

Abstract
Perineuronal nets (PNNs), composed mainly of chondroitin sulfate proteoglycans, are the extracellular matrix that surrounds cell bodies, proximal dendrites, and axon initial segments of adult CNS neurons. PNNs are known to regulate neuronal plasticity, although their physiological roles in cerebellar functions have yet to be elucidated. Here, we investigated the contribution of PNNs to GABAergic transmission from cerebellar Purkinje cells (PCs) to large glutamatergic neurons in the deep cerebellar nuclei (DCN) in male mice by recording IPSCs from cerebellar slices, in which PNNs were depleted with chondroitinase ABC (ChABC). We found that PNN depletion increased the amplitude of evoked IPSCs and enhanced the paired-pulse depression. ChABC treatment also facilitated spontaneous IPSCs and increased the miniature IPSC frequency without changing not only the amplitude but also the density of PC terminals, suggesting that PNN depletion enhances presynaptic GABA release. We also demonstrated that the enhanced GABAergic transmission facilitated rebound firing in large glutamatergic DCN neurons, which is expected to result in the efficient induction of synaptic plasticity at synapses onto DCN neurons. Furthermore, we tested whether PNN depletion affects cerebellar motor learning. Mice having received the enzyme into the interpositus nuclei, which are responsible for delay eyeblink conditioning, exhibited the conditioned response at a significantly higher rate than control mice. Therefore, our results suggest that PNNs of the DCN suppress GABAergic transmission between PCs and large glutamatergic DCN neurons and restrict synaptic plasticity associated with motor learning in the adult cerebellum.
SIGNIFICANCE STATEMENT Perineuronal nets (PNNs) are one of the extracellular matrices of adult CNS neurons and implicated in regulating various brain functions. Here we found that enzymatic PNN depletion in the mouse deep cerebellar nuclei (DCN) reduced the paired-pulse ratio of IPSCs and increased the miniature IPSC frequency without changing the amplitude, suggesting that PNN depletion enhances GABA release from the presynaptic Purkinje cell (PC) terminals. Mice having received the enzyme in the interpositus nuclei exhibited a higher conditioned response rate in delay eyeblink conditioning than control mice. These results suggest that PNNs regulate presynaptic functions of PC terminals in the DCN and functional plasticity of synapses on DCN neurons, which influences the flexibility of adult cerebellar functions.
Keywords: chondroitin sulfate proteoglycan, deep cerebellar nuclei, eyeblink conditioning, GABAergic transmission, motor learning, perineuronal net
Introduction
Perineuronal nets (PNNs) are a specialized form of extracellular matrix (ECM) and surround cell bodies, proximal dendrites, and axon initial segments of adult CNS neurons. Their composition includes chondroitin sulfate proteoglycans (CSPGs), tenascin-R, hyaluronan, and link proteins (Kwok et al., 2011; Soleman et al., 2013; Oohashi et al., 2015). PNNs have been reported to regulate neural plasticity in certain brain areas. In the mouse visual cortex, the degradation of CSPGs reactivates the ocular dominance plasticity (Pizzorusso et al., 2002; Carulli et al., 2010), and the developmental increase in the 4-sulfation/6-sulfation ratio of CSPGs regulates the critical period for ocular dominance plasticity (Miyata et al., 2012). In the amygdala, intact PNNs mediate the formation of erasure-resistant fear memories (Gogolla et al., 2009). The ECMs of hippocampal pyramidal neurons act as lateral diffusion barriers for AMPA receptors and increase the paired-pulse depression of excitatory synaptic transmission (Frischknecht et al., 2009). Hyaluronan not only modulates short-term plasticity but also facilitates the induction of long-term potentiation (LTP) at the hippocampal CA3–CA1 synapses by increasing the activity of L-type voltage-dependent Ca2+ channels (VDCCs; Kochlamazashvili et al., 2010). Removal of hyaluronan before fear conditioning impairs contextual fear memory (Kochlamazashvili et al., 2010).
In the cerebellum, large glutamatergic neurons in the deep cerebellar nuclei (DCN) are enwrapped by PNNs in which aggrecan is the predominant CSPG (Zaremba et al., 1990; Carulli et al., 2006, 2007; Zimmermann and Dours-Zimmermann, 2008; Foscarin et al., 2011; Bekku et al., 2012). Large DCN neurons receive direct GABAergic inhibition from Purkinje cells (PCs) and provide the sole output of the cerebellum to the brainstem and thalamus (Ito et al., 1964; Obata et al., 1967; Chan-Palay, 1977; de Zeeuw and Berrebi, 1996). After the relief of hyperpolarization mediated by PC inhibition, DCN neurons produce postinhibitory rebound firing (Llinás and Mühlethaler, 1988; Aizenman and Linden, 1999; McKay et al., 2005; Baumel et al., 2009; Hoebeek et al., 2010; Bengtsson et al., 2011), which contributes to the induction of synaptic plasticity, including long-term potentiation of excitatory synaptic transmission of mossy fibers to DCN neurons (Racine et al., 1986; Pugh and Raman, 2006, 2008; McElvain et al., 2010; Person and Raman, 2010). The synaptic plasticity between mossy fibers and DCN neurons has been suggested to play a crucial role in cerebellar motor learning in studies on ocular reflex adaptation (Miles and Lisberger, 1981; Kassardjian et al., 2005; Shutoh et al., 2006; Okamoto et al., 2011) and eyeblink conditioning (Krupa et al., 1993; Medina and Mauk, 1999; Christian and Thompson, 2003; Kistler and de Zeeuw, 2003). However, the physiological roles of PNNs, which enwrap PC–DCN neuron synapses in neuronal transmission within the DCN, as well as in cerebellar motor learning, have yet to be elucidated.
In this article, we examined whether PNNs regulate the synaptic physiology of the DCN and motor learning associated with the cerebellar system by combined use of electrophysiological recordings in slices and behavioral analyses. We found that acute enzymatic deletion of PNNs by chondroitinase ABC (ChABC) resulted in the enhancement of GABAergic transmission at synapses between PCs and large glutamatergic DCN neurons in acute slices, indicating persistent suppression of GABAergic transmission by PNNs in the adult DCN. To test whether this impacts cerebellar motor learning, we designed a setup for mouse eyeblink conditioning with eyelid movement detection using a high-speed camera (Heiney et al., 2014). Then we tested whether the depletion of PNNs in the interpositus nuclei, which are the DCN responsible for the delay eyeblink conditioning (Aksenov et al., 2004; Parker et al., 2009; Heiney et al., 2014), affects the behavior. We found that mice that received an interpositus ChABC injection exhibited a conditioned response (CR) at higher rates than in control mice. Thus, our results suggest that PNNs of the DCN suppress GABAergic transmission between PCs and large glutamatergic DCN neurons and impede motor learning in the adult cerebellum.
Materials and Methods
Animals
C57BL/6 male mice were purchased from Japan SLC and housed under a 12 h light/dark cycle. The generation of GAD67-GFP (Δneo) mice has been described previously (Tamamaki et al., 2003), and the heterozygous mice used in the present study were termed GAD67+/GFP mice. The transgenic lines were maintained on a C57BL/6 genetic background at our animal facility. All efforts were made to minimize the number of mice used and their suffering through the course of experiments. All the experimental procedures were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals described by the National Institutes of Health and approved by the Animal Research Committees on the care and use of animals in experiments in the RIKEN Brain Science Institute and Doshisha University.
Electrophysiology
Cerebellar slices from male C57BL/6 mice between postnatal day 18 (P18) and P30 were made, as described previously (Hirono et al., 2015). The mice were treated with CO2 and decapitated, and sagittal slices of the cerebellum 250 μm in thickness were cut using a vibrating microtome (catalog #VT1200S, Leica) in an ice-cold extracellular solution containing the following (in mm): 252 sucrose, 3.35 KCl, 21 NaHCO3, 0.6 NaH2PO4, 9.9 glucose, 0.5 CaCl2, and 10 MgCl2, and gassed with a mixture of 95% O2 and 5% CO2, pH 7.4. The slices were maintained at 30°C for 30 min in a holding chamber, where they were submerged in artificial CSF (ACSF) containing the following (in mm): 138.6 NaCl, 3.35 KCl, 21 NaHCO3, 0.6 NaH2PO4, 9.9 glucose, 2 CaCl2, and 1 MgCl2, bubbled with 95% O2 and 5% CO2 to maintain the pH at 7.4. Some slices were incubated in ACSF with ChABC (0.25 U/ml; catalog #C3667, Sigma-Aldrich) for >3 h before recordings. Individual slices were transferred to a recording chamber attached to the stage of a microscope (catalog #BX51WI, Olympus) and superfused with ACSF. IPSCs were observed by whole-cell voltage-clamp recordings. To isolate IPSCs, a nonselective ionotropic glutamate receptor antagonist, kynurenic acid (1 mm), was added to the ACSF. To detect stimulation-evoked IPSCs (eIPSCs) and spontaneous IPSCs (sIPSCs) as outward current responses, patch pipettes (2–4 MΩ) were filled with an internal solution containing the following (in mm): 140 CsCH3SO3, 5 CsCl, 0.1 CaCl2 1 K-EGTA, 10 Na-HEPES, 10 phosphocreatine, 0.6 QX-314, 3 Mg-ATP, and 0.4 Na-GTP, pH 7.4. The holding potential was set at −10 mV. To observe eIPSCs, focal stimulation with a single electrical pulse (height, 30 V; duration, 0.05 ms) was applied by a concentric bipolar electrode (FHC) to the white matter. Miniature IPSCs (mIPSCs) were recorded using a CsCl-based (140 mm) internal solution, and the holding potential was set at −70 mV in the presence of kynurenic acid (1 mm) and tetrodotoxin (0.5 μm). Synaptic currents were recorded from large DCN neurons with somatic diameters of >20 μm, since most of which are thought to correspond to the glutamatergic projection neurons in the DCN (Telgkamp and Raman, 2002; Uusisaari et al., 2007). Series resistance (10–18 MΩ) was compensated by 70% and monitored using 2 mV hyperpolarizing voltage steps, and experiments were discarded if the value changed by ~20%. Membrane potentials were recorded from large DCN neurons in the current-clamp mode with an internal solution containing the following (in mm): 120 K-gluconate, 9 KCl, 10 KOH, 4 NaCl, 10 Na-HEPES, 17.5 sucrose, 10 phosphocreatine, 3 Mg-ATP, and 0.4 Na-GTP, pH 7.4. Rebound firing of these DCN neurons was examined after the termination of strong hyperpolarization induced by electrical repetitive stimulation of PC axons (100 Hz for 100 ms) through the concentric bipolar electrode positioned in the nearby white matter. To eliminate contributions of the activation of ionotropic glutamate receptors and group I mGluRs to rebound firing of DCN neurons, we bath applied their blockers, 5 μm NBQX, 30 μm dl-APV, and 100 μm 7-(hydroxyimino) cyclopropa[b] chromen-1a-carboxylate ethyl ester (Zheng and Raman, 2011). To keep the prestimulus firing rate between 10 and 20 Hz, constant holding current (−300 to 0 pA) was applied. All experiments were performed at room temperature (24–26°C). The membrane signals were recorded using a MultiClamp 700B amplifier (Molecular Devices) and pCLAMP 10.3 software (Molecular Devices). The current signals, which were filtered at 2–4 kHz, were sampled at 10–20 kHz. Synaptic events were analyzed with a threshold of 10 pA. The potential signals were sampled at 50 kHz.
Immunohistochemistry
Cat-301 labeling.
Under pentobarbital anesthesia (i.p., 60 mg/kg body weight, Nembutal, Dainippon-Sumitomo Pharma), GAD67+/GFP mice (P14 to 3 months) were perfused transcardially with 50 ml of PBS, followed by 50 ml of 4% paraformaldehyde/PBS. Their brains were excised, postfixed, and equilibrated with 20% sucrose in PBS. Frozen sections were prepared at 50 μm thickness using a cryostat microtome (CM3050S, Leica) and mounted on MAS-coated glass slides. The sections were permeabilized with 0.3% Triton X-100 in PBS for 15 min and treated with a blocking buffer, 5% skim milk in PBS, for 1 h. Then the sections were incubated overnight with a mouse monoclonal antibody against aggrecan (clone Cat-301, 1:2000; catalog #MAB5284, Millipore; RRID:AB_2219944; Matthews et al., 2002; Carulli et al., 2007) at 4°C in PBS containing 1% skim milk and then stained with Alexa Fluor 555-conjugated anti-mouse antiserum (1:500; catalog #A-21422, Thermo Fisher Scientific; RRID:AB_2535844) for 3 h at room temperature. For histological observation, a microscope (model BX61, Olympus) and a confocal laser-scanning microscope (model FV1000D, Olympus) were used. Images were generated by stacks of 7–10 captured sections spaced by 1.67 μm.
Wisteria floribunda agglutinin labeling.
Mouse cerebellar acute slices (250 μm thick) were treated with ChABC (0.25 U/ml) and the control solution (ACSF) for 4 h, then fixed with 4% paraformaldehyde/PBS for 2 h at room temperature. The slices were incubated overnight with biotinylated Wisteria floribunda agglutinin (WFA) lectin (1:500; catalog #B-1355, Vector Laboratories; RRID:AB_2336874) at 4°C. Subsequently, the slices were exposed to Alexa Fluor 594-conjugated streptavidin (1:100; catalog #S-11227, Thermo Fisher Scientific; RRID:AB_2313574) for 3 h at room temperature. WFA morphological images were obtained using an epifluorescence microscope (model IX73, Olympus) with a 40×/0.75 numerical aperture (NA) objective lens and an Andor Zyla5.5 camera.
Quantification of size and density of PC terminals
Mouse cerebellar acute slices (250 μm thick) were treated with ChABC (0.25 U/ml) and the control solution (ACSF) for 4–6 h, then fixed with 4% paraformaldehyde/PBS for 2 h at room temperature. The slices were cut at 50 μm thickness using a Vibratome 1000 (Vibratome). The sections were incubated overnight with primary antibodies, mouse SMI32 (1:250; catalog #SMI-32P, Covance Research Products; RRID:AB_2314912) to label DCN projection neurons and rabbit anti-calbindin D-28k (1:4000; catalog #CB38, Swant; RRID:AB_10000340) to label PCs, at 4°C. Subsequently, the sections were incubated with secondary antibodies conjugated to fluorophores, goat anti-rabbit Alexa Fluor 488 (1:500; catalog #A-11034, Thermo Fisher Scientific; RRID:AB_2576217) and anti-mouse Alexa Fluor 594 (1:500; catalog #A-11032, Thermo Fisher Scientific; RRID:AB_2534091) for 3 h at room temperature. After immunofluorescence reaction against SMI32 and calbindin D-28k, the sections were observed using a confocal laser-scanning microscope (model FV1200, Olympus), using a 100× oil-immersion objective lens (NA, 1.35; UPLSAPO 1000XS, Olympus) with appropriate lasers (473 and 559 nm) and filters (the emission window was 490–590 nm for Alexa Fluor 488 or 575–620 nm for Alexa Fluor 594). The z-stack digital images (512 × 94) were captured with the z-step size set at 250 nm.
Surgery
Mice were deeply anesthetized by inhalation of isoflurane (4% induction, 1.5% maintenance; Mylan), and fixed to a stereotaxic apparatus (model SR-6N, Narishige). Each mouse received surgeries, one for injection of drug and another for attachment of a platform for the eyeblink conditioning. Another six male mice at P21–P23 received only surgery for injection and were killed 4–6 d after the single injection for electrophysiology.
Drug injection into the interpositus nuclei in vivo.
ChABC was dissolved in sterile Ringer's solution at 10 U/ml with Alexa Fluor 488 (50 μm; A-10436, Thermo Fisher Scientific) to histologically identify injection site. Under isoflurane anesthesia described above, ChABC was injected into the interpositus nuclei bilaterally (10 U/ml, 0.6 μl/side) through a micropipette (tip diameter, 50–80 μm) made of borosilicate glass via polyethylene tubing filled with Fluorinert (catalog FC-3283, 3M) connected to a 1 μl microsyringe (model 7001 KH, Hamilton). The micropipette was mounted on a standard micromanipulator attached to the stereotaxic apparatus and inserted into the bilateral interpositus nuclei (anteroposterior, 6.0 mm; mediolateral, ±1.8 mm; dorsoventral, −2.7 mm), referring to the atlas of the mouse brain (Paxinos and Franklin, 2001). The flow rate was regulated manually at 0.1 μl/min. As a control, the same amount of Ringer's solution was infused into the bilateral interpositus nuclei. Five mice were injected with ChABC or Ringer's solution under blind conditions, and 6 d after the injection were subjected to delay eyeblink-conditioning experiments. Six young mice (P21–P23) were injected with ChABC or Ringer's solution, and acute cerebellar slices were obtained 4–6 d after the single injection for electrophysiological experiments.
Eyeblink conditioning in head-fixed mice
We trained head-fixed mice for eyeblink conditioning (Koekkoek et al., 2002; Heiney et al., 2014; Boele et al., 2010). Sixteen male C57BL/6 mice (8–12 weeks old) were used. Under isoflurane anesthesia, as described above, a platform for head fixation equipped with one stainless small bolt (diameter, 3 mm; length, 15 mm) was made with synthetic resin (Super-Bond C&B, Sun Medical) on the cranial bone covering the cerebrum under aseptic conditions (Okamoto et al., 2011). No less than 2 d after the operation, the mouse received the habituation training for eyelid conditioning.
The general setup for the conditioning of eyelid response is shown in Figure 1. Mice were allowed to run freely on a running plastic wheel (diameter, 12 cm), which was covered with nonslip sheet. The head was fixed to the bar, which was supported by a pole (diameter, 12 mm; height, 23 cm) with a magnetic stand fixed to a stainless plate table. A 21 gauge (G) needle for the air puff was connected with a small cable to the N2 gas bomb. The tip of the needle was set in front of the anterior dorsal corner of the left eye with a manipulator. An air puff (15–60 psi) of 30 ms duration was applied through a pressure valve Microinjector IM300 (Narishige). The tone signal (frequency, 4 kHz; maximum sound intensity; 80 dB SPL) was generated and amplified through an analog-to-digital/digital-to-analog (A/D–D/A) board (catalog #PCI-6052E, National Instruments) mounted on a Windows personal computer, delivered through an SCB-68 interface box (National Instruments) under the control of LabVIEW software (version 14.0, National Instruments; RRID:SCR_014325), and given to the mouse through the speaker set 5 cm over the mouse head. The eyelid movements were recorded by a high-speed monochrome CCD camera (model MiCAM02-HR, Brain Vision) placed in front of the left eye. Eyelid responses were monitored with 96 × 64 pixel spatial resolution with 2.2 ms interframe interval (IFI). The tone generator and pressure valve for air puff, and the recording of eyelid movement were controlled by the clock pulses generated by the A/D–D/A board mounted on the Windows personal computer.
Figure 1.

Experimental setup for mouse eyeblink conditioning and recording.
First, mice were habituated to the head-fixed condition and placed on the top of the wheel, exposed to a background white noise of 65 dB for 40 min for 3 d. Then, daily sessions of conditioning started. A single daily session consisted of 10 blocks, and each block was composed of 10 trials, which included nine pairs of a conditioned stimulus (CS) and an unconditioned stimulus (US) and one CS alone at the 10th trial. The intertrial interval was randomized at between 20 and 30 s, with the mean being 25 s. The 300 ms duration of tone and air puff of 30 ms duration at 270 ms from the start of the tone were respectively delivered as CS and US. The air-puff pressure was carefully calibrated and increased (from 15 to 60 psi) to elicit an eyeblink and were adjusted daily for first 3 d of sessions. Thereafter, all of these mice were conditioned successively with US pressure (60 psi) for 7 d followed by an additional 3 d for extinction. The startle response to a tone was evaluated 50 ms after CS onset. The extinction of conditioning was examined from day 11 to day 13 by presenting a CS 100 times without a US.
Experimental design and statistical analysis
Throughout the manuscript, all data are expressed as the mean ± SEM. All statistical analyses were performed using Kyplot software version 5.0 (Kyence) or GraphPad Prism software (RRID:SCR_000306). Throughout the data analyses of electrophysiological signals, n refers to the number of recorded neurons, and the numbers of slices and animals are shown as slices/mice (i.e., n = 7 from 7/4 means that 7 neurons were recorded from 7 slices of 4 mice). All the electrical signals were analyzed using Mini Analysis Program 6.0 (Synaptosoft; RRID:SCR_002184), pCLAMP version 10.3 software, and Kyplot software 5.0 (Kyence). Statistical analyses for the intergroup comparison (e.g., ChABC vs control) were performed using an unpaired Student's t test, whereas those for the intragroup comparison were performed using a paired Student's t test. We also used two-way repeated-measures ANOVA for some experiments involving both intergroup and intragroup differences.
For the analysis of synaptic depression to a train of 100 Hz stimuli, the amplitudes of phasic IPSCs were normalized to that of the first IPSC. Because of this, we anticipated that two-way ANOVA would report significant interaction between the control and ChABC groups if the depression really differs, and that this would make the interpretation of group difference difficult. Therefore, we chose to use regression analyses, as we can assume the depression as exponential decay. We used a null hypothesis that a single exponential function with a shared slope and plateau can fit the data from both groups better than separate functions. Rejection of this would therefore lead to the interpretation that two parameters are different between the datasets. The comparison was done with the extra sum-of-squares F test.
To analyze WFA labeling (Fig. 2D), WFA fluorescence intensity in a ChABC-treated slice was normalized to the respective control slice. The intensities were measured from five slice pairs from four mice by ImageJ (RRID:SCR_003070). The background brightness, taken from a nonstained region of the cerebellar molecular layer, was subtracted from the brightness of the DCN (Foscarin et al., 2011). The level of significance was determined by the unpaired Student's t test between groups. Throughout quantification of the size and density of PC terminals (see Fig. 6A–E), the images of PC terminals were analyzed using the free software Fiji (a distribution of ImageJ; RRID:SCR_002285; Schindelin et al., 2012). First, the pixel intensity was adjusted using a window/level function, and noise was removed using Gaussian blur filters and background subtraction. After appropriate thresholding of pixel intensities, the contours of cell bodies and terminals were delineated using an analyze particle function. Each terminal contour was manually checked and corrected, if necessary, to extract a single terminal as a single contour. Perimeters, size, and number of the terminal contours were measured. The number of terminals per cell body was represented as the density of terminals per 10 μm perimeters of the cell body. Statistical comparison between control and ChABC-treated neurons was accomplished by the unpaired Student's t test.
Figure 2.

Expression of PNNs in the mouse DCN. A, Expression of CSPGs in the developing mouse cerebellum. Cat-301 is an antibody against aggrecan, one of the primary CSPGs. Bottom, High-magnification images of the DCN in the top. Red, Cat-301; green, GFP of GAD67+/GFP mouse. B, Large DCN neuron expresses CSPGs. Red, Cat-301; green, GFP of GAD67+/GFP mouse. C, ChABC decreased PNNs labeled by WFA. Mouse cerebellar acute slices with (right) and without (left) 4 h treatment of ChABC (0.25 U/ml). D, Intensity of WFA staining in the slices treated with ChABC for 4 h normalized to the average intensity of control slices. *p < 0.05 using unpaired Student's t test.
Figure 6.
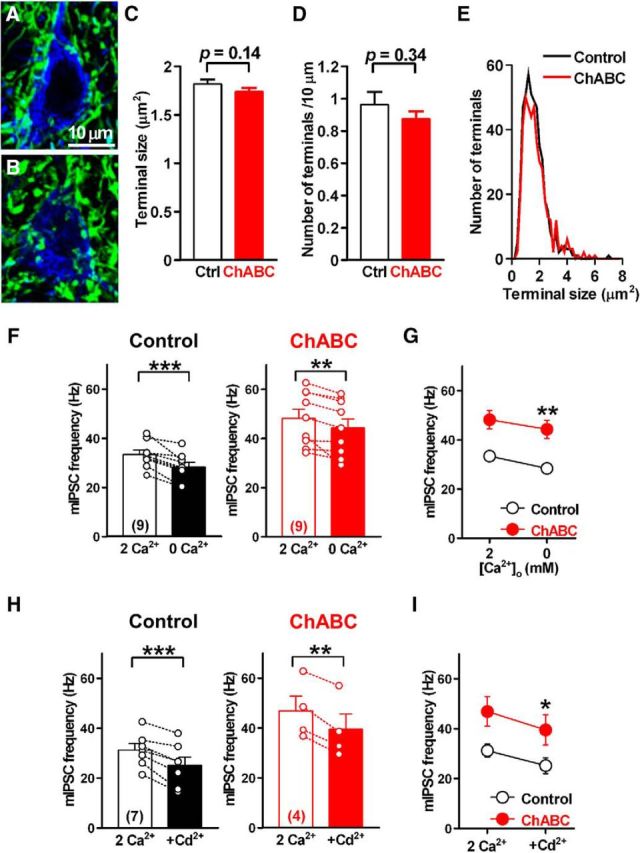
Effects of PNN depletion on PC terminals and extracellular Ca2+ dependency of mIPSCs. A, B, Large DCN neurons and PC terminals in slices treated with normal ACSF (A) and ChABC (B) for 4 h. C, Quantification of the size of PC axon terminals. D, Quantification of the density of PC axon terminals. E, Size distribution of PC axon terminals in slices treated with normal ACSF (black line, n = 428 terminals from 15 cells/3 mice) and ChABC (red line, n = 458 terminals from 15 cells/3 mice) for 4–6 h. F, Effect of ChABC treatment on extracellular Ca2+ dependency of mIPSCs in large DCN neurons. Extracellular Ca2+ concentration was changed from 2 mm to nominal 0 mm, and recordings were made after a 15 min incubation. The mean frequencies of mIPSCs recorded from large DCN neurons with (right) and without (left) ChABC treatment are shown. **p < 0.01, ***p < 0.001 using paired Student's t test. G, Summary of mIPSC frequency data in F. **p < 0.01, two-way repeated-measures ANOVA with Sidak post hoc test. H, Effect of the nonselective voltage-dependent Ca2+ channel blocker cadmium (100 μm) on mIPSC frequencies. The mean frequencies of mIPSCs recorded from large DCN neurons with (right) and without (left) ChABC treatment are shown. **p < 0.01, ***p < 0.001 using paired Student's t test. I, Even in the presence of cadmium, the mean frequency of mIPSCs in ChABC-treated slices was still higher than that in the control slices. *p < 0.05 using two-way repeated-measures ANOVA with Sidak post hoc test. Ctrl, Control.
Eyelid responses were analyzed off-line using MATLAB software (MathWorks; RRID:SCR_001622). The images of the eye were binarized, and the boundary of the open eye was drawn on the first image of each session (see Fig. 9B). The eyelid closure index was calculated by dividing the black area within the boundary in each image by the total area inside the boundary, and subtracting it from 1. The SD of the baseline of the black area was calculated for the 100 ms baseline period before CS onset (0.1–0.2 s), and a threshold for the detection of eyelid closure was set at twice the SD of the baseline area above. Eyelid closure within 50 ms from the CS onset (0.2–0.25 s) was counted as a nonspecific startle response, and that between 50 and 270 ms from CS onset (0.25–0.47 s) was counted as the CR. Large eyelid responses, which occurred sometimes for a 200 ms baseline period before CS onset (0.0–0.2 s), were excluded from the analysis. A two-way repeated-measures ANOVA with Sidak post hoc test was used to examine differences between the ChABC-injected and control mice.
Figure 9.

ChABC injection facilitates learning of the delay eyeblink-conditioned response in mice. A, Eyelid movement of a control mouse, monitored with a high-speed camera during a paired CS–US trial in session 9, a: An open eye before the CS onset. b: A conditioned eyelid response. c: An eyelid closure to the US. B, Example image of an eyelid movement during delay eyeblink conditioning. Each image was obtained at one of the time points indicated in A. Bottom, Images of an example trial binarized by setting a threshold such that the pupil and iris have a value of 0. C, Learning curves of delay eyeblink conditioning. Normal saline or ChABC (10 U/ml) was injected into the interpositus nuclei before a 3 d habituation period. All mice received acquisition training for 10 d (sessions 1–10) and extinction training for 3 d (sessions 11–13). Left, The black trace is the averaged CR% of control mice (n = 7). The gray traces are the CR% for individual control mice. Right, The red trace is the averaged CR% of mice that received ChABC injection (n = 9). The light red traces are the CR% for individual ChABC-injected mice. D, Mean success rates of CR acquisition (circles) and mean rates of startle eyeblink responses (squares) in the trained mice are plotted. Before training (in session 0), basal eyeblink responses were recorded without tone and air puff. The averaged CR% of the mice having received ChABC injection into the interpositus nuclei was higher than that of the control mice. ††p < 0.01 using two-way repeated-measures ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001 using Sidak post hoc test.
Results
Developmental expression of PNNs in the mouse DCN and their removal by ChABC
We hypothesized that PNNs regulate the synaptic transmission between PCs and glutamatergic DCN neurons and sought to test it by removing PNNs in acute cerebellar slices for electrophysiological recordings. First, we determined the age at which PNNs are fully formed around large glutamatergic DCN neurons in mice by immunolabeling of aggrecan, a major component of PNNs in the DCN (Zaremba et al., 1990; Carulli et al., 2006, 2007; Zimmermann and Dours-Zimmermann, 2008; Foscarin et al., 2011; Bekku et al., 2012). We prepared sagittal cerebellar slices from mice at various ages from 14 d to 3 months, and fixed them to label aggrecan. We used GAD67+/GFP mice (Tamamaki et al., 2003) to discriminate between GABAergic and glutamatergic neurons. In the DCN, the expression of aggrecan increased between P14 and P18, and glutamatergic DCN neurons were fully enwrapped by PNNs by P19 (Fig. 2A,B), which is consistent with the findings of a previous report (Carulli et al., 2007). Therefore, we used acute cerebellar slices taken from mice of age P18 to P30 for the subsequent experiments.
Next, to deplete PNNs, we treated cerebellar slices with ChABC (0.25 U/ml), which digests and removes the glycosaminoglycan chains from the core proteins in PNNs (Kwok et al., 2011). Therefore, we applied WFA labeling to recognize PNN expression. The intensity of WFA labeling was decreased by incubation with ChABC, whereas incubating with the control solution (ACSF) did not cause such a reduction in the labeling: the normalized intensity of WFA labeling in the DCN treated with ChABC for 4 h was 0.70 ± 0.04 (n = 5 slice pairs from four mice; t(8) = 2.20, p = 0.030, unpaired Student's t test; Fig. 2C,D). Therefore, we treated acute slices with ChABC for 3–6 h for the electrophysiological experiments.
Effects of PNN depletion on eIPSCs in DCN neurons
To examine the effects of PNN depletion on GABAergic transmission between PCs and glutamatergic DCN neurons, we applied whole-cell voltage-clamp recordings to DCN neurons in acute cerebellar slices incubated with ChABC for 3–6 h. We recorded eIPSCs from large (>20 μm somatic diameter) DCN neurons, which are primarily glutamatergic (Telgkamp and Raman, 2002; Uusisaari et al., 2007), with the voltage clamped at −10 mV by applying paired pulse stimuli to PC axon fibers through a stimulation electrode placed in the white matter near the DCN (Fig. 3A). In the control condition, the mean amplitude of eIPSCs at the first stimulus was 500 ± 112 pA (n = 10 from 10 slices/8 mice), and the average paired-pulse ratio (PPR) was 0.97 ± 0.01 (n = 8 from 8/6). Meanwhile, ChABC treatment increased the amplitude of the first eIPSCs to 956 ± 107 pA (n = 8 from 8/6; t(16) = 2.9, p = 0.011, unpaired Student's t test) and decreased the PPR to 0.86 ± 0.03 (n = 7 from 7/5; t(13) = 3.3, p = 0.0058, unpaired Student's t test) without changing the rise time (0.58 ± 0.02 ms, n = 8 from 8/6 vs 0.54 ± 0.03 ms; 107% of the control, n = 10 from 10/8; t(16) = 1.0, p = 0.33, unpaired Student's t test) or the decay time (6.9 ± 0.5 ms, n = 8 from 8/6 vs 8.5 ± 0.6 ms, n = 10 from 10/8; 81% of the control; t(16) = 1.9, p = 0.072, unpaired Student's t test; Fig. 3A–C). We further examined the effect of ChABC treatment on the depression of eIPSCs evoked by a stimulation of 10 pulses at 100 Hz. ChABC-treated large DCN neurons showed stronger depression (n = 7 from 7/7) compared with the control (n = 8 from 8/6; Fig. 3D,E). Regression analysis using the one-phase exponential decay model indicates that their decay kinetics (slopes and plateaus) are significantly different between the two groups (F(2,144) = 12.86, p < 0.0001). These effects were not observed in intermediate DCN neurons (Uusisaari et al., 2007; Fig. 4), suggesting that the effect of PNN depletion is specific to PC synapses onto large glutamatergic DCN neurons. Together with the increase in eIPSC amplitude and the reduction in the PPR, these results suggest that the depletion of PNNs increases the release probability of GABA at the presynaptic terminals of PCs onto large glutamatergic DCN neurons.
Figure 3.
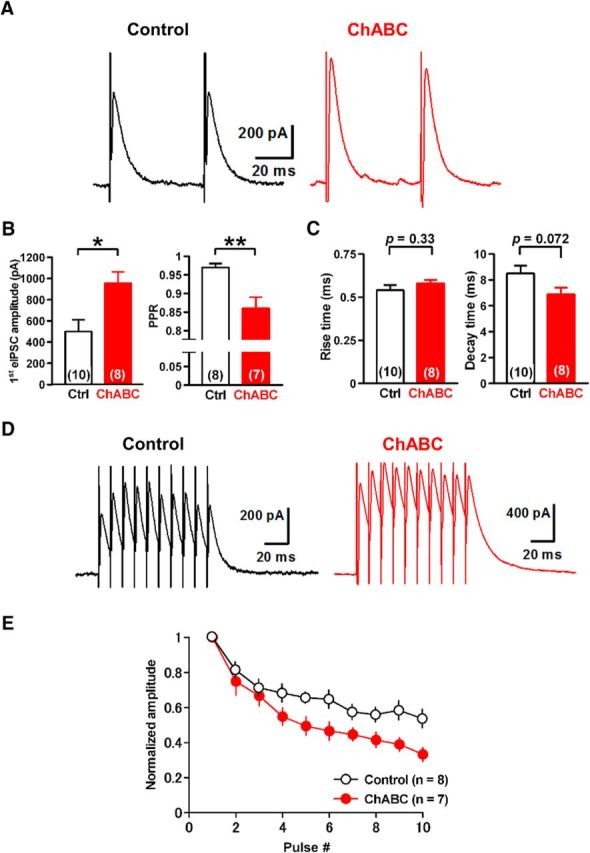
Depletion of PNNs alters GABAergic transmission onto large DCN neurons. A, Representative traces of eIPSCs in large DCN neurons with (right) and without (left) incubation of ChABC (0.25 U/ml). B, ChABC treatment increased the amplitude and reduced the PPR of eIPSCs significantly. *p < 0.05, **p < 0.01 using unpaired Student's t test. C, No effect on rise (left) or decay (right) kinetics of eIPSCs. D, Representative averaged eIPSCs induced by 10 stimuli at 100 Hz in large DCN neurons. E, Mean amplitudes of phasic eIPSCs normalized to the first phasic IPSC in each condition. ChABC treatment enhanced the depression of eIPSCs (p < 0.0001 using regression analysis). Ctrl, Control.
Figure 4.

Depletion of PNNs has no significant effect on eIPSCs in intermediate DCN neurons. A, Representative traces of eIPSCs in intermediate neurons in the DCN with (right) and without (left) incubation of ChABC (0.25 U/ml). B, ChABC treatment did not alter the amplitude (left) or the PPR (right) of eIPSCs in intermediate DCN neurons. C, No effect on the rise (left) or decay (right) kinetics of eIPSCs. D, Representative averaged IPSCs evoked by 10 stimuli at 100 Hz in intermediate DCN neurons with (right) or without (left) ChABC treatment. E, Mean amplitudes of phasic eIPSCs normalized to the first phasic IPSC. ChABC treatment had no effect on the depression of eIPSCs (p = 0.14 using regression analysis). Ctrl, Control.
Effects of PNN depletion on miniature and spontaneous IPSCs in DCN neurons
To further examine whether PNNs regulate the presynaptic release probability, we recorded mIPSCs from large DCN neurons voltage clamped at −70 mV with or without ChABC treatment. ChABC-treated large DCN neurons showed a robust increase in the mIPSC frequency (48.5 ± 2.7 Hz, n = 15 from 14/8 vs 31.7 ± 1.8 Hz, n = 14 from 14/10; 153% of the control; t(27) = 5.1, p = 0.000025, unpaired Student's t test) without significant changes in the mean amplitude (49.8 ± 4.6 pA, n = 15 from 14/8 vs 39.7 ± 1.7 pA, n = 14 from 14/10; 125% of the control; t(27) = 2.0, p = 0.057, unpaired Student's t test; Fig. 5A,B), the rise time (0.51 ± 0.03 ms, n = 15 from 14/8 vs 0.55 ± 0.03 ms, n = 14 from 14/10; 92% of the control; t(27) = 1.2, p = 0.23, unpaired Student's t test), and the decay time of mIPSCs (4.4 ± 0.3 ms, n = 15 from 14/8 vs 3.8 ± 0.3 ms, n = 14 from 14/10; 116% of the control; t(27) = 1.5, p = 0.13, unpaired Student's t test). Together with the decrease in PPR, the increase in mIPSC frequency suggests that PNN depletion increases the release probability from PC presynaptic terminals.
Figure 5.

Depletion of PNNs increases presynaptic release events in large DCN neurons. A, Representative traces of mIPSCs in large DCN neurons with (right) and without (left) treatment of ChABC. B, ChABC treatment increased the mean frequency (left), but not the mean amplitude (right), of mIPSCs. C, Representative traces of sIPSCs in large DCN neurons. ChABC treatment facilitated sIPSCs (right). D, Mean frequency (left) and amplitude (right) of sIPSCs. ChABC increased both the frequency and amplitude of sIPSCs significantly. *p < 0.05, ***p < 0.001 using unpaired Student's t test. Ctrl, Control.
To confirm this in a more physiological context, we analyzed sIPSCs in large DCN neurons voltage clamped at −10 mV after ChABC treatment and found a similar increase in the frequency of sIPSCs (30.6 ± 0.7 Hz, n = 8 from 8/6 vs 24.1 ± 2.6 Hz, n = 10 from 10/8; 127% of the control; t(16) = 2.2, p = 0.041, unpaired Student's t test). In contrast to the amplitude of mIPSCs, however, that of sIPSCs increased in ChABC-treated cells (30.5 ± 4.0 pA, n = 8 from 8/6 vs 21.7 ± 1.9 pA, n = 10 from 10/8; 141% of the control; t(16) = 2.1, p = 0.050, unpaired Student's t test; Fig. 5C,D). Since the mIPSC frequency is greatly increased, and considering the width of a single sIPSC event, we estimate based on the Poisson distribution that the increase in sIPSC amplitude results mainly from the increase in the probability of having overlapping single sIPSCs (from ~0.02 in control to 0.1 in ChABC-treated cells). These results therefore support the hypothesis that PNN depletion increases the release probability at presynaptic terminals without changing postsynaptic GABAergic responses.
Investigating the potential mechanism of presynaptic enhancement by PNN removal
The number of PC terminals in the DCN is reported to be significantly altered in mice deficient in PNN components, including Bral2 and Crtl1 (Bekku et al., 2003, 2012; Foscarin et al., 2011; Stamenkovic et al., 2017). To examine whether the morphological changes in PC terminals contacting large DCN neurons contribute to the ChABC-mediated facilitation of GABA release, we performed immunohistochemical studies of the DCN in the control and ChABC-treated slices and assessed the size and density of PC terminals. ChABC treatment for 4–6 h did not alter the size (1.74 ± 0.03 μm2, n = 458 terminals from 15 cells/3 mice vs 1.82 ± 0.04 μm2, n = 428 terminals from 15 cells/3 mice; 96% of the control; t(894) = 1.5, p = 0.14, unpaired Student's t test) or the density (0.88 ± 0.05 terminals/10 μm, n = 15 cells from 3 mice vs 0.96 ± 0.08 terminals/10 μm, n = 15 cells from 3 mice; 91% of the control; t(28) = 0.96, p = 0.34, unpaired Student's t test) of PC terminals compared with those of the control slices (Fig. 6A–D). This was also evidenced by the similar size distributions of PC axon terminals in control and ChABC-treated slices (Fig. 6E). Therefore, ChABC treatment for 4–6 h did not induce a morphological change in PC terminals in the DCN.
To investigate the mechanism of the increase in release probability due to PNN removal, we examined the effect of ChABC treatment on mIPSCs in a nominally Ca2+-free ACSF. Since PNNs have been shown to act as an extracellular Ca2+ buffer (Hrabetová et al., 2009; Nicholson and Hrabětová, 2017), we speculated that removing PNNs might increase the availability of extracellular Ca2+ to PC presynaptic terminals, thereby increasing the release probability. To test this, we changed the extracellular Ca2+ concentration from the normal 2 mm to a nominal 0 mm. If PNNs act as a Ca2+ buffer, decreasing the Ca2+ concentration would reduce the mIPSC frequency to similar levels in both control and ChABC slices. In the control condition, changing the extracellular Ca2+ concentration to a nominal 0 mm caused a significant decrease in the mIPSC frequency (28.4 ± 1.9 Hz at 0 mm vs 33.4 ± 1.8 Hz at 2 mm, n = 9 from 9/6; t(8) = 5.2, p = 0.00083, paired Student's t test; Fig. 6F). In ChABC-treated slices, reducing the extracellular Ca2+ concentration to 0 mm also resulted in a small decrease in the mIPSC frequency (44.3 ± 3.7 Hz at 0 mm vs 48.2 ± 3.7 Hz at 2 mm, n = 9 from 9/5; t(8) = 3.6, p = 0.0072, paired Student's t test; Fig. 6F). However, this decrease was similar to that observed in control cells (15 ± 3% decrease in control cells vs 8 ± 3% decrease in ChABC-treated cells; t(16) = 3.6, p = 0.084, unpaired Student's t test; Fig. 6G), thereby leaving the mIPSC frequency to remain higher in ChABC-treated slices (t(32) = 3.875, p = 0.001, two-way repeated-measures ANOVA with Sidak post hoc test).
Furthermore, we tested whether blocking Ca2+ influx dissipates the difference in the mIPSC frequencies between control and ChABC-treated neurons using the nonselective VDCC blocker cadmium (100 μm). In both control and ChABC-treated slices, cadmium significantly reduced the mIPSC frequency (25.2 ± 3.2 vs 31.2 ± 2.6 Hz in control slices, n = 7 from 7/4; t(6) = 6.1, p = 0.00087, paired Student's t test; 39.5 ± 6.1 vs 46.9 ± 5.9 Hz in ChABC-treated slices, n = 4 from 4/2; t(3) = 8.2, p = 0.0038, paired Student's t test; Fig. 6H). However, there was still a large difference in the mIPSC frequency between the control and ChABC-treated cells (t(18) = 2.3, p = 0.021, two-way repeated-measures ANOVA with Sidak post hoc test; Fig. 6I). Together, these results suggest that the facilitation of GABA release in the ChABC-treated slices could not be attributed to changes in the density of presynaptic terminals, the availability of extracellular Ca2+, or presynaptic Ca2+ channel conductance. Therefore, we speculate that PNNs regulate the release probability via intracellular Ca2+ stores or the release machinery in PC presynaptic terminals (see Discussion).
Effects of PNN depletion on rebound firing in large DCN neurons
PCs exert powerful inhibition onto DCN neurons (Telgkamp and Raman, 2002; Pedroarena and Schwarz, 2003), and DCN neurons show postinhibitory rebound firing after the termination of strong hyperpolarization induced by PC inhibition (Aizenman and Linden, 1999; McKay et al., 2005; Pugh and Raman, 2006). This rebound firing response is assumed to play a crucial role in synaptic plasticity in the DCN. As we have demonstrated that PNN depletion enhanced GABAergic transmission from PCs to DCN neurons, PNN depletion would be expected to facilitate the postinhibitory rebound firing in DCN neurons.
To test this possibility, we recorded the amplitude of hyperpolarization and the subsequent rebound firing in large DCN neurons elicited by the stimulation of PC axon fibers in slices treated with or without ChABC treatment in the current-clamp mode. There was no significant difference in resting membrane potentials (−58.5 ± 1.1 mV in control, n = 11 from 10/5 vs −59.6 ± 1.2 mV in ChABC-treated, n = 8 from 8/7; t(17) = 0.61, p = 0.55, unpaired Student's t test; corrected for a 15 mV junction potential) or spontaneous action potential frequencies (69.7 ± 12.2 Hz in control, n = 11 from 10/5 vs 57.0 ± 8.5 Hz in ChABC-treated, n = 8 from 8/7; t(17) = 0.79, p = 0.44, unpaired Student's t test) between control and ChABC-treated cells. We applied constant holding currents to keep the prestimulus firing rate at 10–20 Hz to compare the hyperpolarization and rebound firing under near-identical conditions, as previously reported (Mercer et al., 2016). When PC axons in the white matter were stimulated at 100 Hz for 100 ms, large hyperpolarization was recorded in large DCN neurons (Fig. 7A). The amplitude of hyperpolarization (from the baseline) was significantly larger in neurons treated with ChABC (13.7 ± 1.8 mV, n = 5 from 5/5; t(10) = 3.2, p = 0.0094, unpaired Student's t test) than in the control neurons (7.8 ± 0.9 mV, n = 7 from 6/3), as was expected based on the increase in the amplitude of IPSCs induced by ChABC (Fig. 7A,B). This was followed by an immediate increase in the firing rate (i.e., rebound firing) in both conditions. Large DCN neurons treated with ChABC showed rebound firing rates from 15.4 ± 1.8 to 25.3 ± 2.7 Hz (n = 5 from 5/5), while those of control neurons showed rebound firing rates from 16.3 ± 0.8 to 22.9 ± 1.8 Hz (n = 7 from 6/3; Fig. 7A,C). The increase was significantly larger in ChABC-treated than in control neurons (138 ± 8% in control vs 168 ± 8% in ChABC-treated; t(10) = 2.1, p = 0.030, unpaired Student's t test; Fig. 7D).
Figure 7.
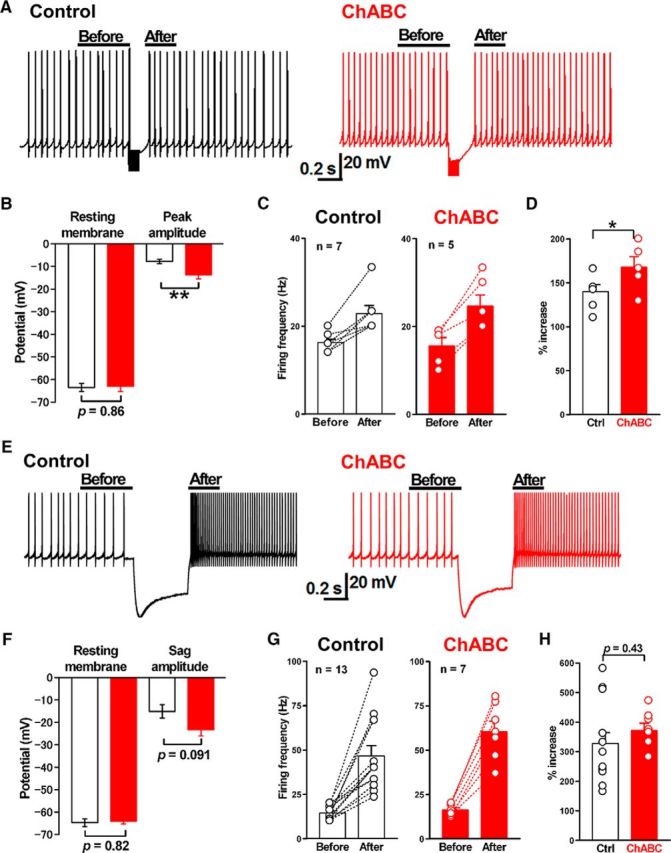
Effects of PNN depletion on rebound firing in large DCN neurons. A, Representative membrane potential and firing before and after repetitive stimulation of GABAergic synapses (100 Hz for 100 ms) in large DCN neurons in control (left) and ChABC-treated neurons (right). Constant holding currents were injected into neurons to keep the prestimulus firing rate similar at 10–20 Hz. B, No difference in the resting membrane potential with the constant holding current between the control (open bar, n = 7) and ChABC-treated neurons (red bar, n = 5). The membrane potential was corrected for a 15 mV junction potential. ChABC treatment increased the mean peak amplitude of IPSP responses during the repetitive stimulation. **p < 0.01 using unpaired Student's t test. C, The effects of ChABC treatment on the mean frequency after the repetitive stimulation. D, The mean increases of rebound firing in large DCN neurons in control (n = 7) and ChABC-treated slices (n = 5) are shown as percentages to the baseline firing rate in each condition. *p < 0.05 using unpaired Student's t test. E, Representative membrane potentials and action potential firings before and after the injection of hyperpolarizing current (400 pA, 500 ms) in the control (left) and ChABC-treated (right) large DCN neurons. Constant holding currents were injected with large DCN neurons to keep the prestimulus firing rate at between 10 and 20 Hz. F, No difference in the resting membrane potential with the constant holding current between the control (open bar, n = 13) and ChABC-treated (red bar, n = 7) neurons. The membrane potential was corrected for a 15 mV junction potential. ChABC treatment had no effect on the amplitude of sag induced by the injection of hyperpolarizing current. G, Changes in the firing rates after the hyperpolarizing current injection in the control neurons (n = 13) and ChABC-treated large DCN neurons (n = 7). H, There is no difference in the percentage increase of rebound firing between the control and ChABC-treated large DCN neurons. Ctrl, Control.
This effect appears to be not postsynaptic, because the rebound firing rate following a hyperpolarizing current injection (400 pA, 500 ms) to DCN neurons did not differ between control and ChABC-treated neurons (372 ± 24% in control, n = 7 from 7/6 vs 328 ± 37% in ChABC-treated, n = 13 from 12/6; t(18) = 0.81, p = 0.43, unpaired Student's t test; Fig. 7E,G,H). Moreover, the amplitude of the voltage sag in response to the current injection plotted against the initial membrane potential (Aizenman and Linden, 1999), which is indicative of the important function of Ih channels in rebound firing, did not differ significantly (15.1 ± 3.0 mV in control neurons, n = 13 from 12/6 vs 23.3 ± 2.7 mV in ChABC-treated neurons, n = 7 from 7/6; t(18) = 1.8, p = 0.091, unpaired Student's t test; Fig. 7E,F). Although chondroitin sulfate solutions are reported to shift the activation curve of Ih in Xenopus photoreceptors (Vigetti et al., 2008), there was no substantial effect of PNN depletion on Ih channels in our experiments. Together, these results suggest that the increased inhibitory synaptic transmission induced by PNN removal results in changes in rebound firing in glutamatergic DCN neurons without changing their spontaneous spiking activity.
Persistent effects of ChABC injected into the DCN in vivo on GABAergic transmission
Since the rebound firing is considered critical for synaptic plasticity in the DCN and cerebellar motor learning, we next sought to test whether PNN removal affects cerebellum-dependent eyeblink conditioning. To this end, we first tested whether the injection of ChABC into the mouse DCN in vivo removes PNNs, whether the potentiation of GABAergic transmission is observed in slices prepared from these animals, and whether the effect persists long enough to enable the behavioral analysis. We injected ChABC bilaterally into age P21 to P23 mouse interpositus nuclei, which are responsible for the eyeblink conditioning among the DCN, and prepared acute cerebellar slices 4–6 d after the single injection.
Indeed, we observed a similar increase in eIPSCs (532 ± 109 pA in saline-injected control, n = 8 from 7/3 vs 1281 ± 291 pA in ChABC-injected, n = 7 from 7/3; t(13) = 2.5, p = 0.025, unpaired Student's t test), a reduction in the PPR (0.94 ± 0.02 in control, n = 8 from 7/3 vs 0.88 ± 0.02 in ChABC-injected, n = 7 from 7/3; t(13) = 2.3, p = 0.040, unpaired Student's t test; Fig. 8A–C), a depression in eIPSCs during a train (with regression analyses comparing slopes and plateaus, F(2,189) = 8.1, p = 0.0004; Fig. 8D,E), and an increase in the frequency of sIPSCs (13.8 ± 2.0 Hz in control, n = 7 from 6/3 vs 31.8 ± 7.6 Hz in ChABC-injected, n = 7 from 7/3; t(12) = 2.3, p = 0.042, unpaired Student's t test; Fig. 8F,G). The frequency of mIPSCs was also increased (4.9 ± 1.0 Hz in control, n = 5 from 5/3 vs 16.7 ± 4.2 Hz in ChABC-injected, n = 4 from 4/3; t(7) = 3.1, p = 0.018, unpaired Student's t test) without changes in their amplitude (13.2 ± 0.4 pA in control, n = 5 from 5/3 vs 15.2 ± 1.0 pA in ChABC-injected, n = 4 from 4/3; t(7) = 2.1, p = 0.071, unpaired Student's t test; Fig. 8H). These results suggest that ChABC injection in vivo into the interpositus nuclei has persistent effects on GABAergic transmission, resulting in the increase in release probability at presynaptic terminals as observed in slices acutely treated with ChABC.
Figure 8.

Persistent effects of ChABC injected into the DCN in vivo on IPSCs. A, Representative traces of eIPSCs in neurons in the mouse DCN injected with control saline (left) and ChABC (right). Acute cerebellar slices were made 4–6 d after Ringer's solution or ChABC (10 U/ml) was injected into the interpositus nuclei of age P21 to P23 mice. B, Large DCN neurons of ChABC-injected mice showed an increase in the mean amplitude of first eIPSCs (left) and the reduction of the mean PPR (right) of eIPSCs. *p < 0.05 using unpaired Student's t test. C, No effect on the rise (left) or decay (right) kinetics of eIPSCs. D, Representative averaged eIPSCs induced by 20 stimuli at 100 Hz in large DCN neurons. E, Mean amplitudes of eIPSCs normalized to the first phasic eIPSC. ChABC injection enhanced the depression of eIPSCs. F, Representative traces of sIPSCs in large DCN neurons. ChABC treatment facilitated sIPSCs (right). G, Mean frequency (left) and amplitude (right) of sIPSCs. ChABC increased the frequency of sIPSC significantly. *p < 0.05 using unpaired Student's t test. H, Mean frequency (left) and amplitude (right) of mIPSCs. ChABC injection in vivo increased the mean frequency but not the mean amplitude of mIPSCs. *p < 0.05 using unpaired Student's t test. Ctrl, Control.
PNN depletion facilitates delay eyeblink conditioning
Since the GABAergic pathway from PCs to DCN neurons is required for eyeblink conditioning (Krupa et al., 1993; Medina and Mauk, 1999; Aksenov et al., 2004; Parker et al., 2009), GABAergic transmission altered by the in vivo injection of ChABC would be expected to affect eyeblink conditioning. With the persistent effect of ChABC in vivo verified, we made up an experimental setup to test its effect on delay eyeblink conditioning. We injected ChABC into the bilateral interpositus nuclei of mice 6 d before the start of conditioning. After the single ChABC injection, the mice were trained for 10 d and tested for 3 additional days for extinction. The mice acquired the CR in the course of the training, as shown in Figure 9, A and B, and individual CR rates and the average CR rate on each training day (CR%) were computed and plotted as a learning curve (Fig. 9 C,D). We also measured the frequencies of startle reflex as indicated by the squares in Figure 9D and the spontaneous eyeblink at session 0. The CR% for control mice with saline injection progressively increased up to the maximum rate of 69% (averaged CR%, 51.1 ± 3.9%; n = 7) during the training period. However, ChABC-injected mice exhibited significantly higher CR% at the end of the training period than control mice (up to 93%, averaged CR%: 72.6 ± 3.6%, n = 9, p = 0.00020, two-way repeated-measures ANOVA with Sidak post hoc test). Two-way repeated-measures ANOVA of the data during the training period revealed a significant overall difference between the control and ChABC-injected animals (F(1,14) = 13.1, p = 0.0028, two-way repeated-measures ANOVA) without significant interaction. In contrast, no difference was observed between the two groups in the extinction period. There was also no significant difference in startle reflex and spontaneous eyeblink frequencies, indicating that the ChABC injection did not alter sensitivity to the stimulus or the basal eyeblink motor function. Together, these results suggest that the depletion of PNNs in the DCN by ChABC, which greatly enhanced the inhibitory synaptic transmission to the DCN, improves the CR rates in delay eyeblink conditioning without significantly affecting the learning extinction in adult mice.
Discussion
This is the first study demonstrating that inhibitory GABAergic transmission is enhanced by PNN removal. Our present data provide evidence for a crucial role of PNNs in modulating GABAergic transmission at PC–DCN neuron synapses. The reduction of PNNs in the DCN by ChABC facilitated GABAergic transmission from PCs onto large glutamatergic DCN neurons via the enhancement of the release probability of GABA from PC axon terminals. The enhanced GABAergic transmission hyperpolarized DCN neurons more strongly and facilitated their rebound firing, which would be expected to result in the efficient induction of synaptic plasticity in the DCN. The characteristics of the facilitated GABAergic transmission, particularly the enhanced depression, are akin to those observed in the DCN of juvenile mice, which exhibit a higher propensity for synaptic plasticity and motor learning than adults. Indeed, our results from delay eyeblink conditioning demonstrated that the mice having received the injection of ChABC into the interpositus nuclei acquired the CR at a higher rate than control animals. These results suggest that the developmental expression of PNNs attenuates GABAergic transmission at synapses between PCs and DCN neurons and prevents unnecessary formation of new memory, which may also protect previously encoded memory in the cerebellum in adulthood.
PNN regulation of GABAergic transmission in the DCN
The physiological roles of PNNs have been investigated and so far have been reported to be involved in synaptic transmission, exclusive of excitation and its plasticity, and memory formation (Frischknecht et al., 2009; Kochlamazashvili et al., 2010; Romberg et al., 2013; Carstens et al., 2016): for example, PNNs regulate hippocampal synaptic plasticity and contribute contextual fear memory (Kochlamazashvili et al., 2010), control long-term object recognition and experience-driven synaptic plasticity in the perirhinal cortex (Romberg et al., 2013), and suppress excitatory synaptic plasticity of CA2 pyramidal neurons (Carstens et al., 2016). In contrast, the present study demonstrates the physiological contribution of PNNs to inhibitory GABAergic transmission.
Expression of PNNs in the DCN increases from P14 (Zaremba et al., 1990; Carulli et al., 2007; Zimmermann and Dours-Zimmermann, 2008) following a timeline similar to those in other brain areas (Carulli et al., 2007; Horii-Hayashi et al., 2015). In mice older than P18, we found that the depletion of PNNs by ChABC treatment increased the amplitude of eIPSCs, decreased the PPR of eIPSCs, augmented the mIPSC frequency, and augmented depression. It has been shown that this synapse exhibits a developmental shift from being strongly depressed in juveniles to weakly depressed in adults (Turecek et al., 2016; Saitow et al., 2018). Our results indicate that the developmental expression of PNNs contributes to this substantial developmental change in GABAergic transmission between PCs and DCN neurons.
Although the aforementioned changes in GABAergic transmission, particularly the increase in mIPSC frequency and the reduction of the PPR, strongly suggest that PNN depletion enhances the release probability of GABA from PC axon terminals, the increase in mIPSC frequency can also be accounted by an increase in the number of PC terminals. Indeed, PNNs in the cerebellum have been reported to restrict the structural plasticity of intact PC axons to maintain normal GABAergic transmission onto DCN neurons, such that ChABC-induced degradation of PNNs induces PC axon sprouting (Corvetti and Rossi, 2005). However, our immunohistochemical analyses demonstrated that there were no significant changes in the size and density of PC terminals in acute slices treated with ChABC. Together with the decrease in PPR, we conclude that the changes in GABAergic transmission by ChABC are due to an increase in the release probability of GABA rather than the formation of new presynaptic terminals by ChABC (de Vivo et al., 2013).
The most plausible mechanism underlying the enhancement of GABA release by ChABC treatment could be that negatively charged CSPGs restrict the availability of extracellular Ca2+ to PC terminals. Hrabetová et al. (2009) proposed that CSPGs regulate the local diffusion properties of Ca2+ in the rat neocortex and hippocampus (Hrabetová et al., 2009; Nicholson and Hrabětová, 2017); they also showed that the ECM slows Ca2+ diffusion in the tissue, with this effect being reduced by the degradation of PNNs with ChABC. Negatively charged CSPGs can buffer Ca2+ through electrical interactions (Crank, 1975) to keep the extracellular Ca2+ concentration in a lower range. If PNNs limit the availability of extracellular Ca2+, reducing PNNs by ChABC would greatly enhance the presynaptic release, as observed in our experiments. However, we found that the increase of mIPSC frequency by ChABC was largely independent of extracellular Ca2+ or VDCCs such that the enhancement was not attenuated with lowered extracellular Ca2+ or in the presence of cadmium. From these findings, we speculate that the reduction of PNNs somehow results in the enhancement of the release machinery (its Ca2+ sensitivity or the intrinsic activity) or drives intracellular Ca2+ release in PC terminals. Further studies are necessary to address the precise mechanism of this newly found regulation of GABA release by PNNs.
Roles of PNNs in eyeblink motor learning
Our results also demonstrated that the enhanced GABAergic transmission greatly increases the magnitude of hyperpolarization, thereby augmenting the rebound firing in DCN neurons. Rebound firing has been implicated in the induction of LTP of excitatory synaptic transmission between mossy fibers and DCN neurons (Miles and Lisberger, 1981; Racine et al., 1986; Pugh and Raman, 2006, 2008; McElvain et al., 2010; Person and Raman, 2010), and mossy fiber LTP, which contributes to the facilitation of action potentials in DCN neurons (Wu and Raman, 2017; Yarden-Rabinowitz and Yarom, 2017), is suggested to be a possible synaptic mechanism underlying cerebellar motor learning (Medina and Mauk 1999; Attwell et al., 2002). Therefore, to translate our findings on the enhanced GABAergic transmission into insights on the cerebellar function, we investigated the effect of ChABC injection into the DCN on a delay eyeblink-conditioning test.
Previous studies reported that the inactivation of the DCN by lesions or pharmacological treatments suppresses CR increase during delay eyeblink training (Yeo et al., 1985; Attwell et al., 2002; Ohyama et al., 2006; Boele et al., 2010; Sakamoto and Endo, 2010). A recent study has further shown that the activation of the interpositus nuclei is necessary for obtaining CRs of trained mice under the head-fixed condition (Heiney et al., 2014). Therefore, we injected ChABC into the interpositus nuclei and tested its effect on this behavior. Our data demonstrated that the conditioning was significantly enhanced in the ChABC-injected mice and suggest that the depletion of PNNs in the interpositus nuclei facilitates the acquisition of the CR. This facilitative effect is similar to what has been observed in other brain areas, in which PNN depletion allows structural synaptic reorganization and promotes memory formation in adult animals (Corvetti and Rossi, 2005; Gogolla et al., 2009; Romberg et al., 2013; de Vivo et al., 2013). In fact, previous studies have shown that the number of PC terminals in the DCN is significantly altered in mice deficient in the components of PNNs, such as Bral2 and Crtl1 (Bekku et al., 2003, 2012; Foscarin et al., 2011; Stamenkovic et al., 2017). It has also been reported that learning itself alters synaptic structures (Kleim et al., 2002; Boele et al., 2013). In contrast to these studies using prolonged or permanent disruption of PNNs, we acutely disrupted PNNs and found that the release probability of GABA in PC terminals was augmented both in slices and in vivo, which resulted in increased rebound firing in large glutamatergic DCN neurons. Therefore, our data suggest a novel role of PNNs in regulating presynaptic functions of PC axons and provide a novel mechanistic insight indicating that not only the structural plasticity, but also the functional plasticity of synapses regulated by PNNs affects behavioral flexibility in adult animals.
Footnotes
The authors declare no competing financial interests.
This work was supported by Takeda Science Foundation (M.H. and Y.Y.), and Japan Society for the Promotion of Science KAKENHI Grants 15K06788 (M.H.); 15K01275 (S.W.); 26350983 and 16H01622 (F.K.); 26290002, 15H01415, and 15H05872 (Y.Y.); and 16K07006 (H.M.). We thank Dr. Shiro Konishi and Dr. Toshitaka Oohashi for their comments on this manuscript; Dr. Toshiro Sakamoto for technical advice regarding stereotaxic injections; and Ms. Moeko Kudoh, Dr. Jun Motoyama, and Dr. Yuki Murakami for help with the initial immunohistochemistry.
References
- Aizenman CD, Linden DJ (1999) Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol 82:1697–1709. 10.1152/jn.1999.82.4.1697 [DOI] [PubMed] [Google Scholar]
- Aksenov D, Serdyukova N, Irwin K, Bracha V (2004) GABA neurotransmission in the cerebellar interposed nuclei: involvement in classically conditioned eyeblinks and neuronal activity. J Neurophysiol 91:719–727. 10.1152/jn.00859.2003 [DOI] [PubMed] [Google Scholar]
- Attwell PJ, Cooke SF, Yeo CH (2002) Cerebellar function in consolidation of a motor memory. Neuron 34:1011–1020. 10.1016/S0896-6273(02)00719-5 [DOI] [PubMed] [Google Scholar]
- Baumel Y, Jacobson GA, Cohen D (2009) Implications of functional anatomy on information processing in the deep cerebellar nuclei. Front Cell Neurosci 3:14. 10.3389/neuro.03.014.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekku Y, Su WD, Hirakawa S, Fässler R, Ohtsuka A, Kang JS, Sanders J, Murakami T, Ninomiya Y, Oohashi T (2003) Molecular cloning of Bral2, a novel brain-specific link protein, and immunohistochemical colocalization with brevican in perineuronal nets. Mol Cell Neurosci 24:148–159. 10.1016/S1044-7431(03)00133-7 [DOI] [PubMed] [Google Scholar]
- Bekku Y, Saito M, Moser M, Fuchigami M, Maehara A, Nakayama M, Kusachi S, Ninomiya Y, Oohashi T (2012) Bral2 is indispensable for the proper localization of brevican and the structural integrity of the perineuronal net in the brainstem and cerebellum. J Comp Neurol 520:1721–1736. 10.1002/cne.23009 [DOI] [PubMed] [Google Scholar]
- Bengtsson F, Ekerot CF, Jörntell H (2011) In vivo analysis of inhibitory synaptic inputs and rebounds in deep cerebellar nuclear neurons. PLoS One 6:e18822. 10.1371/journal.pone.0018822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boele HJ, Koekkoek SK, De Zeeuw CI (2010) Cerebellar and extracerebellar involvement in mouse eyeblink conditioning: the ACDC model. Front Cell Neurosci 3:19. 10.3389/neuro.03.019.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boele HJ, Koekkoek SK, De Zeeuw CI, Ruigrok TJ (2013) Axonal sprouting and formation of terminals in the adult cerebellar during associative motor learning. J Neurosci 33:17897–17907. 10.1523/JNEUROSCI.0511-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carstens KE, Phillips ML, Pozzo-Miller L, Weinberg RJ, Dudek SM (2016) Perineuronal nets suppress plasticity of excitatory synapses on CA2 pyramidal neurons. J Neurosci 36:6312–6320. 10.1523/JNEUROSCI.0245-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carulli D, Rhodes KE, Brown DJ, Bonnert TP, Pollack SJ, Oliver K, Strata P, Fawcett JW (2006) Composition of perineuronal nets in the adult rat cerebellar and the cerebellar origin of their components. J Comp Neurol 494:559–577. 10.1002/cne.20822 [DOI] [PubMed] [Google Scholar]
- Carulli D, Rhodes KE, Fawcett JW (2007) Upregulation of aggrecan, link protein 1, and hyaluronan synthases during formation of perineuronal nets in the rat cerebellum. J Comp Neurol 501:83–94. 10.1002/cne.21231 [DOI] [PubMed] [Google Scholar]
- Carulli D, Pizzorusso T, Kwok JC, Putignano E, Poli A, Forostyak S, Andrews MR, Deepa SS, Glant TT, Fawcett JW (2010) Animals lacking link protein have attenuated perineuronal nets and persistent plasticity. Brain 133:2331–2347. 10.1093/brain/awq145 [DOI] [PubMed] [Google Scholar]
- Chan-Palay V. (1977) Cerebellar dentate nucleus. Organization, cytology, and transmitters. New York: Springer. [Google Scholar]
- Christian KM, Thompson RF (2003) Neural substrates of eyeblink conditioning: acquisition and retention. Learn Mem 10:427–455. 10.1101/lm.59603 [DOI] [PubMed] [Google Scholar]
- Corvetti L, Rossi F (2005) Degradation of chondroitin sulfate proteoglycans induces sprouting of intact Purkinje axons in the cerebellar of the adult rat. J Neurosci 25:7150–7158. 10.1523/JNEUROSCI.0683-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crank J. (1975) The mathematics of diffusion, Ed 2 Oxford: Clarendon. [Google Scholar]
- de Vivo L, Landi S, Panniello M, Baroncelli L, Chierzi S, Mariotti L, Spolidoro M, Pizzorusso T, Maffei L, Ratto GM (2013) Extracellular matrix inhibits structural and functional plasticity of dendritic spines in the adult visual cortex. Nat Commun 4:1484. 10.1038/ncomms2491 [DOI] [PubMed] [Google Scholar]
- de Zeeuw CI, Berrebi AS (1996) Individual Purkinje cell axons terminate on both inhibitory and excitatory neurons in the cerebellar and vestibular nuclei. Ann NY Acad Sci 781:607–610. 10.1111/j.1749-6632.1996.tb15736.x [DOI] [PubMed] [Google Scholar]
- Foscarin S, Ponchione D, Pajaj E, Leto K, Gawlak M, Wilczynski GM, Rossi F, Carulli D (2011) Experience-dependent plasticity and modulation of growth regulatory molecules at central synapses. PLoS One 6:e16666. 10.1371/journal.pone.0016666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frischknecht R, Heine M, Perrais D, Seidenbecher CI, Choquet D, Gundelfinger ED (2009) Brain extracellular matrix affects AMPA receptor lateral mobility and short-term synaptic plasticity. Nat Neurosci 12:897–904. 10.1038/nn.2338 [DOI] [PubMed] [Google Scholar]
- Gogolla N, Caroni P, Lüthi A, Herry C (2009) Perineuronal nets protect fear memories from erasure. Science 325:1258–1261. 10.1126/science.1174146 [DOI] [PubMed] [Google Scholar]
- Heiney SA, Wohl MP, Chettih SN, Ruffolo LI, Medina JF (2014) Cerebellar-dependent expression of motor learning during eyeblink conditioning in head-fixed mice. J Neurosci 34:14845–14853. 10.1523/JNEUROSCI.2820-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirono M, Ogawa Y, Misono K, Zollinger DR, Trimmer JS, Rasband MN, Misonou H (2015) BK channels localize to the paranodal junction and regulate action potentials in myelinated axons of cerebellar Purkinje cells. J Neurosci 35:7082–7094. 10.1523/JNEUROSCI.3778-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoebeek FE, Witter L, Ruigrok TJ, De Zeeuw CI (2010) Differential olovo-cerebellar cortical control of rebound activity in the cerebellar nuclei. Proc Natl Acad Sci U S A 107:8410–8415. 10.1073/pnas.0907118107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horii-Hayashi N, Sasagawa T, Matsunaga W, Nishi M (2015) Developmental and structural variety of the chondroitin sulfate proteoglycans-contained extracellular matrix in the mouse brain. Neural Plast 2015:256389. 10.1155/2015/256389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hrabetová S, Masri D, Tao L, Xiao F, Nicholson C (2009) Calcium diffusion enhanced after cleavage of negatively charged components of brain extracellular matrix by chondroitinase ABC. J Physiol 587:4029–4049. 10.1113/jphysiol.2009.170092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M, Yoshida M, Obata K (1964) Monosynaptic inhibition of the intracerebellar nuclei induced from the cerebellar cortex. Experientia 20:575–576. 10.1007/BF02150304 [DOI] [PubMed] [Google Scholar]
- Kassardjian CD, Tan YF, Chung JY, Heskin R, Peterson MJ, Broussard DM (2005) The site of a memory shifts with consolidation. J Neurosci 25:7979–7985. 10.1523/JNEUROSCI.2215-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kistler WM, De Zeeuw CI (2003) Time windows and reverberating loops: a reverse-engineering approach to cerebellar function. Cerebellum 2:44–54. 10.1080/14734220309426 [DOI] [PubMed] [Google Scholar]
- Kleim JA, Freeman JH Jr, Bruneau R, Nolan BC, Cooper NR, Zook A, Walters D (2002) Synapse formation is associated with memory storage in the cerebellum. Proc Natl Acad Sci U S A 99:13228–13231. 10.1073/pnas.202483399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kochlamazashvili G, Henneberger C, Bukalo O, Dvoretskova E, Senkov O, Lievens PM, Westenbroek R, Engel AK, Catterall WA, Rusakov DA, Schachner M, Dityatev A (2010) The extracellular matrix molecule hyaluronic acid regulates hippocampal synaptic plasticity by modulating postsynaptic l-type Ca2+ channels. Neuron 67:116–128. 10.1016/j.neuron.2010.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koekkoek SK, Den Ouden WL, Perry G, Highstein SM, De Zeeuw CI (2002) Monitoring kinetic and frequency-domain properties of eyelid responses in ice with magnetic distance measurement technique. J Neurophysiol 88:2124–2133. 10.1152/jn.2002.88.4.2124 [DOI] [PubMed] [Google Scholar]
- Krupa DJ, Thompson JK, Thompson RF (1993) Localization of a memory trace in the mammalian brain. Science 260:989–991. 10.1126/science.8493536 [DOI] [PubMed] [Google Scholar]
- Kwok JC, Dick G, Wang D, Fawcett JW (2011) Extracellular matrix and perineuronal nets in CNS repair. Dev Neurobiol 71:1073–1089. 10.1002/dneu.20974 [DOI] [PubMed] [Google Scholar]
- Llinás R, Mühlethaler M (1988) Electrophysiology of guinea-pig cerebellar nuclear cells in the in vivo brain stem-cerebellar preparation. J Physiol 404:241–258. 10.1113/jphysiol.1988.sp017288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews RT, Kelly GM, Zerillo CA, Gray G, Tiemeyer M, Hockfield S (2002) Aggrecan glycoforms contribute to the molecular heterogeneity of perineuronal nets. J Neurosci 22:7536–7547. 10.1523/JNEUROSCI.22-17-07536.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElvain LE, Bagnall MW, Sakatos A, du Lac S (2010) Bidirectional plasticity gated by hyperpolarization controls the gain of postsynaptic firing responses at central vestibular nerve synapses. Neuron 68:763–775. 10.1016/j.neuron.2010.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay BE, Molineux ML, Mehaffey WH, Turner RW (2005) Kv1 K+ channels control Purkinje cell output to facilitate postsynaptic rebound discharge in deep cerebellar neurons. J Neurosci 25:1481–1492. 10.1523/JNEUROSCI.3523-04.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina JF, Mauk MD (1999) Stimulations of cerebellar motor learning: computational analysis of plasticity at the mossy fiber to deep nucleus synapse. J Neurosci 19:7140–7151. 10.1523/JNEUROSCI.19-16-07140.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercer AA, Palarz KJ, Tabatadze N, Woolley CS, Raman IM (2016) Sex differences in cerebellar synaptic transmission and sex-specific responses to autism-liked Gabrb3 mutations in mice. eLife 5:e07596. 10.7554/eLife.07596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles FA, Lisberger SG (1981) Plasticity in the vestibule-ocular reflex: a new hypothesis. Annu Rev Neurosci 4:273–299. 10.1146/annurev.ne.04.030181.001421 [DOI] [PubMed] [Google Scholar]
- Miyata S, Komatsu Y, Yoshimura Y, Taya C, Kitagawa H (2012) Persistent dortical plasticity by upregulation of chondroitin 6-sulfation. Nat Neurosci 15:414–422, S1–2. 10.1038/nn.3023 [DOI] [PubMed] [Google Scholar]
- Nicholson C, Hrabětová S (2017) Brain extracellular space: the final frontier of neuroscience. Biophys J 113:2133–2142. 10.1016/j.bpj.2017.06.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Ito M, Ochi R, Sato N (1967) Pharmacological properties of the postsynaptic inhibition by Purkinje cell axons and the action of γ-aminobutyric acid on deiters neurons. Exp Brain Res 4:43–57. [DOI] [PubMed] [Google Scholar]
- Ohyama T, Nores WL, Medina JF, Riusech FA, Mauk MD (2006) Learning-induced plasticity in deep cerebellar nucleus. J Neurosci 26:12656–12663. 10.1523/JNEUROSCI.4023-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto T, Endo S, Shirao T, Nagao S (2011) Role of cerebellar cortical protein synthesis in transfer of memory trace of cerebellum-dependent motor learning. J Neurosci 31:8958–8966. 10.1523/JNEUROSCI.1151-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oohashi T, Edamatsu M, Bekku Y, Carulli D (2015) The hyaluronan and proteoglycan link proteins: organizers of the brain extracellular matrix and key molecules for neuronal function and plasticity. Exp Neurol 274:134–144. 10.1016/j.expneurol.2015.09.010 [DOI] [PubMed] [Google Scholar]
- Parker KL, Zbarska S, Carrel AJ, Bracha V (2009) Blocking GABAA neurotransmission in the interposed nuclei: effects on conditioned and unconditioned eyeblinks. Brain Res 1292:25–37. 10.1016/j.brainres.2009.07.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ (2001) The mouse brain in stereotaxic coordinates, Ed 2 San Diego, CA: Academic. [Google Scholar]
- Pedroarena CM, Schwarz C (2003) Efficacy and short-term plasticity at GABAergic synapses between Purkinje and cerebellar nuclei neurons. J Neurophysiol 89:704–715. 10.1152/jn.00558.2002 [DOI] [PubMed] [Google Scholar]
- Person AL, Raman IM (2010) Deactivation of L-type ca current by inhibition controls LTP at excitatory synapses in the cerebellar nuclei. Neuron 66:550–559. 10.1016/j.neuron.2010.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizzorusso T, Medini P, Berardi N, Chierzi S, Fawcett JW, Maffei L (2002) Reactivation of ocular dominance plasticity in the adult visual cortex. Science 298:1248–1251. 10.1126/science.1072699 [DOI] [PubMed] [Google Scholar]
- Pugh JR, Raman IM (2006) Potentiation of mossy fiber EPSCs in the cerebellar nuclei by NMDA receptor activation followed by postinhibitory rebound current. Neuron 51:113–123. 10.1016/j.neuron.2006.05.021 [DOI] [PubMed] [Google Scholar]
- Pugh JR, Raman IM (2008) Mechanisms of potentiation of mossy fiber EPSCs in the cerebellar nuclei by coincident synaptic excitation and inhibition. J Neurosci 28:10549–10560. 10.1523/JNEUROSCI.2061-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Racine RJ, Wilson DA, Gingell R, Sunderland D (1986) Long-term potentiation in the interpositus and vestibular nuclei in the rat. Exp Brain Res 63:158–162. [DOI] [PubMed] [Google Scholar]
- Romberg C, Yang S, Melani R, Andrews MR, Horner AE, Spillantini MG, Bussey TJ, Fawcett JW, Pizzorusso T, Saksida LM (2013) Depletion of perineuronal nets enhances recognition memory and long-term depression in the perirhinal cortex. J Neurosci 33:7057–7065. 10.1523/JNEUROSCI.6267-11.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitow F, Nagano M, Suzuki H (2018) Developmental changes in serotonergic modulation of GABAergic synaptic transmission and postsynaptic GABAA receptor composition in the cerebellar nuclei. Cerebellum 17:346–358. 10.1007/s12311-018-0922-9 [DOI] [PubMed] [Google Scholar]
- Sakamoto T, Endo S (2010) Amygdalda, deep cerebellar nuclei and red nucleus contribute to delay eyeblink conditioning in C57BL/6 mice. Eur J Neurosci 32:1537–1551. 10.1111/j.1460-9568.2010.07406.x [DOI] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A (2012) Fiji: an open-source platform for biological-image analysis. Nat Methods 9:676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shutoh F, Ohki M, Kitazawa H, Itohara S, Nagao S (2006) Memory trace of motor learning shifts transsynaptically from cerebellar cortex to nuclei for consolidation. Neuroscience 139:767–777. 10.1016/j.neuroscience.2005.12.035 [DOI] [PubMed] [Google Scholar]
- Soleman S, Filippov MA, Dityatev A, Fawcett JW (2013) Targeting the neural extracellular matrix in neurological disorders. Neuroscience 253:194–213. 10.1016/j.neuroscience.2013.08.050 [DOI] [PubMed] [Google Scholar]
- Stamenkovic V, Stamenkovic S, Jaworski T, Gawlak M, Jovanovic M, Jakovcevski I, Wilczynski GM, Kaczmarek L, Schachner M, Radenovic L, Andjus PR (2017) The extracellular matrix glycoprotein tenascin-C and matrix metalloproteinases modify cerebellar structural plasticity by exposure to an enriched environment. Brain Struct Funct 222:393–415. 10.1007/s00429-016-1224-y [DOI] [PubMed] [Google Scholar]
- Tamamaki N, Yanagawa Y, Tomioka R, Miyazaki J, Obata K, Kaneko T (2003) Green fluorescent protein expression and colocalization with calretinin, parvalbumin, and somatostatin in the GAD67-GFP knock-in mouse. J Comp Neurol 467:60–79. 10.1002/cne.10905 [DOI] [PubMed] [Google Scholar]
- Telgkamp P, Raman IM (2002) Depression of inhibitory synaptic transmission between Purkinje cells and neurons of the cerebellar nuclei. J Neurosci 22:8447–8457. 10.1523/JNEUROSCI.22-19-08447.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turecek J, Jackman SL, Regehr WG (2016) Synaptic specializations support frequency-independent Purkinje cell output from the cerebellar cortex. Cell Rep 17:3256–3268. 10.1016/j.celrep.2016.11.081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uusisaari M, Obata K, Knöpfel T (2007) Morphological and electrophysiological properties of GABAergic and non-GABAergic cells in the deep cerebellar nuclei. J Neurophysiol 97:901–911. 10.1152/jn.00974.2006 [DOI] [PubMed] [Google Scholar]
- Vigetti D, Andrini O, Clerici M, Negrini D, Passi A, Moriondo A (2008) Chondroitin sulfates act as extracellular gating modifiers on voltage-dependent ion channels. Cell Physiol Biochem 22:137–146. 10.1159/000149791 [DOI] [PubMed] [Google Scholar]
- Wu Y, Raman IM (2017) Facilitation of mossy fiber-driven spiking in the cerebellar nuclei by the synchrony of inhibition. J Physiol 595:5245–5264. 10.1113/JP274321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarden-Rabinowitz Y, Yarom Y (2017) In vivo analysis of synaptic activity in cerebellar nuclei neurons unravels the efficacy of excitatory inputs. J Physiol 595:5945–5963. 10.1113/JP274115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo CH, Hardiman MJ, Glickstein M (1985) Classical conditioning of the nictitating membrane response of the rabbit. I. Lesions of the cerebellar nuclei. Exp Brain Res 60:87–98. 10.1007/BF00237022 [DOI] [PubMed] [Google Scholar]
- Zaremba S, Naegele JR, Barnstable CJ, Hockfield S (1990) Neuronal subsets express multiple high-molecular-weight cell-surface glycoconjugates defined by monoclonal antibodies cat-301 and VC1.1. J Neurosci 10:2985–2995. 10.1523/JNEUROSCI.10-09-02985.1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, Raman IM (2011) Prolonged postinhibitory rebound firing in the cerebellar nuclei mediated by group I metabotropic glutamate receptor potentiation of L-type calcium currents. J Neurosci 31:10283–10292. 10.1523/JNEUROSCI.1834-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann DR, Dours-Zimmermann MT (2008) Extracellular matrix of the central nervous system: from neglect to challenge. Histochem Cell Biol 130:635–653. 10.1007/s00418-008-0485-9 [DOI] [PubMed] [Google Scholar]