

Abstract
E3 ubiquitin ligase (UBE3A) levels in the brain need to be tightly regulated, as loss of functional UBE3A protein is responsible for the severe neurodevelopmental disorder Angelman syndrome (AS), whereas increased activity of UBE3A is associated with nonsyndromic autism. Given the role of mPFC in neurodevelopmental disorders including autism, we aimed to identify the functional changes resulting from loss of UBE3A in infralimbic and prelimbic mPFC areas in a mouse model of AS. Whole-cell recordings from layer 5 mPFC pyramidal neurons obtained in brain slices from adult mice of both sexes revealed that loss of UBE3A results in a strong decrease of spontaneous inhibitory transmission and increase of spontaneous excitatory transmission potentially leading to a marked excitation/inhibition imbalance. Additionally, we found that loss of UBE3A led to decreased excitability and increased threshold for action potential of layer 5 fast spiking interneurons without significantly affecting the excitability of pyramidal neurons. Because we previously showed that AS mouse behavioral phenotypes are reversible upon Ube3a gene reactivation during a restricted period of early postnatal development, we investigated whether Ube3a gene reactivation in a fully mature brain could reverse any of the identified physiological deficits. In contrast to our previously reported behavioral findings, restoring UBE3A levels in adult animals fully rescued all the identified physiological deficits of mPFC neurons. Moreover, the kinetics of reversing these synaptic deficits closely followed the reinstatement of UBE3A protein level. Together, these findings show a striking dissociation between the rescue of behavioral and physiological deficits.
SIGNIFICANCE STATEMENT Here we describe significant physiological deficits in the mPFC of an Angelman syndrome mouse model. We found a marked change in excitatory/inhibitory balance, as well as decreased excitability of fast spiking interneurons. A promising treatment strategy for Angelman syndrome is aimed at restoring UBE3A expression by activating the paternal UBE3A gene. Here we find that the physiological changes in the mPFC are fully reversible upon gene reactivation, even when the brain is fully mature. This indicates that there is no critical developmental window for reversing the identified physiological deficits in mPFC.
Keywords: autistic disorder, disease models, ion channel, prefrontal cortex, synaptic transmission, UBE3A
Introduction
Angelman syndrome (AS) is a severe neurodevelopmental disorder that results from mutations affecting the maternally inherited UBE3A gene (Chamberlain and Lalande, 2010). Individuals with AS show severe intellectual disability, lack of speech, motor impairment, seizures, sleep deficits, and behavioral problems, including many features of autism spectrum disorder. Some treatment strategies are aimed at targeting the identified pathophysiological mechanisms (van Woerden et al., 2007; Williams et al., 2010; Egawa et al., 2012; Kaphzan et al., 2013), but perhaps the most promising strategy is aimed at restoring UBE3A levels by reactivating the intact, but silenced, paternal copy of the UBE3A gene (Huang et al., 2011; Meng et al., 2013). For UBE3A reactivation strategies to be successful, it is essential to determine the critical time window during which such therapy can overcome the neurodevelopmental deficits associated with AS (Silva-Santos et al., 2015).
UBE3A is expressed throughout the brain in both glutamatergic and GABA-ergic neurons (Judson et al., 2014, 2016; Silva-Santos et al., 2015; Burette et al., 2017). Previous studies showed that loss of UBE3A leads to synaptic and cellular changes of layer 2/3 neurons of the visual cortex (Wallace et al., 2012; Judson et al., 2016), hippocampal pyramidal neurons (Jiang et al., 1998; Kaphzan et al., 2011, 2013), striatal medium spiny neurons (Hayrapetyan et al., 2014), cerebellar granule cells (Egawa et al., 2012; Bruinsma et al., 2015), and neurons of medial nucleus of the trapezoid body (Wang et al., 2017). Notably, loss of UBE3A appears to have a distinct impact on neurons from different areas of the brain. For instance, hippocampal pyramidal neurons and medial nucleus of the trapezoid body neurons show changes in intrinsic membrane properties, including increased length of the axon initial segment, findings that were not observed in layer 2/3 neurons of the somatosensory cortex (Kaphzan et al., 2011, 2013). Moreover, Purkinje cell functioning appears to be largely unaffected by deletion of UBE3A (Bruinsma et al., 2015).
MRI studies of individuals with AS point to changes in gray mater volume of orbitofrontal areas (Aghakhanyan et al., 2016), which is homologous to the rodent mPFC (Wise, 2008; Balleine and O'Doherty, 2010). In humans, the PFC is involved in coordinating high-level cognitive functions, such as language comprehension, decision making, planning, and reasoning (Miller, 2000; Miller and Cohen, 2001). Moreover, motor learning and anxiety behavior are dependent on striatum, thalamus, and amygdala (Zikopoulos and Barbas, 2013), which are downstream targets of mPFC layer 5 pyramidal neurons (Morishima and Kawaguchi, 2006; Hunnicutt et al., 2016; Zhang et al., 2016); hence, these cells are critically poised to shape the behavioral output of the animal. Alterations in PFC function have also been frequently implicated in neurodevelopmental disorders, including autism spectrum disorder, for which genetic lesions involving UBE3A show a strong association (Rudie et al., 2012; Zikopoulos and Barbas, 2013; Schubert et al., 2014; Bicks et al., 2015; Lee et al., 2015; Shafritz et al., 2015). Notably, increased activity of UBE3A leads to nonsyndromic autism (Hogart et al., 2010; Urraca et al., 2013; Elgersma, 2015; Yi et al., 2015). Thus, we hypothesize that PFC dysfunction may be a shared feature of both AS and Dup15q pathophysiology.
We previously described an AS mouse model (Silva-Santos et al., 2015) that recapitulates many of the AS behavioral phenotypes. Some of these phenotypes may be at least partially dependent on PFC function, including the motor deficits (Chersi et al., 2013; Santini et al., 2013; Shepherd, 2013; Kupferschmidt et al., 2017) and changes in repetitive behavior and anxiety (Slattery et al., 2011; Warden et al., 2012; Adhikari et al., 2015; Dwyer et al., 2015). Using this mouse model, which allows Cre-dependent activation of the maternal Ube3a allele at any time point during neurodevelopment, we identified distinct critical neurodevelopmental windows during which UBE3A restoration can rescue behavioral phenotypes. Importantly, we found that Ube3a gene reinstatement in adult animals did not rescue any of the behavioral defects. Here we investigated whether layer 5 mPFC neurons are affected in the AS mouse model and explored whether there is also a critical developmental window to reverse these physiological deficits.
Materials and Methods
Mouse model
We used our previously generated conditional AS mouse model (Ube3aStop), which allows us to control the reactivation of the Ube3a gene upon Cre-mediated deletion of a floxed transcriptional stop cassette inserted within intron 3 by homologous recombination (Silva-Santos et al., 2015). We showed previously that the transcriptional stop cassette is highly efficient in blocking Ube3a expression, whereas its Cre-mediated deletion either early embryonic (Ube3amStop/p+;Cre+ mouse) or later during development, and upon tamoxifen treatment (Ube3amStop/p+;CreERT+ mouse) reliably restored UBE3A levels (Silva-Santos et al., 2015).
To test whether there are physiological changes in layer 5 pyramidal neurons resulting from the loss of UBE3A, we crossed female Ube3am+/pStop mice (maintained in the 129S2/SvPasCrl background [Charles River] with the mutation at the paternal allele) with TgCAG-Cre mice in the C57BL/6J background (Charles River; referred as Cre+ mice) to generate heterozygous Ube3amStop/p+;Cre+ and Ube3amStop/p+;Cre− mutants and Ube3am+/p+;Cre+ and Ube3am+/p+;Cre− littermate controls in the F1 hybrid 129S2-C57BL/6 background. Because Ube3am+/p+;Cre+ and Ube3am+/p+;Cre− were indistinguishable, we pooled that data to increase statistical power. For clarity, we used an abbreviated nomenclature for these genotypes as follows (Table 1): Ube3am+/p+;Cre+ and Ube3am+/p+;Cre− animals: wild-type (WT), Ube3amStop/p+;Cre− animals: AS, and Ube3amStop/p+;Cre+ animals in which the Ube3a gene is embryonically reactivated: AS;Cre.
Table 1.
Mouse lines and groups abbreviation
| Mouse line | Groups | Abbreviation | Maternal Ube3a gene expressed? |
|---|---|---|---|
| Ube3amStop/p+; Cre+ | Ube3am+/p+; Cre− and Ube3am+/p+; Cre+ | WT | Yes |
| Ube3amStop/p+; Cre− | AS | No | |
| Ube3amStop/p+; Cre+ | AS;Cre | Yes (starting at early development) | |
| Ube3amStop/p+; CreERT+ | Ube3am+/p+; CreERT+, treated with vehicle | WT–VEH | Yes |
| Ube3am+/p+; CreERT+, treated with tamoxifen | WT–TAM | Yes | |
| Ube3amStop/p+; CreERT+, treated with vehicle | AS–VEH | No | |
| Ube3amStop/p+; CreERT+, treated with tamoxifen | AS–TAM | Yes (starting at time of injection) |
To test whether Ube3a reinstatement in adult animals rescued the identified phenotypes, we crossed the female Ube3am+/pStop mice with homozygous Tg(CAG-cre/Esr1*)5Amc/J (The Jackson Laboratory) (referred to as CreERT+) in the C57BL/6J background to generate heterozygous Ube3amStop/p+;CreERT+ mutants and Ube3am+/p+;CreERT+ littermate controls in the F1 hybrid 129S2-C57BL/6 background. For clarity, we used an abbreviated nomenclature for these genotypes as follows (Table 1): Ube3am+/p+;CreERT+ treated with vehicle: WT-VEH; Ube3am+/p+;CreERT+ treated with tamoxifen: WT-TAM; Ube3amStop/p+;CreERT+ treated with vehicle: AS-VEH; and Ube3amStop/p+;CreERT+ treated with tamoxifen: AS-TAM.
Tamoxifen injection
To induce gene reinstatement in adult mutants, we injected 14-week-old mutant and WT control mice (both males and females) with tamoxifen. Each mouse received 0.10 mg tamoxifen per gram body weight, via daily intraperitoneal injection for 7 consecutive days. Tamoxifen was diluted in sunflower oil at a concentration of 20 mg/ml. The control group was injected at 14 weeks old with vehicle (each mouse received 0.5 μl of sunflower oil per gram body weight, via daily intraperitoneal injections for 7 consecutive days). Upon tamoxifen injection, the levels of UBE3A in the PFC were restored to WT levels (see Fig. 5B,C), thus confirming our previously published results that showed reliable gene reinstatement in both cortical and hippocampal tissue (Silva-Santos et al., 2015).
Figure 5.
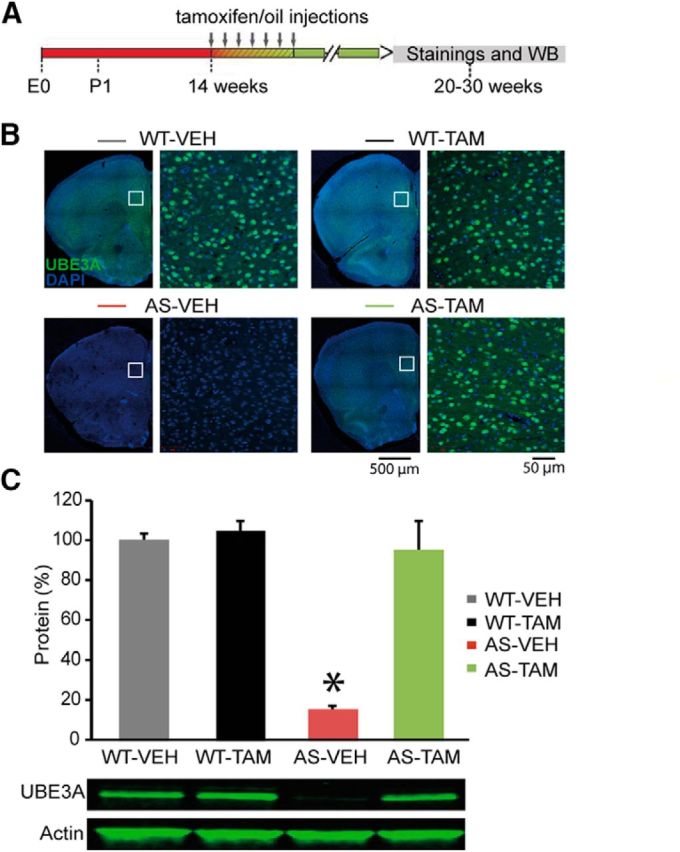
Adult Ube3a reinstatement restores UBE3A levels in mouse mPFC. A, Schematic representation of Ube3a reactivation during mouse embryonic development and time point of staining and Western blot analysis. B, UBE3A and DAPI stainings of PFC coronal sections from Ube3amStop/p+ and WT littermates crossed with inducible Cre (CreERT+) line treated with either vehicle or tamoxifen. Left, Overview of a coronal PFC section, acquired by tiled images obtained at 5× magnification. Right, Zoomed-in image of the white square on the left image, obtained at 20× magnification. There is a presence of UBE3A in the majority of neurons in WT-VEH/TAM mice and a lack of UBE3A in AS-VEH. Tamoxifen treatment restores UBE3A in AS-TAM mice. C, Western blot analysis of UBE3A protein expression in frontal cortex. Average data representing mean ± SE. Number of animals included in each group: WT-VEH, N = 3 mice; WT-TAM, N = 2; AS-VEH, N = 4 mice; AS-TAM, N = 2 mice. Two-way ANOVA (genotype × treatment) showed a significant interaction between genotype (WT;CreERT+ and Ube3amStop/p+;CreERT+) and treatment (vehicle or tamoxifen), (F(1,10) = 44.845, p = 0.0001). Post hoc Bonferroni: AS-VEH against WT-VEH (p = 0.001), AS-VEH against WT-TAM (p = 0.0001), and AS-VEH against AS-TAM (p = 0.001). *indicates statistically significant differences for p < 0.05.
Immunohistochemistry
Adult mice were deeply anesthetized with an overdose of pentobarbital (60 mg/ml) and perfused with 4% PFA in 0.1 m phosphate buffer (PB). Brains were postfixed with PFA for 1 h. After embedding in 12% gelatin/12% sucrose, coronal frontal cortex sections were cut on a freezing microtome (Leica; SM 2000r) at a thickness of 40 μm. The brain sections were washed in PBS and placed in blocking solution (10% normal horse serum, 0.5% Triton X-100 in PBS) for 1 h. Sections were incubated overnight at room temperature in PBS-containing primary antibodies, 2% normal horse serum, 0.5% Triton X-100. The following primary antibodies were used: rabbit anti-Pv (Swant, PV27, 1:1000 dilution) and mouse anti-E6AP (Sigma-Aldrich, E8655; 1:750 dilution). After incubation with primary antibodies, the sections were washed in PBS and incubated with secondary antibodies AlexaFluor-488-labeled donkey anti-mouse (Jackson ImmunoResearch Laboratories, 715-545-150, 1:400 dilution) and donkey anti-rabbit Cy3 (Jackson ImmunoResearch Laboratories, 711-165-152, 1:400 dilution) in PBS buffer containing 2% normal horse serum, 0.5% Triton X-100. After a 3 h incubation at room temperature, the sections were washed with PBS and 0.1 m PB, followed by 10 min DAPI (Invitrogen, D3571, 1:10.000 dilution). Sections were washed with 0.1 m PB and mounted using mowiol-DABCO (Sigma-Aldrich) mounting medium. Confocal images were acquired with the LSM700 confocal microscope (Carl Zeiss).
Western blot analysis
To collect tissue for Western blot analysis, frontal cortex tissue was dissected from adult mice and immediately frozen in liquid nitrogen. The lysates were prepared by adding lysis buffer (10 mm Tris-HCl, pH 6.8, 2.5% SDS) supplemented with protease inhibitor mixture (#P8340, Sigma-Aldrich, 1:100 dilution) to the tissue, and homogenization was achieved by sonication. After centrifugation (6000 rpm for 5 min), supernatants were collected and concentration was measured using the BCA protein assay kit (Thermo Fisher Scientific). Lysate concentrations were adjusted to 1 mg/ml. A total of 20 μg of each sample was loaded on the gel, and a semidry transfer was performed (Trans-Blot Turbo Transfer System, Bio-Rad). The blotted nitrocellulose membrane was probed with antibodies directed against E6AP (Sigma-Aldrich, E8655, 1:1000 dilution) and actin (Millipore, MAB1501r, 1:20,000 dilution). A fluorophore-conjugated secondary goat anti-mouse antibody (Westburg, IRDye 800CW, 926-32210, 1:15.000) was used, and the protein was detected using Odyssey Scanner system (LI-COR Biosciences). Quantification was done using Odyssey 3.0 software (LI-COR Biosciences).
Electrophysiology
Slices.
All electrophysiological experiments were performed on animals of both sexes between 20 and 30 weeks of age. Mice were decapitated under isoflurane anesthesia, and brains quickly removed and immersed in ice-cold, modified ACSF containing the following (in mm): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 7 MgSO4, and 0.5 CaCl2, pH 7.3–7.4, when bubbled with 95% O2-5% CO2. Next, coronal PFC slices (300 μm) were made and immediately placed in an incubation chamber filled with normal ACSF containing the following (in mm): 125 NaCl, 3 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 glucose, 1 MgSO4, and 2 CaCl2, pH 7.3–7.4, when bubbled with 95% O2-5% CO2. Slices were incubated for ∼10–15 min at 35°C followed by stabilization at room temperature, in the same solution, for at least 60 min before they were transferred to the recording chamber. The recording chamber was superfused at a flow rate of 2 ml/min with normal ACSF. Chamber temperature was adjusted to 28°C–30°C.
Cells were visualized using a Nikon microscope equipped with infrared illumination and differential interference contrast video microscopy. Whole-cell recordings were obtained from layer 5 pyramidal cells and interneurons in the infralimbic, or prelimbic regions of the mouse medial frontal cortex, here collectively referred to as the mPFC. Recordings were obtained using Multiclamp 700B amplifiers (Molecular Devices). Signals were low-pass filtered at 4 kHz and digitized at 20 kHz using Digidata acquisition interfaces. Data acquisition and analysis were performed using Clampex or Minianalysis software.
Voltage clamp.
The pipette capacitance was compensated, and series resistance was continuously monitored but was not compensated. Only recordings with a stable series resistance of <20 mΩ were used for analysis. Patch electrodes (3–5 mΩ) were backfilled with two types of internal solution, depending on whether we recorded inhibitory or excitatory events. To record inhibitory transmission, we used internal solution with high concentration of KCl, containing the following (in mm): K-gluconate 77, KCl 77, HEPES 10, EGTA 0.2, MgATP 4.5, NaGTP 0.3, and Na-phosphocreatine 10. To record excitatory transmission, we used internal solution containing the following (in mm): K-gluconate 125, NaCl 10, HEPES 10, EGTA 0.2, MgATP 4.5, NaGTP 0.3, and Na-phosphocreatine 10. Both internal solutions were adjusted for pH to 7.2–7.4 using KOH. sIPSCs, mIPSCs, sEPSCs, and mEPSCs were recorded in gap-free protocol in Clampex for 10 min while the cells were voltage-clamped at −70 mV. To isolate the inhibitory and excitatory events, we added either CNQX (10 mm) or bicuculline (10 mm), respectively, to the normal ACSF perfusate. To record miniature events, we further added TTX (1 μm) to block action potential (AP) mediated synaptic transmission.
Current clamp.
For current-clamp recording, patch electrodes were filled with the same solution used for recording the excitatory transmission. Series resistance and pipette capacitance were monitored and cancelled using bridge and capacitance neutralization. To obtain the firing pattern of neurons, we recorded voltage responses from neurons in current clamp and held at ∼−65 mV, while we injected a family of 500 ms long square pulses starting from −250 pA with 10 pA increments, delivered at 0.2 Hz.
Electrophysiology data analysis
IPSC and EPSC data analysis.
sIPSCs, mIPSCs, sEPSCs, and mEPSCs recorded at −70 mV were detected using Mini analysis software (Synaptosoft). To analyze the frequency, events were counted over 5 min of recording. To obtain the average events, for each cell, at least 100 nonoverlapping events were detected and averaged. The peak amplitude of the average sIPSCs and sEPSCs as well as the average mIPSCs and mEPSCs was measured relative to the baseline current. The decay kinetics was quantified by fitting a double exponential function and computing a weighted decay time constant (τw) as follows:
 |
where Aslow, Afast, τslow, and τfast are the amplitudes and decay time constants of slow and fast IPSC/EPSC decay components.
Neuron classification.
Pyramidal cells were identified visually, based on large soma size and the presence of apical dendrites. For interneurons, we focused on patching layer 5 cells with small round soma and without an apical dendrite. In our dataset, we obtained a mix of putative interneurons. Here we included only fast spiking (FS) neurons that were distinguished from the non-FS interneurons by their typical response to depolarizing current steps, which included high firing rates, with low AP adaption and narrow spike half-width (Rotaru et al., 2011). Single AP properties of FS neurons, which included threshold to fire an AP (AP threshold), AP amplitude, AP duration at half-maximal amplitude (AP half-width), afterhyperpolarization (AHP) amplitude, maximum rise slope, and maximum decay slope, were determined using two different stimulation protocols. First, we assessed AP properties from spikes elicited by 500-ms-long current injections at rheobase (minimum current injection used to evoke at least one spike). Second, we assessed AP after injecting short current pulses with amplitude of 1 nA and duration of 1 ms, to evoke one single AP in FS interneurons. The AP threshold was the voltage where dV/dt = 30 mV/ms. The AP amplitude represented the difference between the AP threshold and the peak depopulation of the AP. The AHP amplitude was estimated as the difference between the AP threshold and the voltage at the AHP trough. The maximum rise and decay slope represented maximum dV/dt value on the rising and decay phase, respectively, of the AP.
Chemicals
CNQX, bicuculline, and TTX were obtained from Invitrogen; all other chemicals and reagents were obtained from Sigma-Aldrich.
Experimental design and statistical analysis
We used AS, AS;Cre, and WT control littermates mice to test whether there are physiological changes in layer 5 pyramidal neurons resulting from the loss of UBE3A (see Figs. 1–4). In each figure, results are expressed as mean ± SEM. We used a one-way ANOVA with three genotypes (WT, AS, and AS;Cre) for each measured physiological variable (sIPSC, mIPSC, sEPSC, and mEPSC frequency, amplitude, and rise and decay time). To identify specific differences between groups, we used post hoc Bonferroni.
Figure 1.
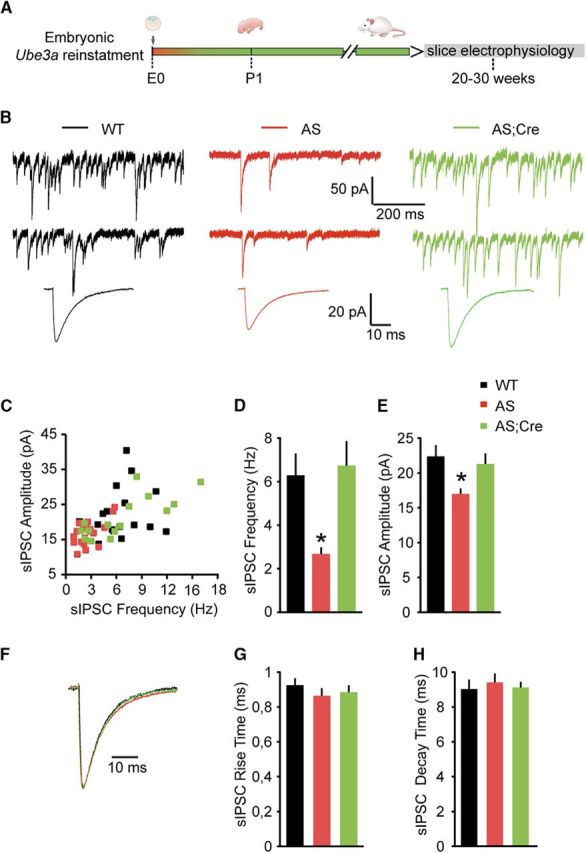
Embryonic reactivation of Ube3a expression rescues the spontaneous inhibitory neurotransmission in pyramidal neurons (PN) from mouse layer 5 PFC. A, Schematic representation of Ube3a reactivation during mouse embryonic development and time point of electrophysiological recordings. B, Top, Representative voltage-clamp recordings of sIPSCs from layer 5 PN obtained by clamping the neurons at −70 mV, in the presence of CNQX (10 μm), in Ube3amStop/p+ and WT littermates crossed with an embryonically active Cre line. Bottom, Average sIPSCs obtained by averaging at least 100 nonoverlapping individual events. AS mice show a clear decrease in both sIPSC frequency (B, top) and amplitude (B), which can be rescued by embryonic reactivation of the Ube3a gene in the AS;Cre mice. C, Raster plot of all the cells included in the analysis. Each dot indicates one cell, showing the amplitude of averaged sIPSCs and the sIPSC frequency. D, E, Average data representing mean ± SE, for sIPSC frequency and amplitude, respectively, in layer 5 PN. Number of neurons included in each group: WT, N = 18 neurons/6 mice; AS, N = 20 neurons/6 mice; AS;Cre, N = 17 neurons/6 mice. A one-way ANOVA shows a significant effect of the genotype on both frequency (F(2,52) = 9.182, p = 0.0001) and amplitude (F(2,52) = 5.63, p = 0.006). Post hoc Bonferroni: AS against WT (p = 0.002 for frequency, and p = 0.012 for amplitude), AS against AS;Cre (p = 0.002 for frequency, and p = 0.028 for amplitude), and WT against AS;Cre (p = 1 for frequency, and p = 1 for amplitude). F, Examples of scaled averaged sIPSCs from B (bottom), pointing to no changes in kinetic properties. G, H, Average data representing mean ± SE, for sIPSC 10–90 rise time and decay time, respectively, in layer 5 PN. No significant effect of genotype on either rise time (F(2,52) = 0.76, p = 0.4) or decay time (F(2,52) = 0.21, p = 0.8) is observed (one-way ANOVA). *indicates statistically significant differences for p < 0.05.
Figure 4.
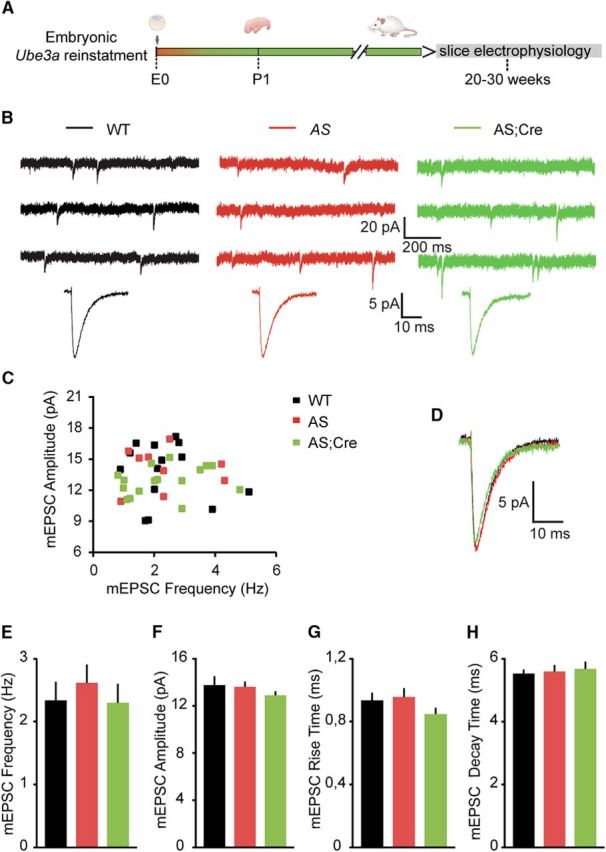
Loss of Ube3a expression does not affect the miniature excitatory neurotransmission of pyramidal neurons from mouse mPFC. A, Schematic representation of Ube3a reactivation during mouse embryonic development and time point of electrophysiological recordings. B, Top, Representative voltage-clamp recordings of mEPSCs from layer 5 PN obtained by clamping the neurons at −70 mV, in the presence of bicuculine (10 μm) and TTX (1 μm), in Ube3amStop/p+ and WT littermates crossed with an embryonically active Cre line. Bottom, Average mEPSCs obtained by averaging at least 100 nonoverlapping individual events. No difference for any of the measured parameters is observed between groups. C, Raster plot of all the cells included in the analysis. Each dot indicates one cell, showing the amplitude of averaged mEPSC and the mEPSC frequency. D, Overlapping averaged mEPSCs from B. Bottom, No changes in amplitude or kinetic properties. E–H, Average data representing mean ± SE, for mEPSC frequency, amplitude, rise and decay time constant, in layer 5 PN. Number of neurons included in each group: WT, N = 14 neurons/3 mice; AS, N = 16 neurons/4 mice; AS;Cre, N = 16 neurons/3 mice. No significant effect of the genotype on frequency (F(2,43) = 0.34, p = 0.71), amplitude (F(2,43) = 0.76, p = 0.47), rise time (F(2,43) = 1.42, p = 0.25), or decay time (F(2,43) = 0.12, p = 0.88) is observed (one-way ANOVA).
To test whether Ube3a reinstatement rescued the previously identified phenotypes, we used WT and AS mice treated either with vehicle (oil) or tamoxifen (see Figs. 5–10). For statistical significance, we used two-way ANOVA with genotype (WT, AS) and treatment (vehicle, tamoxifen) as independent variable and Bonferroni as post hoc test. The results of the tests are specified in each figure legend.
Figure 10.

Loss of UBE3A does not affect the excitability of pyramidal neurons from mouse mPFC. A, Schematics representing Ube3a reactivation achieved by tamoxifen administration (gray arrows) and time point of electrophysiological recordings. B, Representative firing patterns from layer 5 pyramidal neurons, obtained by delivering 180 pA depolarizing square pulses of 500 ms duration in AS and WT littermates treated with either vehicle or tamoxifen. No difference can be observed between genotypes. C, D, Averages of input-output curves from pyramidal neurons in layer 5 and layer 3, respectively, pointing to a lack of significant effect between groups in both layers. Number of neurons included in each group: Layer 5: WT-VEH, N = 26 neurons/6 mice; WT-TAM, N = 20 neurons/5 mice; AS-VEH, N = 32 neurons/7 mice; AS-TAM, N = 24 neurons/8 mice. Two-way ANOVA (genotype × treatment) showed no significant interaction between genotype (WT and AS) and treatment (vehicle and tamoxifen), for firing frequency (F(1,100) = 0.16, p = 0.68; two-way ANOVA). Layer 3: WT-VEH, N = 32 neurons/6 mice; WT-TAM, N = 24 neurons/3 mice; AS-VEH, N = 35 neurons/5 mice; AS-TAM, N = 22 neurons/3 mice. A two-way ANOVA showed also no significant interaction between genotype (WT and AS) and treatment (vehicle and tamoxifen), for firing frequency (F(1,100) = 0.19, p = 0.66).
Results
sIPSCs in layer 5 pyramidal neurons are significantly decreased by the loss of UBE3A
Previous data suggested that loss of UBE3A leads to hyperexcitable neuronal circuits (Kaphzan et al., 2011; Wallace et al., 2012, 2017), which may be responsible for some of the behavioral deficits observed in AS mice. A hyperexcitable circuit is often the result of an increased excitation to inhibition ratio (Isaacson and Scanziani, 2011). In AS mouse models, it was shown that layer 2/3 pyramidal neurons of the visual cortex receive less inhibition (Wallace et al., 2012; Judson et al., 2016). Additionally, excitatory neurons in hippocampus are more excitable due to a decrease in spike threshold (Kaphzan et al., 2011, 2013). Moreover, in vivo recordings from visual cortex pyramidal neurons showed increased firing rates of these cells (Wallace et al., 2017). To test whether layer 5 pyramidal neurons in the PFC are similarly affected by the loss of UBE3A, we first assessed the inhibitory synaptic properties in AS/AS;Cre and WT mice. We showed previously that UBE3A is not expressed in AS mice due to the presence of a floxed transcriptional stop cassette inserted in intron 3 of the Ube3a gene. Upon Cre-mediated deletion of the stop cassette, UBE3A levels in AS;Cre mice are restored to WT levels (Silva-Santos et al., 2015).
We recorded sIPSCs from PFC layer 5 pyramidal neurons from AS/AS;Cre and WT littermates. The sIPSCs were recorded using a high chloride internal solution while holding the cells at −70 mV in the presence of CNQX (10 μm) to block AMPA-mediated synaptic transmission (see Materials and Methods). Using this approach, we found that the frequency of sIPSCs onto layer 5 pyramidal neurons was reduced by more than half in AS (2.67 ± 0.31 Hz) mice compared with WT (6.29 ± 0.64 Hz) littermates (Fig. 1B–D), and that this deficit was accompanied by a significant reduction in sIPSC amplitude (Fig. 1B,C,E) (AS: 16.99 ± 0.77 pA; WT: 22.35 ± 1.62 pA). These results suggest that layer 5 pyramidal neurons in mPFC receive a lower level of inhibitory transmission. In contrast to sIPSC frequency and amplitude, the kinetic properties of sIPSC, including rise time (WT: 0.93 ± 0.04 ms; AS: 0.86 ± 0.04 ms; AS;Cre: 0.86 ± 0.04 ms) and decay time constant (WT: 9.03 ± 0.53 ms; AS: 9.41 ± 0.51 ms; AS;Cre: 9.46 ± 0.39 ms), were not changed (Fig. 1F–H), suggesting that there are no significant changes in the postsynaptic GABAA receptor (GABAAR) composition (Dixon et al., 2014; Labrakakis et al., 2014). Moreover, both sIPSC frequency (6.26 ± 1.05 Hz) and amplitude (21.68 ± 1.36 pA) were normalized to WT levels in AS;Cre littermates (Fig. 1B–E), demonstrating that embryonic Ube3a gene reinstatement fully rescues the decrease of inhibitory transmission onto layer 5 pyramidal neurons.
mIPSCs in layer 5 pyramidal neurons are not affected by the loss of UBE3A
sIPSCs in brain slices are postsynaptic responses to presynaptic GABA release following either spontaneous AP firing of inhibitory interneurons or the AP-independent release of single synaptic vesicles from GABAergic synapses, also known as mIPSCs (Auger and Marty, 2000; Bories et al., 2013). The axon of one GABAergic neuron may establish up to 10 synaptic contacts onto a postsynaptic pyramidal neuron (Somogyi et al., 1998; Branco and Staras, 2009); thus, AP-mediated sIPSCs are thought to involve synchronous GABA release across multiple synaptic contacts arising from a common axon. Therefore, the observed decrease in sIPSC frequency and amplitude in AS mice could reflect a reduction of GABAergic interneuron AP firing (Hua et al., 2010). Alternatively, the sIPSC deficits could also result from fewer and/or weaker GABAergic synapses onto layer 5 pyramidal neurons in AS mice. To differentiate between these two possibilities, we recorded mIPSCs in the presence of TTX (1 μm) to block AP-mediated GABA release. As expected (Bories et al., 2013), blocking AP-mediated inhibitory transmission resulted in a substantial decrease in the frequency and amplitude of events in all experimental groups (Figs. 1B–E, 2B–F), suggesting that more than half of the sIPSCs are the result of AP-mediated transmission. However, in contrast to the results of sIPSCs, analysis of mIPSCs revealed no differences in frequency (WT: 1.46 ± 0.18 Hz; AS: 1.36 ± 0.13 Hz; AS;Cre: 1.34 ± 0.12 Hz) (Fig. 2B,C,E) or amplitude (WT: 14.32 ± 0.58 pA; AS: 13.52 ± 0.45 pA; AS;Cre: 13.23 ± 0.51 pA) (Fig. 2B–D,F). The kinetic properties of mIPSCs, including rise time (WT: 0.63 ± 0.03 ms; AS: 0.61 ± 0.04 ms; AS;Cre: 0.56 ± 0.03 ms) and the decay time constant (WT: 8.69 ± 0.67 ms; AS: 8.20 ± 0.64 ms; AS;Cre: 8.05 ± 0.75 ms), were also statistically indistinguishable (Fig. 2D,G,H). Together, these data support a model whereby presynaptic deficits of AP-mediated inhibitory neurotransmission underlie the sIPSC phenotypes, whereas postsynaptic GABAAR composition is not significantly affected by loss of UBE3A in layer 5 pyramidal neurons (Dixon et al., 2014; Labrakakis et al., 2014).
Figure 2.
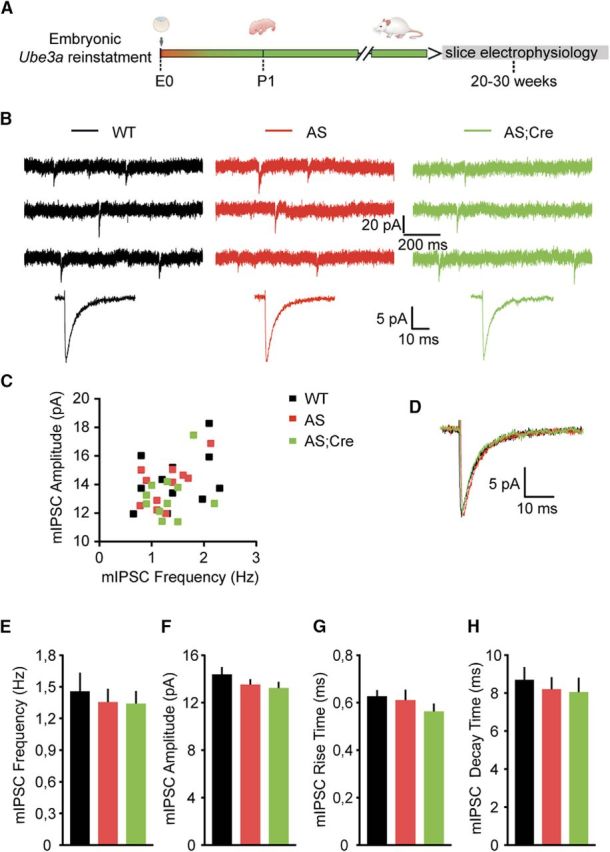
Loss of Ube3a expression does not affect the miniature inhibitory neurotransmission of pyramidal neurons from mouse mPFC. A, Schematic representation of Ube3a reactivation during mouse embryonic development and time point of electrophysiological recordings. B, Top, Representative voltage-clamp recordings of mIPSCs from layer 5 PN obtained by clamping the neurons at −70 mV, in the presence of CNQX (10 μm) and TTX (1 μm), in AS and WT littermates crossed with an embryonically active Cre line. Bottom, Average mIPSCs obtained by averaging at least 100 nonoverlapping individual events. No difference for any of the measured parameters is observed between groups. C, Raster plot of all the cells included in the analysis. Each dot indicates one cell, showing the amplitude of averaged mIPSCs and the mIPSC frequency. D, Overlapping averaged mIPSCs from B. Bottom, No changes in amplitude or kinetic properties. E–H, Average data representing mean ± SE, for mIPSC frequency, amplitude, rise and decay time constant, in layer 5 PN. Number of neurons included in each group: WT, N = 11 neurons/3 mice; AS, N = 11 neurons/3 mice; AS;Cre, N = 11 neurons/3 mice. No significant effect of genotype on frequency (F(2,30) = 0.36, p = 0.7), amplitude (F(2,30) = 1.19, p = 0.3), rise time (F(2,30) = 1.4, p = 0.2), or decay time (F(2,30) = 0.29, p = 0.74) is observed (one-way ANOVA).
sEPSCs in layer 5 pyramidal neurons are significantly increased by the loss of UBE3A
If the change in inhibitory transmission is a primary deficit resulting from the loss of UBE3A, then compensatory decreases in excitatory neurotransmission may occur to maintain a proper excitation to inhibition ratio (Xue et al., 2014; Wefelmeyer et al., 2016; Gainey and Feldman, 2017). Alternatively, an independent UBE3A-mediated primary deficit may lead to an increase in excitatory transmission. To test for any of these possibilities, we recorded sEPSCs from layer 5 pyramidal neurons, by holding the cells at −70 mV in the presence of bicuculline (10 μm) to block GABAAR-mediated transmission. We found a significant increase of sEPSC frequency and amplitude in AS mice (sEPSC frequency: 4.90 ± 0.33 Hz, Fig. 2B–D; amplitude: 21.49 ± 1.27 pA, Fig. 2B,C,E) compared with WT (sEPSC frequency: 3.62 ± 0.26 Hz; amplitude: 17.44 ± 0.80 pA). Reinstating Ube3a during early embryonic development in AS;Cre mice rescued the functional phenotype (sEPSC frequency: 3.43 ± 0.26 Hz; amplitude: 14.69 ± 0.69 pA) mice (Fig. 3B–E). The kinetic properties of sEPSCs, including rise time (WT: 1.16 ± 0.06 ms; AS: 1.31 ± 0.08 ms; AS;Cre: 1.08 ± 0.07 ms) and the decay time constant (WT: 5.76 ± 0.16 ms; AS: 5.92 ± 0.20 ms; AS;Cre: 5.91 ± 0.28 ms), were not affected by the loss of UBE3A or by gene reinstatement at early embryonic development (Fig. 3F- H), indicating that the composition of postsynaptic AMPA-containing receptors is preserved (Stincic and Frerking, 2015).
Figure 3.
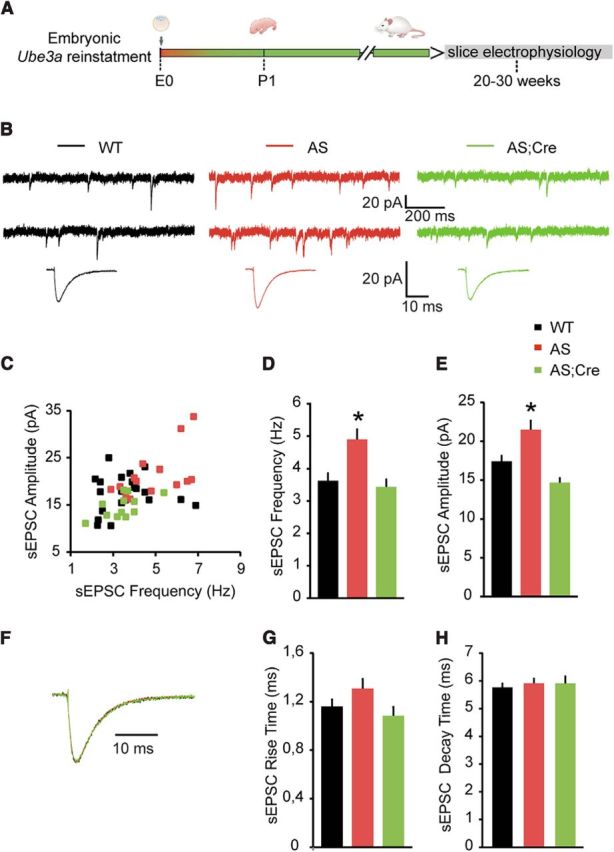
Embryonic reactivation of Ube3a expression rescues the spontaneous excitatory neurotransmission of pyramidal neurons from mouse mPFC. A, Schematic representation of Ube3a reactivation during mouse embryonic development and time point of electrophysiological recordings. B, Top, Representative voltage-clamp recordings of sEPSCs from layer 5 PN obtained by clamping the neurons at −70 mV, in the presence of bicuculine (10 μm), in Ube3amStop/p+ and WT littermates crossed with an embryonically active Cre line. B, Bottom, Average sEPSCs obtained by averaging at least 100 nonoverlapping individual events. AS mice show a clear increase in both sEPSC frequency (B, top) and amplitude (B), which can be rescued by embryonic reactivation of the Ube3a gene in the AS;Cre mice. C, Raster plot of all the cells included in the analysis. Each dot indicates one cell, showing the amplitude of averaged sEPSC and the sEPSC frequency. D, E, Average data representing mean ± SE, for sEPSC frequency and amplitude in layer 5 PN. Number of neurons included in each group: WT, N = 22 neurons/5 mice; AS, N = 15 neurons/4 mice; AS;Cre, N = 12 neurons/4 mice. A one-way ANOVA shows a significant effect of the genotype on both frequency (F(2,46) = 6.8, p = 0.003) and amplitude (F(2,46) = 10.47, p = 0.0001). Post hoc Bonferroni: AS against WT (p = 0.007 for frequency, and p = 0.01 for amplitude), AS against AS;Cre (p = 0.007 for frequency, and p = 0.0001 for amplitude), and WT against AS;Cre (p = 1 for frequency, and p = 0.17 for amplitude). F, Examples of scaled averaged sEPSCs from B. Bottom, No changes in kinetic properties. G, H, Average data representing mean ± SE, for sIPSCs 10–90 rise time and decay time, respectively, in layer 5 PN. No significant effect of the genotype on either rise time (F(2,46) = 0.93, p = 0.4) or decay time (F(2,46) = 0.516, p = 0.6) is observed (one-way ANOVA). *indicates statistically significant differences for p < 0.05.
mEPSCs in layer 5 pyramidal neurons are not affected by the loss of UBE3A
Similarly to sIPSCs, sEPSCs represent a mix of AP-mediated and mEPSCs (Bories et al., 2013). To test whether the increase in excitatory transmission was dependent on the firing of glutamatergic axons, we recorded mEPSCs in the presence of TTX (1 μm). As expected, compared with sEPSCs (Figs. 3, 4), mEPSC frequency and amplitude were decreased (Figs. 3, 4), confirming that a portion of the sEPSCs arise from AP firing of glutamatergic axons. However, mEPSC properties themselves were similar across the experimental groups: frequency (WT: 2.34 ± 0.30 Hz; AS: 2.62 ± 0.29 Hz; AS;Cre: 2.30 ± 0.30 Hz), amplitude (WT: 13.77 ± 0.76 pA; AS: 13.61 ± 0.46 pA; AS;Cre: 12.90 ± 0.34 pA), rise time (WT: 0.93 ± 0.05 ms; AS: 0.96 ± 0.06 ms; AS;Cre: 0.85 ± 0.04 ms), and the decay time constant (WT: 5.53 ± 0.12 ms; AS: 5.60 ± 0.20 ms; AS;Cre: 5.68 ± 0.23 ms) (Fig. 4). Combined with our other findings (Figs. 1–3), these results suggest that loss of UBE3A does not significantly affect either excitatory or inhibitory synaptic inputs onto layer 5 pyramidal neurons per se, but rather significantly impairs AP-mediated neurotransmission. These AP-corresponding decreases in spontaneous inhibitory transmission and increases in spontaneous excitatory transmission onto layer 5 output neurons reinforce the idea that cortical circuits may be hyperexcitable in AS mouse models. Moreover, they imply that homeostatic circuit mechanisms for balancing excitation and inhibition are impaired in AS mice.
Adult Ube3a reinstatement restores UBE3A protein levels and rescues the changes in spontaneous inhibitory and excitatory neurotransmission in PFC
We previously identified distinct neurodevelopmental windows during which Ube3a reinstatement can rescue AS-relevant behavioral phenotypes. In particular, we showed that motor deficits could still be rescued by Ube3a reinstatement in adolescent mice, whereas anxiety, repetitive behavior, and seizure threshold were only rescued when Ube3a was reinstated early in development (Silva-Santos et al., 2015). In contrast, hippocampal synaptic long-term potentiation was rescued by adult Ube3a reinstatement (Silva-Santos et al., 2015), suggesting a potential dissociation between the role of UBE3A in behavior relative to the single-cell deficits. To investigate the extent to which the critical period for rescuing physiological changes in the mPFC reflects the observed critical period for behavioral rescue, we used our previously described inducible AS mouse line (Silva-Santos et al., 2015), and induced Ube3a gene expression when the mice were 14 weeks old, an age at which the brain is considered to be fully mature (Hammelrath et al., 2016).
We previously showed that, in our AS-inducible mouse line, the transcriptional stop cassette is highly efficient in blocking Ube3a expression, whereas its Cre-mediated deletion restores the levels of UBE3A in the cortex and hippocampus (Silva-Santos et al., 2015). Here we wanted to further validate our AS mouse line, by focusing specifically on the PFC. Using immunohistochemistry, we showed the presence of UBE3A in the majority of neurons in PFC in WT-VEH/TAM mice (Fig. 5B) and the lack of UBE3A in these neurons in the AS-VEH mice (Fig. 5B). Upon tamoxifen treatment, UBE3A levels were restored in neurons from mPFC (Fig. 5B). Next, we used Western blot analysis to investigate that UBE3A protein levels in AS-TAM mice are restored up to nearly WT-VEH control levels upon tamoxifen-mediated Cre activation. In contrast, AS-VEH mice do not show significant Ube3a gene reinstatement (Fig. 5C). These findings confirm our previously published results that were obtained for the hippocampus using the same mouse model (Silva-Santos et al., 2015).
Next, we recorded both sIPSCs and sEPSCs in mPFC layer 5 pyramidal neurons, from AS and WT mice, treated either with vehicle or tamoxifen. To ensure that maximum levels of UBE3A are reached and the cells have some time to adapt, we performed these electrophysiological experiments at least 6 weeks after the first tamoxifen injection was delivered (Fig. 6A; see Fig. 7A). As we found for AS mice from Ube3amStop/p+;Cre line (Figs. 1, 3), sIPSCs were less frequent and the amplitude of the averaged sIPSCs was reduced in AS-VEH mice (sIPSC frequency: 3.06 ± 0.36 Hz; amplitude: 20.17 ± 1.12 pA) compared with WT-VEH mice (sIPSC frequency: 8.22 ± 1.01 Hz; amplitude: 27.04 ± 1.86 pA) (Fig. 6B–E). Tamoxifen-mediated Ube3a reinstatement in AS-TAM mice at 14 weeks of age rescued both frequency and amplitude (sIPSC frequency: 8.38 ± 0.96 Hz; amplitude: 30.27 ± 2.29 pA), whereas tamoxifen treatment did not significantly alter sIPSCs in WT-TAM mice (sIPSC frequency: 8.92 ± 1.33 Hz; amplitude: 28.03 ± 1.95 pA) (Fig. 6B–E). No differences were observed for either sIPSC rise time (WT-VEH: 0.98 ± 0.04 ms; AS-VEH: 0.84 ± 0.03 ms; WT-TAM: 0.88 ± 0.03 ms; AS-TAM: 0.90 ± 0.03 ms) or the decay time constant (WT-VEH: 9.50 ± 0.64 ms; AS-VEH: 9.51 ± 0.24 ms; WT-TAM: 9.19 ± 0.38 ms; AS-TAM: 9.18 ± 0.50 ms). For sEPSCs, AS-VEH mice showed a significant increase in frequency and amplitude (sEPSC frequency: 7.42 ± 0.65 Hz; amplitude: 22.15 ± 1.39 pA) compared with WT-VEH (sEPSC frequency: 4.30 ± 0.55 Hz; amplitude: 17.07 ± 1.40 pA) (Fig. 7B–E). Tamoxifen treatment at 14 weeks of age rescued these phenotypes in AS-TAM mice (sEPSC frequency: 4.15 ± 0.36 Hz; amplitude: 17.40 ± 0.66 pA) but did not affect sEPSC frequency and amplitude in WT-TAM as compared with vehicle-treated controls (sEPSC frequency: 4.17 ± 0.37 Hz; amplitude: 17.25 ± 0.72 pA) (Fig. 7B,C). No changes in the rise time (WT-VEH: 1.26 ± 0.08 ms; AS-VEH: 1.39 ± 0.07 ms; WT-TAM: 1.25 ± 0.07 ms; AS-TAM: 1.19 ± 0.05 ms) and the decay time constant (WT-VEH: 5.90 ± 0.45 ms; AS-VEH: 6.10 ± 0.29 ms; WT-TAM: 6.13 ± 0.24 ms; AS-TAM: 6.04 ± 0.18 ms) were observed.
Figure 6.

Adult reactivation of Ube3a expression rescues the spontaneous inhibitory neurotransmission of pyramidal neurons from mouse mPFC. A, Schematics representing Ube3a reactivation achieved by tamoxifen administration (gray arrows) and time point of electrophysiological recordings. B, Top, Representative voltage-clamp recordings of sIPSCs from layer 5 PN obtained by clamping the neurons at −70 mV, in the presence of CNQX (10 μm), in Ube3amStop/p+ and WT littermates crossed with inducible Cre (CreERT+) line treated with either vehicle or tamoxifen. Bottom, Average sIPSCs obtained by averaging at least 100 nonoverlapping individual events. Ube3amStop/p+;CreERT+ mice treated with vehicle show a clear decrease in both sIPSC frequency and amplitude, which can be rescued by adult reactivation of the Ube3a gene in the Ube3amStop/p+;CreERT+ mice treated with tamoxifen. C, Raster plot of all the cells included in the analysis. Each dot indicates one cell, showing the amplitude of averaged sIPSCs and the sIPSC frequency. D, E, Average data representing mean ± SE, for sIPSC frequency and amplitude in layer 5 PN. Number of neurons included in each group: WT-VEH, N = 21 neurons/4 mice; WT-TAM, N = 18 neurons/6 mice; AS-VEH, N = 24 neurons/4 mice; AS-TAM, N = 19 neurons/6 mice. Two-way ANOVA (genotype × treatment) showed a significant interaction between genotype (WT;CreERT+ and Ube3amStop/p+;CreERT+) and treatment (vehicle or tamoxifen), for both frequency (F(1,78) = 5.96, p = 0.017) and amplitude (F(1,78) = 6.05, p = 0.016) (two-way ANOVA). Post hoc Bonferroni: AS-VEH against WT-VEH (p = 0.001 for frequency, and p = 0.043 for amplitude), AS-VEH against WT-TAM (p = 0.0001 for frequency, and p = 0.02 for amplitude), and AS-VEH against AS-TAM (p = 0.001 for frequency, and p = 0.001 for amplitude). *indicates statistically significant differences for p < 0.05.
Figure 7.

Adult reactivation of Ube3a expression rescues the spontaneous excitatory neurotransmission of pyramidal neurons from mouse mPFC. A, Schematics representing Ube3a reactivation achieved by tamoxifen administration (gray arrows) and time point of electrophysiological recordings. B, Top, Representative voltage-clamp recordings of sEPSCs from layer 5 PN obtained by clamping the neurons at −70 mV, in the presence of bicuculline (10 μm), in AS and WT littermates treated with either vehicle or tamoxifen. Bottom, Average sEPSCs obtained by averaging at least 100 nonoverlapping individual events. AS-VEH show a clear increase in both sEPSC frequency and amplitude, which can be rescued by adult reactivation of the Ube3a gene in the AS-VEH mice treated with tamoxifen. C, Raster plot of all the cells included in the analysis. Each dot indicates one cell, showing the amplitude of averaged sEPSC and the sEPSC frequency. D, E, Average data representing mean ± SE, for sIPSC frequency and amplitude in layer 5 PN. Number of neurons included in each group: WT-VEH, N = 18 neurons/5 mice; WT-TAM, N = 22 neurons/7 mice; AS-VEH, N = 22 neurons/7 mice; AS-TAM, N = 20 neurons/5 mice. Two-way ANOVA (genotype × treatment) showed a significant interaction between genotype (WT and AS) and treatment (vehicle and tamoxifen); for both frequency (F(1,78) = 9.88, p = 0.002) and amplitude (F(1,78) = 5.30, p = 0.024; two-way ANOVA). Post hoc Bonferroni: AS-VEH against WT-VEH (p = 0.0001 for frequency, and p = 0.009 for amplitude), AS-VEH against WT-TAM (p = 0.0001 for frequency, and p = 0.007 for amplitude), and AS-VEH against AS-TAM (p = 0.0001 for frequency, and p = 0.013 for amplitude). *indicates statistically significant differences for p < 0.05.
The restoration of the inhibitory and excitatory transmission deficits parallels the reexpression of UBE3A protein
Our previous data showed that, in the hippocampus of AS-TAM mice, UBE3A levels gradually return to ∼80% of the WT-VEH/TAM mice over a period of 3 weeks after the first tamoxifen injection (Silva-Santos et al., 2015). To test whether reinstatement of UBE3A in the cortex shows similar kinetics, we performed Western blot analysis on cortical tissue collected at several time points after the initiation of Ube3a gene reinstatement (Fig. 8A,B). UBE3A levels in AS-TAM mice gradually returned to WT-TAM levels over a period of 4 weeks from the first tamoxifen injection, pointing toward a slight delay in restoring cortical UBE3A protein level compared with hippocampus. To investigate the kinetics of electrophysiological changes, we recorded sIPSCs at 2 and 4 weeks after the first tamoxifen injection. Notably, the rescue of sIPSC frequency and amplitude closely paralleled the recovery of cortical UBE3A levels (Fig. 8B–D). Thus, in AS-TAM mice, at 2, 4, and 6–16 weeks after the first tamoxifen injection, sIPSC frequency was restored to 50%, 78%, and 91%, respectively, and amplitude to 78%, 91%, and 100%, respectively, compared with WT-TAM levels (sIPSC frequency and amplitude: AS-TAM 2 weeks: 4.83 ± 0.36 Hz and 22.63 ± 1.40 pA; AS-TAM 4 weeks: 6.92 ± 0.74 Hz and 25.01 ± 1.11 pA; AS-TAM 6–16 weeks: 8.39 ± 0.96 Hz and 30.27 ± 2.29 pA; WT-TAM 6–16 weeks: 8.92 ± 1.33 Hz and 28.03 ± 1.95 pA). Recordings of sEPSC frequency and amplitude at 4 weeks after the first tamoxifen injection show a partial recovery of the glutamatergic transmission (Fig. 8E,F), whereas a full recovery of the glutamatergic transmission is achieved after 6 weeks from the first tamoxifen injection (sEPSC frequency and amplitude, AS-TAM 4 weeks: 6.29 ± 0.96 Hz and 19.45 ± 1.59 pA; AS-TAM 6–16 weeks: 4.15 ± 0.36 Hz and 17.40 ± 0.66 pA; WT-TAM 6–16 weeks: 4.17 ± 0.38 Hz and 17.25 ± 0.73 pA).
Figure 8.
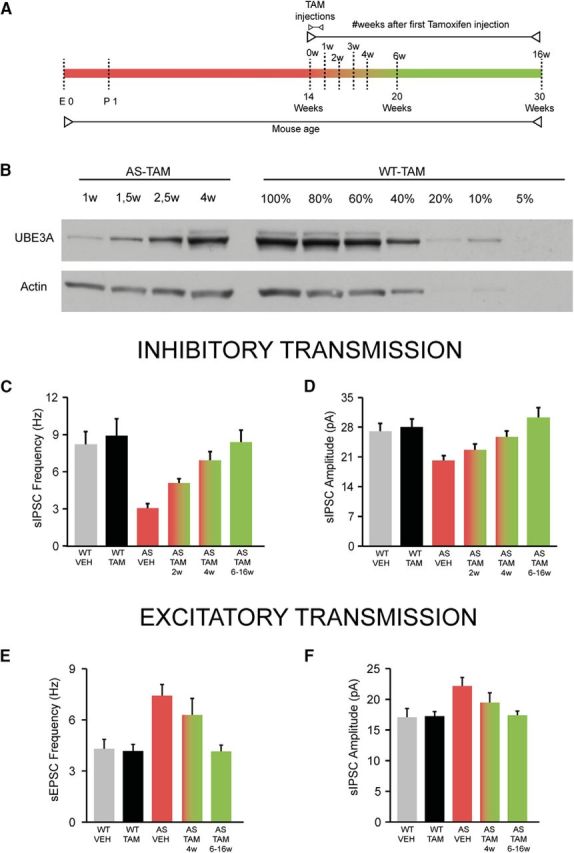
The restoration of the in inhibitory and excitatory transmission parallels the restoration of the UBE3A levels. A, Schematics representing Ube3a reactivation achieved by tamoxifen administration (TAM injections, lasting 1 week) and time point of Western blot analysis and electrophysiological recordings. Color gradients reflect the gradual restoration of UBE3A levels, spanning over a period of 4–6 weeks. B, Representative Western blot showing UBE3A expression following gene reinstatement (weeks following the first tamoxifen injection) in AS-TAM mice and a comparative gradient of protein levels (amount of protein loaded per lane in percent) in WT-TAM. C, D, Timeline of the recovery for inhibitory transmission. Average data representing mean ± SE, for sIPSC frequency and amplitude in layer 5 PN. For comparison, the same data presented in Figure 6D, E were also included in these bar graphs, along with two other groups of experiments obtained at 2 weeks (AS-TAM 2w) and at 4 weeks (AS-TAM 4w) after the first tamoxifen injection in the AS-TAM mice. The AS-TAM 6–16w group is identical with the AS-TAM group in Figure 6D, E. Number of neurons included in each group: identical with Figure 6D, E: WT-VEH, N = 21 neurons/4 mice; WT-TAM, N = 18 neurons/6 mice; AS-VEH, N = 24 neurons/4 mice; AS-TAM 2w, N = 21 neurons/3 mice; AS-TAM 4w, N = 25 neurons/3 mice; AS-TAM 6–16w (same group as AS-TAM in Fig. 6D,E), N = 19 neurons/6 mice one-way ANOVA (genotype) showed a significant effect for both frequency (F(5,116) = 6.04, p = 0.0001) and amplitude (F(1,116) = 5.43, p = 0.0001). Post hoc Bonferroni: AS-VEH against WT-VEH (p = 0.002 for frequency, and p = 0.05 for amplitude), AS-VEH against WT-TAM (p = 0.001 for frequency, and p = 0.02 for amplitude), and AS-VEH against AS-TAM (p = 0.002 for frequency, and p = 0.001 for amplitude). E, F, Timeline of recovery for excitatory transmission. Average data representing mean ± SE, for sEPSC frequency and amplitude in layer 5 PN. For comparison, the same data presented in Figure 7D, E were also included in these bar graphs, along with one other group of experiments obtained at 4 weeks (AS-TAM 4w) after the first tamoxifen injection in the AS-TAM mice. The AS-TAM 6–16w group is identical with the AS-TAM group in Figure 7. Number of neurons included in each group: identical with Figure 7D, E: WT-VEH, N = 18 neurons/5 mice; WT-TAM, N = 22 neurons/7 mice; AS-VEH, N = 22 neurons/7 mice; AS-TAM; AS-TAM 4w, N = 14 neurons/2 mice; and AS-TAM 6–16w (same group as AS-TAM in Fig. 7D, E), N = 20 neurons/5 mice. One-way ANOVA (genotype) showed a significant effect for both frequency (F(4,91) = 7.13, p = 0.0001) and amplitude (F(4,91) = 5.30, p = 0.006). Post hoc Bonferroni: AS-VEH against WT-VEH (p = 0.002 for frequency, and p = 0.02 for amplitude), AS-VEH against WT-TAM (p = 0.001 for frequency, and p = 0.019 for amplitude), and AS-VEH against AS-TAM (p = 0.001 for frequency, and p = 0.032 for amplitude).
FS interneurons are less excitable in AS mice, which can be rescued by adult Ube3a reinstatement
Our results show that loss of UBE3A leads to a significant decrease of sIPSC frequency and amplitude, whereas mIPSCs remain unchanged. Thus, the main changes in inhibitory transmission are the result of changes in AP-mediated vesicle release, potentially resulting from a decrease in spontaneous firing of APs of the inhibitory interneurons from PFC brain slices. A decrease in the spontaneous firing of APs from inhibitory interneurons may be the result of decreased intrinsic excitability (Holm et al., 2009). Hence, we next tested the impact of loss of UBE3A on the excitability of mPFC inhibitory interneurons, and if so, whether reinstating Ube3a in adult mice would rescue their excitability. We found that UBE3A is expressed in parvalbumin-positive interneurons in frontal cortex in WT-VEH/TAM mice (Fig. 9B) and confirmed the lack of UBE3A in these neurons in the AS-VEH mice (Fig. 9B). Upon tamoxifen treatment, UBE3A levels were restored in both pyramidal neurons and parvalbumin interneurons (Fig. 9B). Because of the challenges of space clamp, our pyramidal cell recordings are strongly biased to preferentially record inhibitory synapses in close proximity to the soma, for which FS interneurons provide the dominant contribution (Tremblay et al., 2016). Parvalbumin is a well-known marker for FS interneurons (Kawaguchi and Kubota, 1997). We therefore focused our recordings on FS interneurons, which are easily identifiable by their characteristic firing pattern in response to depolarizing current pulses (Rotaru et al., 2011). Compared with WT-VEH mice (at 300 pA current injection AP frequency: 114.11 ± 4.85 Hz), the firing frequency of FS neurons in AS-VEH mice was significantly decreased with increasingly depolarizing current steps (at 300 pA current injection AP frequency: 87.33 ± 10.03 Hz) (Fig. 7C,D). This effect was ameliorated by tamoxifen-mediated Ube3a gene reinstatement at 14 weeks of age (AS-TAM FS neurons at 300 pA current injection AP frequency: 121.11 ± 4.86 Hz; WT-TAM FS neurons at 300 pA current injection AP frequency: 123.28 ± 5.90 Hz) (Fig. 7C,D). Although passive membrane properties of FS cells were not changed across groups (Table 2), analysis of the first AP triggered by the 500-ms-long current injections, at rheobase current level, revealed several statistically significant changes, including a slower maximum rise slope, an increase in the half-width, and a smaller AP amplitude in the AS-VEH mice, compared with WT-VEH mice (Table 3). Tamoxifen treatment of AS-TAM mice at 14 weeks of age rescued these changes, whereas tamoxifen treatment in WT-TAM mice did not significantly affect the AP properties (Table 3). Although we found a decrease in the average AP threshold in AS-VEH mice, this did not reach statistical significance. Nevertheless, the decrease in the maximum rise slope and the AP amplitude in AS-VEH mice could suggest a possible impairment of Na channels with impact on the AP threshold. Because we measured the AP properties on traces obtained at rheobase level, which varied among cells, the initial subthreshold depolarization may have contributed to the inactivation of some Na channels. To control for this variable, we triggered single AP using current injection with short duration (1 ms) and large amplitude (1nA). Using this approach, we found a significant decrease in the AP threshold in AS-VEH mice, compared with WT-VEH (Fig. 9E,F). Additionally, the AP amplitude, maximum rise and decay slopes were also significantly decreased, whereas the AP half-width was significantly prolonged in AS-VEH mice, compared with WT-VEH (Fig. 9E–J). Tamoxifen treatment restored all these changes in AS-TAM mice and had no effect on the WT-TAM mice (Table 3). These findings suggest that a decrease in currents through Na channels could underlie the decrease in the maximum rise slope and the AP amplitude of AS mice.
Figure 9.
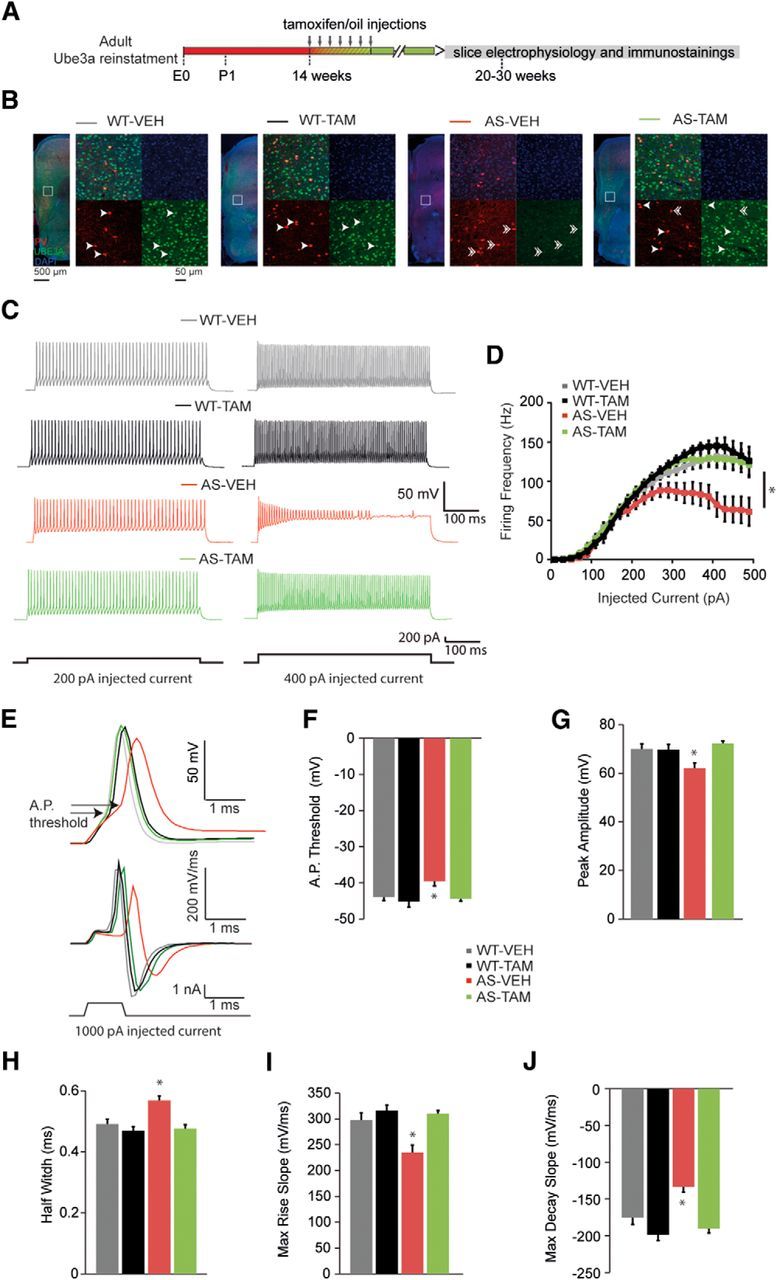
Adult reactivation of Ube3a expression rescues the excitability of FS interneurons from mouse PFC. A, Schematics representing Ube3a reactivation achieved by tamoxifen administration (gray arrows) and time point of electrophysiological recordings and immunostainings. B, UBE3A, parvalbumin (PV), and DAPI stainings of PFC coronal sections from Ube3amStop/p+ and WT littermates crossed with inducible Cre (CreERT+) line treated with either vehicle or tamoxifen. Left, Overview of a coronal mPFC section, acquired by tiled images obtained at 5× magnification. Right, Zoomed-in image, (white square on left), obtained at 20× magnification containing 4 individual images showing the following: stainings for PV+UBE3A+DAPI (top left), DAPI (top right), PV (bottom left), UBE3A (bottom right). Arrows indicate PV+ interneurons that costain for UBE3A. Double arrows indicate PV+ interneurons that lack UBE3A. There is a presence of UBE3A in PV interneurons from WT-VEH/TAM, and the lack of UBE3A in staining in PV interneurons from AS-VEH. Tamoxifen treatment restores UBE3A in PV interneurons from AS-TAM mice. C, Representative firing patterns from layer 5 FS interneurons, obtained by delivering 200 pA (left) and 400 pA (right) depolarizing square pulses of 500 ms duration in AS and WT littermates crossed with treated with either vehicle or tamoxifen. AS-VEH show a clear decrease in the firing frequency with 400 pA current injection. The firing frequency can be rescued by adult reactivation of the Ube3a gene in the AS-TAM. D, Averages of input-output curves showing a clear drop in firing frequency in AS-VEH with current injection >300 pA. This decrease is rescued by adult reactivation of the Ube3a gene in the AS-TAM. Number of neurons included in each group: WT-VEH, N = 17 neurons/5 mice; WT-TAM, N = 14 neurons/3 mice; AS-VEH, N = 15 neurons/6 mice; AS-TAM, N = 25 neurons/5 mice. Two-way ANOVA (genotype × treatment) showed a significant interaction between genotype (WT and AS) and treatment (vehicle and tamoxifen), for firing frequency (F(1,68) = 4.98, p = 0.029). Post hoc Bonferroni: AS-VEH against WT-VEH (p = 0.01), AS-VEH against WT-TAM (p = 0.002), and AS-VEH against AS-TAM (p = 0.001). FS neurons from AS-VEH responded with significantly less spikes when current injection was >300 pA. E, Overlapping examples of single AP (top) and their first derivative (dV/dt) (middle), triggered by a current injection of 1 ms and 1 nA (bottom). There is a more depolarized threshold as well as smaller amplitude of AP and in AS-VEH mice compared with WT-VEH/TAM, which are rescued by gene reinstatement in the AS-TAM. Additionally, the first derivative of the AP trace shows decreased maximum rise and decay slopes in rate of depolarization AS-VEH mice. F–J, Averages of AP threshold, amplitude, half-width, maximum rise slope, and maximum decay slope indicate significant differences (*) between AS-VEH mice compared with WT-VEH/TAM, which is rescued by gene reinstatement in the AS-TAM. Number of neurons included in each group, as well as statistical tests used, are included in Table 3.
Table 2.
Passive membrane properties of neurons in PFC
| Cell type | Genotype | Capacitance (pF) | Membrane resistance (mΩ) | Rest (mV) | Pipette resistance (mΩ) | Access resistance (mΩ) |
|---|---|---|---|---|---|---|
| Fast spiking interneurons | WT–VEH | 29.94 ± 1.32 | 254.58 ± 19.53 | 65.17 ± 0.72 | 3.77 ± 0.16 | 15.7 ± 0.86 |
| WT–TAM | 28.71 ± 1 | 242.64 ± 23.08 | 67.53 ± 1.02 | 3.9 ± 0.16 | 15 ± 0.79 | |
| AS–VEH | 30.2 ± 2.1 | 254.2 ± 15.92 | 66.07 ± 0.83 | 4.01 ± 0.14 | 16.86 ± 1.54 | |
| AS–TAM | 27.69 ± 0.85 | 245.84 ± 26.75 | 66.72 ± 0.75 | 3.71 ± 0.1 | 14.64 ± 10.48 | |
| Layer 5 pyramidal neurons | WT–VEH | 138.84 ± 5.91 | 185.4 ± 17.96 | 65.05 ± 0.44 | 3.76 ± 0.13 | 14.78 ± 0.60 |
| WT–TAM | 132.35 ± 7.08 | 153.2 ± 7.08 | 66.02 ± 0.98 | 3.67 ± 0.12 | 15.63 ± 7.08 | |
| AS–VEH | 141.96 ± 5.08 | 200.35 ± 16.75 | 65.79 ± 0.63 | 3.58 ± 0.16 | 14.64 ± 0.63 | |
| AS–TAM | 122.13 ± 5.98 | 161.77 ± 11.72 | 65.05 ± 0.77 | 4.15 ± 0.15 | 16.45 ± 0.84 | |
| Layer 3 pyramidal neurons | WT–VEH | 101.84 ± 5.10 | 308.37 ± 25.57 | 69.74 ± 0.77 | 3.33 ± 0.13 | 15.78 ± 1.37 |
| WT–TAM | 118.54 ± 7.6 | 323.20 ± 29.3 | 68.94 ± 0.65 | 3.85 ± 0.09 | 14.33 ± 1.16 | |
| AS–VEH | 103.6 ± 0.22 | 328.51 ± 34.88 | 69.06 ± 1.25 | 3.71 ± 0.22 | 14.74 ± 2.14 | |
| AS–TAM | 102.9 ± 5.7 | 313.76 ± 35.2 | 67.68 ± 0.75 | 3.89 ± 0.15 | 14.52 ± 1.23 |
Table 3.
AP properties of FS neurons in PFC
| Stimulation protocol | Genotype | No. of neurons/mice | AHP (mV) | Threshold (mV) | AP peak amplitude (mV) | AP half-width (ms) | Maximum rise slope (mV/ms) | Maximum decay slope (mV/ms) |
|---|---|---|---|---|---|---|---|---|
| Current injection of 500 ms at rheobase | WT–VEH | 17/5 | 22.55 ± 0.75 | −38.13 ± 0.51 | 61.46 ± 1.57 | 0.50 ± 0.01 | 255.83 ± 11.37 | −159.38 ± 7.06 |
| WT–TAM | 14/3 | 24.17 ± 0.74 | −37.56 ± 1.18 | 60.98 ± 1.95 | 0.48 ± 0.01 | 248.11 ± 10.68 | −179.13 ± 7.84 | |
| AS–VEH | 15/6 | 22.75 ± 1.25 | −36.54 ± 0.65 | 55.19 ± 1.74 | 0.61 ± 0.02 | 183.39 ± 13.65 | −118.98 ± 8.02 | |
| AS–TAM | 25/5 | 22.57 ± 0.59 | −37.50 ± 0.66 | 61.20 ± 1.40 | 0.51 ± 0.01 | 226.44 ± 9.73 | −158.6 ± 5.57 | |
| Statistics | ||||||||
| Two-way ANOVA (genotype × treatment) F/p values | F(1,68) = 0.83, p = 0.36 | F(1,68) = 0.38, p = 0.35 | F(1,68) = 4.51, p = 0.03 | F(1,68) = 6.93, p = 0.01 | F(1,68) = 4.31, p = 0.04 | F(1,68) = 1.57 p = 0.21 | ||
| Post-hoc Bonferroni p values | ||||||||
| AS–VEH versus WT–VEH | 0.0001 | 0.001 | 0.0001 | |||||
| AS–VEH versus WT–TAM | 0.0001 | 0.004 | 0.0001 | |||||
| AS–VEH versus AS–TAM | 0.0001 | 0.0001 | 0.05 | |||||
| Current injection of 1 ms and 1 nA | WT–VEH | 17/5 | 19.39 ± 0.94 | −43.92 ± 0.97 | 69.96 ± 2.15 | 0.49 ± 0.01 | 297.42 ± 14.46 | −175.15 ± 9.23 |
| WT–TAM | 14/3 | 18.76 ± 1.01 | −45.10 ± 1.30 | 69.65 ± 2.13 | 0.46 ± 0.01 | 316.21 ± 10.87 | −198.49 ± 8.18 | |
| AS–VEH | 15/6 | 21.60 ± 1.33 | −39.53 ± 1.57 | 62.09 ± 2.77 | 0.56 ± 0.01 | 234.81 ± 14.54 | −133.58 ± 7.06 | |
| AS–TAM | 25/5 | 20.17 ± 0.67 | −44.42 ± 0.63 | 72.22 ± 1.05 | 0.47 ± 0.01 | 310.29 ± 6.24 | −190.11 ± 6.22 | |
| Statistics | ||||||||
| Two-way ANOVA (genotype × treatment) F/p values | F(1,68) = 0.13, p = 0.70 | F(1,68) = 5.78, p = 0.019 | F(1,68) = 10.30, p = 0.002 | F(1,68) = 8.08, p = 0.006 | F(1,68) = 9.64, p = 0.003 | F(1,68) = 8.06 p = 0.006 | ||
| Post-hoc Bonferroni p values | ||||||||
| AS–VEH versus WT–VEH | 0.03 | 0.005 | 0.0001 | 0.0001 | 0.0001 | |||
| AS–VEH versus WT–TAM | 0.011 | 0.035 | 0.0001 | 0.0001 | 0.0001 | |||
| AS–VEH versus AS–TAM | 0.001 | 0.001 | 0.0001 | 0.0002 | 0.0003 |
Excitability of pyramidal neurons in PFC is not changed in AS mice
We found enhancements in excitatory transmission onto layer 5 pyramidal neurons, which opposed inhibitory deficits and which were dependent on AP-mediated glutamatergic transmission. Layer 5 pyramidal neurons receive excitatory inputs from a large number of sources, including inputs from local collaterals of other layer 5 pyramidal neurons, layer 3 pyramidal neurons from mPFC as well as other cortical regions, glutamatergic neurons from the mediodorsal thalamic inputs, and amygdala (Lübke et al., 2003; Douglas and Martin, 2004; DeNardo et al., 2015; Gerfen et al., 2016). We hypothesized that at least one of these glutamatergic populations might become hyperexcitable following loss of UBE3A. Focusing on local layer 3 and layer 5 pyramidal neurons within mPFC, we recorded both passive (Table 2) and active (Fig. 10) membrane properties in response to a range of hyperpolarizing and depolarizing current injections. We observed no significant differences on these measures between experimental groups, suggesting that loss of UBE3A does not lead to significant changes in the excitability of pyramidal neurons in mPFC.
Discussion
Here we used a conditional Ube3a mouse model to identify physiological changes in layer 5 neurons of the mPFC, and investigated whether these changes are rescued by embryonic and adult Ube3a gene reinstatement. Our results indicate that synaptic transmission onto layer 5 pyramidal neurons is significantly affected by the loss of UBE3A. These cells receive less inhibition and more excitation pointing toward a marked excitatory/inhibitory imbalance. The physiological changes do not appear to arise from changes at a single synapse level, but potentially from a decreased excitability of FS interneurons. Our findings are in line with other studies showing GABA-ergic deficits in the PFC of patients with neurodevelopmental disorders as well as mouse models of these diseases (Le Magueresse and Monyer, 2013; Wamsley and Fishell, 2017). Surprisingly, and in contrast to our previously reported behavioral phenotypes, Ube3a gene reinstatement in the mature brain fully rescued all the physiological phenotypes in layer 5 neurons. The recovery of synaptic function developed over a period of 6 weeks from the beginning of gene reactivation. The significance and implications of these findings will be discussed below.
Changes in inhibitory transmission
We identified a decrease of inhibition in our mouse model, although the mechanisms underlying this dysfunction appear different relative to previous data. Despite strong effects on both inhibitory and excitatory spontaneous transmission, we found no significant changes in miniature events. Although previous studies did not assess changes in sIPSCs or sEPSCs, it was found that mIPSCs and mEPSCs are both decreased in layer 2/3 pyramidal neurons from visual cortex (Wallace et al., 2012), possibly resulting from an increase of clathrin-coated vesicles in presynaptic boutons of both excitatory and inhibitory neurons. Although we did not directly analyze release probability in our mouse models, we did not find changes in the miniature events, which suggests that the release probability may not be impaired in layer 5 mPFC of AS mice. Our data may reflect differences between visual cortex and mPFC neurons as well as layer-specific differences. Alternatively, the differences may result from using different animal models, as the decrease in the frequency of mIPSCs observed by Wallace et al. (2012) when using the Ube3am−/p+ mouse model were not observed by Judson et al. (2016) when using the Ube3amStop/p+ mouse model.
We show, for the first time, that the excitability of FS interneurons is decreased by the loss of UBE3A. Although previous studies linked the changes in FS excitability with sIPSCs changes (Holm et al., 2009), here we do not investigate whether a causal link between the decreased FS excitability and the decreased sIPSC frequency and amplitude exists; thus, it remains to be addressed in future studies. Notably, the profile of FS neurons in our mouse model resembles the profile of immature FS neurons from PFC (Miyamae et al., 2017), suggesting that these cells may fail to properly mature (Stanurova et al., 2016). Previous data from visual cortex did not find significant changes in the excitability of FS neurons (Wallace et al., 2012); therefore, our findings may again point to changes between different layers and brains areas (Wang et al., 2016). We show changes in the AP properties in FS neurons from AS mice, including increased threshold, decreased amplitude, and lower maxim rise and decay slopes. These data suggest that a decrease in currents through Na channels most like the Nav1.1 subtype may lead to lower excitability of FS interneurons in AS mice (Yu et al., 2006; Ogiwara et al., 2007; von Schoubye et al., 2018).
Changes in excitatory transmission
The increase in excitation observed here is also dependent on AP firing, as no effects on mEPSCs were observed. Previous data showed decreased spine density in the visual cortex, cerebellum, and hippocampus of the Ube3am−/p+ AS mouse model (Dindot et al., 2008; Yashiro et al., 2009; Sato and Stryker, 2010; Kim et al., 2016; Sun et al., 2016). A decreased spine density would tend to result in a decreased frequency of mEPSCs. Such a decrease was previously reported for the visual cortex layer 3 pyramidal cells (Wallace et al., 2012). Here we did not find significant differences in the frequency of mEPSCs in layer 5 pyramidal neurons of the mPFC, suggesting that the excitatory synapses onto these neurons are not affected by the loss of UBE3A. Although we cannot rule out that our mouse model may be less severely affected than the Ube3am−/p+ AS mouse model in which these changes have been described, it is also possible that the developmental silencing of the paternal Ube3a gene differentially affects neurons of the cortical layers. For instance, it is possible that silencing of paternal Ube3a affects the later born neurons, such as neurons in layer 2–3 of the cortex, more severely compared with layer 5–6 neurons, which are born at an earlier time point. However, previous data suggest that Ube3a is silenced in mature (NeuN+) neurons (Judson et al., 2014; Ehlen et al., 2015; Jones et al., 2016), suggesting that neuronal maturity, rather than developmental age, dictates Ube3a silencing.
We found that increased frequency and amplitude of sEPSCs of mPFC pyramidal neurons are not due to changes in excitability, as pyramidal neurons from both layer 2–3 and layer 5 show no significant changes in excitability. Nevertheless, there could be changes in axonal properties, such as changes in Kv1 channels, which may be responsible for increased axonal firing without an impact on somatic excitability (Kole et al., 2007; Li et al., 2011). Kv1 channels are highly expressed at the axon initial segment (Inda et al., 2006; Kole et al., 2007; Van Wart et al., 2007), a subregion of the axon shown to be enlarged in AS mouse models and potentially responsible for increased excitability of neurons in hippocampus (Kaphzan et al., 2011, 2013) and medial nucleus of the trapezoid body (Wang et al., 2017).
How does loss of UBE3A cause changes in neuronal excitability and synaptic transmission?
The precise contribution of UBE3A and its downstream targets to the regulation of neuronal excitability and synaptic transmission remains elusive. Previous studies reported direct interaction between UBE3A and Arc (Greer et al., 2010; Yi et al., 2017; but see Kühnle et al., 2013), as well as changes in CAMK2 activity (van Woerden et al., 2007), but the precise impact of the deregulation of these proteins on synaptic transmission and excitability remains to be shown. Notably, UBE3A has also been shown to interact with the SK2 channel (Sun et al., 2015) and the GABA (GAT1) transporter (Egawa et al., 2012). However, membrane proteins are regulated and degraded via the endocytic pathway rather than through the proteasome pathway, a process that requires K63-linked ubiquitination of these proteins (Schwarz and Patrick, 2012; Laeremans et al., 2013; Babst, 2014; Foot et al., 2017). Currently, there is no evidence that UBE3A can perform this type of ubiquitination. The observed electrophysiological phenotypes could also be the result of the deregulation of many proteins, either through direct or indirect effects resulting from the loss of UBE3A. Nevertheless, our experiments make the important point that the observed electrophysiological changes can be surprisingly quickly reversed and closely follow UBE3A protein reexpression.
What is determining the critical period for therapeutic reversal?
Surprisingly, Ube3a reactivation in the fully mature mouse brain rescued all of the identified physiological phenotypes in mPFC neurons. The reversal of the synaptic phenotypes was gradual and completed 6 weeks after initiation of gene reactivation. This recovery was rather unexpected, given the lack of a behavioral rescue. Moreover, it has been shown, for primary sensory areas and their associated subcortical areas, that they are shaped during the critical period for an optimal neural representation of the environment (Hensch, 2004; Nabel and Morishita, 2013; Takesian and Hensch, 2013).
Our findings of full reversibility of the cellular deficits of mPFC neurons in adult mice are in marked contrast to AS-related behavioral phenotypes, which could not be reversed in adult mice. There are at least two possible explanations for the observed mismatch between the time course of reversibility for the cellular and behavioral deficits in the AS mouse model:
The previously observed behavioral deficits in AS mice might be independent of mPFC functioning and entirely rely on other brain areas, such as striatum and/or amygdala. We consider this unlikely because layer 5 pyramidal neurons of the PFC provide downstream excitatory inputs to several brain areas important in mediating our previously described behavioral deficits in AS mice, which include motor learning deficits and anxiety behavior (Silva-Santos et al., 2015). Layer 5 pyramidal neurons from PFC are involved in motor learning behavior (Kupferschmidt et al., 2017), via their inputs into the dorsal striatum (Morishima and Kawaguchi, 2006; Hunnicutt et al., 2016; Zhang et al., 2016) and nucleus accumbens (Rothwell et al., 2014) as well as in anxiety behavior, via inputs into amygdala (Thompson-Schill et al., 2005; Yu et al., 2008; Mátyás et al., 2014; Zhang et al., 2016).
The observed behavioral deficits indeed depend (at least in part) on mPFC functioning, but certain aspects of mPFC functioning (not identified in this study) are not rescued by adult gene reinstatement. We think that this is a plausible scenario. In particular, the proper formation of long-range projections from PFC onto its target areas may be affected (Brenhouse et al., 2008; Cressman et al., 2010; Johnson et al., 2016). Moreover, loss of UBE3A leads to a global impairment of axonal morphology and white matter development (Judson et al., 2017) with potential impairments in long-range mPFC efferent output that have not been captured in our recordings. Additionally, because of space clamp technical issues, we may have missed changes in synaptic transmission onto distal dendrites of pyramidal neurons (Williams and Mitchell, 2008).
Together, our findings highlight the utility of our Ube3a-inducible AS mouse model to study the pathophysiology of AS, to identify biomarkers and treatments, and to establish the optimal period for therapeutic intervention. Our data further highlight the need to identify physiological changes that are directly related to the behavioral changes. Moreover, we believe there is a great need to obtain detailed insight into the precise mechanisms that underlie the therapeutic critical period. It is possible that the hitherto largely unsuccessful therapeutic clinical trials for neurodevelopmental disorders (van der Vaart et al., 2015) are to some extent caused by administering interventions outside the boundaries of the therapeutic window.
Footnotes
This work was supported by Simons Foundation SFARI Award 275234 and The Netherlands Organization for Scientific Research NWO-ZoN-MW Award 91216045 to Y.E.
The authors declare no competing financial interests.
References
- Adhikari A, Lerner TN, Finkelstein J, Pak S, Jennings JH, Davidson TJ, Ferenczi E, Gunaydin LA, Mirzabekov JJ, Ye L, Kim SY, Lei A, Deisseroth K (2015) Basomedial amygdala mediates top-down control of anxiety and fear. Nature 527:179–185. 10.1038/nature15698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aghakhanyan G, Bonanni P, Randazzo G, Nappi S, Tessarotto F, De Martin L, Frijia F, De Marchi D, De Masi F, Kuppers B, Lombardo F, Caramella D, Montanaro D (2016) From cortical and subcortical grey matter abnormalities to neurobehavioral phenotype of Angelman syndrome: a voxel-based morphometry study. PLoS One 11:e0162817. 10.1371/journal.pone.0162817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auger C, Marty A (2000) Quantal currents at single-site central synapses. J Physiol 526:3–11. 10.1111/j.1469-7793.2000.t01-3-00003.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M. (2014) Quality control: quality control at the plasma membrane: one mechanism does not fit all. J Cell Biol 205:11–20. 10.1083/jcb.201310113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine BW, O'Doherty JP (2010) Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology 35:48–69. 10.1038/npp.2009.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bicks LK, Koike H, Akbarian S, Morishita H, Must A, Kroener S, Yizhar O, Bicks LK, Koike H, Akbarian S, Morishita H (2015) Prefrontal cortex and social cognition in mouse and man. Front Psychol 6:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bories C, Husson Z, Guitton MJ, De Koninck Y (2013) Differential balance of prefrontal synaptic activity in successful versus unsuccessful cognitive aging. J Neurosci 33:1344–1356. 10.1523/JNEUROSCI.3258-12.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco T, Staras K (2009) The probability of neurotransmitter release: variability and feedback control at single synapses. Nat Rev Neurosci 10:373–383. 10.1038/nrn2634 [DOI] [PubMed] [Google Scholar]
- Brenhouse HC, Sonntag KC, Andersen SL (2008) Transient D1 dopamine receptor expression on prefrontal cortex projection neurons: relationship to enhanced motivational salience of drug cues in adolescence. J Neurosci 28:2375–2382. 10.1523/JNEUROSCI.5064-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinsma CF, Schonewille M, Gao Z, Aronica EM, Judson MC, Philpot BD, Hoebeek FE, van Woerden GM, De Zeeuw CI, Elgersma Y (2015) Dissociation of locomotor and cerebellar deficits in a murine Angelman syndrome model. J Clin Invest 125:4305–4315. 10.1172/JCI83541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burette AC, Judson MC, Burette S, Phend KD, Philpot BD, Weinberg RJ (2017) Subcellular organization of UBE3A in neurons. J Comp Neurol 525:233–251. 10.1002/cne.24063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamberlain SJ, Lalande M (2010) Angelman syndrome, a genomic imprinting disorder of the brain. J Neurosci 30:9958–9963. 10.1523/JNEUROSCI.1728-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chersi F, Mirolli M, Pezzulo G, Baldassarre G (2013) A spiking neuron model of the cortico-basal ganglia circuits for goal-directed and habitual action learning. Neural Netw 41:212–224. 10.1016/j.neunet.2012.11.009 [DOI] [PubMed] [Google Scholar]
- Cressman VL, Balaban J, Steinfeld S, Shemyakin A, Graham P, Parisot N, Moore H (2010) Prefrontal cortical inputs to the basal amygdala undergo pruning during late adolescence in the rat. J Comp Neurol 518:2693–2709. 10.1002/cne.22359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo LA, Berns DS, DeLoach K, Luo L (2015) Connectivity of mouse somatosensory and prefrontal cortex examined with trans-synaptic tracing. Nat Neurosci 18:1687–1697. 10.1038/nn.4131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dindot SV, Antalffy BA, Bhattacharjee MB, Beaudet AL (2008) The Angelman syndrome ubiquitin ligase localizes to the synapse and nucleus, and maternal deficiency results in abnormal dendritic spine morphology. Hum Mol Genet 17:111–118. 10.1093/hmg/ddm288 [DOI] [PubMed] [Google Scholar]
- Dixon C, Sah P, Lynch JW, Keramidas A (2014) GABA A receptor α and γ subunits shape synaptic currents via different mechanisms. J Biol Chem 289:5399–5411. 10.1074/jbc.M113.514695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas RJ, Martin KA (2004) Neuronal circuits of the neocortex. Annu Rev Neurosci 27:419–451. 10.1146/annurev.neuro.27.070203.144152 [DOI] [PubMed] [Google Scholar]
- Dwyer JM, Maldonado-Avilés JG, Lepack AE, DiLeone RJ, Duman RS (2015) Ribosomal protein S6 kinase 1 signaling in prefrontal cortex controls depressive behavior. Proc Natl Acad Sci U S A 112:6188–6193. 10.1073/pnas.1505289112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa K, Kitagawa K, Inoue K, Takayama M, Takayama C (2012) Decreased tonic inhibition in cerebellar granule cells causes motor dysfunction in a mouse model of Angelman syndrome. Sci Transl Med 4:163ra157. 10.1126/scitranslmed.3004655 [DOI] [PubMed] [Google Scholar]
- Ehlen JC, Jones KA, Pinckney L, Gray CL, Burette S, Weinberg RJ, Evans JA, Brager AJ, Zylka MJ, Paul KN, Philpot BD, DeBruyne JP (2015) Maternal Ube3a loss disrupts sleep homeostasis but leaves circadian rhythmicity largely intact. J Neurosci 35:13587–13598. 10.1523/JNEUROSCI.2194-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elgersma Y. (2015) A molecular tightrope. Nature 526:50–51. 10.1038/526050b [DOI] [PubMed] [Google Scholar]
- Foot N, Henshall T, Kumar S (2017) Ubiquitination and the regulation of membrane proteins. Physiol Rev 97:253–281. 10.1152/physrev.00012.2016 [DOI] [PubMed] [Google Scholar]
- Gainey MA, Feldman DE (2017) Multiple shared mechanisms for homeostatic plasticity in rodent somatosensory and visual cortex. Philos Trans R Soc B Biol Sci 372:20160157. 10.1098/rstb.2016.0157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Economo MN, Chandrashekar J (2016) Long distance projections of cortical pyramidal neurons. J Neurosci Res 96:1467–1475. 10.1002/jnr.23978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greer PL, Hanayama R, Bloodgood BL, Mardinly AR, Lipton DM, Flavell SW, Kim TK, Griffith EC, Waldon Z, Maehr R, Ploegh HL, Chowdhury S, Worley PF, Steen J, Greenberg ME (2010) The Angelman syndrome protein Ube3A regulates synapse development by ubiquitinating arc. Cell 140:704–716. 10.1016/j.cell.2010.01.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammelrath L, Škokić S, Khmelinskii A, Hess A, van der Knaap N, Staring M, Lelieveldt BP, Wiedermann D, Hoehn M (2016) Morphological maturation of the mouse brain: an in vivo MRI and histology investigation. Neuroimage 125:144–152. 10.1016/j.neuroimage.2015.10.009 [DOI] [PubMed] [Google Scholar]
- Hayrapetyan V, Castro S, Sukharnikova T, Yu C, Cao X, Jiang YH, Yin HH (2014) Region-specific impairments in striatal synaptic transmission and impaired instrumental learning in a mouse model of Angelman syndrome. Eur J Neurosci 39:1018–1025. 10.1111/ejn.12442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensch TK. (2004) Critical period regulation. Annu Rev Neurosci 27:549–579. 10.1146/annurev.neuro.27.070203.144327 [DOI] [PubMed] [Google Scholar]
- Hogart A, Wu D, LaSalle JM, Schanen NC (2010) The comorbidity of autism with the genomic disorders of chromosome 15q11.2-q13. Neurobiol Dis 38:181–191. 10.1016/j.nbd.2008.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm MM, Nieto-Gonzalez JL, Vardya I, Vaegter CB, Nykjaer A, Jensen K (2009) Mature BDNF, but not proBDNF, reduces excitability of fast-spiking interneurons in mouse dentate gyrus. J Neurosci 29:12412–12418. 10.1523/JNEUROSCI.2978-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua Y, Sinha R, Martineau M, Kahms M, Klingauf J (2010) A common origin of synaptic vesicles undergoing evoked and spontaneous fusion. Nat Neurosci 13:1451–1453. 10.1038/nn.2695 [DOI] [PubMed] [Google Scholar]
- Huang HS, Allen JA, Mabb AM, King IF, Miriyala J, Taylor-Blake B, Sciaky N, Dutton JW Jr, Lee HM, Chen X, Jin J, Bridges AS, Zylka MJ, Roth BL, Philpot BD (2011) Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature 481:185–189. 10.1038/nature10726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunnicutt BJ, Jongbloets BC, Birdsong WT, Gertz KJ, Zhong H, Mao T (2016) A comprehensive excitatory input map of the striatum reveals novel functional organization. Elife 5:e19103. 10.7554/eLife.19103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inda MC, DeFelipe J, Muñoz A (2006) Voltage-gated ion channels in the axon initial segment of human cortical pyramidal cells and their relationship with chandelier cells. Proc Natl Acad Sci U S A 103:2920–2925. 10.1073/pnas.0511197103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Scanziani M (2011) How inhibition shapes cortical activity. Neuron 72:231–243. 10.1016/j.neuron.2011.09.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YH, Armstrong D, Albrecht U, Atkins CM, Noebels JL, Eichele G, Sweatt JD, Beaudet AL (1998) Mutation of the Angelman ubiquitin ligase in mice causes increased cytoplasmic p53 and deficits of contextual learning and long-term potentiation. Neuron 21:799–811. 10.1016/S0896-6273(00)80596-6 [DOI] [PubMed] [Google Scholar]
- Johnson CM, Loucks FA, Peckler H, Thomas AW, Janak PH, Wilbrecht L (2016) Long-range orbitofrontal and amygdala axons show divergent patterns of maturation in the frontal cortex across adolescence. Dev Cogn Neurosci 18:113–120. 10.1016/j.dcn.2016.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones KA, Han JE, DeBruyne JP, Philpot BD (2016) Persistent neuronal Ube3a expression in the suprachiasmatic nucleus of Angelman syndrome model mice. Sci Rep 6:28238. 10.1038/srep28238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Sosa-Pagan JO, Del Cid WA, Han JE, Philpot BD (2014) Allelic specificity of Ube3a expression in the mouse brain during postnatal development. J Comp Neurol 522:1874–1896. 10.1002/cne.23507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Wallace ML, Sidorov MS, Burette AC, Gu B, van Woerden GM, King IF, Han JE, Zylka MJ, Elgersma Y, Weinberg RJ, Philpot BD (2016) GABAergic neuron-specific loss of Ube3a causes Angelman syndrome-like EEG abnormalities and enhances seizure susceptibility. Neuron 90:56–69. 10.1016/j.neuron.2016.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Judson MC, Burette AC, Thaxton CL, Pribisko AL, Shen MD, Rumple AM, Del Cid WA, Paniagua B, Styner M, Weinberg RJ, Philpot BD (2017) Decreased axon caliber underlies loss of fiber tract integrity, disproportional reductions in white matter volume, and microcephaly in Angelman syndrome model mice. J Neurosci 37:7347–7361. 10.1523/JNEUROSCI.0037-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Jung JI, Rasband MN, Klann E (2011) Alterations in intrinsic membrane properties and the axon initial segment in a mouse model of Angelman syndrome. J Neurosci 31:17637–17648. 10.1523/JNEUROSCI.4162-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaphzan H, Buffington SA, Ramaraj AB, Lingrel JB, Rasband MN, Santini E, Klann E (2013) Genetic reduction of the α1 subunit of Na/K-ATPase corrects multiple hippocampal phenotypes in Angelman syndrome. Cell Rep 4:405–412. 10.1016/j.celrep.2013.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawaguchi Y, Kubota Y (1997) GABAergic cell subtypes and their synaptic connections in rat frontal cortex. Cereb Cortex 7:476–486. 10.1093/cercor/7.6.476 [DOI] [PubMed] [Google Scholar]
- Kim H, Kunz PA, Mooney R, Philpot BD, Smith SL (2016) Maternal loss of Ube3a impairs experience-driven dendritic spine maintenance in the developing visual cortex. J Neurosci 36:4888–4894. 10.1523/JNEUROSCI.4204-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kole MH, Letzkus JJ, Stuart GJ (2007) Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron 55:633–647. 10.1016/j.neuron.2007.07.031 [DOI] [PubMed] [Google Scholar]
- Kühnle S, Mothes B, Matentzoglu K, Scheffner M (2013) Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein arc. Proc Natl Acad Sci U S A 110:8888–8893. 10.1073/pnas.1302792110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupferschmidt DA, Juczewski K, Cui G, Johnson KA, Lovinger DM (2017) Parallel, but dissociable, processing in discrete corticostriatal inputs encodes skill learning. Neuron 96:476–489.e5. 10.1016/j.neuron.2017.09.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrakakis C, Rudolph U, De Koninck Y (2014) The heterogeneity in GABAA receptor-mediated IPSC kinetics reflects heterogeneity of subunit composition among inhibitory and excitatory interneurons in spinal lamina II. Front Cell Neurosci 8:424. 10.3389/fncel.2014.00424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laeremans A, Van de Plas B, Clerens S, Van den Bergh G, Arckens L, Hu TT (2013) Protein expression dynamics during postnatal mouse brain development. J Exp Neurosci 7:61–74. 10.4137/JEN.S12453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Chung C, Ha S, Lee D, Kim DY, Kim H, Kim E (2015) Shank3-mutant mice lacking exon 9 show altered excitation/inhibition balance, enhanced rearing, and spatial memory deficit. Front Cell Neurosci 9:94. 10.3389/fncel.2015.00094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Magueresse C, Monyer H (2013) GABAergic interneurons shape the functional maturation of the cortex. Neuron 77:388–405. 10.1016/j.neuron.2013.01.011 [DOI] [PubMed] [Google Scholar]
- Li KX, Lu YM, Xu ZH, Zhang J, Zhu JM, Zhang JM, Cao SX, Chen XJ, Chen Z, Luo JH, Duan S, Li XM (2011) Neuregulin 1 regulates excitability of fast-spiking neurons through Kv1.1 and acts in epilepsy. Nat Neurosci 15:267–273. 10.1038/nn.3006 [DOI] [PubMed] [Google Scholar]
- Lübke J, Roth A, Feldmeyer D, Sakmann B (2003) Morphometric analysis of the columnar innervation domain of neurons connecting layer 4 and layer 2/3 of juvenile rat barrel cortex. Cereb Cortex 13:1051–1063. 10.1093/cercor/13.10.1051 [DOI] [PubMed] [Google Scholar]
- Mátyás F, Lee J, Shin HS, Acsády L (2014) The fear circuit of the mouse forebrain: connections between the mediodorsal thalamus, frontal cortices and basolateral amygdala. Eur J Neurosci 39:1810–1823. 10.1111/ejn.12610 [DOI] [PubMed] [Google Scholar]
- Meng L, Person RE, Huang W, Zhu PJ, Costa-Mattioli M, Beaudet AL (2013) Truncation of Ube3a-ATS unsilences paternal Ube3a and ameliorates behavioral defects in the Angelman syndrome mouse model. PLoS Genet 9:e1004039. 10.1371/journal.pgen.1004039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller EK. (2000) The prefrontal cortex and cognitive control. Nat Rev Neurosci 1:59–65. 10.1038/35036228 [DOI] [PubMed] [Google Scholar]
- Miller EK, Cohen JD (2001) An integrative theory of prefrontal cortex function. Annu Rev Neurosci 24:167–202. 10.1146/annurev.neuro.24.1.167 [DOI] [PubMed] [Google Scholar]
- Miyamae T, Chen K, Lewis DA, Gonzalez-Burgos G (2017) Distinct physiological maturation of parvalbumin-positive neuron subtypes in mouse prefrontal cortex. J Neurosci 37:4883–4902. 10.1523/JNEUROSCI.3325-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morishima M, Kawaguchi Y (2006) Recurrent connection patterns of corticostriatal pyramidal cells in frontal cortex. J Neurosci 26:4394–4405. 10.1523/JNEUROSCI.0252-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nabel EM, Morishita H (2013) Regulating critical period plasticity: insight from the visual system to fear circuitry for therapeutic interventions. Front Psychiatry 4:146. 10.3389/fpsyt.2013.00146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogiwara I, Miyamoto H, Morita N, Atapour N, Mazaki E, Inoue I, Takeuchi T, Itohara S, Yanagawa Y, Obata K, Furuichi T, Hensch TK, Yamakawa K (2007) Nav1.1 localizes to axons of parvalbumin-positive inhibitory interneurons: a circuit basis for epileptic seizures in mice carrying an Scn1a gene mutation. J Neurosci 27:5903–5914. 10.1523/JNEUROSCI.5270-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotaru DC, Yoshino H, Lewis DA, Ermentrout GB, Gonzalez-Burgos G (2011) Glutamate receptor subtypes mediating synaptic activation of prefrontal cortex neurons: relevance for schizophrenia. J Neurosci 31:142–156. 10.1523/JNEUROSCI.1970-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothwell PE, Fuccillo MV, Maxeiner S, Hayton SJ, Gokce O, Lim BK, Fowler SC, Malenka RC, Südhof TC (2014) Autism-associated neuroligin-3 mutations commonly impair striatal circuits to boost repetitive behaviors. Cell 158:198–212. 10.1016/j.cell.2014.04.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudie JD, Shehzad Z, Hernandez LM, Colich NL, Bookheimer SY, Iacoboni M, Dapretto M (2012) Reduced functional integration and segregation of distributed neural systems underlying social and emotional information processing in autism spectrum disorders. Cereb Cortex 22:1025–1037. 10.1093/cercor/bhr171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Huynh TN, MacAskill AF, Carter AG, Pierre P, Ruggero D, Kaphzan H, Klann E (2013) Exaggerated translation causes synaptic and behavioural aberrations associated with autism. Nature 493:411–415. 10.1038/nature11782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Stryker MP (2010) Genomic imprinting of experience-dependent cortical plasticity by the ubiquitin ligase gene Ube3a. Proc Natl Acad Sci U S A 107:5611–5616. 10.1073/pnas.1001281107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert D, Martens GJ, Kolk SM (2014) Molecular underpinnings of prefrontal cortex development in rodents provide insights into the etiology of neurodevelopmental disorders. Mol Psychiatry 20:795–809. 10.1038/mp.2014.147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz LA, Patrick GN (2012) Ubiquitin-dependent endocytosis, trafficking and turnover of neuronal membrane proteins. Mol Cell Neurosci 49:387–393. 10.1016/j.mcn.2011.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shafritz KM, Bregman JD, Ikuta T, Szeszko PR (2015) Neural systems mediating decision-making and response inhibition for social and nonsocial stimuli in autism. Prog Neuropsychopharmacol Biol Psychiatry 60:112–120. 10.1016/j.pnpbp.2015.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd GM. (2013) Corticostriatal connectivity and its role in disease. Nat Rev Neurosci 14:278–291. 10.1038/nrn3469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva-Santos S, van Woerden GM, Bruinsma CF, Mientjes E, Jolfaei MA, Distel B, Kushner SA, Elgersma Y (2015) Ube3a reinstatement identifies distinct developmental windows in a murine Angelman syndrome model. J Clin Invest 125:2069–2076. 10.1172/JCI80554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slattery DA, Neumann ID, Cryan JF (2011) Transient inactivation of the infralimbic cortex induces antidepressant-like effects in the rat. J Psychopharmacol 25:1295–1303. 10.1177/0269881110368873 [DOI] [PubMed] [Google Scholar]
- Somogyi P, Tamás G, Lujan R, Buhl EH (1998) Salient features of synaptic organisation in the cerebral cortex. Brain Res Brain Res Rev 26:113–135. 10.1016/S0165-0173(97)00061-1 [DOI] [PubMed] [Google Scholar]
- Stanurova J, Neureiter A, Hiber M, de Oliveira Kessler H, Stolp K, Goetzke R, Klein D, Bankfalvi A, Klump H, Steenpass L (2016) Angelman syndrome-derived neurons display late onset of paternal UBE3A silencing. Sci Rep 6:30792. 10.1038/srep30792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stincic TL, Frerking ME (2015) Different AMPA receptor subtypes mediate the distinct kinetic components of a biphasic EPSC in hippocampal interneurons. Front Synaptic Neurosci 7:7. 10.3389/fnsyn.2015.00007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Zhu G, Liu Y, Standley S, Ji A, Tunuguntla R, Wang Y, Claus C, Luo Y, Baudry M, Bi X (2015) UBE3A regulates synaptic plasticity and learning and memory by controlling SK2 channel endocytosis. Cell Rep 12:449–461. 10.1016/j.celrep.2015.06.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Ikrar T, Davis MF, Gong N, Zheng X, Luo ZD, Lai C, Mei L, Holmes TC, Gandhi SP, Xu X (2016) Neuregulin-1/ErbB4 signaling regulates visual cortical plasticity. Neuron 92:160–173. 10.1016/j.neuron.2016.08.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takesian AE, Hensch TK (2013) Balancing plasticity/stability across brain development. Prog Brain Res 207:3–34. 10.1016/B978-0-444-63327-9.00001-1 [DOI] [PubMed] [Google Scholar]
- Thompson-Schill SL, Bedny M, Goldberg RF (2005) The frontal lobes and the regulation of mental activity. Curr Opin Neurobiol 15:219–224. 10.1016/j.conb.2005.03.006 [DOI] [PubMed] [Google Scholar]
- Tremblay R, Lee S, Rudy B (2016) GABAergic interneurons in the neocortex: from cellular properties to circuits. Neuron 91:260–292. 10.1016/j.neuron.2016.06.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urraca N, Cleary J, Brewer V, Pivnick EK, McVicar K, Thibert RL, Schanen NC, Esmer C, Lamport D, Reiter LT (2013) The interstitial duplication 15q11.2-q13 syndrome includes autism, mild facial anomalies and a characteristic EEG signature. Autism Res 6:268–279. 10.1002/aur.1284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Vaart T, Overwater IE, Oostenbrink R, Moll HA, Elgersma Y (2015) Treatment of cognitive deficits in genetic disorders: a systematic review of clinical trials of diet and drug treatments. JAMA Neurol 72:1052–1060. 10.1001/jamaneurol.2015.0443 [DOI] [PubMed] [Google Scholar]
- Van Wart A, Trimmer JS, Matthews G (2007) Polarized distribution of ion channels within microdomains of the axon initial segment. J Comp Neurol 500:339–352. 10.1002/cne.21173 [DOI] [PubMed] [Google Scholar]
- van Woerden GM, Harris KD, Hojjati MR, Gustin RM, Qiu S, de Avila Freire R, Jiang YH, Elgersma Y, Weeber EJ (2007) Rescue of neurological deficits in a mouse model for Angelman syndrome by reduction of alphaCaMKII inhibitory phosphorylation. Nat Neurosci 10:280–282. 10.1038/nn1845 [DOI] [PubMed] [Google Scholar]
- von Schoubye NL, Frederiksen K, Kristiansen U, Petersen AV, Dalby NO, Grunnet M, Jensen HS, Jespersen T, Sohal VS, Perrier JF (2018) The sodium channel activator lu AE98134 normalizes the altered firing properties of fast spiking interneurons in Dlx5/6+/− mice. Neurosci Lett 662:29–35. 10.1016/j.neulet.2017.10.004 [DOI] [PubMed] [Google Scholar]
- Wallace ML, Burette AC, Weinberg RJ, Philpot BD (2012) Maternal loss of Ube3a produces an excitatory/inhibitory imbalance through neuron type-specific synaptic defects. Neuron 74:793–800. 10.1016/j.neuron.2012.03.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace ML, van Woerden GM, Elgersma Y, Smith SL, Philpot BD (2017) Ube3a loss increases excitability and blunts orientation tuning in the visual cortex of Angelman syndrome model mice. J Neurophysiol 118:634–646. 10.1152/jn.00618.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wamsley B, Fishell G (2017) Genetic and activity-dependent mechanisms underlying interneuron diversity. Nat Rev Neurosci 18:299–309. 10.1038/nrn.2017.30 [DOI] [PubMed] [Google Scholar]
- Wang B, Ke W, Guang J, Chen G, Yin L, Deng S, He Q, Liu Y, He T, Zheng R, Jiang Y, Zhang X, Li T, Luan G, Lu HD, Zhang M, Zhang X, Shu Y (2016) Firing frequency maxima of fast-spiking neurons in human, monkey, and mouse neocortex. Front Cell Neurosci 10:239. 10.3389/fncel.2016.00239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, van Woerden GM, Elgersma Y, Borst JG (2017) Enhanced transmission at the calyx of held synapse in a mouse model for Angelman syndrome. Front Cell Neurosci 11:418. 10.3389/fncel.2017.00418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warden MR, Selimbeyoglu A, Mirzabekov JJ, Lo M, Thompson KR, Kim SY, Adhikari A, Tye KM, Frank LM, Deisseroth K (2012) A prefrontal cortex-brainstem neuronal projection that controls response to behavioural challenge. Nature 492:428–432. 10.1038/nature11617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wefelmeyer W, Puhl CJ, Burrone J (2016) Homeostatic plasticity of subcellular neuronal structures: from inputs to outputs. Trends Neurosci 39:656–667. 10.1016/j.tins.2016.08.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CA, Driscoll DJ, Dagli AI (2010) Clinical and genetic aspects of Angelman syndrome. Genet Med 12:385–395. 10.1097/GIM.0b013e3181def138 [DOI] [PubMed] [Google Scholar]
- Williams SR, Mitchell SJ (2008) Direct measurement of somatic voltage clamp errors in central neurons. Nat Neurosci 11:790–798. 10.1038/nn.2137 [DOI] [PubMed] [Google Scholar]
- Wise SP. (2008) Forward frontal fields: phylogeny and fundamental function. Trends Neurosci 31:599–608. 10.1016/j.tins.2008.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Atallah BV, Scanziani M (2014) Equalizing excitation-inhibition ratios across visual cortical neurons. Nature 511:596–600. 10.1038/nature13321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Riday TT, Condon KH, Roberts AC, Bernardo DR, Prakash R, Weinberg RJ, Ehlers MD, Philpot BD (2009) Ube3a is required for experience-dependent maturation of the neocortex. Nat Neurosci 12:777–783. 10.1038/nn.2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Berrios J, Newbern JM, Snider WD,Philpot BD, Hahn KM, Zylka MJ (2015) An autism-linked mutation disables phosphorylation control of UBE3A. Cell 162:795–807. 10.1016/j.cell.2015.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JJ, Paranjape SR, Walker MP, Choudhury R, Wolter JM, Fragola G, Emanuele MJ, Major MB, Zylka MJ (2017) The autism-linked UBE3A T485A mutant E3 ubiquitin ligase activates the Wnt/β-catenin pathway by inhibiting the proteasome. J Biol Chem 292:12503–12515. 10.1074/jbc.M117.788448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Mantegazza M, Westenbroek RE, Robbins CA, Kalume F, Burton KA, Spain WJ, McKnight GS, Scheuer T, Catterall WA (2006) Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat Neurosci 9:1142–1149. 10.1038/nn1754 [DOI] [PubMed] [Google Scholar]
- Yu J, Anderson CT, Kiritani T, Sheets PL, Wokosin DL, Wood L, Shepherd GM (2008) Local-circuit phenotypes of layer 5 neurons in motor-frontal cortex of YFP-H mice. Front Neural Circuits 2:6. 10.3389/neuro.04.006.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Xu M, Chang WC, Ma C, Hoang Do JP, Jeong D, Lei T, Fan JL, Dan Y (2016) Organization of long-range inputs and outputs of frontal cortex for top-down control. Nat Neurosci 19:1733–1742. 10.1038/nn.4417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H (2013) Altered neural connectivity in excitatory and inhibitory cortical circuits in autism. Front Hum Neurosci 7:609. 10.3389/fnhum.2013.00609 [DOI] [PMC free article] [PubMed] [Google Scholar]