Exogenous application of GFLs sensitizes single mechanically activated bone afferent neurons
To further test whether each of the GFLs could sensitize single Aδ bone afferent neurons to noxious mechanical stimulation, we assessed their threshold for activation and their discharge frequency during responses to a ramp-and-hold pressure stimulus (300 mmHg) before and after application of each of the GFLs (Fig. 4). Both GDNF and neurturin sensitized one-third of the single units tested (n = 5 animals each; N = 4 of 12 units each), and artemin sensitized two-thirds of the units tested (n = 4 animals; N = 8 of 12 units; Fig. 4B). For each of the GFLs, sensitization was evident at 15 min after application. The GDNF-sensitized units had a reduced threshold for activation (ANOVA: F = 32.63, p < 0.0001) and increased discharge frequency (ANOVA: F = 11.26, p = 0.0006) in response to the ramp-and-hold pressure stimulus at 15 min postapplication; the neurturin-sensitized units had a reduced threshold for activation (ANOVA: F = 22.18, p < 0.0001) and increased discharge frequency (ANOVA: F = 12.98, p = 0.0003) at 15 min postapplication; and the artemin-sensitized units had a reduced threshold for activation (ANOVA: F = 33.65, p < 0.0001) and increased discharge frequency (ANOVA: F = 19.47, p < 0.0001) at 15 min postapplication (Fig. 4C). Saline injection had no effect on activation threshold or discharge frequency in any of the single units tested (n = 4 animals; N = 10 units; Fig. 4C).
Figure 4.
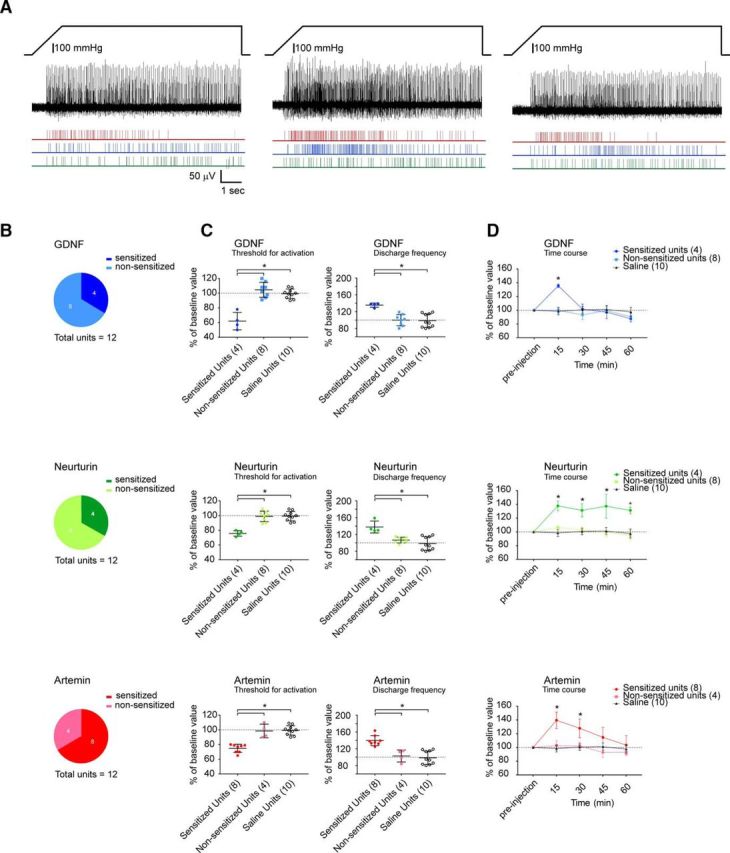
GDNF, neurturin, and artemin applied directly to the marrow cavity sensitizes mechanically activated bone afferent neurons. A, Example of a whole-nerve recording and rasters of single-unit activity in response to a 300 mmHg ramp-and-hold pressure stimulus before (left), and 15 min (middle) and 30 min (right) after the application of artemin to the marrow cavity. There was a clear reduction in threshold for activation and an increase in discharge frequency in single units isolated from this recording after the application of artemin. B, Pie charts representing the proportion of units that were sensitized by each of the GFLs. Sensitization was defined as a 20% increase in discharge frequency, as outlined in Materials and Methods. GDNF and neurturin each sensitized 4 of 12 single units tested (n = 5 animals; N = 4 of 12 units), and artemin sensitized 8 of 12 units tested (n = 4 animals; N = 8 of 12 units). C, Threshold for activation (left panels) and the discharge frequency (right panels) of single mechanically activated units expressed as a percentage of preinjection values at 15 min after the injection of each GFL (n = 12 units for each) or saline (n = 4 animals; N = 10 units). At 15 min, the threshold for activation was significantly decreased and discharge frequency was significantly increased in GDNF-, neurturin-, or artemin-sensitized units, relative to nonsensitized units and the saline control (Bonferroni's post hoc test, *p < 0.05). D, Time course of GFL-induced sensitization. GDNF-sensitized units had significantly increased discharge frequency only at 15 min (Bonferroni's post hoc test, *p < 0.05), neurturin-sensitized units had significantly increased discharge frequency at each of the time-points tested (Bonferroni's post hoc test, *p < 0.05), and artemin-sensitized units had increased discharge frequency at 15 and 30 min (Bonferroni's post hoc test, *p < 0.05). Error bars in C and D represent the mean ± SEM.
The time course of GDNF-induced sensitization was shorter than the time course for artemin- and neurturin-induced sensitization (Fig. 4D). Two-way ANOVA with repeated measures showed significant treatment effects for neurturin and artemin relative to saline (neurturin: F(2,19) = 11.65, p = 0.0005; artemin: F(2,19) = 8.821, p = 0.002). Neurturin-induced sensitization was evident at each of the time-points we tested, while artemin-induced sensitization peaked at 15 min and resolved by 45 min (Fig. 4D). While two-way ANOVA with repeated measures did not reveal a significant treatment effect for GDNF (F(2,19) = 1.089, p = 0.3567), there was a significant interaction effect between treatment and time (F(8,76) = 3.42, p = 0.0021), and GDNF-induced sensitization was observed only at the 15 min time-point (Fig. 4D).
GFLs induce acute changes in pain behavior at times consistent with sensitization of bone afferent neurons
To test whether artemin, neurturin, or GDNF injected into the bone marrow can produce pain behavior in conscious animals, we applied each of these ligands to the marrow cavity of anesthetized rats and, following recovery of consciousness, determined the percentage of total weight bearing on the injected hindlimb. Two-way ANOVA with repeated measures revealed significant treatment effects in animals injected with GDNF (F(1,11) = 14.68, p = 0.0028, n = 6 animals), neurturin (F(1,11) = 16.08, p = 0.0021, n = 6 animals), and artemin (F(1,11) = 24.86, p = 0.0004, n = 6 animals) relative to those injected with saline (n = 7 animals). There was significantly reduced weight bearing within 15 min of GDNF, neurturin, and artemin injection (Fig. 5B–D). The reduced weight bearing was prolonged, lasting for up to 5 h for GDNF (Fig. 5B), and 2 h for neurturin (Fig. 5C) and artemin (Fig. 5D).
Figure 5.

GDNF, neurturin, and artemin applied directly to the marrow cavity results in altered weight bearing. A, Weight bearing was assessed using an incapacitance meter that measures the distribution of weight bearing across each hindlimb. B–D, There was a significant reduction in weight bearing on the injected hindlimb, relative to the uninjected hindlimb, at 15 min and 5 h after application GDNF (B; Bonferroni's post hoc test, *p < 0.05; n = 6 animals) and for up to 1 h after the application of neurturin (C; Bonferroni's post hoc test, *p < 0.05; n = 6 animals) and artemin (D; Bonferroni's post hoc test, *p < 0.05; n = 6 animals).
Sequestering GFLs prevents the induction of pain behavior in an animal model of inflammatory bone pain
To determine whether there is a role for endogenous GFL signaling in the induction of inflammatory bone pain, we explored weight-bearing behavior in animals after sequestering artemin, neurturin, or GDNF, with antibodies applied at the same time that CFA was injected to induce experimental inflammation. To validate the model of CFA-induced inflammatory bone pain, we first determined whether there was altered weight-bearing behavior in animals injected with CFA alone (n = 14 animals), relative to those injected with saline (n = 10 animals). Two-way ANOVA with repeated measures revealed significant treatment effects in animals injected with CFA relative to those injected with saline (F(1,23) = 8.359, p = 0.0048; Fig. 6A). The percentage reduction in ipsilateral hindlimb weight bearing induced by CFA peaked at 4 d when it was significantly lower than the values for saline-injected controls (Fig. 6A). Sequestration of artemin (n = 12 animals) and neurturin (n = 10 animals), but not GDNF (n = 12 animals), completely abrogated the CFA-induced pain behavior (Fig. 6B–D). Two-way ANOVA with repeated measures demonstrated that injection of anti-neurturin antibodies (F(1,23) = 4.583, p = 0.0431) and anti-artemin antibodies (F(1,24) = 4.539, p = 0.0436) reduced CFA-induced pain behavior relative to injection of their isotype control antibodies (n = 14 animals each). Significant reductions in pain behavior were detected at days 4 and 7 for neurturin (Fig. 6C) and at day 4 for artemin (Fig. 6D). There was no difference in CFA-induced pain behavior following sequestration with anti-GDNF relative to injection of its isotype control antibody (F(1,24) = 0.2496, p = 0.6219; Fig. 6B).
Figure 6.

Sequestration of GDNF, neurturin, and artemin with antibodies applied to the marrow cavity abrogates altered weight-bearing behavior in an animal model of CFA-induced inflammatory bone pain. A, Time course of CFA-induced inflammatory bone pain. The percentage of ipsilateral hindlimb weight bearing at the peak of CFA-induced pain (day 4; n = 14 animals) was significantly lower than the values for saline-injected controls at this time-point (Bonferroni's post hoc test *p < 0.05; n = 10 animals). B–D, Sequestration of artemin and neurturin, but not GDNF, completely abrogated the CFA-induced pain-like behavior. B, There was no difference in CFA-induced pain behavior in animals injected with the anti-GDNF sequestering antibody (n = 12 animals) relative to those injected with its isotype control antibody (n = 14 animals). C, There was a significant reduction in pain behavior in animals injected with the neurturin-sequestering antibody (n = 10 animals) relative to those injected with its isotype control antibody (n = 14 animals) at days 4 and 7 (Bonferroni's post hoc test, *p < 0.05). D, There was a significant reduction in pain behavior in animals injected with the artemin-sequestering antibody (n = 12 animals) relative to those injected with its isotype control antibody (n = 14 animals) at day 4 (Bonferroni's post hoc test, *p < 0.05).
Altered expression of GFL receptors is not associated with inflammatory bone pain
To determine whether inflammatory bone pain is associated with changes in the expression of GFRα1–3 receptors, we compared the expression of each of these in the soma of retrograde-labeled bone afferent neurons in animals that had CFA-induced reductions in weight bearing (inflammatory bone pain) with those that had no reduction in weight bearing following saline injection (control). There were no differences in the proportions of retrograde-labeled bone afferent neurons that expressed GFRα1 to GFRα3 in animals that had CFA-induced reductions in weight bearing (n = 4 animals) relative to those that were injected with saline (n = 4 animals; Mann–Whitney test, p > 0.05; Table 3). There were also no significant differences in the intensity of immunolabeling for GFRα1 to GFRα3 in the retrograde-labeled cells (Mann–Whitney test, p > 0.05; Table 3). This suggests that the upregulation of the GFL receptors, or their internalization and retrograde transport to the soma of bone afferent neurons, does not occur in this model of inflammatory bone pain. We were unable to test whether inflammation affected the expression of other pain-signaling molecules (TRPV1, Nav1.8, or Nav1.9) in bone afferent neurons that express GFRα1 to GFRα3 because in both treatment groups only a relatively small number of retrograde-labeled bone afferent neurons coexpressed the GFRα1 to GFRα3 receptors and TRPV1, Nav1.8, or Nav1.9 at the 4 d time-point when pain peaked. When the number of cells used to calculate proportions is low, small changes in expression produce too much variability to produce meaningful comparisons between CFA- and saline-injected animals.
Table 3.
Expression of GFL receptors in inflamed versus control animals
| Saline |
CFA |
Significant difference between saline- and CFA-injected animals (y/n) | |||||
|---|---|---|---|---|---|---|---|
| Number of animals | Number of retrograde-labeled bone afferent neurons | % | Number of animals | Number of retrograde-labeled bone afferent neurons | % | ||
| Proportion | |||||||
| GFRα1 | 4 | 336 | 26 ± 6.7 | 4 | 126 | 36 ± 4.5 | n |
| GFRα2 | 4 | 274 | 6.7 ± 4.8 | 4 | 141 | 5 ± 1.6 | n |
| GFRα3 | 4 | 257 | 41 ± 0.8 | 4 | 122 | 47 ± 4.9 | n |
| Intensity | |||||||
| GFRα1 | 4 | 336 | 1.2 ± 0.1 | 4 | 126 | 1.45 ± 0.06 | n |
| GFRα2 | 4 | 274 | 1.1 ± 0.03 | 4 | 141 | 1.1 ± 0.01 | n |
| GFRα3 | 4 | 257 | 1.2 ± 0.03 | 4 | 122 | 1.3 ± 0.06 | n |
n, No; y, yes. Values are the mean ± SEM, unless otherwise indicated.
Discussion
We have shown the first evidence of interactions for GDNF, neurturin, and artemin with bone afferent neurons, and that preventing some of these interactions influences pain behavior. Our findings suggest that GFL signaling pathways are involved in the pathogenesis of bone pain and could be targets for pharmacological manipulations to treat it.
Inflammatory bone pain involves activation and sensitization of nonpeptidergic bone afferent neurons via GDNF/GFRα1 and neurturin/GFRα2 signaling pathways
GFRα1 and GFRα2 expression in peripheral nociceptors is confined mostly to those that are nonpeptidergic and not sensitive to NGF (Bennett et al., 1998; Zwick et al., 2002; Malin et al., 2006). However, few studies have confirmed expression profiles in peripheral nociceptors that innervate specific targets. GFRα1 is expressed in approximately half of all retrograde-labeled muscle and cutaneous afferent neurons, and GFRα2 is expressed in half of all retrograde-labeled cutaneous but only a quarter of retrograde-labeled muscular afferent neurons (Malin et al., 2011). Here we also show the expression of GFRα1 or GFRα2 in ∼20% of bone afferent neurons, and that bone afferent neurons that express GFRα1 and GFRα2 are mostly nonpeptidergic and do not express the receptor for NGF. Thus, GDNF and neurturin signaling through nonpeptidergic sensory neurons is likely to be important for bone pain.
There have been only a few studies that have explored direct roles for GDNF in modulating responses of peripheral nociceptors to noxious mechanical stimulation. Intramuscular injection of GDNF induces mechanical hyperalgesia that is associated with sensitization of Aδ-fiber but not C-fiber muscle afferents (Alvarez et al., 2012; Murase et al., 2014). In contrast, hindpaw injection of GDNF induces mechanical hyperalgesia in skin that involves, at least in part, sensitization of cutaneous C-fiber nociceptors (Bogen et al., 2008). Together, these findings suggest that the response of peripheral nociceptors to GDNF differs in superficial versus deep tissues. In the present study, we have shown that GDNF transiently sensitizes Aδ bone afferent neurons to mechanical stimulation, and that it activates C bone afferent neurons, but not Aδ bone afferent neurons. This highlights the possibility that there are differences in GDNF signaling in afferents innervating different tissue types, and even in deep tissues such as bone and muscle.
While neurturin applied exogenously to the mouse hindpaw induces thermal hypersensitivity assayed using Hargreaves behavioral testing apparatus (Malin et al., 2006), there is no previous direct evidence that it causes physiological activation or sensitization of peripheral nociceptors in vivo. One study (Malin et al., 2006) has shown that neurturin potentiates calcium responses to capsaicin in dissociated peripheral sensory neurons. Other studies have used transgenic approaches to implicate neurturin/GFRα2 signaling in pain. Knockout of GFRα2 alters heat, but not mechanical responses, in dissociated sensory neurons (Stucky et al., 2002) and reduces formalin-induced inflammatory pain behaviors without altering responses to mechanical stimulation (Lindfors et al., 2006). Overexpression of neurturin in skin increases behavioral responsiveness to mechanical stimulation (Wang et al., 2013). It also alters the response of A-fiber nociceptors to mechanical stimulation and polymodal C-fiber afferents to thermal stimulation (Jankowski et al., 2017). However, transgenic overexpression of neurturin may result in altered responses through the switching of neuronal phenotypes during development. Our study revealed that exogenous neurturin applied acutely to the bone marrow activates and increases the sensitivity of bone afferent neurons to noxious mechanical stimulation. The changes we observed are too fast to be accounted for by phenotypic switching. Importantly, both GDNF and neurturin applied exogenously to bone marrow produce pain behavior in response to weight bearing. The early response we have observed coincides with a rapid and acute change in mechanical sensitivity of Aδ bone afferent neurons induced by both GDNF and neurturin, suggesting that Aδ bone afferent neurons contribute to the acute phase of GDNF- and neurturin-induced bone pain.
At the cellular level, changes in expression profiles of ion channels and receptors and/or their internalization and retrograde transport to the soma of peripheral nociceptors can contribute to peripheral sensitization (Ji et al., 2002; Amaya et al., 2004; Elitt et al., 2006). These changes are important to understand as they provide targets for the development of novel therapeutics to treat persistent pain of the sort experienced in many bony pathologies. However, we failed to show changes in the expression or retrograde transport of GFRα1 or GFRα2 in peripheral bone afferent neurons in the present study, or other pain-signaling molecules (including TrkA, TRPV1, Nav1.8, and Nav1.9) in our previous studies (Nencini et al., 2017). This suggests that these mechanisms do not contribute to inflammatory bone pain, at least in the model we have used in our study.
Inflammatory bone pain involves activation and sensitization of peptidergic, NGF-sensitive neurons via artemin/GFRα3 signaling pathways
Almost all GFRα3-expressing neurons in the mouse and rat DRGs are NGF-sensitive peptidergic neurons (Orozco et al., 2001; Elitt et al., 2006; Malin et al., 2006). There is substantial literature that documents the innervation of bone by NGF-sensitive sensory neurons (Jimenez-Andrade et al., 2010; Castañeda-Corral et al., 2011; Mantyh, 2014), and this has formed the impetus for the development of NGF sequestration therapy to treat bone pain. Our data show that GFRα3 is expressed in a greater proportion (∼40%) of bone afferent neurons than GFRα1 and GFRα2, and that GFRα3 expressing bone afferent neurons almost always coexpress TrkA and are therefore likely to be NGF-sensitive peptidergic nociceptors. Thus, the GFRα3-expressing bone afferent neurons are likely the same neurons that respond to NGF and NGF sequestration therapy.
Intraplantar injection of recombinant artemin induces mechanical and heat hyperalgesia within 4 h of the injection in mice (Malin et al., 2006; Thornton et al., 2013), and both mechanical and thermal hyperalgesia can persist for days if repeated injections are made (Ikeda-Miyagawa et al., 2015). Here we have shown that exogenous artemin applied directly to bone also produces pain behavior and sensitizes Aδ bone afferent neurons to mechanical stimulation, having effects that begin within minutes and last for up to half an hour, a much shorter time course than we observed for sensitization of bone afferent neurons by GDNF and neurturin. In addition, we have also shown that exogenous application of artemin induces rapid and transient increases in the activity of both C and Aδ bone afferent neurons. Thus, the presence of artemin at peripheral nerve terminals significantly enhances the activity and sensitivity of bone afferent neurons and contributes to bone pain.
The expression of GFRα3 mRNA is increased in the lumbar DRG subsequent to CFA-induced inflammation of the hindpaw (Malin et al., 2006), and in the trigeminal ganglia in an animal model of migraine (Shang et al., 2016). However, we did not find any change in the proportion of bone afferent neurons that expressed GFRα3 protein or in the intensity of labeling for GFRα3 in these neurons in the present study. These findings suggest that the upregulation of GFRα3 expression and/or retrograde transport of artemin/GFRa3 does not occur in the CFA-induced inflammatory bone pain model used in this study.
Sequestration of neurturin and artemin to treat inflammatory bone pain
We have provided the first evidence that the sequestration of neurturin may help to resolve inflammatory pain. Our findings suggest that this is at least relevant to the induction of inflammatory bone pain, because coadministration of the antibody used to sequester neurturin with the CFA used to induce inflammatory bone pain prevents the development of pain behavior. Interestingly, sequestration of GDNF did not prevent the induction of inflammation-induced pain behavior in our study. This is consistent with findings that GDNF sequestration affects the maintenance but not the induction of delayed onset muscle pain (Murase et al., 2013), suggesting that GDNF may contribute to maintenance, but not induction of pain in deep tissues. Antibody sequestration of artemin transiently reduces CFA-induced mechanical hyperalgesia when applied subcutaneously 24 h after the inflammatory agent is administered, suggesting endogenous artemin is also important for maintenance of inflammatory pain in skin (Thornton et al., 2013). In addition, anti-artemin therapy has been shown to block the development of, and reverse already established bladder pain (DeBerry et al., 2015). We have now also shown that sequestering artemin prevents the development of inflammatory bone pain, providing evidence that endogenous artemin is also involved in the induction of inflammatory pain from bone. These findings for both neurturin and artemin provide proof of principle that sequestration may constitute a therapeutic approach relevant to treating inflammatory bone pain.
Conclusions
Our findings show that GFL signaling pathways are involved in the pathogenesis of bone pain and could be targets for pharmacological manipulations to treat it. We have shown that inflammatory bone pain involves the activation and sensitization of NGF-sensitive peptidergic neurons via artemin/GFRα3 signaling pathways, and that sequestering artemin might be useful to prevent inflammatory bone pain derived from the activation of the NGF-sensitive bone afferent neurons. We have also shown that inflammatory bone pain involves the activation and sensitization of nonpeptidergic neurons via GDNF/GFRα1 and neurturin/GFRα2 signaling pathways, and that the sequestration of neurturin, but not GDNF, might be useful to treat inflammatory bone pain derived from the activation of GDNF-sensitive nonpeptidergic bone afferent neurons. These findings provide proof-of-principle that both neurturin and artemin sequestration may constitute a therapeutic approach relevant to treating inflammatory bone pain.
Footnotes
This work was supported by funding from the Australian National Health and Medical Research Council, a Clive and Vera Ramaciotti Foundation Establishment Grant, and a Medical Research and Technology Grant from Australian and New Zealand Charitable Trustees. We thank Associate Professor James Brock for his comments on the manuscript.
The authors declare no competing financial interests.
References
- Alamri A, Bron R, Brock JA, Ivanusic JJ (2015) Transient receptor potential cation channel subfamily V member 1 expressing corneal sensory neurons can be subdivided into at least three subpopulations. Front Neuroanat 9:71. 10.3389/fnana.2015.00071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez P, Chen X, Bogen O, Green PG, Levine JD (2012) IB4(+) nociceptors mediate persistent muscle pain induced by GDNF. J Neurophysiol 108:2545–2553. 10.1152/jn.00576.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaya F, Shimosato G, Nagano M, Ueda M, Hashimoto S, Tanaka Y, Suzuki H, Tanaka M (2004) NGF and GDNF differentially regulate TRPV1 expression that contributes to development of inflammatory thermal hyperalgesia. Eur J Neurosci 20:2303–2310. 10.1111/j.1460-9568.2004.03701.x [DOI] [PubMed] [Google Scholar]
- Bennett A. (1988) The role of biochemical mediators in peripheral nociception and bone pain. Cancer Surv 7:55–67. [PubMed] [Google Scholar]
- Bennett DL, Michael GJ, Ramachandran N, Munson JB, Averill S, Yan Q, McMahon SB, Priestley JV (1998) A distinct subgroup of small DRG cells express GDNF receptor components and GDNF is protective for these neurons after nerve injury. J Neurosci 18:3059–3072. 10.1523/JNEUROSCI.18-08-03059.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenbaum F. (2013) Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage 21:16–21. 10.1016/j.joca.2012.11.012 [DOI] [PubMed] [Google Scholar]
- Bespalov MM, Saarma M (2007) GDNF family receptor complexes are emerging drug targets. Trends Pharmacol Sci 28:68–74. 10.1016/j.tips.2006.12.005 [DOI] [PubMed] [Google Scholar]
- Bjurholm A, Kreicbergs A, Brodin E, Schultzberg M (1988) Substance P- and CGRP-immunoreactive nerves in bone. Peptides 9:165–171. 10.1016/0196-9781(88)90023-X [DOI] [PubMed] [Google Scholar]
- Bogen O, Joseph EK, Chen X, Levine JD (2008) GDNF hyperalgesia is mediated by PLCgamma, MAPK/ERK, PI3K, CDK5 and Src family kinase signaling and dependent on the IB4-binding protein versican. Eur J Neurosci 28:12–19. 10.1111/j.1460-9568.2008.06308.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bove SE, Flatters SJ, Inglis JJ, Mantyh PW (2009) New advances in musculoskeletal pain. Brain Res Rev 60:187–201. 10.1016/j.brainresrev.2008.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castañeda-Corral G, Jimenez-Andrade JM, Bloom AP, Taylor RN, Mantyh WG, Kaczmarska MJ, Ghilardi JR, Mantyh PW (2011) The majority of myelinated and unmyelinated sensory nerve fibers that innervate bone express the tropomyosin receptor kinase A. Neuroscience 178:196–207. 10.1016/j.neuroscience.2011.01.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrastil J, Sampson C, Jones KB, Higgins TF (2013) Postoperative opioid administration inhibits bone healing in an animal model. Clin Orthop Relat Res 471:4076–4081. 10.1007/s11999-013-3232-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerry JJ, Saloman JL, Dragoo BK, Albers KM, Davis BM (2015) Artemin immunotherapy is effective in preventing and reversing cystitis-induced bladder hyperalgesia via TRPA1 regulation. J Pain 16:628–636. 10.1016/j.jpain.2015.03.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolatshad NF, Saffrey MJ (2007) Differential expression of glial cell line-derived neurotrophic factor family receptor alpha-2 isoforms in rat urinary bladder and intestine. Neurosci Lett 415:215–218. 10.1016/j.neulet.2007.01.024 [DOI] [PubMed] [Google Scholar]
- Elitt CM, McIlwrath SL, Lawson JJ, Malin SA, Molliver DC, Cornuet PK, Koerber HR, Davis BM, Albers KM (2006) Artemin overexpression in skin enhances expression of TRPV1 and TRPA1 in cutaneous sensory neurons and leads to behavioral sensitivity to heat and cold. J Neurosci 26:8578–8587. 10.1523/JNEUROSCI.2185-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everaerts W, Sepúlveda MR, Gevaert T, Roskams T, Nilius B, De Ridder D (2009) Where is TRPV1 expressed in the bladder, do we see the real channel? Naunyn Schmiedebergs Arch Pharmacol 379:421–425. 10.1007/s00210-008-0391-7 [DOI] [PubMed] [Google Scholar]
- Furusawa S. (1970) A neurophysiological study on the sensibility of the bone marrow. Nippon Seikeigeka Gakkai Zasshi 44:365–370. [PubMed] [Google Scholar]
- George L, Kasemeier-Kulesa J, Nelson BR, Koyano-Nakagawa N, Lefcort F (2010) Patterned assembly and neurogenesis in the chick dorsal root ganglion. J Comp Neurol 518:405–422. 10.1002/cne.22248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldring MB. (2000) Osteoarthritis and cartilage: the role of cytokines. Curr Rheumatol Rep 2:459–465. 10.1007/s11926-000-0021-y [DOI] [PubMed] [Google Scholar]
- Haegerstam GA. (2001) Pathophysiology of bone pain: a review. Acta Orthop Scand 72:308–317. 10.1080/00016470152846682 [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Ito T, Fukumitsu H, Nomoto H, Furukawa Y, Furukawa S (2005) Stimulation of production of glial cell line-derived neurotrophic factor and nitric oxide by lipopolysaccharide with different dose-responsiveness in cultured rat macrophages. Biomed Res 26:223–229. 10.2220/biomedres.26.223 [DOI] [PubMed] [Google Scholar]
- Hochberg MC. (2015) Serious joint-related adverse events in randomized controlled trials of anti-nerve growth factor monoclonal antibodies. Osteoarthritis Cartilage 23 [Suppl. 1]:S18–S21. 10.1016/j.joca.2014.10.005 [DOI] [PubMed] [Google Scholar]
- Ikeda-Miyagawa Y, Kobayashi K, Yamanaka H, Okubo M, Wang S, Dai Y, Yagi H, Hirose M, Noguchi K (2015) Peripherally increased artemin is a key regulator of TRPA1/V1 expression in primary afferent neurons. Mol Pain 11:8. 10.1186/s12990-015-0004-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanusic JJ. (2009) Size, neurochemistry, and segmental distribution of sensory neurons innervating the rat tibia. J Comp Neurol 517:276–283. 10.1002/cne.22160 [DOI] [PubMed] [Google Scholar]
- Ivanusic JJ, Mahns DA, Sahai V, Rowe MJ (2006) Absence of large-diameter sensory fibres in a nerve to the cat humerus. J Anat 208:251–255. 10.1111/j.1469-7580.2006.00519.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowski MP, Baumbauer KM, Wang T, Albers KM, Davis BM, Koerber HR (2017) Cutaneous neurturin overexpression alters mechanical, thermal, and cold responsiveness in physiologically identified primary afferents. J Neurophysiol 117:1258–1265. 10.1152/jn.00731.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeon SM, Lee KM, Park ES, Jeon YH, Cho HJ (2008) Monocyte chemoattractant protein-1 immunoreactivity in sensory ganglia and hindpaw after adjuvant injection. Neuroreport 19:183–186. 10.1097/WNR.0b013e3282f3c781 [DOI] [PubMed] [Google Scholar]
- Ji RR, Samad TA, Jin SX, Schmoll R, Woolf CJ (2002) p38 MAPK activation by NGF in primary sensory neurons after inflammation increases TRPV1 levels and maintains heat hyperalgesia. Neuron 36:57–68. 10.1016/S0896-6273(02)00908-X [DOI] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Martin CD, Koewler NJ, Freeman KT, Sullivan LJ, Halvorson KG, Barthold CM, Peters CM, Buus RJ, Ghilardi JR, Lewis JL, Kuskowski MA, Mantyh PW (2007) Nerve growth factor sequestering therapy attenuates non-malignant skeletal pain following fracture. Pain 133:183–196. 10.1016/j.pain.2007.06.016 [DOI] [PubMed] [Google Scholar]
- Jimenez-Andrade JM, Mantyh WG, Bloom AP, Xu H, Ferng AS, Dussor G, Vanderah TW, Mantyh PW (2010) A phenotypically restricted set of primary afferent nerve fibers innervate the bone versus skin: therapeutic opportunity for treating skeletal pain. Bone 46:306–313. 10.1016/j.bone.2009.09.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keast JR, Forrest SL, Osborne PB (2010) Sciatic nerve injury in adult rats causes distinct changes in the central projections of sensory neurons expressing different glial cell line-derived neurotrophic factor family receptors. J Comp Neurol 518:3024–3045. 10.1002/cne.22378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koewler NJ, Freeman KT, Buus RJ, Herrera MB, Jimenez-Andrade JM, Ghilardi JR, Peters CM, Sullivan LJ, Kuskowski MA, Lewis JL, Mantyh PW (2007) Effects of a monoclonal antibody raised against nerve growth factor on skeletal pain and bone healing after fracture of the C57BL/6J mouse femur. J Bone Miner Res 22:1732–1742. 10.1359/jbmr.070711 [DOI] [PubMed] [Google Scholar]
- Kumar V, Mahal BA (2012) NGF—the TrkA to successful pain treatment. J Pain Res 5:279–287. 10.2147/JPR.S33408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefcort F, Clary DO, Rusoff AC, Reichardt LF (1996) Inhibition of the NT-3 receptor TrkC, early in chick embryogenesis, results in severe reductions in multiple neuronal subpopulations in the dorsal root ganglia. J Neurosci 16:3704–3713. 10.1523/JNEUROSCI.16-11-03704.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindfors PH, Võikar V, Rossi J, Airaksinen MS (2006) Deficient nonpeptidergic epidermis innervation and reduced inflammatory pain in glial cell line-derived neurotrophic factor family receptor α2 knock-out mice. J Neurosci 26:1953–1960. 10.1523/JNEUROSCI.4065-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsay RM, Yancopoulos GD (1996) GDNF in a bind with known orphan: accessory implicated in new twist. Neuron 17:571–574. 10.1016/S0896-6273(00)80189-0 [DOI] [PubMed] [Google Scholar]
- Ma QP, Woolf CJ (1997) The progressive tactile hyperalgesia induced by peripheral inflammation is nerve growth factor dependent. Neuroreport 8:807–810. 10.1097/00001756-199703030-00001 [DOI] [PubMed] [Google Scholar]
- Mach DB, Rogers SD, Sabino MC, Luger NM, Schwei MJ, Pomonis JD, Keyser CP, Clohisy DR, Adams DJ, O'Leary P, Mantyh PW (2002) Origins of skeletal pain: sensory and sympathetic innervation of the mouse femur. Neuroscience 113:155–166. 10.1016/S0306-4522(02)00165-3 [DOI] [PubMed] [Google Scholar]
- Malin SA, Davis BM (2008) Postnatal roles of glial cell line-derived neurotrophic factor family members in nociceptors plasticity. Sheng Li Xue Bao 60:571–578. [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Molliver DC, Koerber HR, Cornuet P, Frye R, Albers KM, Davis BM (2006) Glial cell line-derived neurotrophic factor family members sensitize nociceptors in vitro and produce thermal hyperalgesia in vivo. J Neurosci 26:8588–8599. 10.1523/JNEUROSCI.1726-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin S, Molliver D, Christianson JA, Schwartz ES, Cornuet P, Albers KM, Davis BM (2011) TRPV1 and TRPA1 function and modulation are target tissue dependent. J Neurosci 31:10516–10528. 10.1523/JNEUROSCI.2992-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantyh PW. (2004) A mechanism-based understanding of bone cancer pain. Novartis Found Symp 261:194–214. [PubMed] [Google Scholar]
- Mantyh PW. (2014) The neurobiology of skeletal pain. Eur J Neurosci 39:508–519. 10.1111/ejn.12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molliver DC, Radeke MJ, Feinstein SC, Snider WD (1995) Presence or absence of TrkA protein distinguishes subsets of small sensory neurons with unique cytochemical characteristics and dorsal horn projections. J Comp Neurol 361:404–416. 10.1002/cne.903610305 [DOI] [PubMed] [Google Scholar]
- Murase S, Terazawa E, Hirate K, Yamanaka H, Kanda H, Noguchi K, Ota H, Queme F, Taguchi T, Mizumura K (2013) Upregulated glial cell line-derived neurotrophic factor through cyclooxygenase-2 activation in the muscle is required for mechanical hyperalgesia after exercise in rats. J Physiol 591:3035–3048. 10.1113/jphysiol.2012.249235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase S, Kato K, Taguchi T, Mizumura K (2014) Glial cell line-derived neurotrophic factor sensitized the mechanical response of muscular thin-fibre afferents in rats. Eur J Pain 18:629–638. 10.1002/j.1532-2149.2013.00411.x [DOI] [PubMed] [Google Scholar]
- Nencini S, Ivanusic J (2017) Mechanically sensitive Aδ nociceptors that innervate bone marrow respond to changes in intra-osseous pressure. J Physiol 595:4399–4415. 10.1113/JP273877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nencini S, Ringuet M, Kim DH, Chen YJ, Greenhill C, Ivanusic JJ (2017) Mechanisms of nerve growth factor signaling in bone nociceptors and in an animal model of inflammatory bone pain. Mol Pain 13:1744806917697011. 10.1177/1744806917697011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okragly AJ, Niles AL, Saban R, Schmidt D, Hoffman RL, Warner TF, Moon TD, Uehling DT, Haak-Frendscho M (1999) Elevated tryptase, nerve growth factor, neurotrophin-3 and glial cell line-derived neurotrophic factor levels in the urine of interstitial cystitis and bladder cancer patients. J Urol 161:438–441. 10.1016/S0022-5347(01)61915-3 [DOI] [PubMed] [Google Scholar]
- Orozco OE, Walus L, Sah DW, Pepinsky RB, Sanicola M (2001) GFRalpha3 is expressed predominantly in nociceptive sensory neurons. Eur J Neurosci 13:2177–2182. 10.1046/j.0953-816x.2001.01596.x [DOI] [PubMed] [Google Scholar]
- Peleshok JC, Ribeiro-da-Silva A (2011) Delayed reinnervation by nonpeptidergic nociceptive afferents of the glabrous skin of the rat hindpaw in a neuropathic pain model. J Comp Neurol 519:49–63. 10.1002/cne.22500 [DOI] [PubMed] [Google Scholar]
- Pierchala BA, Milbrandt J, Johnson EM Jr (2006) Glial cell line-derived neurotrophic factor-dependent recruitment of ret into lipid rafts enhances signaling by partitioning ret from proteasome-dependent degradation. J Neurosci 26:2777–2787. 10.1523/JNEUROSCI.3420-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pountos I, Georgouli T, Calori GM, Giannoudis PV (2012) Do nonsteroidal anti-inflammatory drugs affect bone healing? A critical analysis. Sci World J 2012:606404 10.1100/2012/606404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakowicz WP, Staples CS, Milbrandt J, Brunstrom JE, Johnson EM Jr (2002) Glial cell line-derived neurotrophic factor promotes the survival of early postnatal spinal motor neurons in the lateral and medial motor columns in slice culture. J Neurosci 22:3953–3962. 10.1523/JNEUROSCI.22-10-03953.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah DW, Ossipov MH, Rossomando A, Silvian L, Porreca F (2005) New approaches for the treatment of pain: the GDNF family of neurotrophic growth factors. Curr Top Med Chem 5:577–583. 10.2174/1568026054367593 [DOI] [PubMed] [Google Scholar]
- Schröpel A, von Schack D, Dechant G, Barde YA (1995) Early expression of the nerve growth factor receptor ctrkA in chick sympathetic and sensory ganglia. Mol Cell Neurosci 6:544–566. 10.1006/mcne.1995.0006 [DOI] [PubMed] [Google Scholar]
- Seidel MF, Wise BL, Lane NE (2013) Nerve growth factor: an update on the science and therapy. Osteoarthritis Cartilage 21:1223–1228. 10.1016/j.joca.2013.06.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seike W. (1976) Electrophysiological and histological studies on the sensibility of the bone marrow nerve terminal. Yonago Acta Med 20:192–211. [PubMed] [Google Scholar]
- Sevcik MA, Ghilardi JR, Peters CM, Lindsay TH, Halvorson KG, Jonas BM, Kubota K, Kuskowski MA, Boustany L, Shelton DL, Mantyh PW (2005) Anti-NGF therapy profoundly reduces bone cancer pain and the accompanying increase in markers of peripheral and central sensitization. Pain 115:128–141. 10.1016/j.pain.2005.02.022 [DOI] [PubMed] [Google Scholar]
- Shang HQ, Wang Y, Mao YY, Kong LG, Sun GY, Xu L, Zhang DG, Han YC, Li JF, Wang HB, Fan ZM (2016) Expression of artemin and GFRα3 in an animal model of migraine: possible role in the pathogenesis of this disorder. J Headache Pain 17:81. 10.1186/s10194-016-0673-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snider WD, McMahon SB (1998) Tackling pain at the source: new ideas about nociceptors. Neuron 20:629–632. 10.1016/S0896-6273(00)81003-X [DOI] [PubMed] [Google Scholar]
- Starr AM, Wessely MA, Albastaki U, Pierre-Jerome C, Kettner NW (2008) Bone marrow edema: pathophysiology, differential diagnosis, and imaging. Acta Radiol 49:771–786. 10.1080/02841850802161023 [DOI] [PubMed] [Google Scholar]
- Stucky CL, Rossi J, Airaksinen MS, Lewin GR (2002) GFR alpha2/neurturin signalling regulates noxious heat transduction in isolectin B4-binding mouse sensory neurons. J Physiol 545:43–50. 10.1113/jphysiol.2002.027656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton P, Hatcher JP, Robinson I, Sargent B, Franzén B, Martino G, Kitching L, Glover CP, Anderson D, Forsmo-Bruce H, Low CP, Cusdin F, Dosanjh B, Williams W, Steffen AC, Thompson S, Eklund M, Lloyd C, Chessell I, Hughes J (2013) Artemin-GFRalpha3 interactions partially contribute to acute inflammatory hypersensitivity. Neurosci Lett 545:23–28. 10.1016/j.neulet.2013.04.007 [DOI] [PubMed] [Google Scholar]
- Urch C. (2004) The pathophysiology of cancer-induced bone pain: current understanding. Palliat Med 18:267–274. 10.1191/0269216304pm887ra [DOI] [PubMed] [Google Scholar]
- Võikar V, Rossi J, Rauvala H, Airaksinen MS (2004) Impaired behavioural flexibility and memory in mice lacking GDNF family receptor alpha2. Eur J Neurosci 20:308–312. 10.1111/j.1460-9568.2004.03475.x [DOI] [PubMed] [Google Scholar]
- von Boyen GB, Steinkamp M, Geerling I, Reinshagen M, Schäfer KH, Adler G, Kirsch J (2006) Proinflammatory cytokines induce neurotrophic factor expression in enteric glia: a key to the regulation of epithelial apoptosis in Crohn's disease. Inflamm Bowel Dis 12:346–354. 10.1097/01.MIB.0000219350.72483.44 [DOI] [PubMed] [Google Scholar]
- Wang T, Jing X, DeBerry JJ, Schwartz ES, Molliver DC, Albers KM, Davis BM (2013) Neurturin overexpression in skin enhances expression of TRPM8 in cutaneous sensory neurons and leads to behavioral sensitivity to cool and menthol. J Neurosci 33:2060–2070. 10.1523/JNEUROSCI.4012-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanigasekara Y, Airaksinen MS, Heuckeroth RO, Milbrandt J, Keast JR (2004) Neurturin signalling via GFRalpha2 is essential for innervation of glandular but not muscle targets of sacral parasympathetic ganglion neurons. Mol Cell Neurosci 25:288–300. 10.1016/j.mcn.2003.10.019 [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Safieh-Garabedian B, Ma QP, Crilly P, Winter J (1994) Nerve growth factor contributes to the generation of inflammatory sensory hypersensitivity. Neuroscience 62:327–331. 10.1016/0306-4522(94)90366-2 [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Allchorne A, Safieh-Garabedian B, Poole S (1997) Cytokines, nerve growth factor and inflammatory hyperalgesia: the contribution of tumour necrosis factor alpha. Br J Pharmacol 121:417–424. 10.1038/sj.bjp.0701148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Hoff AO, Wimalawansa SJ, Cote GJ, Gagel RF, Westlund KN (2001) Arthritic calcitonin/alpha calcitonin gene-related peptide knockout mice have reduced nociceptive hypersensitivity. Pain 89:265–273. 10.1016/S0304-3959(00)00378-X [DOI] [PubMed] [Google Scholar]
- Zwick M, Davis BM, Woodbury CJ, Burkett JN, Koerber HR, Simpson JF, Albers KM (2002) Glial cell line-derived neurotrophic factor is a survival factor for isolectin B4-positive, but not vanilloid receptor 1-positive, neurons in the mouse. J Neurosci 22:4057–4065. 10.1523/JNEUROSCI.22-10-04057.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]