

Abstract
Inositol hexakisphosphate kinases (IP6Ks) regulate various biological processes. Among pyrophosphates generated by IP6Ks, diphosphoinositol pentakisphosphate (IP7), and bis-diphosphoinositol tetrakisphosphate have been extensively characterized. IP7 is produced in mammals by a family of inositol hexakisphosphate kinases, IP6K1, IP6K2, and IP6K3, which have distinct biological functions. We report that IP6K2 binds protein 4.1.N with high affinity and specificity. Nuclear translocation of 4.1N, which is required for its principal functions, is dependent on IP6K2. Both of these proteins are highly expressed in granule cells of the cerebellum where their interaction regulates Purkinje cell morphology and cerebellar synapses. The deletion of IP6K2 in male/female mice elicits substantial defects in synaptic influences of granule cells upon Purkinje cells as well as notable impairment of locomotor function. Moreover, the disruption of IP6K2–4.1N interactions impairs cell viability. Thus, IP6K2 and its interaction with 4.1N appear to be major determinants of cerebellar disposition and psychomotor behavior.
SIGNIFICANCE STATEMENT Inositol phosphates are produced by a family of inositol hexakisphosphate kinases (IP6Ks)—IP6K1, IP6K2, and IP6K3. Of these, the physiological roles of IP6K2 in the brain have been least characterized. In the present study, we report that IP6K2 binds selectively to the neuronal protein 4.1N. Both of these proteins are highly expressed in granule cells of the cerebellum. Using IP6K2 knock-out (KO) mice, we establish that IP6K2–4.1N interactions in granule cells regulate Purkinje cell morphology, the viability of cerebellar neurons, and psychomotor behavior.
Keywords: 4.1N, cerebellum, IP6K2, synapse
Introduction
Inositol phosphates act as molecular messengers regulating diverse cellular functions throughout the animal and plant kingdoms. Inositol 1,4,5-trisphosphate (IP3), which releases intracellular calcium, has been studied in depth (Irvine and Schell, 2001; Saiardi et al., 2002, 2005; Luo et al., 2003; York et al., 2005). Inositol pyrophosphates are derivatives that contain an energetic pyrophosphate bond analogous to ATP. Best characterized among these are diphosphoinositol pentakisphosphate (IP7) whose 5′-diphosphate has energetic pyrophosphate properties (Saiardi et al., 1999; Chakraborty et al., 2011a). While other isomers of IP7 have been reported, the predominant mammalian form contains a 5′ diphosphate pyrophosphate moiety and is known as diphosphoinositol pentakisphosphate (5-PP-IP5), which is also designated as IP7. IP7 is generated by three inositol hexakisphosphate kinases (IP6Ks) that are the products of three separate genes (Shears, 2007). Another isomer of IP7, 3-PP-IP5, is synthesized by a distinct enzyme, Vip1, and regulates cell shape, cell growth, and phosphate disposition of yeast (Lee et al., 2007; Mulugu et al., 2007).
Functions for the different forms of IP6K vary. Thus, IP6K1 impacts weight gain and insulin resistance (Bhandari et al., 2008). IP7 formed by IP6K1 inactivates the protein kinase Akt, enhances glycogen synthase kinase β and inhibits the mammalian target of rapamycin pathway (Chakraborty et al., 2010). IP6K2 was first reported (Morrison et al., 2001) as a proapoptotic gene so that mice with deleted IP6K2 are resistant to ionizing radiation and display enhanced tumor formation. Cell survival associated with heat shock protein 90 involves its binding IP6K2 to inhibit catalytic activity so that mutations blocking this binding activate IP6K2 and elicit cell death (Chakraborty et al., 2008). The death of cells associated with activated IP6K2 is predominately mediated by p53. IP6K2 acts by binding directly to p53 and decreasing the expression of proarrest gene targets such as the cyclin-dependent kinase inhibitor p21 (Koldobskiy et al., 2010). Casein kinase-2, which elicits cell survival by phosphorylating and degrading IP6K2 is upregulated during tumor formation (Chakraborty et al., 2011b). IP6K3 regulates the disposition of cerebellar Purkinje cells via interactions with spectrin and adducin (Fu et al., 2015).
Heretofore, the biosynthesis of inositol phosphates in the brain has been best characterized for IP6K1 and IP6K3, with negligible investigation of IP6K2. We now report that IP6K2 plays a major role in the disposition of cerebellar granule cells in which it is highly enriched. This pool of IP6K2 regulates Purkinje cell morphology as well as motor coordination. The influence of IP6K2 upon cerebellar disposition involves protein 4.1N, which is also highly expressed in cerebellar granule cells. Protein 4.1N is a neuronal-selective isoform of the erythrocyte membrane cytoskeleton protein 4.1R. The protein 4.1 family comprises a group of cytoskeletal proteins that include 4.1R (erythrocyte), 4.1G (general type), 4.1B (brain type), and 4.1N (neuron type). The proteins are structurally related to erythroid membrane skeletal protein 4.1R, which plays a critical role in determining the morphology and mechanical stability of the red blood cell plasma membrane. The 4.1 proteins are characterized by the presence of three main conserved structural and functional domains. The FERM domain (F, 4.1; E, ezrin; R, radixin; M, moesin), a 30 kDa N-terminal membrane-binding domain, possesses binding sites for the cytoplasmic tails of integral membrane proteins such as band 3, glycophorin C, and CD44 (Parra et al., 2000). The 4.1N protein has previously been shown to mediate the antiproliferative influences of nerve growth factor by binding to the nuclear mitotic apparatus protein (NuMA; Ye et al., 1999).
In the present study, we report the physiologic association of 4.1N and IP6K2, whose disruption impacts cerebellar disposition.
Materials and Methods
Reagents.
Anti-IP6K1, IP6K2, IP6K3, 4.1N, 4.1R, 4.1B, and 4.1G antibodies were purchased from Sigma-Aldrich. VGlut1 and VGlut2 antibodies were purchased from Millipore. Antibodies against calbindin, calretinin, Arc, BDNF, Ras-related protein 5 (Rab 5), sorting nexin 27 (SNX-27), Rheb, Fos-B, c-Fos, nerve growth factor (NGF), and synaptophysin as well as rabbit and mouse IgG antibodies, were procured from Santa Cruz Biotechnology. Alexa Fluor 488 goat anti-rabbit and anti-mouse and Alexa Fluor 568 goat anti-mouse and anti-rabbit IgG were purchased from Life Technologies. IP6K2 shRNA, 4.1N, and control shRNA plasmids were from Santa Cruz Biotechnology. All other chemicals used were either purchased from Sigma-Aldrich or Thermo Fisher Scientific.
Animals.
Eight-week-old C57BL/6 male/female mice were used for all the animal-based experiments. Animal breeding and procedures were conducted in strict accordance with the NIH Guide for the Care and Use of Laboratory Animals. Animal experiments were approved by the Johns Hopkins University Animal Care and Use Committee. Animals were kept on a 12 h light/dark cycle and were provided food and water ad libitum.
Coimmunoprecipitation.
Cell lysates or mouse brain/cerebellar tissue lysates were homogenized in lysis buffer (50 mm Tris, pH 7.4, 150 mm NaCl, 0.5% Triton X-100, 4 μg/ml leupeptin, 2 μg/ml, 2 μg/ml chymotrypsin, 2 μg/ml pepstatin, 1 mm PMSF) on ice for 10 min. Equal amounts of protein were incubated with the respective antibodies for 4 h and immunoprecipitated using protein AG agarose beads. Beads were washed (3×) with lysis buffer for 5 min each. Coimmunoprecipitates were then resolved by SDS-PAGE and immunoblotted through Western blotting. Anti-IgG antibodies were used as controls. Resolved bands were also silver stained (using Silver Stain for Mass Spectrometry, Pierce) and analyzed through liquid chromatography-tandem mass spectrometry (LC-MS/MS).
LC-MS/MS analysis.
In-gel proteins were trichloroacetic acid/acetone precipitated and proteolyzed with trypsin. In-solution trypsin digestion was performed as per standard protocols (Nagpal and Panda, 2015). Peptides were desalted on Oasis u-HLB plates (Waters) and eluted with 60% acetonitrile/0.1% trifluoroacetic acid and dried. Desalted, digested peptides were analyzed by LC-MS/MS on QExactive Plus using a 60 min total gradient, resolution at 200 m/z, 70,000 for full MS and 35,000 for MS/MS, and higher-energy collisional dissociation of 27. MS/MS spectra were processed by Proteome Discoverer (version 1.4; Thermo Fisher Scientific) in three ways, using the following three nodes: common; Xtract (spectra are extracted, charge state deconvoluted, and deisotoped using Xtract option, at a resolution of 50,000 at 400 Da); and an MS2 processor. MS2 spectra from three nodes were analyzed with Mascot version 2.5.1 (Matrix Science) with a concatenated decoy database, specifying mammalian species, trypsin as an enzyme used for cleavage, precursor mass tolerance of 10 ppm, and fragment mass tolerance of 0.02 Da.
Cell culture and transfection conditions.
N2A cells were grown in a humid atmosphere of 5% CO2 at 37°C in DMEM supplemented with 10% FBS, l-glutamine (2 mm), penicillin (100 U/ml), and streptomycin (100 μg/ml). PC-12 cells were maintained in DMEM supplemented with 10% FBS and 5% horse serum. Differentiation was initiated by the addition of 50 ng/ml NGF with culture medium changed to DMEM containing 2% horse serum and 1% FBS. Cells were transfected with Lipofectamine LTX and incubated for 10–12 h before the transfection medium was replaced with serum-containing medium.
Double immunofluorescence staining and confocal microscopy. Cells were grown in chamber slides, fixed using 4% paraformaldehyde (PFA), and permeabilized by incubation with 0.01% Triton X-100 at 4°C for 5 min (Nagpal et al., 2013). Nonspecific sites were then blocked by incubation with 5% goat serum. Subsequently, the cells were incubated with the primary antibodies (1:100) following which the cells were stained with respective fluorescent secondary antibodies (1:300). Nuclei were counterstained with DAPI. Images were captured and analyzed using a confocal microscope (LSM 700, Zeiss).
For immunofluorescence staining with brain sections, mice were perfused with 4% PFA, and brain slices were cut in sagittal sections using a cryostat (Leica Biosystems). The sections were treated as described above.
Golgi staining and hematoxylin and eosin staining.
The 150-μm-thick brain sections were cut using a cryostat at −20°C. FD Rapid GolgiStain Kit (FD Neurotechnologies) was used to perform the Golgi staining. The hematoxylin and eosin (H&E) stain protocol followed was as described before (Chakraborty et al., 2008).
Electron microscopy.
For electron microscopy, mice were perfused with 2% glutaraldehyde and 2% PFA, and the samples were embedded into EPON resin. Sections were then stained with uranyl acetate and lead citrate and viewed using transmission electron microscopy. Synapses were counted from at least six different fields of view.
Cell viability assay.
Cell viability was measured using 2,3-Bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide (XTT) assay following protocols supplied by the manufacturer (Thermo Fisher Scientific). N2A cells were seeded in 96-well plates and transfected with IP6K2 and K2 (Δ202-261) shRNA. Absorbance was measured at 450 nm to determine cell viability.
In vivo cell proliferation assay.
To compare cell proliferation in wild-type (WT) and IP6K2 knock-out mice, 100 μg of 5-ethynyl-2′-deoxyuridine (EdU) in PBS was injected intraperitoneally into adult mice (8 weeks old) and brain tissues were harvested 96 h after the injection. The brain tissues were then fixed in 4% PFA in PBS at 4°C for 24 h. The tissue was then rinsed in PBS and was processed according to the manufacturer protocols. Brain cerebellar sections were stained using Click-iT EdU imaging kit (Invitrogen) with Alexa Fluor 488 azide for 30 min. The sections were then imaged under a confocal microscope.
Mouse behavioral tests.
The behavior tests were conducted on 8-week-old mice at the Johns Hopkins University Brain Science Institute Behavioral Core.
Rotarod test.
Motor learning and coordination were examined in IP6K2-KO mice by the rotarod test (Rotamex-5, Columbus Instruments). Tests were conducted over a 3 d period where each mouse was given five trials per day and latency to fall off the rotarod (speed as well as time) was measured as it accelerated from 4 to 99 rpm over a 5 min period. Before the start of the test, each mouse was given a habituation trial by placing them on the rotarod rotating at a constant speed of 4 rpm for 10 min. The highest and lowest latencies of the trials were discarded and the average was taken from those remaining.
Open field tests.
General locomotor activity of IP6K2-KO mice was assessed by open field analysis (Photobeam Activity System, San Diego Instruments). Mice were placed in the center of an enclosed acrylic chamber and allowed to move freely for 20 min. The total peripheral and central activities of the mice were recorded and analyzed.
Gait analysis.
The CatWalk XT System (Noldus Information Technologies) was used to test or study walking patterns of IP6K2-KO mice through already established protocols (Chakraborty et al., 2008).
Image quantification and statistical analysis.
Images were quantified with ImageJ software. For Western blots, the expression changes of the proteins were evaluated by normalizing their recorded band intensities against their input band (β-actin or GAPDH). Ponceau membrane stain was also used as the loading control for cytosolic fractions, and histone-H3A was used as a loading control for nuclear fraction intensities as shown in the supporting histograms. Data are presented as the mean ± SD from at least three independent experiments. The p values were calculated by two-tailed Student's t test or one-way ANOVA.
Results
IP6K2 binds 4.1N
In an effort to identify proteins that physiologically interact with IP6K2, we immunoprecipitated IP6K2 from brain lysates seeking proteins that coprecipitate. A particularly prominent interactor was protein 4.1N, hereafter designated as 4.1N (Fig. 1A). Supporting an association of 4.1N with IP6K2 was our identification of 17 discrete 4.1N peptides from IP6K2 immunoprecipitates using LC-MS/MS (Fig. 1B, Tab. 1-1).
Figure 1.

IP6K2 binds 4.1N in the brain. A, Silver staining of SDS-PAGE of IP6K2 immunoprecipitates (lane 2) from wild-type mouse brain lysate showed the presence of 4.1N protein (130 kDa) as a potential interacting partner of IP6K2 against IgG (lane1) as a control. B, Seventeen (17) 4.1N peptides (highlighted in yellow) were identified from IP6K2 immunoprecipitates using LC-MS/MS to confirm the identity of the 4.1N protein. C, Coimmunoprecipitates of IP6K2 from WT (lane 2) and IP6K2-knockout (K2-KO; lane 3) mouse brain lysates immunoblotted (IB) with 4.1N antibody confirmed the binding to 4.1N protein against IgG immunoprecipitate (IP) as a control (lane 1). D, Coimmunoprecipitates of IP6K2 blotted against 4.1N in neuronal cells also revealed its association with the 4.1N protein. Lane 1, IgG immunoprecipitate; lane 2, IP6K2 immunoprecipitates from N2A cell lysates; lane 3, IP6K2 immunoprecipitates from PC-12 cell lysates. E, GST-DNA constructs of different fragments of IP6K2 were transfected into N2A cells. Coimmunoprecipitates of such IP6K2 constructs expressing cell lysates were blotted against 4.1N antibody. This revealed the binding site of IP6K2 with 4.1N to be present between residues 202 and 261 of IP6K2. F, Coimmunoprecipitation of IP6K2 from wild-type mouse brain lysates (lane 1) and IP6K2-KO mice brain lysates (lane 2, control) showed the binding of 4.1N and 4.1B to IP6K2. However, 4.1G and 4.1R isoforms of the 4.1 family of proteins did not bind to IP6K2. G, Coimmunoprecipitates of 4.1N from mouse brain lysates were blotted against all three isoforms of IP6K (K1, K2, and K3). Corresponding immunoblots against IgG immunoprecipitates as a control (lane 2) showed selective binding of the 4.1N protein to IP6K2 and not to the IP6K1 or IP6K3 isoforms. H, Full-length IP6K2 and IP6K2 kinase dead mutants (K222A) were transfected into N2A cells. Immunoblots of the lysates from both cells showed confirmatory binding of 4.1N (lane 1) when compared with their IgG controls (lane 2) indicating that the binding of 4.1N to IP6K2 is not dependent on the kinase activity of the latter. Data are representative of three independent experiments performed under identical conditions. 4.1N peptides detected through LC/MS-MS with their respective peptide identification probability, mascot ion score and mascot identity score, Table 1-1.
Seventeen (17) 4.1N peptides detected through LC/MS-MS with their respective peptide identification probability, mascotion score and mascot identity score. Download Table 1-1, DOC file (39.5KB, doc)
We also observed direct binding of IP6K2 to 4.1N (Fig. 1C,D). We examined various fragments of IP6K2, observing binding to 4.1N most robustly with IP6K2 (202–261), whereas negligible binding occurred with IP6K2 (144–201; Fig. 1E).
Subtypes of protein 4.1 have been reported (Peters et al., 1998). 4.1N is largely neuronal, 4.1R is most concentrated in red blood cells, 4.1G is ubiquitously distributed, while 4.1B is most highly expressed in the brain. IP6K2 bound strongly to 4.1N and 4.1B and much less to 4.1R and 4.1G, indicating a relatively selective association with neuronal forms of 4.1N (Fig. 1F). The selective nature of the IP6K2–4.1N association was evident in our failure to detect any binding of 4.1N to IP6K1 or IP6K3 (Fig. 1G).
A mutant form of IP6K2 wherein lysine-222 is transformed to alanine, lacks catalytic activity (Saiardi et al., 2001). IP6K2-K222A as well as wild-type IP6K2 bound to 4.1N (Fig. 1H). Thus, the binding of these two proteins is not dependent upon IP6K2 catalytic activity.
Major functions of 4.1N require its translocation to the nucleus (Ye et al., 1999, 2000). Thus, the antiproliferative action of NGF requires nuclear translocation of 4.1N in association with NuMA (Ye et al., 1999). We wondered whether IP6K2 participates in this nuclear translocation process. In subcellular fractionation of mouse brain homogenates, IP6K2 was equally distributed between nuclear and cytosolic fractions (Fig. 2A,C). In IP6K2-deleted preparations, the proportion of nuclear 4.1N was decreased by ∼60%, indicating that IP6K2 is responsible, at least in part, for the nuclear disposition of 4.1N (Fig. 2B,C). NGF elicited nuclear translocation of 4.1N, which was abolished in IP6K2 knock-out preparations (Fig. 2D,E). NGF also greatly enhanced the nuclear localization of IP6K2 (Fig. 2D,F).
Figure 2.
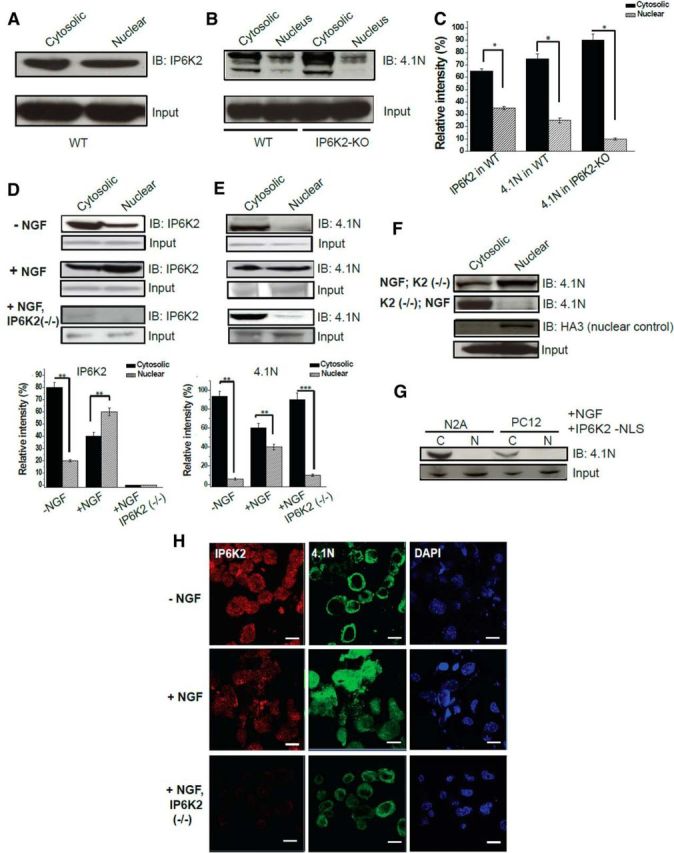
Nuclear translocation of 4.1N requires IP6K2 facilitation. A, Western blots show endogenous expression of IP6K2 in the cytosolic and nuclear fractions of wild-type mouse brain lysates. B, Corresponding comparisons of 4.1N expression in the wild-type brain lysates and IP6K2-KO brain lysates. C, The relative levels of the protein bands in the immunoblot depicted in A and B estimated through normalization against their relevant inputs are shown in the histogram. D, E, Immunoblots of N2A cell lysates demonstrated that both 4.1N as well as IP6K2 translocate into the nucleus of N2A cells in the presence of NGF. However, in the absence of IP6K2, 4.1N failed to transfer into the nucleus of N2A cells. F, Experimental evidence that IP6K2 is a prime facilitator for nuclear translocation of 4.1N along with NGF in N2A cells. G, Immunoblots establish that NLS of IP6K2 is essential for the translocation of 4.1N into the nucleus, as 4.1N fails to translocate into the nucleus when N2A cells are transfected with IP6K2 lacking NLS. H, Double-immunofluorescence staining of N2A cells against IP6K2 (green) and 4.1N (red) reveals significant nuclear translocation of 4.1N in the presence of NGF and no nuclear expression of 4.1N in absence of IP6K2. Scale bar, 10 μm. Data are representative of three independent experiments performed under identical conditions. Data are presented as the mean ± SD: ***p < 0.001, **p < 0.01, *p < 0.05, analyzed by one-way ANOVA.
A critical role for IP6K2 in regulating the disposition of 4.1N was evident in our finding that nuclear translocation of 4.1N was abolished in N2A cells containing IP6K2 that lacks a nuclear localization signal (NLS; Fig. 2G). Double-immunofluorescence staining of N2A cells confirmed the absence of nuclear pools of 4.1N in preparations lacking IP6K2 (Fig. 2H).
To develop further insight into linkages of 4.1N and IP6K2 in the brain, we immunoprecipitated IP6K2-deleted and wild-type mouse brain lysates with antibodies to 4.1N (Fig. 3A) and identified two prominent interactors, Rab 5 and SNX-27 (Fig. 3B,C). Direct binding studies revealed robust binding of 4.1N to Rab 5 and to SNX-27. To assess possible physiologic relevance for these interactions, we monitored levels of Rab 5 and SNX-27 in IP6K2-deleted brains, observing a decrease of ∼50% in both Rab 5 and SNX-27 expression in the mutants (Fig. 3D,E).
Figure 3.

Rab 5 and SNX-27 form a ternary complex with IP6K2–4.1N. A, Workflow for screening proteins that bind to the IP6K2–4.1N complex. B, Immunoprecipitates (IPs) of Rab 5 proteins blotted with IP6K2 and 4.1N antibodies, respectively, revealed the binding of Rab 5 to both IP6K2 and 4.1N in wild-type mouse brain lysates. C, Immunoblot (IB) showed that SNX-27 binds to 4.1N and not to IP6K2. D, Western blot of wild-type and IP6K2-KO mice brain lysates showed reduced the protein expression of Rab5 and SNX-27 in IP6K2-KO mouse brain lysates. E, Quantitation of the relative expression of Rab 5 and SNX-27 depicted in D through normalization against the relative expression of the relevant input. F, Western blots depict reduced expression of some prominent Immediate Early Gene (IEG)-encoded proteins like Arc, Fos B, NGF, and Synaptophysin (Syn) in the IP6K2-KO mice brain lysates. G, Quantitation of the depicted. Western blots in F through normalization against the input is shown in the histogram. Data were statistically analyzed by paired Student's t test. Significant differences (*p < 0.05) were observed in comparison with WT controls. Data are presented as the mean ± SD and are representative of three independent experiments done under identical conditions.
To ascertain further neural roles for IP6K2, we monitored levels of synaptically relevant proteins in IP6K2 knock-out brain (Fig. 3F,G). We observed substantial decreases in levels of synaptophysin, NGF, Fos-B, and Arc in IP6K2 knock-out brains (Fig. 3F,G).
Relationship of IP6K2 and 4.1N in cerebellar granule cells
We examined the localizations of IP6K2 and 4.1N by immunofluorescence in the cerebellum. Both IP6K2 and 4.1N were localized almost exclusively to cerebellar granule cells (Fig. 4A). To ascertain the influence of IP6K2 upon the disposition of various cerebellar cell types, we monitored calretinin, a marker for granule cells, and calbindin, which labels Purkinje cells (Fig. 4B). Levels of both calretinin and calbindin were substantially decreased in IP6K2 mutant preparations. Consistent with the loss of these cellular markers, the width of the molecular layer was reduced ∼30% in IP6K2 knock-outs (Fig. 4C). However, the overall morphology of the WT and K2-KO mice brains as well as the structure of the hippocampus and the cerebellum showed no significant influence of IP6K2 deletion (Fig. 4-1). Relative numbers of granule cells were diminished ∼25% in IP6K2 knockouts (Fig. 4D).
Figure 4.

Lack of IP6K2 and 4.1N, highly expressed granule cells proteins, leads to diminished proliferation of cerebellar cells. A, Immunofluorescent staining of wild-type mouse brain cerebellar sections showed localization of IP6K2 and 4.1N in granule cells of the cerebellum. B, Western blots for calbindin and calretenin, respective markers for Purkinje and granule cells, showed reduced expression of both proteins in mouse brain lysates lacking IP6K2. C, Histological comparison through H&E staining of WT and IP6K2 KO brain (IP6K2-KO; 8-week-old) sections showed substantial decreases in the molecular layer compared with the WT. GCL, Granule cell layer; PCL, Purkinje cell layer; ML, molecular layer. D, H&E-stained sections of IP6K2-KO mouse also showed substantially fewer granule cells in the cerebellar region of mutant mice compared with WT. E, F, Mice injected with EdU showed fewer proliferating cells (marked with white arrow heads) in IP6K2-KO mice compared with the WT. G, XTT assay performed with knockdown of K2 as well as K2 lacking binding site for 4.1N (202–261) in N2A cells also revealed lower cell viability compared with control shRNA. Scale bar, 50 μm. The data are presented as the mean ± SD: ***p < 0.001, **p < 0.01, analyzed by one-way ANOVA. The H&E staining of wild type (WT) and IP6K2-Knockout (KO) sagittal brain sections (A), Hippocampus (B) and Cerebellum (C) is shown in Figure 4-1.
H&E staining of wild type (WT) and IP6K2 knockout (KO) sagittal brain sections (A), Hippocampus (B) and Cerebellum (C). The overall morphology do not show any significant difference. Data are representative of three independent experiments done under identical conditions. Download Figure 4-1, EPS file (2MB, eps)
We examined whether these aberrations stem from altered cellular development. We monitored the proliferation of cerebellar cells in 8-week-old mice injected with EdU (Fig. 4E,F). The proportion of EdU-labeled cells was decreased ∼45% in IP6K2 mutants, which could account for the fewer adult neurons in the mutant mice.
To elucidate regulatory mechanisms, we examined a continuous cell line, N2A, in which we depleted IP6K2 by shRNA or transfected IP6K2 lacking binding sites for 4.1N (Fig. 4G). Cell viability was reduced ∼25% in preparations with depletion of IP6K2 and 35% with IP6K2 lacking binding site for 4.1N. Accordingly, it appears that both 4.1N and IP6K2 are required for the optimal development of cerebellar neurons.
Parallel fiber–Purkinje cell synapses are depleted with loss of IP6K2
As Purkinje cells are the principal targets of granule cells, which are enriched in IP6K2, we explored the disposition of Purkinje cells in IP6K2 mutants. The density of spines in Purkinje cells as well as cellular volume were diminished 50–65% in IP6K2 knock-out mice (Fig. 5A). We did not observe any significant change in other cell types of the IP6K2-KO mouse brain (Fig. 5-1).
Figure 5.
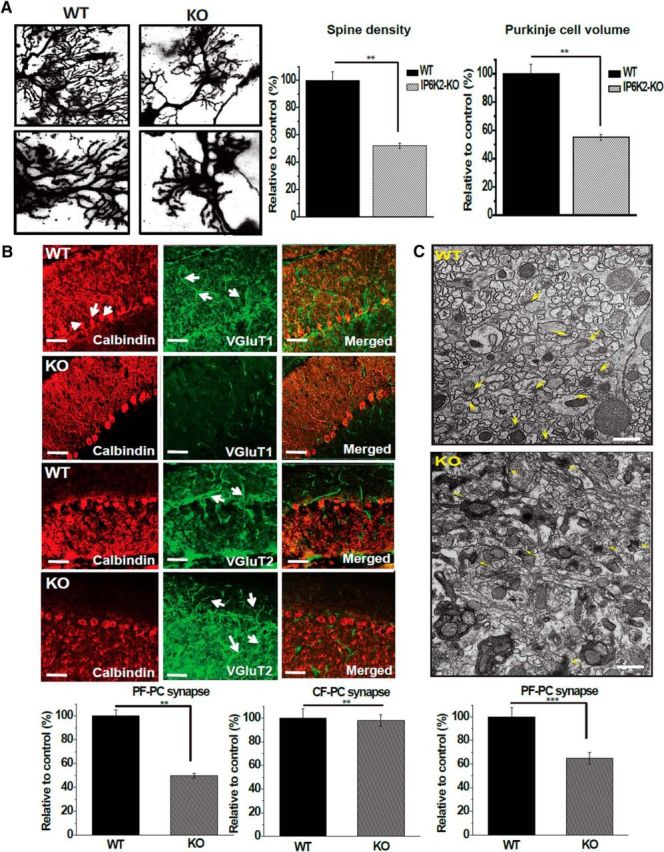
IP6K2 and 4.1N expressed in the granule cells of cerebellum affect Purkinje cell morphology and parallel fiber–Purkinje cell synapses. A, Purkinje cell volume, spine number, and spine density were significantly decreased in IP6K2-KO mice when compared with their WT counterparts. (top, 60× magnification; bottom, 100× magnification). B, Immunostaining of VGlut1/calbindin and VGlut2/calbindin in cerebellar Purkinje cells from WT and IP6K2-KO mice showed that the density of VGlut1 was markedly reduced in IP6K2-KO mice, indicating that knock-out mouse brain sections contained fewer parallel fiber–Purkinje cell synapses compared with WT mouse brain. However, there was no difference in the climbing fiber–Purkinje cell synapses of IP6K2-KO mouse cerebellum compared with wild type. Scale bar, 20 μm. C, Electron microscopic analysis of the cerebellar molecular layer from IP6K2-KO mouse brain (8 weeks old) also revealed fewer synapses compared with WT mouse brain. However, the ultrastructure of synapses in the cerebellum was relatively normal in the IP6K2-KO. Scale bar, 500 nm. Data were statistically analyzed by paired Student's t test. Significant differences (***p < 0.001, **p < 0.01) were observed in comparison with wild-type controls. Data are presented as the mean ± SD and are representative of three independent experiments performed under identical conditions. Golgi staining of IP6K2-knockout (KO) mice cerebellum with no significant morphological changes in pyramidal cells, granule cells, stellate cells and Golgi cells when compared to their wild-type (WT) counterparts, as shown in Figure 5-1.
Golgi staining of IP6K2-knockout (KO) mice cerebellum (8 weeks old) shows no morphological changes in Pyramidal cells, granule cells, stellate cells and Golgi cells when compared to their wild-type (WT) counterparts. However, Purkinje cell number were reduced in IP6K2-KO mice. Data are representative of three independent experiments done under identical conditions. Download Figure 5-1, EPS file (2.4MB, eps)
We counted the numbers of synapses upon Purkinje cells. In IP6K2 knock-out mice, the density of parallel fiber-Purkinje cell synapses was decreased by ∼50%. By contrast, there was no alteration in numbers of climbing fiber-Purkinje cell synapses (Fig. 5B). Electron microscopic analysis of cerebellar brain sections provided similar findings, namely a 35% decrease of parallel fiber-Purkinje cell synapses in the IP6K2 mutants (Fig. 5C). However, the ultrastructure of synapses was relatively normal in the knockouts.
How 4.1N influences the morphology and function of Purkinje cells has not been characterized in detail. Conceivably, sites on IP6K2 that recognize 4.1N may also mediate interactions with other proteins that regulate Purkinje cells. A screen of such interacting proteins broader than that performed in the present study is beyond the scope of this investigation.
Defects in motor coordination of IP6K2 knock-out mice
We monitored the motor coordination of the mice using the rotarod test (Fig. 6A). Both male and female IP6K2 knock-out mice displayed a substantially reduced latency to fall indicating a defect in motor coordination (Fig. 6A). In open field tests, both central and peripheral activity were reduced in IP6K2 mutants (Fig. 6B). We also monitored gait, revealing a significant decrease in stride length and speed of movement in the IP6K2 knock-out mice (Fig. 6C).
Figure 6.

IP6K2-KO mice show deficiencies in motor coordination and locomotor activity. A, Motor learning and coordination were compared between the IP6K2-KO and WT mice by the rotarod test. This test revealed reduced motor learning and coordination in the IP6K2-KO mice compared with WT mice. B, General locomotor activity in IP6K2-KO mice was assessed by the open field analysis. Such behavioral analysis also revealed reduced peripheral as well as central activity in IP6K2-KO mice when compared with WT mice. C, Gait analysis in IP6K2-KO mice also showed significant decreases in stride length and average speed of movement with respect to WT mice. However, there was no significant change between the paw print width of the WT and K2-KO mice. Data shown are representative of three independent experiments done under identical conditions with the mean ± SD: ***p < 0.001, **p < 0.01.
Discussion
Among the three principal isoforms of IP6K, IP6K2 has been least characterized. IP6K2 has been regarded as a predominantly p53-dependent proapoptotic enzyme, whose expression increases cell death, and whose functional or genetic abrogation restores cell survival (Morrison et al., 2001; Koldobskiy et al., 2010). Deletion of the IP6K2 gene results in a predisposition to cancer and tumor formation (Rao et al., 2014, 2015). However, little is known about the functions of IP6K2 in the brain, although it is selectively localized to neuronal cells. Our finding that IP6K2 regulates cerebellar influences on motor behavior establishes a notable role for inositol pyrophosphates in motor coordination.
IP7/bis-diphosphoinositol tetrakisphosphate (IP8) can also be generated by members of the mammalian vasoactive intestinal peptide (VIP) family (Fridy et al., 2007). Difference in function of inositol pyrophosphates generated by VIPs rather than IP6 kinases have not yet been characterized in depth. The study by Fridy et al. (2007) contrasted yeast VIPs with human IP6/IP7 kinases in terms of their comparative formation of IP7/IP8.
Proteins of the 4.1 family are differentially distributed, suggesting that its subtypes serve different functions depending on their specific tissue localization (Peters et al., 1998). Neuronal localizations of 4.1N are consistent with its influences upon synaptic proteins. 4.1N binds AMPA-glutamate receptors and facilitates their synaptic trafficking (Shen et al., 2000; Lin et al., 2009). It also impacts the disposition of dopamine receptors and appears to facilitate their anchoring to the cytoskeleton (Binda et al., 2002). Whether IP6K2 affects these processes via 4.1N is unclear. 4.1N may also influence calcium disposition through its binding to IP3 receptors (Fiedler and Nathanson, 2011). How IP3 and IP7 generated by IP6K2, interface is also not known. We previously demonstrated that NGF induces interactions between the protein PIKE (PI3Kinase Enhancer) and 4.1N, leading to nuclear translocation of 4.1N (Ye et al., 2000). Our present findings support a dominant role for IP6K2 in eliciting such NGF-induced nuclear translocation of 4.1N, while NGF also enhances the nuclear localization of IPK62.
We explored potential functions of IP6K2 by seeking binding partners and observed robust interactions of IP6K2 with 4.1N, which, like IP6K2, is highly expressed in cerebellar granule cells (Peters et al., 1998). This binding was selective, as it was not evident for IP6K1 or IP6K3. Moreover, nuclear translocation of 4.1N, which is required for its principal functions, was also dependent on IP6K2. Apart from 4.1N, the interactome of IP6K2 revealed two other proteins: SNX-27 and Rab 5. SNX-27, a brain-enriched PDZ domain protein, has been implicated in AMPA receptor trafficking (Loo et al., 2014), which is consistent with a similar role for 4.1N (Saiardi et al., 2001). SNX27 also facilitates excitatory synaptic function (Wang et al., 2013) as well as ependymal cell differentiation and ciliogenesis (Wang et al., 2016). Rab 5 controls synaptic functioning in Caenorhabditis elegans (Sann et al., 2012; Sasidharan et al., 2012).
Particularly striking was the impact of IP6K2, localized to granule cells, upon Purkinje cells in the cerebellum. The deletion of IP6K2 led to substantial defects in synaptic influences of granule cells upon Purkinje cells with a drastic decrease in the density of parallel fiber-Purkinje cell synapses. Reduced expression of proteins involved in synaptic transmission was also observed in the brains of IP6K2 knock-out mice, consistent with the observed loss in synaptic functions. The notable defects of motor coordination, a major cerebellar function, in the IP6K2 knock-out mice presumably reflect diminution of IP6K2 and its decreased association with 4.1N. Cerebellar neuronal cell viability was also reduced in both IP6K2 and 4.1N-deficient neuronal cells, indicating that both of these factors are essential for optimal development of cerebellar neurons.
How the three isoforms of IP6K interface is unclear. In preliminary experiments, we conducted functional epistatic analysis using LC-MS/MS techniques, which failed to reveal the influences of IP6K1 or IP6K3 upon IP6K2–4.1N interactions.
In summary, our study reveals a linkage of IP6K2 with 4.1N, which contributes to Purkinje cell morphology, the viability of cerebellar neurons, and the synaptic and locomotor functions controlled by the cerebellum.
Footnotes
This work was supported by US Public Health Service Grant MH18501. We thank Lauren K. Albacarys and Roxanne Barrow for their excellent technical support. We also thank Tatiana N. Boronina, Mass Spectrometry and Proteomics Facility, The Johns Hopkins University School of Medicine, for helping with the mass spectrometric protein detection. In addition, we thank Barbara Smith, Cell Biology Imaging Facility, The Johns Hopkins University School of Medicine, for helping with sample preparation and analysis of electron microscopy; and the members of the S.H.S. laboratory for reagents and discussions.
The authors declare no competing financial interests.
References
- Bhandari R, Juluri KR, Resnick AC, Snyder SH (2008) Gene deletion of inositol hexakisphosphate kinase 1 reveals inositol pyrophosphate regulation of insulin secretion, growth, and spermiogenesis. Proc Natl Acad Sci U S A 105:2349–2353. 10.1073/pnas.0712227105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binda AV, Kabbani N, Lin R, Levenson R (2002) D2 and D3 dopamine receptor cell surface localization mediated by interaction with protein 4.1N. Mol Pharmacol 62:507–513. 10.1124/mol.62.3.507 [DOI] [PubMed] [Google Scholar]
- Chakraborty A, Koldobskiy MA, Sixt KM, Juluri KR, Mustafa AK, Snowman AM, van Rossum DB, Patterson RL, Snyder SH (2008) HSP90 regulates cell survival via inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A 105:1134–1139. 10.1073/pnas.0711168105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Koldobskiy MA, Bello NT, Maxwell M, Potter JJ, Juluri KR, Maag D, Kim S, Huang AS, Dailey MJ, Saleh M, Snowman AM, Moran TH, Mezey E, Snyder SH (2010) Inositol pyrophosphates inhibit akt signaling, thereby regulating insulin sensitivity and weight gain. Cell 143:897–910. 10.1016/j.cell.2010.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Kim S, Snyder SH (2011a) Inositol pyrophosphates as mammalian cell signals. Sci Signal 4:re1. 10.1126/scisignal.2001958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Werner JK Jr, Koldobskiy MA, Mustafa AK, Juluri KR, Pietropaoli J, Snowman AM, Snyder SH (2011b) Casein kinase-2 mediates cell survival through phosphorylation and degradation of inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A 108:2205–2209. 10.1073/pnas.1019381108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiedler MJ, Nathanson MH (2011) The type I inositol 1, 4, 5-trisphosphate receptor interacts with protein 4.1N to mediate neurite formation through intracellular ca waves. Neurosignals 19:75–85. 10.1159/000324507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridy PC, Otto JC, Dollins DE, York JD (2007) Cloning and characterization of two human VIP1-like inositol hexakisphosphate and diphosphoinositol pentakisphosphate kinases. J Biol Chem 282:30754–30762. 10.1074/jbc.M704656200 [DOI] [PubMed] [Google Scholar]
- Fu C, Xu J, Li RJ, Crawford JA, Khan AB, Ma TM, Cha JY, Snowman AM, Pletnikov MV, Snyder SH (2015) Inositol hexakisphosphate kinase-3 regulates the morphology and synapse formation of cerebellar Purkinje cells via spectrin/adducin. J Neurosci 35:11056–11067. 10.1523/JNEUROSCI.1069-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine RF, Schell MJ (2001) Back in the water: the return of the inositol phosphates. Nat Rev Mol Cell Biol 2:327–338. 10.1038/35073015 [DOI] [PubMed] [Google Scholar]
- Koldobskiy MA, Chakraborty A, Werner JK Jr, Snowman AM, Juluri KR, Vandiver MS, Kim S, Heletz S, Snyder SH (2010) p53-mediated apoptosis requires inositol hexakisphosphate kinase-2. Proc Natl Acad Sci U S A 107:20947–20951. 10.1073/pnas.1015671107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Mulugu S, York JD, O'Shea EK (2007) Regulation of a cyclin-CDK-CDK inhibitor complex by inositol pyrophosphates. Science 316:109–112. 10.1126/science.1139080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin DT, Makino Y, Sharma K, Hayashi T, Neve R, Takamiya K, Huganir RL (2009) Regulation of AMPA receptor extrasynaptic insertion by 4.1N, phosphorylation and palmitoylation. Nat Neurosci 12:879–887. 10.1038/nn.2351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo LS, Tang N, Al-Haddawi M, Dawe GS, Hong W (2014) A role for sorting nexin 27 in AMPA receptor trafficking. Nat Commun 5:3176. 10.1038/ncomms4176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo HR, Huang YE, Chen JC, Saiardi A, Iijima M, Ye K, Huang Y, Nagata E, Devreotes P, Snyder SH (2003) Inositol pyrophosphates mediate chemotaxis in dictyostelium via pleckstrin homology domain-PtdIns (3,4,5) P3 interactions. Cell 114:559–572. 10.1016/S0092-8674(03)00640-8 [DOI] [PubMed] [Google Scholar]
- Morrison BH, Bauer JA, Kalvakolanu DV, Lindner DJ (2001) Inositol hexakisphosphate kinase 2 mediates growth suppressive and apoptotic effects of interferon-beta in ovarian carcinoma cells. J Biol Chem 276:24965–24970. 10.1074/jbc.M101161200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulugu S, Bai W, Fridy PC, Bastidas RJ, Otto JC, Dollins DE, Haystead TA, Ribeiro AA, York JD (2007) A conserved family of enzymes that phosphorylate inositol hexakisphosphate. Science 316:106–109. 10.1126/science.1139099 [DOI] [PubMed] [Google Scholar]
- Nagpal L, Panda K (2015) Characterization of calmodulin-free murine inducible nitric-oxide synthase. PLoS One 10:e0121782. 10.1371/journal.pone.0121782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagpal L, Haque MM, Saha A, Mukherjee N, Ghosh A, Ranu BC, Stuehr DJ, Panda K (2013) Mechanism of inducible nitric-oxide synthase dimerization inhibition by novel pyrimidine imidazoles. J Biol Chem 288:19685–19697. 10.1074/jbc.M112.446542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parra M, Gascard P, Walensky LD, Gimm JA, Blackshaw S, Chan N, Takakuwa Y, Berger T, Lee G, Chasis JA, Snyder SH, Mohandas N, Conboy JG (2000) Molecular and functional characterization of protein 4.1B, a novel member of the protein 4.1 family with high level, focal expression in brain. J Biol Chem 275:3247–3255. 10.1074/jbc.275.5.3247 [DOI] [PubMed] [Google Scholar]
- Peters LL, Weier HU, Walensky LD, Snyder SH, Parra M, Mohandas N, Conboy JG (1998) Four paralogous protein 4.1 genes map to distinct chromosomes in mouse and human. Genomics 54:348–350. 10.1006/geno.1998.5537 [DOI] [PubMed] [Google Scholar]
- Rao F, Cha J, Xu J, Xu R, Vandiver MS, Tyagi R, Tokhunts R, Koldobskiy MA, Fu C, Barrow R, Wu M, Fiedler D, Barrow JC, Snyder SH (2014) Inositol pyrophosphates mediate the DNA-PK/ATM-p53 cell death pathway by regulating CK2 phosphorylation of Tti1/Tel2. Mol Cell 54:119–132. 10.1016/j.molcel.2014.02.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao F, Xu J, Fu C, Cha JY, Gadalla MM, Xu R, Barrow JC, Snyder SH (2015) Inositol pyrophosphates promote tumor growth and metastasis by antagonizing liver kinase B1. Proc Natl Acad Sci U S A 112:1773–1778. 10.1073/pnas.1424642112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiardi A, Erdjument-Bromage H, Snowman AM, Tempst P, Snyder SH (1999) Synthesis of diphosphoinositol pentakisphosphate by a newly identified family of higher inositol polyphosphate kinases. Curr Biol 9:1323–1326. 10.1016/S0960-9822(00)80055-X [DOI] [PubMed] [Google Scholar]
- Saiardi A, Nagata E, Luo HR, Snowman AM, Snyder SH (2001) Identification and characterization of a novel inositol hexakisphosphate kinase. J Biol Chem 276:39179–39185. 10.1074/jbc.M106842200 [DOI] [PubMed] [Google Scholar]
- Saiardi A, Sciambi C, McCaffery JM, Wendland B, Snyder SH (2002) Inositol pyrophosphates regulate endocytic trafficking. Proc Natl Acad Sci U S A 99:14206–14211. 10.1073/pnas.212527899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saiardi A, Resnick AC, Snowman AM, Wendland B, Snyder SH (2005) Inositol pyrophosphates regulate cell death and telomere length through phosphoinositide 3-kinase related protein kinases. Proc Natl Acad Sci U S A 102:1911–1914. 10.1073/pnas.0409322102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sann SB, Crane MM, Lu H, Jin Y (2012) Rabx-5 regulates RAB-5 early endosomal compartments and synaptic vesicles in C. elegans. PLoS One 7:e37930. 10.1371/journal.pone.0037930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasidharan N, Sumakovic M, Hannemann M, Hegermann J, Liewald JF, Olendrowitz C, Koenig S, Grant BD, Rizzoli SO, Gottschalk A, Eimer S (2012) RAB-5 and RAB-10 cooperate to regulate neuropeptide release in Caenorhabditis elegans. Proc Natl Acad Sci U S A 109:18944–18949. 10.1073/pnas.1203306109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shears SB. (2007) Understanding the biological significance of diphosphoinositol polyphosphates (“inositol pyrophosphates”). Biochem Soc Symp 74:211–221. 10.1042/BSS2007c18 [DOI] [PubMed] [Google Scholar]
- Shen L, Liang F, Walensky LD, Huganir RL (2000) Regulation of AMPA receptor GluR1 subunit surface expression by a 4.1N-linked actin cytoskeletal association. J Neurosci 20:7932–7940. 10.1523/JNEUROSCI.20-21-07932.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao Y, Zhang X, Badie H, Zhou Y, Mu Y, Loo LS, Cai L, Thompson RC, Yang B, Chen Y, Johnson PF, Wu C, Bu G, Mobley WC, Zhang D, Gage FH, Ranscht B, Zhang YW, Lipton SA, Hong W, Xu H (2013) Loss of sorting nexin 27 contributes to excitatory synaptic dysfunction by modulating glutamate receptor recycling in Down's syndrome. Nat Med 19:473–480. 10.1038/nm.3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhou Y, Wang J, Tseng IC, Huang T, Zhao Y, Zheng Q, Gao Y, Luo H, Zhang X, Bu G, Hong W, Xu H (2016) SNX27 deletion causes hydrocephalus by impairing ependymal cell differentiation and ciliogenesis. J Neurosci 36:12586–12597. 10.1523/JNEUROSCI.1620-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Compton DA, Lai MM, Walensky LD, Snyder SH (1999) Protein 4.1N binding to nuclear mitotic apparatus protein in PC12 cells mediates the antiproliferative actions of nerve growth factor. J Neurosci 19:10747–10756. 10.1523/JNEUROSCI.19-24-10747.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye K, Hurt KJ, Wu FY, Fang M, Luo HR, Hong JJ, Blackshaw S, Ferris CD, Snyder SH (2000) Pike. A nuclear gtpase that enhances PI3kinase activity and is regulated by protein 4.1N. Cell 103:919–930. 10.1016/S0092-8674(00)00195-1 [DOI] [PubMed] [Google Scholar]
- York SJ, Armbruster BN, Greenwell P, Petes TD, York JD (2005) Inositol diphosphate signaling regulates telomere length. J Biol Chem 280:4264–4269. 10.1074/jbc.M412070200 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Seventeen (17) 4.1N peptides detected through LC/MS-MS with their respective peptide identification probability, mascotion score and mascot identity score. Download Table 1-1, DOC file (39.5KB, doc)
H&E staining of wild type (WT) and IP6K2 knockout (KO) sagittal brain sections (A), Hippocampus (B) and Cerebellum (C). The overall morphology do not show any significant difference. Data are representative of three independent experiments done under identical conditions. Download Figure 4-1, EPS file (2MB, eps)
Golgi staining of IP6K2-knockout (KO) mice cerebellum (8 weeks old) shows no morphological changes in Pyramidal cells, granule cells, stellate cells and Golgi cells when compared to their wild-type (WT) counterparts. However, Purkinje cell number were reduced in IP6K2-KO mice. Data are representative of three independent experiments done under identical conditions. Download Figure 5-1, EPS file (2.4MB, eps)