

Abstract
Action potential-evoked neurotransmitter release is impaired in knock-out neurons lacking synaptic cell-adhesion molecules α-neurexins (αNrxns), the extracellularly longer variants of the three vertebrate Nrxn genes. Ca2+ influx through presynaptic high-voltage gated calcium channels like the ubiquitous P/Q-type (CaV2.1) triggers release of fusion-ready vesicles at many boutons. α2δ Auxiliary subunits regulate trafficking and kinetic properties of CaV2.1 pore-forming subunits but it has remained unclear if this involves αNrxns. Using live cell imaging with Ca2+ indicators, we report here that the total presynaptic Ca2+ influx in primary hippocampal neurons of αNrxn triple knock-out mice of both sexes is reduced and involved lower CaV2.1-mediated transients. This defect is accompanied by lower vesicle release, reduced synaptic abundance of CaV2.1 pore-forming subunits, and elevated surface mobility of α2δ-1 on axons. Overexpression of Nrxn1α in αNrxn triple knock-out neurons is sufficient to restore normal presynaptic Ca2+ influx and synaptic vesicle release. Moreover, coexpression of Nrxn1α together with α2δ-1 subunits facilitates Ca2+ influx further but causes little augmentation together with a different subunit, α2δ-3, suggesting remarkable specificity. Expression of defined recombinant CaV2.1 channels in heterologous cells validates and extends the findings from neurons. Whole-cell patch-clamp recordings show that Nrxn1α in combination with α2δ-1, but not with α2δ-3, facilitates Ca2+ currents of recombinant CaV2.1 without altering channel kinetics. These results suggest that presynaptic Nrxn1α acts as a positive regulator of Ca2+ influx through CaV2.1 channels containing α2δ-1 subunits. We propose that this regulation represents an important way for neurons to adjust synaptic strength.
SIGNIFICANCE STATEMENT Synaptic transmission between neurons depends on the fusion of neurotransmitter-filled vesicles with the presynaptic membrane, which subsequently activates postsynaptic receptors. Influx of calcium ions into the presynaptic terminal is the key step to trigger vesicle release and involves different subtypes of voltage-gated calcium channels. We study the regulation of calcium channels by neurexins, a family of synaptic cell-adhesion molecules that are essential for many synapse properties. Using optical measurements of calcium influx in cultured neurons and electrophysiological recordings of calcium currents from recombinant channels, we show that a major neurexin variant facilitates calcium influx through P/Q-type channels by interacting with their α2δ-1 auxiliary subunits. These results propose a novel way how neurons can modulate the strength of distinct synapses.
Keywords: calcium channels, cell adhesion molecules, live cell imaging, neurexins, patch-clamp electrophysiology, synaptic transmission
Introduction
Membrane depolarization leads to transient activation of high-threshold voltage-gated calcium channels (VGCCs) and subsequent Ca2+ influx that drives numerous essential cellular processes, including transmitter release in neurons (Catterall, 2000). In neurons, synaptic strength and synchronous release depend on the subtype, number, activity and topography of VGCCs (Sheng et al., 2012; Nakamura et al., 2015). Action potential-evoked vesicle release from synaptic boutons is often triggered by rapid Ca2+ influx through CaV2.1 (P/Q-type) channels but their abundance and activity differ greatly among synapses (Bucurenciu et al., 2010; Ermolyuk et al., 2013).
VGCCs generally consist of three major subunits: the pore-forming α1 subunit that defines the type of Ca2+ current conducted (α1A in CaV2.1) and at least two auxiliary subunits, an intracellular β and an extracellular α2δ subunit, which influence trafficking and kinetic properties of the pore-forming subunits (Dolphin, 2012; Zamponi et al., 2015). Four different α2δ subunits have been described (α2δ-1 to α2δ-4), which may interact with α1 at different degrees of association (Cassidy et al., 2014; Voigt et al., 2016). In this study, we focus on α2δ-1 and α2δ-3 because they differ with respect to their MIDAS domain (Cantí et al., 2005) and gabapentin binding (Hendrich et al., 2008; Tran-Van-Minh and Dolphin, 2010) but are both abundant in the mammalian hippocampus (Klugbauer et al., 1999; Schlick et al., 2010).
Several aspects of the regulation of neuronal CaV2.1 channels by α2δ are incompletely understood, including the role of additional interaction partners of α2δ (Eroglu et al., 2009). The importance of additional interaction partners of α2δ for regulating calcium channels is obvious from their discrepant behavior in heterologous cells and neurons: in recombinant expression systems, α2δ subunits generally enhance Ca2+ currents through VGCCs (Dolphin, 2012), whereas overexpression of α2δ in neurons inexplicably leads to reduced synaptic Ca2+ influx (Hoppa et al., 2012). A parsimonious explanation for this discrepancy could be that partner molecules modify the function or distribution of α2δ and VGCCs in neurons. Possible candidates for a collaboration include neurexins (Nrxns) because Ca2+-dependent synaptic release is impaired in absence of Nrxns (Missler et al., 2003; L. Y. Chen et al., 2017).
Nrxns are polymorphic synaptic cell adhesion molecules, for which multiple presynaptic and postsynaptic ligands exist (Reissner et al., 2013; Südhof, 2017). Vertebrates express three neurexin genes (Nrxn1–3), each encoding extracellularly longer α-neurexins (αNrxns) and shorter β-neurexins (βNrxns), which sustain diverse aspects of synapse differentiation and neurotransmission (Dean et al., 2003; Missler et al., 2003; Graf et al., 2004; Aoto et al., 2013; Anderson et al., 2015; Born et al., 2015; L. Y. Chen et al., 2017). Evidence for involvement of Nrxns in Ca2+ channel function has accumulated mostly from knock-out studies: the Ca2+-dependent component of α-latrotoxin-triggered glutamate release is decreased in synaptosomes from Nrxn1α KO (Geppert et al., 1998); Ca2+ channel-dependent evoked release is reduced at excitatory and inhibitory synapses of various brain regions of mice lacking all α-neurexins [triple knock-out, (TKO); Missler et al., 2003; Zhang et al., 2005]; baclofen-induced GABAB receptor-mediated inhibition of Ca2+ currents is ineffective in TKO and Ca2+-dependent exocytosis of secretory granules from pituitary gland cells is reduced (Dudanova et al., 2006); Ca2+ transients in synapses of somatostatin-positive interneurons are reduced in a conditional KO model lacking all αNrxns and βNrxn isoforms (L. Y. Chen et al., 2017); and Nrxn1α is found in CaV2 proteomes (Müller et al., 2010). Despite this wealth of information, available evidence does not sufficiently elaborate if and how Nrxns regulate Ca2+ influx through CaV2.1 (P/Q-type) channels.
Here, we investigated total and CaV2.1-mediated presynaptic Ca2+ transients in wild-type and mutant hippocampal neurons. In addition, we analyzed Ca2+ currents through defined recombinant CaV2.1 channels in heterologous cells upon expression of distinct combinations of Nrxn1α and α2δ subunits. Our results establish a key role for Nrxn1α in Ca2+ influx and uncover a specific facilitation of CaV2.1 by Nrxn1α in combination with α2δ-1. These findings offer a new perspective on how neurons might increase synaptic strength at distinct boutons.
Materials and Methods
Animals.
Wild-type and mutant mice of either sex were used for neuronal cultures derived from timed-pregnant dams at E17. Genotyping of αNrxn-deficient mice was as published (Missler et al., 2003). Animal experiments were performed at the University of Münster in accordance with government regulations for animal welfare and approved by the Landesamt für Natur, Umwelt und Verbraucherschutz (LANUV, NRW, Germany), license numbers 84-02.05.20.11.209 and 84-02.04.2015.A423.
Cell culture.
Dissociated primary neurons were prepared in HBSS from hippocampi as described previously (Neupert et al., 2015). Briefly, cell suspensions obtained after 0.25% trypsin and trituration were plated onto 18 mm glass coverslips (Menzel-Glaeser) coated with poly-l-lysine (Sigma-Aldrich) at a density of 55,000 cells/coverslip. After 4 h at 37°C in plating medium (MEM, 10% horse serum, 0.6% glucose, 1 mm sodium pyruvate), coverslips were inverted onto a 70–80% confluent monolayer of astrocytes grown in 12-well plates (Falcon), and incubated in neurobasal medium supplemented with B27, 0.5 mm glutamine, and 12.5 μm glutamate. After 3 d, media were refreshed with neurobasal medium supplemented with B27, 0.5 mm glutamine, and 5 μm AraC. Cultures were maintained at 37°C in a humidified incubator with an atmosphere of 95% air and 5% CO2. Neurons were transfected at 14 d in vitro (DIV) using Lipofectamine (ThermoFisher Scientific), and experiments performed between 17–21 DIV.
tsA201 cells were grown in DMEM supplemented with 10% fetal bovine serum and penicillin/streptomycin at 37°C/5% CO2 tsA201 cells were plated onto 16 mm glass coverslips coated with poly-l-lysine 1 d before transient transfection with a 2:1:1 ratio of α1–β3–α2δ subunits using calcium-phosphate method. For additional expression of Nrxn or other proteins the transfection mix contained cDNA in a ratio of 2:1:1:2 (α1–β3–α2δ–cotransfected cDNA). Twenty-four hours post-transfection, cells were provided with fresh medium supplemented with 2% fetal bovine serum and penicillin/streptomycin and maintained at 37°C for another 24–48 h before recordings.
Expression vectors.
For Ca2+ imaging, we generated synaptophysin-GCaMP6f driven by the synapsin promotor (synGCaMP6f). synEGFP was obtained by replacing CMV in pEGFP-C (Clontech) with human synapsin 1 promoter. Synaptophysin was amplified from pAAV2-hSyn-SYP1-mSO (Addgene, 50971) using MM14-37 (5′-GGG GCT AGC GTG AGC AAG GGC GAG GAG AAT AAC ATG GCC ATC ATC-3′), MM14-34 (5′-ACC AAG CTT GCT GCC GCC ACC ACT GC-3′) to replace a NheI/HindIII fragment containing EGFP in synEGFP-C. The newly generated HindIII site was finally used to insert the genetically encoded indicator GCaMP6f (T. W. Chen et al., 2013) from pGP-CMV-GCaMP6f (Addgene, 40755) using oligos MM14-35 (5′-AGC AAG CTT GGT TCT CAT CAT CAT CAT CAT CAT GGT ATG GC-3′) and MM14-36 (5′-TGC AAG CTT CGC GGC CGC TCA CTT CGC T-3′). Constructs expressing N-terminal EGFP-tagged calcium channels α1A (CACNA1A, human), auxiliary subunits β3 (CACNB3, rat), and HA-tagged α2δ-1 (CACNA2D1, rabbit) were a gift by Gerald Obermair and Bernhard Flucher (Obermair et al., 2004; Schneider et al., 2015). α2δ-3 (CACNA2D3, mouse) plasmid was a gift by Norbert Klugbauer (Klugbauer et al., 1999) and a double HA-tag was inserted after the predicted signal peptide similar to the α2δ-1 construct. pSyn5-Nrxn vectors were described previously (Neupert et al., 2015). Novel synNChNA was generated by replacing NheI flanked GFP in synNENA (Neupert et al., 2015), with mCherry from pcDNA3.1/hChR2-mCherry using oligos MM10-78 (CGA CGA GCT AGC AAG CTT CCA GGC CCG ATG GTG AGC AAG GGC GAG GAG, +HindIII, +NheI) and MM10–79 (CGA CGA GCT AGC CGG GCC TGG CTT GTA CAG CTC GTC CAT GCC GC, +NheI). For better identification of neuronal morphology, we used pMH4-SYN-tdimer2-RFP (T. Oertner, Center for Molecular Neurobiology, Hamburg). SynCAM (mouse, Cadm1), vGluT1-pHluorin, pHluorin(SEP)-tagged Nrxn1α (Neupert et al., 2015) and untagged Nrxn1α (pCMV-L2) were published previously. In biochemical studies, we used GFP-antibody-sensitive Venus-VE-Cad (Addgene, 56340), GFP-E-Cad (Addgene, 28009) and GFP-Nlgn1 (Nils Brose, Max Planck-Institute of Experimental Medicine, Göttingen) as control proteins. The proteolytic cutoff from Fc-tagged Nrxn1α required the insertion a HRV 3C protease cutting site LEVLFQ↓GP between Nrxn1α signal peptide and human Fc-tag in our Fc-control vector (Reissner et al., 2008) using QuikChange primers (MM-1741, 5′-GCG GCC GCT CTA GAG CTC GAG GTA CTA TTC CAG GGA CCG GAT CCC GAT CCC GAG-3′). The same cutting site was added to splice insert 4-free variant of Nrxn1α::Fc (Ichtchenko et al., 1995) to obtain Nrxn1α-pFc using primers (MM13-40, 5′-CTA TGA CAA CTG AGT CGA CGC TCG AGG TAC TAT TCC AGG GAC CGG ATC CCG ATC CCG AGG GTG AG-3′). The extracellular domain of α2δ-3 (ending at … CGGAS-STOP, CA2D3_MOUSE) was made by deleting the transmembrane region and adding a STOP signal to HA-tagged α2δ-3 using primers (MM15-28, 5′-GGA GAA TGC AAG AGA GTG TGG GGG TGC TAG CTG ACA CTA ACT AAG GGG ATG-3′). Finally, an HA-tagged mature neurexophilin 1 (Nxph1-HA) was cloned by adding HA-tag-STOP sequence between Nxph1 and Fc-tag of pCMVIgNxph1mat (Reissner et al., 2014) using primers (MM09-75, 5′-CTT ACT TCC CCT CCG GAT ACC CAT ATG ATG TTC CAG ATT ACG CTT AGT AGG GAC GCG AC-3′).
Ca2+ imaging and vesicle exocytosis.
To determine presynaptic Ca2+ influx, primary neurons were transfected at 14 DIV with synaptophysin-conjugated GCaMP6f by use of Lipofectamine, following the suppliers protocol (ThermoFisher Scientific). As indicated, additional plasmids like α2δ subunits or Nrxn were cotransfected. 3–5 d post-transfection, neurons growing on glass coverslips were placed in a recording chamber mounted to an inverted microscope (Observer.A1, Zeiss) and superfused at 1.0–1.5 ml/min with bath solution at room temperature (≈22°C), containing the following (in mm): 145 NaCl, 3 KCl, 1.5 MgCl2, 1.5 CaCl2, 11 glucose, 10 HEPES, pH 7.3 adjusted with NaOH; to suppress postsynaptic signaling, 10 μm 6-cyano-7-nitroquinoxaline-2,3-dione, 25 μm d,l-2-amino-5-phosphonovaleric acid, and 10 μm bicuculline were added. All chemicals were obtained from Sigma-Aldrich, except calcium channel blockers (Alomone Labs), and Fluo5 and Fura-2 (Invitrogen, ThermoFisher Scientific). A stimulation electrode, built by two platinum wires of 10 mm length in 10 mm distance was positioned with a micromanipulator (MPC-200, Sutter Instruments) and neurons were stimulated with 50 Hz trains of 1, 3, or 10 current pulses (1 ms, 55 mA). Ca2+ transients were visualized and recorded (10 ms exposure time, frame rate 100 Hz) with a CMOS camera (Orca Flash4.0, Hamamatsu), a LED-light source (SpectraX, Lumencor) using the green channel (excitation at 470 ± 20 nm) and controlled by VisiView software (Visitron Systems). The CaV2.1 Ca2+ channel antagonist (ω-agatoxin) was added by droplet application: during halted perfusion, a 10 μl drop of stock solution (40 μm) was applied to the bath solution (volume ≈1 ml) above the recording area 3–5 min before recording, leading to a calculated final concentration of 400 nm agatoxin.
As an alternative method to measure Ca2+ transients in presynaptic boutons, single neurons were recorded by patch-clamp technique in current-clamp mode (resting potential −70 ± 1 mV by current injection, if necessary) with the calcium indicator dye Fluo5 (300 μm) added to the intracellular solution, containing the following (in mm): 140 K-gluconate, 1.5 MgCl2, 10 HEPES, 4 Na-ATP; 0.5 Na-GTP, 0.5 EGTA; 0.05 AlexaFluor 594, pH 7.3 with KOH. After loading the cells for 8–10 min, neuronal processes were identified by the AlexaFluor 594 fluorescence with red light (560 ± 20 nm excitation, emission 595–660 nm). When axonal regions with synaptic boutons were identified by their thin processes and bead-like protrusions, short (1 ms) depolarizing somatic current injections induced action potentials in the recorded neuron (1 or 10 APs, 50 Hz) and Fluo5 fluorescence (green channel) was imaged (10 ms exposure time, frame rate 100 Hz). In addition, somatic Ca2+ dynamics in single neurons were investigated with Fura-2, delivered during patch-clamp recordings via the pipette solution (as above, but containing 100 μm Fura-2 instead of Fluo5 and AlexaFluor 594). Fura-2 was excited alternatingly at 360 and 380 nm by a polychromator (Visitron) with 5 Hz, and VGCCs were activated during voltage-clamp by a 100 ms depolarization to +20 mV. Changes of somatic Ca2+ concentrations were evaluated by the ratio of the fluorescence (360/380 nm) taken from regions-of-interest (ROIs) placed on a somatic region near to a distal process.
Vesicular release was quantified with vGluT-conjugated pHluorin (gift from Jürgen Klingauf, Institute of Medical Physics and Biophysics, Münster) to image vesicle fusion. vGluT-pHluorin was transfected at 14 DIV before imaging neurons between 17–19 DIV. Optical measurements were performed using the same laminar-flow perfusion and stimulation chamber as for Ca2+ transients. Release was evoked by trains of 10 action potentials (10 Hz) and images acquired at a frame rate of 10 Hz with an exposure time of 100 ms. Two recordings with at least 2 min recovery in between were averaged to reduce noise.
Data analysis of imaging recordings of Ca2+ transients or vesicle release was done with ImageJ (National Institute of Health) and IgorPro (WaveMetrics). Up to 36 ROIs per measurement area were drawn around active boutons as indicated by stimulation with a train of 3 AP for experiments with synGCaMP6f (or 10 AP for vGluT-pHluorin recordings). Single AP responses were analyzed after averaging four consecutive recordings, and for amplitude analysis, their traces were smoothed (coefficient 5) to improve signal-to-noise ratio. Before quantification, background subtraction (rolling ball, 10 μm diameter, ImageJ) was used. Twenty frames were recorded before the stimulus train and for each ROI the average value of frames 10–20 was taken as baseline control (F0). Changes were calculated as the change of fluorescence intensity divided by the control (ΔF/F0) for each ROI. For presentation of ΔF fluorescence images of isolated active regions (Fig. 1A), the average of frames number 10–20 (before stimulation) was subtracted from the average of 11 consecutive frames around the maximal response. In a subset of experiments (Fig. 2E,F), the Ca2+ ionophore ionomycin (10 μm) was applied after halting the perfusion at the end of the recording to saturate the Ca2+ indicator. For each ROI, the maximum of the stimulation-induced Ca transients was compared with the maximal fluorescence, obtained with ionomycin, to calculate the relative fluorescence increase.
Figure 1.
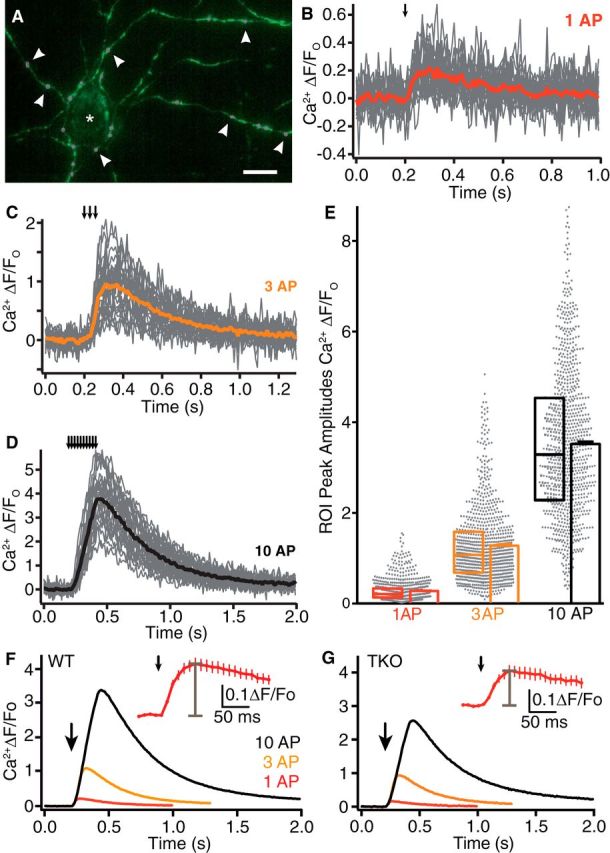
Monitoring presynaptic Ca2+ influx with synGCaMP6f. A, Axonal branches and putative presynaptic boutons in a hippocampal culture transfected with synGCaMP6f (green) during a stimulation with 10 AP. ROIs were drawn around synaptic boutons (circles, magenta, some are indicated by arrowheads) for evaluation of presynaptic Ca2+ transients. Asterisk, nontransfected neuron. Scale bar, 20 μm. B, Exemplary experiment of fluorescence changes to stimulation by a single action potential (1 AP; arrow indicates stimulation) in a recording from WT neurons transfected with synGCaMP6f; recordings of individual ROIs (gray lines), and averaged response (red line). C, Exemplary fluorescence changes as in B to stimulation by a train of 3 AP (averaged response, orange). D, Exemplary fluorescence changes as in B to stimulation by a train of 10 AP (averaged response, black). E, Maxima of all ROIs from WT neurons corresponding to single synaptic boutons (n = 887 from 35 independent experiments; scatter diagram, gray dots). Overlaid box plots represent median and 25–75% percentiles; bar diagrams show average of ROI maxima (mean ± SEM), colors as in B–D). Note that the mean of ROI maxima is not identical to the maximum of averaged traces due to noise of individual ROI traces and different time points of maxima. F, Traces of Ca2+ fluorescence changes determined by transfected synGCaMP6f from WT neurons averaged across 887 boutons (35 experiments) in response to 1 (red), 3 (yellow), and 10 (black) APs (arrow: start of stimulation train). Inset, The initial response to single AP on an enlarged time scale, gray bar indicates peak amplitude. The recording frequency (100 Hz) is adequate for determination of the peak of the synGCaMP6f fluorescence signal. G, Fluorescence changes of boutons as in F from neurons lacking αNrxns (TKO, n = 750/28).
Figure 2.
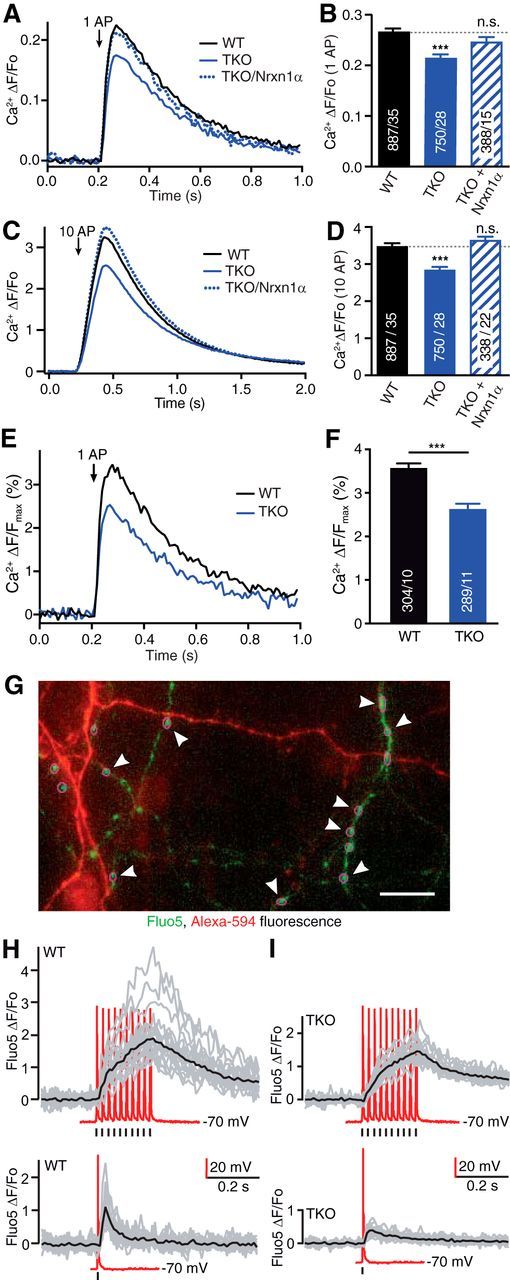
α-Neurexins are required for normal presynaptic Ca2+ influx in primary hippocampal neurons. A, Comparison of averaged presynaptic Ca2+ traces (n given in corresponding bars in B) from single AP responses of WT, TKO and TKO transfected with Nrxn1α. B, Summary of mean peak synGCaMP6f signals (ΔF/Fo) of Ca2+ transients after single AP stimulation of WT boutons compared with TKO, and TKO transfected with Nrxn1α. Data are mean ± SEM n = ROIs/neurons (in bars), differences to WT are indicated (dotted line); ***p < 0.001; n.s. not significant, p = 0.221; one-way ANOVA, F(2,2040) = 14.6. C, Comparison of presynaptic Ca2+ traces as in A using 10 AP trains for stimulation. Arrow indicates start of stimulation train. D, Analysis as in B with stimulation by 10 AP trains; ***p < 0.001; n.s. not significant, p = 0.182; one-way ANOVA, F(2,2040) = 51.3. E, Traces from WT neurons and from neurons lacking all αNrxns (TKO) that were stimulated with 1 AP (arrow) and normalized offline to the maximal synGCaMP6f fluorescence, as measured by saturating internal Ca2+ after application of the Ca2+-ionophore ionomycin (10 μm) at the end of each recording. F, For each ROI, the maximum of a Ca2+ transient induced by 1 AP was compared with the maximal fluorescence seen in presence of ionomycin. Data are mean ± SEM; n = number of ROIs/neurons, shown in bars. ***p < 0.001 by unpaired t test, t(587) = 5.37. G, Fluorescence image of neurites from WT hippocampal neurons loaded with Fluo5 (green) and AlexaFluor 568 (red) in a combined patch-clamp and imaging experiment. Magenta circles (some are highlighted by arrowheads) indicate ROIs around putative presynaptic boutons. Scale bar, 20 μm. H, Fluorescence changes of Fluo5 recorded from individual WT synaptic boutons (gray lines) and their averaged response (black) to 10 AP (top) or 1 AP (bottom) stimulation. Current traces of depolarization-induced somatic APs recorded in current-clamp (red); position of 1 and 10 AP stimulations indicated by marker bars. I, Individual and averaged responses as in H but from recordings of neurons lacking all αNrxns (TKO). Representative samples from at least three independent experiments per genotype are shown.
Single-particle tracking analysis.
Hemagglutinin (HA)-tagged α2δ were labeled with Quantum dots (QD; QD-655) bound to monoclonal anti-HA antibodies (Roche, catalog #11867423001). QD-655, goat F(ab′)2 anti-rat IgG Conjugate (H+L) highly cross-adsorbed (0.1 μm, ThermoFisher Scientific, catalog #Q-11621MP) were precoated with rat anti-HA (0.1 μg) in 10 μl PBS for 30 min and blocked with casein for 15 min. Transfected neurons were incubated with 0.1–0.05 nm QD for 5 min at 37°C, washed in HEPES buffered physiological solution (in mm: 145 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 10 HEPES, and 10 d-glucose, pH 7.4, and 0.5% BSA), transferred to an open Ludin chamber (Life Imaging Services) and imaged. Recordings of anti-HA QDs labeled α2δ were conducted at a spinning disc Axio Observer-Z1 (Visitron) equipped with an EMCCD camera (ImageEM C9100–13, Hamamatsu) using a 100 × 1.46 NA Plan-Apochromat oil-immersion objective (Zeiss). Fluorescence of QDs was excited by a 561 nm Laser (100 mW; Visitron). Emitted fluorescence was acquired through an ET Bandpass 690/50. Recordings of QDs were obtained with an integration time of 30 ms for up to 1000 consecutive frames. QD-labeled α2δ were followed on randomly selected axonal regions for up to 20 min. QDs fixed to the coverslip allowed to compensate for mechanical drifts of the stage. All recordings of molecular mobility were performed at 36–37°C.
Single QDs were tracked using custom-made software (Groc et al., 2007). Trajectories of single QDs were reconstructed with a point accuracy of 50–60 nm and subtrajectories reconnected when the displacement between two frames was 1–2 pixels (maximal dark period of 25 frames). For motion parameters of single molecules, mean squared displacement (MSD) were calculated and plotted over time for reconnected trajectories of at least 100 frames. Based on MSD curves from immobilized QDs on the coverslip the resolution limit for diffusion was 0.001 μm2 · s−1. Diffusion coefficients were calculated by linear fit of the first 4 points of the MSD plots versus time.
Ca2+ currents.
Recordings of recombinant VGCCs were performed using a fixed-stage inverted microscope (Observer.A1) equipped with a 40× objective (C-Apochromat). Patch-clamp recordings from transfected tsA-201 cells were done 3–5 d after plating by transferring the coverslip into a custom-made chamber superfused at 1.0–1.5 ml/min with bath solution at 32°C, containing the following (in mm): 115 NaCl, 3 CaCl2, 1 MgCl2, 10 HEPES, glucose, 20 TEA-Cl, pH 7.4 (300 ± 5 mOsm/kg osmolality). Patch pipettes (borosilicate glass, 1.5 mm outer diameter; Hilgenberg) were pulled by a two-stage electrode puller (PIP 6, HEKA Elektronik), showing resistances of 2–4 MΩ when filled with pipette solution containing the following (in mm): 125 Cs-methane sulfonate, 20 TEA-Cl, 5 EGTA, 2 MgCl2, 10 HEPES, 4 Na2-ATP, 0.5 Na-GTP, pH 7.4 (285 ± 5 mOsm/kg osmolality). Whole-cell calcium currents were recorded with an EPC 10 USB Double patch-clamp amplifier and Patchmaster software (HEKA Elektronik). Signals were filtered at 3 kHz and digitized at 10 kHz except where mentioned otherwise. Cells were held at −80 mV in whole-cell configuration, series resistance and membrane capacitance determined and compensated online. Leak currents were subtracted online using a P/5 protocol. Recordings for each condition were done on cells from at least three independent experiments.
Current–voltage (I–V) relationships were obtained by 50 ms voltage pulses from a holding potential of −80 mV to voltages between −40 mV and +70 mV in 10 mV increments with 6 s intervals. I–V traces from individual cells were fit with a modified Boltzmann equation as follows:
 |
where Gmax is the maximum slope conductance, Vrev is the reversal potential, V1/2 act is the half-activation potential, and kact is the slope factor. Current densities were calculated as currents normalized to whole-cell capacitance. Steady-state inactivation properties were measured by evoking currents with a 500 ms test pulse to +20 mV after 2 s voltage displacement (pre-pulse) from +20 mV to −80 mV in 10 mV increments. Amplitudes of currents evoked by the test pulses were normalized to the maximum current and plotted against the pre-pulse potential. The data from individual cells were fit with a Boltzmann equation as follows:
 |
where A1 and A2 are the non-inactivating and inactivating fractions, respectively, V1/2 inact is the half-inactivation potential, and kinact is the slope factor.
Analysis of activation-deactivation properties was done on tail currents (Yarotskyy et al., 2009). The voltage dependency of channel activation was measured from tail currents after repolarization to −40 mV from voltages ranging from −40 to +70 mV. Tail current amplitudes were plotted versus step voltage and fitted with a Boltzmann equation:
 |
which yielded half-activation voltage (V1/2) and slope factor kact. For additional tail current analysis, calcium currents were activated by a 10 ms step from the holding potential to +10 mV, followed by a 10 ms repolarizing pulse ranging from −100 to +10 mV in 10 mV increments. Data were analyzed by averaging for 0.2 ms, beginning 0.3 ms after the repolarizing pulse. Currents were digitized at a sample interval of 50 μs (20 kHz). To obtain an estimate for the mean open time (τdeact) tail currents were fit at −20 mV by a single-exponential function only recordings where the single-exponential fit nicely overlaid the trace were included in evaluation.
Immunocytochemistry.
For labeling of hippocampal cultures, 21 DIV neurons were fixed with 4% paraformaldehyde/ 4% sucrose for 8 min, washed with PBS, blocked in 10% normal goat serum (NGS), 0.1% Triton X-100/PBS for 1 h, and incubated with primary antibodies overnight at 4°C: mouse anti-vGluT1 (1:1000; Synaptic Systems, catalog #135511), rabbit anti-P/Q-type calcium channels (1:500; Synaptic Systems, catalog #152203), mouse anti-HA Clone 16B12 (BioLegend), diluted in 10% NGS, 0,1% Triton X-100/PBS or used for surface staining as described below. After washing, cells were incubated with the following secondary antibodies: AlexaFluor 488 goat-anti-mouse IgG, AlexaFluor 647 goat-anti-mouse IgG, AlexaFluor 647 goat-anti-rabbit IgG (Invitrogen), AlexaFluor 647 goat-anti-chicken IgG (Invitrogen), diluted 1:500 in 5% NGS/PBS for 1 h at RT. After additional washings in PBS, coverslips were embedded in mounting medium (Dako). Imaging of cells was performed with a confocal spinning disc Axio Observer-Z1 (Visitron) with an EMCCD camera (ImagEM 512 CCD, Hamamatsu), using 40× or 63× Plan-Neofluar oil-immersion objectives. 0.5 μm Z-stacks were acquired, with a maximal distance of 1.5 μm from the focus plane. All images were acquired with the same laser power, exposure time and camera gain settings. Using ImageJ software, maximum intensity projections of Z-Stacks were generated for the analysis. For fluorescence intensity quantification of Cav2.1 calcium channels, WT and TKO neurons were transfected either with the cytosolic marker t-dimer-RFP alone or in combination with GFP-tagged Nrxn1α (synNENA). Twenty-micrometer-long axonal regions were selected and only punctae that resided on t-dimer-RFP axons and colabeled for the presynaptic marker vGlut1 and Cav2.1 were analyzed with ImageJ software tools.
For quantification of synaptic localization of HA-tagged α2δ-1 or HA-tagged α2δ-3, ROIs were drawn around synGCaMP6f-positive puncta (used as reference of active presynaptic boutons) and copied over the fluorescent images correspondent to anti-HA-specific channel (AlexaFluor 647 goat-anti-mouse IgG) or Nrxn1α::mCherry fluorescent signal. Each copied ROI was considered as positive when its fluorescent intensity in the selected channel was higher than twice the amount of the background. Percentages of double-positive synGCaMP6f/anti-HA and triple-positive synGCaMP6f/anti-HA/Nrxn1α::mCherry were then extracted. For surface staining of HA-tagged α2δ-1 or HA-tagged α2δ-3, the experiments were performed as reported in (Aoto et al., 2013); briefly, neurons were quickly washed in PBS supplemented with 0.5 mm CaCl2, 1 mm MgCl2, and 4% sucrose (PBS-MC). Anti-HA antibody was incubated for 10 min at 37°C. After a quick wash in cold PBS-MC, cells were fixed 15 min with 4% paraformaldehyde/4% sucrose and processed for routine immunocytochemistry as described above. The percentages quantifications of synGCaMP6f/surface HA-tagged α2δ-1 or surface HA-tagged α2δ-3 double-positive puncta were performed with same criteria used above for synaptic localization.
For immunolabeling of heterologous cells, tsA-201 cells were fixed in 4% PFA/PBS for 10 min at RT, washed with PBS and blocked in 5% normal goat serum/PBS for 30 min at RT. Primary and secondary antibodies were diluted in blocking solution. Primary antibody mouse anti-HA (BioLegend) was applied 1:250 overnight at 4°C. After washing, secondary antibody AlexaFluor 647 goat-anti-mouse IgG (Life Technologies) was applied 1:500 for 1 h at RT. After additional washing, coverslips were embedded in mounting medium (Dako). Images were taken with epifluorescence microscopes (Axio Imager.Z2, Zeiss) equipped with a 63× 1.4 numerical aperture Plan-Apochromat objective (Zeiss) and CCD camera (Spot Xplorer and Apotome, Visitro). Z-stacks (0.5 μm) were acquired, with a maximal distance of 1.5 μm from the focus plane. All images were acquired with the same laser power, exposure time and camera gain settings. Using ImageJ software, maximum intensity projections of Z-Stacks were generated for analysis.
Immunoprecipitations and protein biochemistry.
Forty-eight hours after tsA-201 cells were transfected at 60–70% confluence, cells were washed once in cold PBS buffer, scraped and lysed in 1% Triton X-100, 20 mm HEPES, 150 mm NaCl, 2 mm MgCl2, 0.1 mm EDTA and protease inhibitor cOmplete (Roche). Lysates were centrifuged (16,000 × g for 10 min, 4°C) and supernatant precleared by incubating 5 μl Protein-G beads (50% slurry) for 30 min. 1 μg of anti-HA antibody (16b12, BioLegend) was added for immunoprecipitation (IP) of HA-tagged α2δ subunits and incubated overnight at 4°C. After 4 washings with lysis buffer, IPs were eluted in 2× loading buffer and subjected to immunoblot analysis. Briefly, total lysates (input) and IPs were run on 6% acrylamide/bis-acrylamide gels and transferred to PVDF membranes (Roth). After blocking with BSA, membranes were incubated overnight with the following antibodies: anti-HA (16b12, BioLegend) 1:1000, anti-GFP (ab290, Abcam) 1:1000, anti-HSP70 (3A3, Dianova) 1:1000. Membranes were washed in TBS 0.3% Tween, incubated with HRP-conjugated secondary antibodies and developed using an ECL system (GE Healthcare).
For co-secretion and proteolytic cleavage, Fc-tagged Nrxn1α was harvested 3 d after transfection of HEK cells: 50 μl of protein A beads were added to 100 ml of each medium to bind the Fc-tag of co-secreted proteins α2δ-3::HA/Nrxn1α::Fc, α2δ-3::HA/pFc or Nxph1-HA/Nrxn1α::Fc. Protein-bound beads were washed 3-times with Tris-buffer (50 mm Tris, pH7.4, 2 mm MgCl2, 150 mm NaCl) and split in two aliquots. The first was used for immunoblot using anti-HA (1:500, 16b12) and the second aliquot was used for proteolytic overnight digest with 5 units of HRV 3C protease (Novagen) in 500 μl Tris-buffer. The pellet of centrifuged (50 s, 11,000 × g) probes contained Fc-bound proteins. His-tagged protease was removed from supernatant with 10 μl of Ni-NTA beads. The final supernatant that contained the Nrxn1α-bound proteins was concentrated to 50 μl using Pierce Concentrator (10,000 MW cutoff (ThermoFisher Scientific). Pellet and supernatant were diluted in 2× sample buffer, boiled for 5 min and applied to nUView Precast gradient gel 4–20% (NuSep) to visualize Fc-fragment and Nrxn1αECD by UV light and for an immunoblot to label HA-tagged proteins.
For immunoblots of wild-type and TKO cultures, protein samples were extracted from 21–22 DIV primary hippocampal neurons with 2×SDS lysis buffer (125 mm Tris-HCl, pH 6.8, 20% glycerol, 4% SDS, 2% β-mercaptoethanol). Extracts were pooled from 330,000 cells per genotype. Briefly, 25 μl of hot 2× SDS lysis buffer were added to each coverslips and cells were scraped to collect protein extracts. To remove DNA and RNA aggregates, samples were passed six times through 1 ml syringes (30Gx1/2 inch) and subsequently subjected to Western blot analysis as described before, using the following antibodies: anti-Cav2.1 P/Q type (Synaptic Systems, catalog #152203) 1:1000, anti-α2δ-1 (Alomone Labs, catalog #ACC-015) 1:500, anti-α2δ-3 (Alomone Lab, catalog #ACC-103) 1:500, anti-Actin (Sigma-Aldirch, catalog #A50-60) 1:2000, and anti-Nrxn123 (Synaptic Systems, catalog #175003) 1:1000.
Statistical analysis.
No statistical methods were engaged to predetermine sample size, instead we based our experimental design on numbers reported in previous studies. The experiments were not randomized, and investigators were only partially blinded during experiments and analyses. Statistical tests were performed with Prism (GraphPad Software). If samples met criteria for normality, we used a Student's t test to compare two groups, and a one-sided ANOVA for more than two groups. If ANOVAs were significant, we used a post hoc Tukey's multiple-comparisons test to compare groups. Exact p values are given between 0.001 and 0.99. For analysis of diffusion coefficients, we used a Kruskal–Wallis test followed by a Dunn's test. Data are presented as mean ± SEM. Significance levels were as indicated in figures: *p < 0.05, **p < 0.01, and ***p < 0.001.
Results
αNrxns are required for normal presynaptic Ca2+ influx
To dissect the role of αNrxn in presynaptic Ca2+ influx, we first investigated whether Ca2+ transients of boutons differ between cultured hippocampal wild-type and αNrxn TKO neurons. Using the genetically encoded Ca2+ indicator GCaMP6f (T. W. Chen et al., 2013) fused to synaptophysin (synGCaMP6f; Fig. 1A), we monitored Ca2+ influx induced by single action potentials (APs). Single APs reliably led to traces of Ca2+ transients with variable responses across synaptic boutons (Fig. 1B) as expected (Koester and Sakmann, 2000). In addition to 1 AP stimuli, we recorded traces elicited by 3 and 10 APs (Fig. 1C,D) to also include trains of activity that are physiologically relevant (Rozov et al., 2001). Maxima of Ca2+ transients from synaptic ROIs were normalized to baseline fluorescence and reached mean peak amplitudes of 0.27 ± 0.01 ΔF/F0 at 1 AP, 1.26 ± 0.03 at 3 AP and 3.36 ± 0.07 at 10 AP in wild-type neurons (n = 887 ROIs from ≥35 neurons; Fig. 1E). Compared with wild-type boutons (Figs. 1F, 2A–D), Ca2+ transients in TKO showed lower averaged traces (Figs. 1G, 2A–D), and reached reduced mean peak amplitudes of 0.22 ± 0.01 ΔF/F0 at 1 AP, 1.02 ± 0.02 at 3 AP and 2.87 ± 0.05 at 10 AP (n = 750 ROIs from ≥28 neurons), which indicated a reduction by almost 20% of the Ca2+ transients at 1 AP in TKO compared with WT. To support this critical observation, we performed two additional experiments: first, we used an alternative procedure to normalize Ca2+ transients to the maximal response obtained by application of 10 μm ionomycin at the end of each recording. This produced a similar difference between wild-type and TKO Ca2+ influx (26% less in TKO; Fig. 2E,F), but had the disadvantage that less experiments could be performed on cultures of triple mutants. Second, we measured presynaptic Ca2+ signals of wild-type and TKO with an alternative fluorescent indicator, Fluo5 (Fig. 2G–I) applied through a patch-pipette. This indicator showed fast, reliable Ca2+ transients, which also confirmed the reduction in TKO terminals compared with wild-type (Fig. 2H,I). These data demonstrate that the smaller presynaptic Ca2+ influx into TKO terminals is a reproducible and robust finding.
To prove the specificity of the finding, we then tried to rescue the TKO phenotype by expressing an abundant hippocampal αNrxn variant, Nrxn1α (Aoto et al., 2013; Fuccillo et al., 2015), in our cultured neurons from this region. Transfection of a single Nrxn1α into TKO was sufficient to reverse the reduced Ca2+ transient traces to a mean peak amplitude of 0.25 ± 0.01 ΔF/F0 at 1 AP, which was like wild-type influx (Fig. 2A,B). Moreover, the reduction of Ca2+ transients in TKO and its rescue in TKO/Nrxn1α synapses were present across stimulation protocols with single APs (Fig. 2A,B), 3 AP (data not shown) and 10 AP trains (Fig. 2C,D). To probe whether the reduced Ca2+ transients in TKO affected presynaptic vesicle release in our model system of primary neurons, we measured exocytosis in the same cultures (Fig. 3A). For this, we used a pH-dependent indicator (pHluorin, fused to the luminal side of the vesicular glutamate transporter; vGluT_pH) and a similar 10 AP train as described for Ca2+ transients above. Stimulation increased the fluorescence by 0.292 ± 0.009 ΔF/F0 (n = 278 ROIs from ≥8 neurons; Fig. 3B). Monitoring vGluT_pH fluorescence changes in TKO neurons revealed a reduced release activity compared with wild-type terminals with only 0.217 ± 0.007 ΔF/F0 increase (n = 463 ROIs from ≥17 neurons; p < 0.001; Fig. 3B,C), that is 26% less release as in WT. As for the presynaptic Ca2+ influx, expression of Nrxn1α could ameliorate the release phenotype in TKO neurons (TKO/Nrxn1α: 0.278 ± 0.008 ΔF/F0; n = 482 ROIs from ≥14 neurons; p < 0.001 compared with TKO, p = 0.59 compared with wild-type; Fig. 3B,C). Together, these data demonstrate the reliability, specificity, and physiological relevance of our finding that deletion of αNrxns leads to reduced presynaptic Ca2+ influx in cultured hippocampal neurons and a subsequently reduced neurotransmitter release.
Figure 3.

Synaptic vesicle release is reduced in neurons lacking all α-neurexins. A, ΔF fluorescence image of vGlut_pHluorin (green) from a 100 AP stimulation, overlayed on DIC image of WT neurons. Scale bar, 20 μm. B, Exocytotic response of vGluT_pHluorin averaged across multiple synapses; comparison of WT, TKO, and TKO transfected with Nrxn1α (N given in corresponding bars in C). C, Summary of mean peak vGluT_pHluorin signals (ΔF/Fo) from conditions as in B. Data are mean ± SEM; n = ROIs/neurons (in bars), differences to WT are indicated. ***p < 0.001, n.s. = not significant, p = 0.454, by one-way ANOVA, F(2,1222) = 25.4.
CaV2.1-mediated Ca2+ influx and channel abundance depend on αNrxns
The strength of evoked neurotransmitter release from hippocampal synapses is controlled efficiently by high-voltage activated CaV2.1 (P/Q-type) calcium channels (Cao et al., 2004; Li et al., 2007; Bucurenciu et al., 2010; Holderith et al., 2012). We therefore asked whether a reduced contribution of CaV2.1 was involved in the diminished presynaptic Ca2+ influx in αNrxn TKO neurons. To test this idea, we recorded presynaptic Ca2+ transients before and after addition of the specific CaV2.1 inhibitor ω-agatoxin IVA (Fig. 4A,B). Stimulations with 10 AP trains were applied to ensure quantifiable responses from just one of several VGCC subtypes that contribute to Ca2+ transients in hippocampal neurons (Li et al., 2007; Ermolyuk et al., 2013). We compared averaged CaV2.1-specific traces of presynaptic Ca2+ transients between wild-type, TKO and TKO with transfected Nrxn1α (Fig. 4A). Traces were then normalized to the total Ca2+ influx, revealing that CaV2.1 mediated >40% of Ca2+ transients in wild-type boutons (41.1 ± 1.3%; n = 468 ROIs from ≥20 neurons; Fig. 4B). In TKO boutons, the relative contribution of CaV2.1 was strongly reduced to 25.9 ± 1.1% (n = 561 ROIs from ≥9 neurons; p < 0.001; Fig. 4B). Interestingly, Nrxn1α in TKO did not fully restore the relative contribution of CaV2.1 (TKO/Nrxn1α: 30.2 ± 1.3%; n = 389 ROIs from ≥17 neurons; p < 0.001 compared with wild-type, p = 0.042 compared with TKO; Fig. 4A,B), constituting a partial rescue. This contrasts with the expression of a single Nrxn1α variant in TKO shown above that was able to enhance the total presynaptic Ca2+ influx to normal amounts (Fig. 2A–D). The discrepancy is interesting because other VGCC subtypes such as CaV2.2, CaV2.3 or even members of the CaV1.x subfamily could also be upregulated by overexpression of Nrxn1α and may overproportionally compensate the reduction in total presynaptic Ca2+ influx. In addition, the partial rescue of CaV2.1 could possibly indicate that expression of one neurexin isoform alone is not sufficient to secure normal numbers of activatable α1A pore-forming subunits in the synapse.
Figure 4.
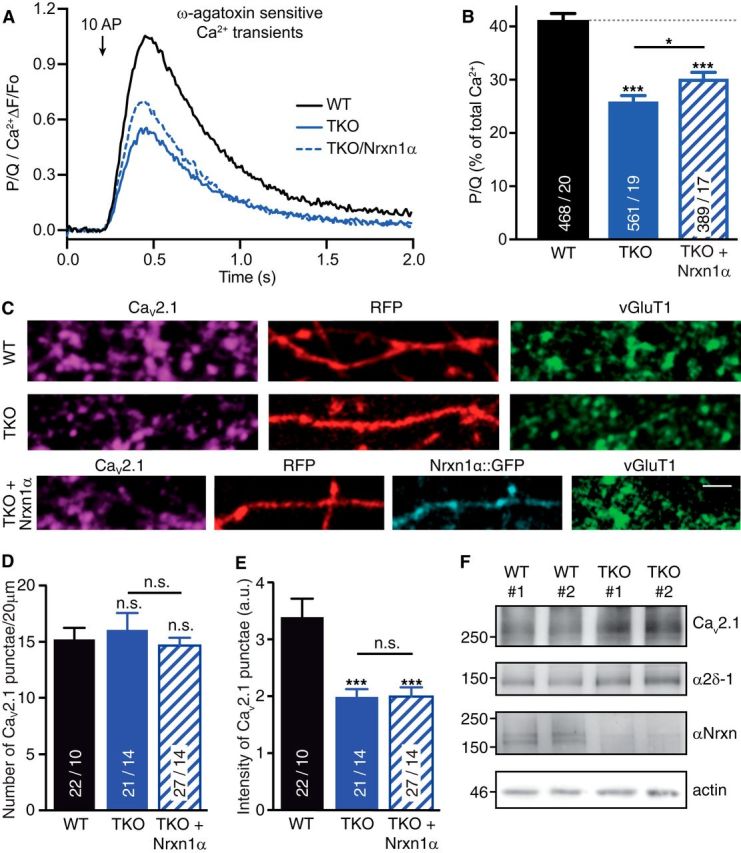
Deletion of αNrxns leads to reduced CaV2.1-mediated presynaptic Ca2+ influx and channel abundance. A, Isolated ω-agatoxin IVA-sensitive traces of Ca2+ transients through CaV2.1 (P/Q-type) channels derived from subtraction of transients before and after addition of the CaV2.1 blocker. Traces are recorded by synGCaMP6f and averaged across multiple boutons of WT, TKO, and TKO neurons transfected with Nrxn1α (TKO+Nrxn1α). B, Summary histogram of CaV2.1 channel contributions (in percentage of total Ca2+ transients) of WT boutons compared with TKO, and TKO+Nrxn1α. Data are mean ± SEM; n = ROIs/neurons (in bars), differences to WT (dotted line) are indicated above columns; ***p < 0.001; *p = 0.042, by one-way ANOVA, F(2,1415) = 42.6. C, Representative images of immunofluorescence with antibodies against endogenous α1A of CaV2.1 (magenta), colabeled against vesicular glutamate transporter (vGluT1, green) in WT and TKO neurons transfected with RFP (red) alone (WT, TKO) or in combination with Nrxn1α::GFP (cyan, TKO+Nrxn1α). Scale bar, 2.5 μm. D, Quantification of the number of α1A-positive puncta that colocalize with presynaptic vGluT1 along RFP-filled axons of WT, TKO and TKO+Nrxn1α neurons. E, Quantification of immunofluorescence intensity of α1A-positive puncta colocalizing with vGluT1. Data in D and E are mean ± SEM; n = number of axonal segments (shown in bars) from at least three independent experiments. **p < 0.01; ***p < 0.001, n.s. = not significant, by one-way ANOVA. F, Immunoblots of total protein lysates from two independent WT and TKO cultures, each representing ∼300,000 hippocampal cells. Blots were probed with antibodies against the α1A pore forming and α2δ-1 auxiliary subunits of CaV2.1, and against all αNrxn variants that are deleted in TKO; actin = loading control.
We therefore tested whether the difficult-to-reverse reduction reflected a diminished abundance of the α1A pore-forming subunit at TKO and TKO/Nrxn1α synapses. We immunolabeled endogenous α1A-subunits in presynaptic boutons of wild-type, TKO, and TKO/Nrxn1α neurons transfected by RFP alone or in combination with Nrxn1α. Axonal regions of 20 μm length were selected and overlapping boutons identified by colocalization with presynaptic marker vGluT1 (Fig. 4C). Although the number of α1A-positive boutons was not altered (WT: 15.18 ± 1.056, n = 22/10; TKO: 15.95 ± 1.616, n = 21/14; p = 0.947; TKO/Nrxn1α: 14.44 ± 0.867, n = 27/14; pwt = 0.979, ptko = 0.773; Fig. 4D), the fluorescence intensity of α1A-positive puncta (Fig. 4E) was diminished in TKO neurons. The reduction of α1A fluorescence intensity by 42% (WT: 3.37 ± 0.336, n = 22/10; TKO: 1.962 ± 0.156, n = 21/14; p < 0.001) could indicate a lower abundance of pore-forming subunits of CaV2.1 channels at presynaptic boutons lacking all αNrxns, consistent with the reduced overall presynaptic Ca2+ influx and vesicle release (Figs. 2, 3). This reduction was not due, however, to an overall lower expression of the channel subunit because immunoblotting of wild-type and TKO cultures revealed similar amounts of protein (Fig. 4F). Similar to the partial rescue of CaV2.1-mediated presynaptic Ca2+ transients (Fig. 4A,B), expression of Nrxn1α alone was not sufficient to increase α1A fluorescence intensity (TKO/Nrxn1α: 1.94 ± 0.193, n = 27/14; pwt < 0.001, ptko > 0.99). However, in this immunolabeling experiment we can neither distinguish between active and non-active boutons nor between activatable and non-activatable α1A subunits, possibly leading to an underestimation of the functional rescue. In contrast, the synGCaMP6f measurements included only responsive boutons with stimulated changes of Ca2+ transients (Fig. 4A).
Lower abundance of activatable presynaptic CaV2.1 pore-forming subunits (Fig. 4A,B) could additionally imply that more functional CaV2.1 channels are present outside synapses. To test whether a subpopulation of channels were ectopically enriched in TKO, we measured Ca2+ influx into the soma. We loaded Fura-2 with a patch-clamp pipette into individual neurons (Fig. 5A) that were depolarized under voltage-clamp condition for 100 ms to +20 mV (Fig. 5B). In contrast to presynaptic Ca2+ influx (Fig. 2), a comparison of mean peak amplitudes of total somatic Ca2+ transients showed an increase in TKO (wild-type: 0.026 ± 0.003 ΔF360nm/380nm, n = 14; TKO: 0.036 ± 0.004 ΔF360nm/380nm, n = 23, p = 0.040). This elevation was specific because it could be reversed by expression of Nrxn1α to a level indistinguishable from WT (TKO/Nrxn1α: 0.022 ± 0.002 ΔF360nm/380nm; n = 8; p = 0.79). We then analyzed the relative contribution of CaV2.1 channels to total Ca2+ transients by application of ω-agatoxin to wild-type and TKO neurons. The relative contribution of CaV2.1 to somatic Ca2+ transients was small in wild-type (8.8 ± 1.8%; n = 8 neurons; Fig. 5C) as expected. However, Ca2+ influx through somatic CaV2.1 increased considerably in TKO neurons (20.3 ± 3.6%; n = 7, p = 0.012; Fig. 5C), possibly representing an ectopic presence of pore-forming units on the soma. Expression of Nrxn1α in TKO neurons revealed a tendency to reverse the increased somatic CaV2.1 contribution (TKO/Nrxn1α: 13.6 ± 2.1%; n = 7; Fig. 5C). While this level was statistically similar to wild-type (p = 0.395), the reduction lacked significance compared with TKO (p = 0.191), which indicates a clear tendency to rescue similar to presynaptic Ca2+ influx (Fig. 4A, B). More importantly, the increased somatic Ca2+ transients in TKO show that deletion of αNrxns does not interfere with the trafficking of CaV2.1 to the plasma membrane in general. This is consistent with abundant surface delivery of Ca2+ channels in heterologous expression systems that do not contain αNrxns (Dolphin, 2012). Together, the presynaptically reduced agatoxin-sensitive Ca2+ transients and diminished α1A immunofluorescence, along with increased somatic agatoxin-sensitive Ca2+ transients suggest that localization of functional α1A pore-forming subunits of CaV2.1 is altered in absence of αNrxns. This defect raises the question how an αNrxn variant can affect the α1A.
Figure 5.

Monitoring Ca2+ influx in the somata of neurons with Fura-2. A, Representative images of a WT neuron loaded with Fura-2 via patch pipette and excited at 380 nm before stimulation (A1). Ca2+ transient by 100 ms depolarization to +20 mV visualized by the 360/380 nm ratio (A2). The oval indicates a ROI for evaluation; Scale bar, 20 μm. B, Sample traces of fluorescence changes within the ROI marked in A at 360 and 380 nm excitation (green). The Ca2+ transient expressed as 360/380 nm ratio is shown below (red). C, Summary of mean peak amplitudes (ΔF/Fo) at 100 ms depolarization, comparing untransfected WT neurons (black bars), TKO neurons (blue) and TKO neurons transfected with Nrxn1α. Bars indicate somatic Ca2+ transients through ω-agatoxin IVA-sensitive P/Q-channels, analyzed in relation to the total Ca2+-transient before wash-in of the blocker. The part of P/Q-channels in somatic Ca2+-transients (in WT: 8.8 ± 1.8%) was increased in TKO (20.3 ± 3.6%; *p = 0.012) and showed a tendency to rescue (not significant: p = 0.191) in TKO-expressing Nrxn1α (13.6 ± 2.1%), this value is not significantly different to WT (p = 0.395). Data are mean ± SEM. n = neurons, as indicated in bars; p values by one-way ANOVA, F(2,19) = 5.17.
α2δ-1 and α2δ-3 modulate Ca2+ influx differently in presence of Nrxn1α
α2δ Auxiliary subunits of VGCCs were described as partners of α1A and other pore-forming subunits during trafficking (Cantí et al., 2005) and at the cell surface (Cassidy et al., 2014). Interestingly, overexpression of α2δ subunits increased synaptic abundance of CaV2.1 but not Ca2+ influx (Kurshan et al., 2009; Hoppa et al., 2012), opening the possibility that α2δ need to cooperate with sufficiently large amounts of αNrxns to affect Ca2+ influx.
To determine whether α2δ-1 or α2δ-3 subunits are able to enhance presynaptic Ca2+ transients together with an abundant amount of a defined αNrxn variant, we cotransfected α2δ-1 or α2δ-3 with and without Nrxn1α into TKO neurons, and applied 1, 3, and 10 AP stimuli (Fig. 6A,B). Strikingly, cotransfection of α2δ-1 together with Nrxn1α into TKO not only rescued, as shown for Nrxn1α alone (Figs. 2A–D, 6C) but facilitated Ca2+ transients 21.7% beyond wild-type amounts at single AP (wild-type control: 0.267 ± 0.007, n = 887/35; cotransfection of α2δ-1/Nrxn1α: 0.325 ± 0.012, n = 437/17; p < 0.001; Fig. 6C). Expression of α2δ-1 alone failed to restore normal Ca2+ influx in TKO (Fig. 6C; p = 0.978), suggesting that presence of an αNrxn is necessary for the facilitation. We then transfected α2δ-3 in combination with Nrxn1α and alone into TKO neurons. In contrast to α2δ-1, coexpression of α2δ-3 together with Nrxn1α in TKO did not facilitate Ca2+ influx beyond wild-type in response to 1 AP (α2δ-3/Nrxn1α: 0.255 ± 0.009, n = 443/17, p = 0.027 compared with TKO; p = 0.966 compared with wild-type; Fig. 6C). Like α2δ-1, α2δ-3 overexpression alone failed to restore normal Ca2+ influx during stimulation (0.236 ± 0.008; n = 544/22, p = 0.506 compared with TKO, p = 0.057 compared with wild-type; Fig. 6C). These data presumably reveal two implications: first, presence of an αNrxn might be necessary for the full potential of α2δ-1 subunits to promote presynaptic Ca2+ influx; and second, presence of Nrxn1α may diversify the function of distinct α2δ subunits in neurons because together with Nrxn1α, α2δ-1 causes a much stronger facilitation than α2δ-3.
Figure 6.

α2δ-1 auxiliary subunits together with Nrxn1α facilitate presynaptic Ca2+ influx in TKO neurons. A, Traces of Ca2+ fluorescence changes determined from TKO neurons cotransfected with Nrxn1α and α2δ-1 subunits. Ca2+ transients indicated by synGCaMP6f are averaged across multiple boutons in response to 1 (red), 3 (yellow), and 10 (black) APs (arrow: start of stimulation train). Inset, Initial response to a single AP on an enlarged time scale. B, Fluorescence changes of boutons as in A from TKO neurons expressing α2δ-3 subunits together with Nrxn1α. C, Summary of mean peak synGCaMP6f signals (ΔF/Fo) of Ca2+ transients after single AP stimulation of neurons transfected with different proteins. Data are mean ± SEM. n = ROIs/neurons (in bars), differences to WT and TKO are indicated (dotted lines); significance is given compared with WT above columns (black) and compared with TKO (blue; above dashed line). ***p < 0.001, *p < 0.05, n.s. = not significant, by one-way ANOVA with Tukey's multiple-comparisons test (F(6,4034) = 17.79); exact p values are given in gray. D, Immunofluorescent images of Ca2+ indicator synGCaMP6f, mCherry-tagged Nrxn1α and HA-tagged α2δ-1 subunits (top) or HA-tagged α2δ-3 (bottom), labeled by an antibody against the HA moiety, cotransfected into TKO neurons. Scale bars: E, F, 5 μm. E, Similar experiment to D but without expression of mCherry-tagged Nrxn1α. F, Quantification of colocalization of α2δ-1/Nrxn1α and α2δ-3/Nrxn1α with synGCaMP6f-positive puncta as in E, and of colocalization between α2δ-1 or α2δ-3 with synGCaMP6f-positive puncta as in F. Data are mean ± SEM. n = puncta/neurons (in bars); n.s. = not significant, by unpaired t test.
To exclude that these differences between α2δ-1 and α2δ-3 in αNrxn TKO were merely due to differences in the localization or abundance of the transfected molecules, we performed additional immunolabeling experiments. Fluorescence of synGCaMP6f in combination with antibodies recognizing the HA-tagged α2δ-1 or α2δ-3 subunits with (Fig. 6D) and without (Fig. 6E) additional mCherry-tagged Nrxn1α was analyzed in TKO neurons. We determined the percentage of colocalization of transfected α2δ subunits with synGCaMP6f-positive terminals but found no difference in synaptic localization between α2δ-1 or α2δ-3 (Fig. 6F).
Nrxn1α together with α2δ-1 facilitates Ca2+ currents through recombinant CaV2.1
Because any biophysical analysis of the combined effect by Nrxn1α/α2δ-1 on CaV2.1, the key finding in our study, is potentially confounded by presence of other variants in neurons, we finally addressed this important aspect by recordings from recombinant CaV2.1 channels. We expressed core complexes of CaV2.1 channels (P/Q-type: α1A and β3) alone and in combination with either α2δ-1 or α2δ-3 subunits in heterologous tsA201 cells. Using patch-clamp electrophysiology, we recorded whole-cell Ca2+ currents with or without cotransfected Nrxn1α, and compared representative current traces (Fig. 7A), averaged current–voltage relationships (I–V curves; Fig. 7B,C) and peak amplitudes of current densities (Fig. 7D). Although the CaV2.1 core complex alone produced only moderate current densities (9.4 ± 1.3 pA/pF; n = 16), we found that cotransfection of α1A/β3, the CaV2.1 core complex, with α2δ-1 (41.4 ± 7.8 pA/pF; n = 14; p < 0.01; Fig. 7D, full red bar) or α2δ-3 (63.8 ± 8.8 pA/pF, n = 17; p < 0.001; Fig. 7D, full blue bar) robustly and significantly increased currents. No changes of current densities were observed when we cotransfected Nrxn1α with α1A/β3 (CaV2.1 core) without presence of an α2δ subunit (12.9 ± 1.9 pA/pF; n = 12; Fig. 7D), suggesting that Nrxn1α does not act directly on pore-forming subunits.
Figure 7.
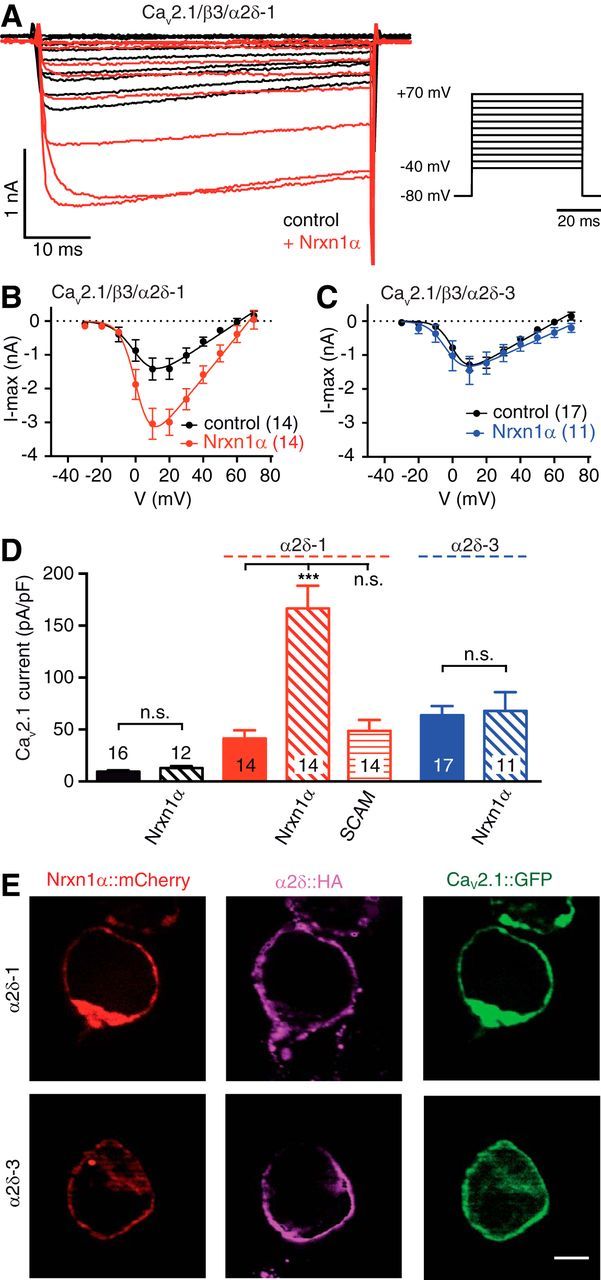
Nrxn1α in combination with α2δ-1 facilitates Ca2+ currents through recombinant CaV2.1 channels. A, Representative CaV2.1-mediated Ca2+ current traces recorded from heterologous tsA201 cells expressing α1A, β3 and α2δ-1 subunits alone (black) or together with Nrxn1α (red). Step potentials as shown (right) were used to elicit Ca2+ currents. B, I–V relationships of CaV2.1/β3/α2δ-1 alone (black) or in combination with Nrxn1α (red). C, Similar analysis as in B but using α2δ-3; trace in combination with Nrxn1α in blue. D, Summary of maximum current densities for cells expressing CaV2.1(α1A/β3) without an α2δ (black bars), with α2δ-1 (red) or with α2δ-3 (blue), and additionally with Nrxn1α or SynCAM1 (SCAM) as indicated below bars. Data are mean ± SEM. n = number of cells as indicated in bars from at least four independent experiments. ***p < 0.001, n.s. = not significant (all p > 0.99), by one-way ANOVA, F(6,91) = 20.1. E, Immunofluorescence images of transfected tsA201 cells showing Nrxn1α fused to mCherry (red, Nrxn1α::mCherry), α1A pore-forming subunit fused to EGFP (green, CaV2.1::GFP), and colabeling with antibodies against HA-tagged α2δ (magenta, α2δ::HA). β3 auxiliary subunits were coexpressed in all conditions. Scale bar, 5 μm. Res, residues.
When we cotransfected Nrxn1α with the distinct α2δ subunits, however, we discovered remarkable changes of Ca2+ currents. Nrxn1α in combination with α2δ-1 facilitated Ca2+ currents from CaV2.1 channels (Fig. 7A,B), and elevated current densities almost fourfold (402%) compared with α2δ-1 alone (α2δ-1: 41.4 ± 7.8 pA/pF vs Nrxn1α/α2δ-1: 166.6 ± 21.9 pA/pF, n = 14; p < 0.001; Fig. 7D). In contrast, Nrxn1α together with α2δ-3 had no effect on CaV2.1-mediated currents (Fig. 7C), with current densities almost unchanged compared with α2δ-3 alone (α2δ-3: 63.8 ± 8.8 pA/pF, n = 17, vs Nrxn1α /α2δ-3: 67.9 ± 17.9, n = 11; p > 0.99; Fig. 7D). These results appeared specific because an unrelated synaptic cell-adhesion molecule SynCAM1 (SCAM; together with α2δ-1: 48.7 ± 10.6 pA/pF, n = 14; Fig. 7D) failed to produce significant changes of CaV2.1 currents (p > 0.99). Our data also appeared reliable because the use of different epitope-tagged or untagged versions of Nrxn1α reproduced similar changes (data not shown). Furthermore, the observed changes of Ca2+ currents were likely not due to simple failures in expression or targeting of α2δ-1 or α2δ-3 subunits because they were delivered similarly to the cell surface along with the pore-forming subunit of CaV2.1 and Nrxn1α (see Fig. 9E). Thus, our results in heterologous cells confirm a specific interaction of Nrxn1α with α2δ-1, but not with α2δ-3, which facilitates Ca2+ currents through CaV2.1 channels. We conclude that this regulation is responsible for the difference of presynaptic CaV2.1-mediated Ca2+ influx, which we observed in wild-type and TKO neurons (Fig. 4).
Figure 9.

Nrxn1α does not engage in stable complexes with α2δ subunits. A, IP of cotransfected α2δ subunits and Nrxn1α or control membrane proteins from HEK293 cell lysates (top). IPs of HA-tagged α2δ-1 and α2δ-3 enrich Nrxn1α::GFP (lanes 4, 5) similar to the controls neuroligin-1 (Nlgn1), E-cadherin (E-Cad) and VE-cadherin (VE-Cad; lanes 6–8). Single transfections served as control for antibody specificity (lanes 1–3). Endogenous HSP70 indicates equal amounts of lysates used (botto). B, Co-secretion of extracellular domains of α2δ-3 (α2δ-3ECD::HA) and Nrxn1α (Nrxn1α::Fc) into HEK293 cell medium with subsequent binding of the Fc moiety to protein A beads. Lysates of cells show α2δ-3ECD::HA (lanes 1–2). Whereas the positive control, Nxph1-HA, is hardly detectable in cell lysates (lane 3, bottom), it is enriched with Nrxn1α::Fc (lane 6, bottom). α2δ-3ECD::HA is enriched similarly with Nrxn1α::Fc (lane 5, top) but also with the Fc-tag alone. C, Diagram of the cleavage experiment using HRV 3C protease to release the Nrxn1αECD from Fc-beads (immunoblot data in D). Left, Nxph1 (magenta) is bound to Nrxn1αECD (green) as expected. Right, α2δ-3ECD (cyan) remains on Fc-coupled beads (orange) but does not interact with Nrxn1αECD. D, Immunoblot of the cleavage experiment (C) that starts from the precipitated samples in B. After addition of protease, α2δ-3ECD remains on Fc-tag bound to beads (lanes 7, 8) but is not found on Nrxn1αECD in the supernatant (lane 11). The positive control, Nxph1, is bound to the released Nrxn1αECD (lane 12). α2δ-3 and Nph1 are shown by immunoblot, Nrxn1α and Fc proteins are visualized by UV light.
The reduced abundance of CaV2.1 in synaptic boutons and concomitant upregulation in the somata of TKO neurons (Figs. 4, 5) suggested impaired synaptic targeting of VGCCs as one reason for the phenotype in absence of αNrxn. To exclude that changes of channel gating or kinetic properties of CaV2.1 channels also contribute to the reduced Ca2+ currents (Klugbauer et al., 1999), we analyzed biophysical channel properties that are observable in whole-cell recordings such as steady-state inactivation, tail current activation and inactivation to estimate the mean open time. However, we found no alteration of the voltage dependence of steady-state inactivation of CaV2.1 by cotransfection of Nrxn1α, independent of the presence α2δ-1 or α2δ-3 (Fig. 8A,B). To evaluate the Ca2+ channel activation-deactivation properties with and without Nrxn1α at higher resolution, we then analyzed tail currents from CaV2.1-expressing cells (Fig. 8C). The voltage dependence of channel activation for α2δ-1/CaV2.1 was independent of the presence of Nrxn1α because slope factor (Fig. 8D; p = 0.204) as well as half-activation voltage (Fig. 8E; p = 0.812) showed no significant differences. As seen with voltage step protocols, the I–V curves of tail currents reflected the facilitation of CaV2.1 by Nrxn1α together with α2δ-1 (Fig. 8F). However, the changes of these currents could not be explained by changes in mean open time of the channels as we found no significant differences in their deactivation time constants (Fig. 8G; p = 0.199), which can serve as a proxy of channel open time (Yarotskyy et al., 2012). Together, our findings in heterologous cells suggest a preferential functional association of α2δ-1 with CaV2.1 that depends on Nrxn1α, consistent with our experiments in neurons. Because the association did not affect channel properties like mean open time or activation/inactivation kinetics, the mechanism likely involves the number of activatable CaV2.1 channels at the cell surface, also in line with our results in presynaptic boutons.
Figure 8.
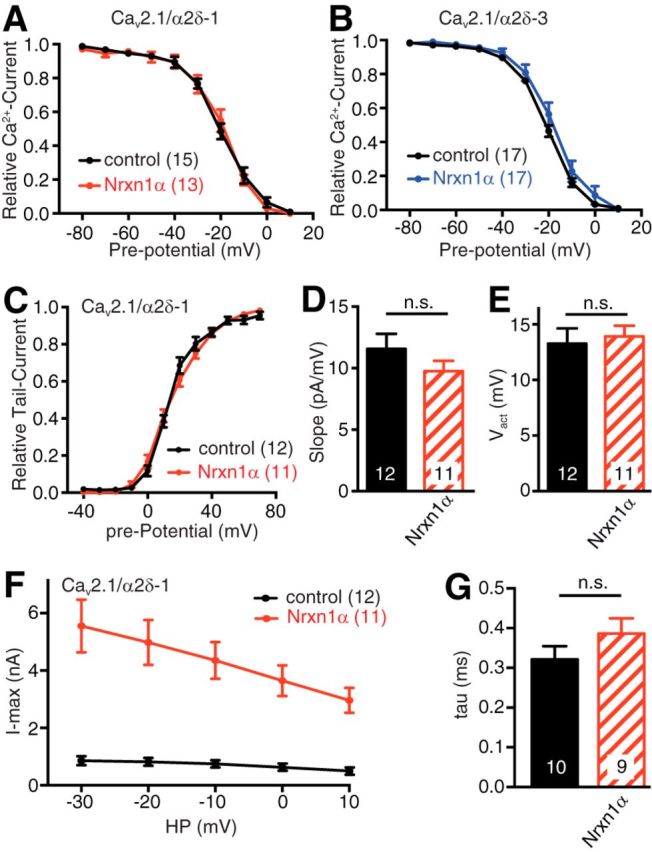
Biophysical properties of recombinant CaV2.1 are not altered by Nrxn1α. A, Voltage dependence of steady-state inactivation of CaV2.1 channels tested by a pre-pulse protocol in tsA201 cells expressing α1A, β3, and α2δ-1 subunits alone (black) or together with Nrxn1α (red). B, Analysis as in A expressing α1A, β3, and α2δ-3 subunits alone (black) or together with Nrxn1α (blue). C, Tail current amplitude at −40 mV after a 10 ms voltage step to the given pre-potential, recorded in tsA201 cells expressing CaV2.1/α2δ-1 without (black) or with Nrxn1α (red). D, Slope factor of the voltage dependence of the channel activation of CaV2.1/α2δ-1 without (black) or with Nrxn1α (red); n.s. = not significant, p = 0.204, by unpaired t test, t(21) = 1.32. E, Half-activation voltage of the voltage dependence of activation of CaV2.1/α2δ-1 tail current (as given in C) without (black) or with Nrxn1α (red); n.s. = not significant, p = 0.812, by unpaired t test, t(21) = 0.24. F, I–V curves of tail currents of CaV2.1/α2δ-1 without (black) or with Nrxn1α (red). G, Analysis of tail current deactivation time constant at −20 mV of CaV2.1/α2δ-1 without (black) or with Nrxn1α (red); n.s. = not significant, p = 0.199 by unpaired t test, t(17) = 1.34. Data are mean ± SEM. N = number of cells as shown in bars or in brackets from at least four independent experiments.
Extracellular domains of α2δ and Nrxn1α do not form stable complexes
The specific requirement of α2δ-1 subunits for the effect of Nrxn1α on CaV2.1 channels shown above might entail that Nrxn1α forms a stable complex with α2δ. To clarify this aspect, we cotransfected HA-tagged α2δ-1 or α2δ-3 and GFP-tagged Nrxn1α in HEK293 cells, and then performed IP with an anti-HA antibody (Fig. 9A). Pulldown of α2δ-1 (Fig. 9A, lane 4) or α2δ-3 (lane 5) showed coprecipitation of Nrxn1α, similar to a recent study on CaV2.2 channels and α2δ-3 (Tong et al., 2017). However, the observed binding was not specific because transfected neuroligin-1 and even completely unrelated membrane proteins such E-cadherin or VE-cadherin could interact equally well with α2δ-1 (Fig. 9A, lanes 6–8). This apparent lack of binding specificity of α2δ-1 and the discrepancy to another study (Tong et al., 2017) prompted us to perform more control experiments. First, additional IPs with HA-tagged α2δ-1 from membrane fractions of HEK293 cells revealed nonspecific binding also to endogenous membrane proteins, for example to β-integrin (data not shown). Second, to overcome the pitfall of these IPs, we probed binding between pure recombinant proteins without contamination from other membrane proteins. Because α2δ subunits have no cytosolic domain (Ellis et al., 1988), any putative interaction should involve the extracellular domains of α2δ and Nrxn1α. To probe this idea, we expressed the extracellular domains (ECD) of HA-tagged α2δ-3ECD and Fc-tagged Nrxn1αECD in HEK293 cultures. α2δ-3ECD contained the cysteine bridge between the von Willebrand factor-A domain (α2-chain) and the δ-chain (Calderon-Rivera et al., 2012), but excluded the transmembrane region (res. 1069–1091). Both proteins were co-secreted into the culture medium and their binding tested subsequently by precipitating Nrxn1α::Fc with protein A-beads (Fig. 9B). Although a complex of α2δ-3ECD and Nrxn1α::Fc could be found (Fig. 9B, lane 5), the binding was again not specific as the Fc-tag alone also precipitated α2δ-3ECD equally well (lane 4). To test whether at least some of the α2δ-3ECD was able to bind to the Nrxn1αECD, in addition to the unspecific interaction with the Fc-tag, we engineered a 3C-protease cutting site between the Nrxn1αECD and Fc moieties to release any protein complexed to Nrxn1αECD into the supernatant, whereas Fc-bound proteins remained on the protein A beads (Fig. 9C). The tight complex of Nxph1 with the Nrxn1αECD (Reissner et al., 2014) served as control for successful proteolytic cleavage (Fig. 9C). As expected, Nxph1 was bound to the released Nrxn1αECD after protease cleavage (lane 12), however, α2δ-3ECD remained bound exclusively to the Fc-fraction on beads (Fig. 9D, lanes 7,8). These biochemical assays show that α2δ and Nrxn1α are not likely to engage in stable protein-protein binding.
αNrxns affect surface mobility of α2δ-1 and α2δ-3 subunits differentially
Despite the lack of stable complex formation (Fig. 9), the cooperative role of Nrxn1α and α2δ-1 in facilitating CaV2.1-mediated Ca2+ transients (Fig. 6) and Ca2+ currents (Fig. 7) could indicate that the molecules engage in weak, transient interactions in neurons. We have previously used single particle tracking methods to analyze the surface mobility of Nrxns, α1A, and α2δ subunits, and found that changes in mobility are useful as a highly sensitive readout for an altered association with partner molecules (Biermann et al., 2014; Neupert et al., 2015; Schneider et al., 2015; Voigt et al., 2016). To start to explore weak and transient interactions, we asked whether the surface diffusion of α2δ subunits is altered on the axonal/ synaptic membrane of αNrxn TKO neurons. First, we performed live labeling of neurons with antibodies recognizing a transfected HA-tagged α2δ-1 and observed reliable punctate labeling on both wild-type and TKO neurons (Fig. 10A). We determined the percentage of colocalization of surface populations of transfected α2δ-1 subunits with synGCaMP6f-positive terminals but found no difference (Fig. 10B). Moreover, no alterations in fluorescence intensity were apparent (WT: 11,137 ± 2498 A.U., n = 13; TKO: 14,565 ± 3516 A.U., n = 19; p = 0.433). These data indicate that plasma membrane trafficking and synaptic localization of the α2δ subunit was independent of αNrxns.
Figure 10.
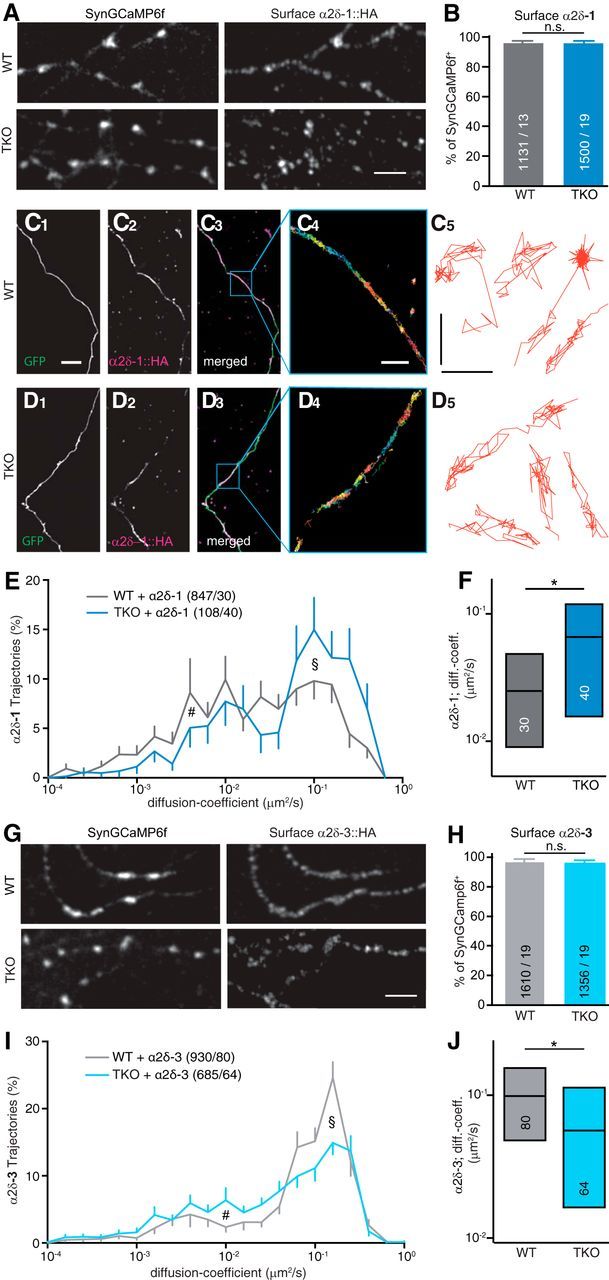
αNrxn modulates surface mobility of α2δ-1 and α2δ-3 auxiliary subunits differentially. A, Representative immunofluorescent images of surface α2δ-1 enriched in synaptic boutons, visualized by an antibody against the HA moiety of α2δ-1::HA cotransfected with synGCaMP6f into WT neurons (top) or TKO neurons (bottom). Scale bar, 5 μm. B, Quantification of colocalization between synGCaMP6f and surface α2δ-1-positive puncta in WT and TKO. Data are mean ± SEM; n = synGCaMP6f-positive puncta/neurons from three to four independent experiments per condition; n.s. = not significant (p = 0.433) by unpaired t test. C, Labeling of the surface population of HA-tagged α2δ-1 (C1) transfected into WT neurons using an antibody specific to the HA moiety. EGFP was cotransfected to visualize neurites (C2), merged images (C3) and an overlay of all trajectories of QD-tracked single α2δ-1 molecules in a subfield as indicated (C4); sample trajectories of QD-tracked single α2δ-1 molecules (C5). Scale bars: C1–D3, 10 μm; C4, D4, 2 μm; C5, D5, 0.5 μm. D, Labeling of surface α2δ-1 as in C using TKO neurons. E, Logarithmic distribution of diffusion coefficients for α2δ-1 on axons of WT and TKO neurons, showing more trajectories of higher mobility in TKO (see §) and fewer low mobility trajectories (see #); n = trajectories/cells; error bars (SEM) shown only in outward direction. F, Median and IQR (25–75%) of diffusion coefficients of α2δ-1 shown in E. Numbers of cells from four independent experiments (in bars). *p = 0.0277, by Kruskal–Wallis test with Dunn's post-test. G, Immunofluorescent images of surface α2δ-3 in synaptic boutons as in A. Scale bar, 5 μm. H, Quantification of colocalization between synGCaMP6f and surface α2δ-3-positive puncta in WT and TKO. Data are mean ± SEM. n = synGCaMP6f-positive puncta/neurons from three to four independent experiments per condition; n.s. = not significant (p = 0.4835), by unpaired t test. I, Logarithmic distribution of diffusion coefficients as in E but for α2δ-3. With α2δ-3, more trajectories of higher mobility occurred in WT (see §), indicating a reverse effect when compared with α2δ-1 (E). J, Median and IQR (25–75%) of diffusion coefficients of α2δ-3 shown in I. Numbers of cells from four independent experiments (in bars). *p = 0.0347, by Kruskal–Wallis test with Dunn's post-test.
We then investigated the hypothesis that αNrxns affect the surface mobility of α2δ-1 since these auxiliary subunits can associate tightly with VGCC pore-forming subunits at the plasma membrane (Cassidy et al., 2014). To determine diffusion, we transfected HA-tagged α2δ-1 in wild-type (Fig. 10C) or αNrxn TKO (Fig. 10D) neurons. We used single-molecule tracking with QD-coupled antibodies (Neupert et al., 2015) to determine trajectories of single α2δ-1 molecules on the axonal surface (Fig. 10C4,C5). Trajectories of QD tracks (Fig. 10C4–D5) and the distribution of diffusion coefficients (Fig. 10E) demonstrated that almost all α2δ-1 molecules were mobile (D > 10−3 μm2/s). Quantification of diffusion coefficients (Fig. 10F) showed that α2δ-1 became even more mobile when αNrxns are lacking in TKO neurons [wild-type: median 0.03811 μm2/s, interquartile range (IQR) 0.00736/0.1158, n = 847 trajectories/30 cells; TKO: median 0.09235 IQR 0.02173/0.1979, n = 108/40; p = 0.0277 for cell-based comparison, p < 0.001 for trajectories]. Our data suggest that the synaptic cell adhesion molecules αNrxns may be required to limit surface diffusion of α2δ-1.
To test whether the effect of αNrxns on the surface mobility was specific for α2δ-1, we explored localization and diffusion of another α2δ subunit, α2δ-3, which has different structural properties (Hendrich et al., 2008). Repeating the antibody labeling of live neurons as for α2δ-1 (Fig. 10A), we observed no changes in the percentage of colocalization of surface α2δ-3 with synGCaMP6f-positive terminals between wild-type and TKO (Fig. 10G). Similarly, we found no differences in surface abundance as shown by comparison of fluorescence intensity (WT: 9881 ± 1882 A.U., n = 19; TKO: 7665 ± 2541, n = 19; p = 0.4835; Fig. 10H). Then, we probed whether the surface diffusion of α2δ-3 is enhanced by deletion of αNrxns, as seen with α2δ-1 (Fig. 10E). Transfection of α2δ-3 into wild-type or TKO and comparison of diffusion coefficients (Fig. 10I,J) revealed two surprising findings: first, α2δ-3 showed a higher mobility than α2δ-1 in wild-type neurons (α2δ-3 in WT: median 0.0768 μm2/s, IQR 0.00952/0.1609; n = 930/80; Fig. 10J); and second, compared with their diffusion in wild-type, α2δ-3 subunits became slower by deletion of αNrxns in TKO (α2δ-3 in TKO: 0.0394 μm2/s, IQR 0.007055/0.1261; n = 685/64; p = 0.035 for cell-based comparison, p < 0.001 for trajectories; Fig. 10I,J), reaching values similar to α2δ-1 mobility in wild-type (Fig. 10F). Together, these data might implicate that deletion of αNrxns alters the surface diffusion of α2δ-1 and α2δ-3 in opposite ways, possibly reflecting their different ability to enhance Ca2+ transients in hippocampal neurons (Fig. 6) and Ca2+ currents in heterologous tsA201 cells (Fig. 7) through CaV2.1 channels.
Discussion
Here, we propose that a member of the neurexin family of presynaptic cell adhesion molecules, Nrxn1α, facilitates the amount of Ca2+ influx through CaV2.1 channels by acting together with α2δ-1 auxiliary subunits. This addresses an important aspect because modulating Ca2+ influx through CaV2.1 into presynaptic boutons is an effective way of setting synaptic strength due to the high cooperativity of Ca2+ in triggering release of transmitter-filled vesicles (Schneggenburger and Neher, 2000). We studied this key process in primary hippocampal neurons, a standard model for superior visibility and accessibility of mammalian CNS synapses and complemented our analysis by dissection of recombinant CaV2.1 channels in heterologous cells. Although loss-of-function studies of αNrxns investigated synaptic transmission in the same conventional knock-out mice used here (Missler et al., 2003), presynaptic Ca2+ influx has not been probed in this mouse model. The reduced amount of presynaptic Ca2+ influx (Fig. 2), and diminished vesicle exocytosis (Fig. 3), is in accordance with reduced release probability and evoked postsynaptic responses found at excitatory synapses lacking one (Etherton et al., 2009; Born et al., 2015) or more αNrxn genes (Missler et al., 2003; Zhang et al., 2005; Dudanova et al., 2006; Sons et al., 2006). Our current study provides justification for the hypothesis that αNrxn couples the function of presynaptic Ca2+ channels to transmission (Missler et al., 2003) by demonstrating that the coupling involves specific α2δ auxiliary subunits.
Our study relies on a well characterized indicator of neuronal Ca2+ transients, GCaMP6f (T. W. Chen et al., 2013; Lin and Schnitzer, 2016; Yang et al., 2018) that we and others have fused to a synaptic marker protein (Fig. 1), frequently used to monitor presynaptic and postsynaptic Ca2+ influx (Grauel et al., 2016; Hannan et al., 2016; Reese and Kavalali, 2016; Glebov et al., 2017; de Juan-Sanz et al., 2017). To validate an important finding in our study, we also used Fluo5 as an alternative indicator with different properties that is often applied to monitor presynaptic Ca2+ influx (Hoppa et al., 2012; Wang et al., 2016), essentially giving identical results (Fig. 2). Although we are acutely aware of the limitations of any fluorescent indicator as a proxy of actual Ca2+ currents, we strongly believe in the reliability of our imaging data. For example, coexpression of α2δ-1/Nrxn1α leads to strongly increased synGCaMP6f Ca2+ transients in neurons (Fig. 6) and Ca2+ currents in heterologous cells (Fig. 7), whereas α2δ-3/Nrxn1α fails to do so in both conditions. Moreover, the effect of αNrxns on presynaptic Ca2+ influx appears specific because expression of Nrxn1α was able to restore total Ca2+ transients in knock-out neurons (Fig. 2). This result is consistent with the improvement of impaired evoked release when transgenic mice overexpressing Nrxn1α were crossed into TKO (Missler et al., 2003; Zhang et al., 2005). However, these 'rescue mice' still showed the same lethality as TKO, suggesting that not all relevant parameters were restored. We now discovered at least one reason for the incomplete rescue because Nrxn1α was not able to restore the normal contribution of CaV2.1 channels to presynaptic Ca2+ influx (Fig. 4). Furthermore, we found that α2δ-1 together with Nrxn1α increased presynaptic Ca2+ influx much stronger in TKO neurons than Nrxn1α or α2δ-1 alone (Fig. 6). In extension of an earlier investigation of α2δ overexpression (Hoppa et al., 2012), we uncovered a distinct behavior for α2δ-1 and α2δ-3 when coexpressed with Nrxn1α in TKO neurons (Fig. 6), because α2δ-1/Nrxn1α increased presynaptic Ca2+ influx much stronger than α2δ-3/Nrxn1α.
More evidence for a specified function of α2δ-1 and α2δ-3 subunits comes from our analysis of Ca2+ currents through recombinant CaV2.1 channels that showed a facilitating effect of Nrxn1α on CaV2.1, which depended on α2δ-1 (Fig. 7). We observed that the coexpression of Nrxn1α with α2δ-1 does not cause additional modifications of Cav2.1 channel parameters like activation/inactivation kinetics or mean open time (Fig. 8), whereas α2δ-1 itself may increase the inactivation rate of α1A channels (Felix et al., 1997). Thus, our data suggest that the Nrxn1α-induced effect, which requires α2δ-1, rather alters the surface presence of activatable Ca2+ channels. In fact, this is in line with the changes of presynaptic α1A abundance (Fig. 4), which could not simply be explained on basis of a modulation of their biophysical properties. In addition, the different behavior of α2δ-1 and α2δ-3 in our experiments (Figs. 6, 7) may reflect differences in their molecular structure (Klugbauer et al., 1999) and in their distribution in the brain, which is only partially overlapping (Schlick et al., 2010). The Nrxn1α-mediated facilitation of the effect of α2δ-1 on CaV2.1 discovered here corroborates the lower evoked release observed at hippocampal Schaffer collateral synapses of CaV2.1 knock-out mice (Mallmann et al., 2013). It is further consistent with a correlation of α2δ-1 surface expression and excitatory mini frequencies at glutamatergic terminals (Cordeira et al., 2014) as well as the reduced Ca2+ influx after gabapentin application in neocortical neurons, which is α2δ-1 dependent (Fink et al., 2002).
The finding that Nrxn1α cooperates with α2δ-1 to modulate Ca2+ influx raises the question of the nature of their association. A quantitative proteomic study showed that the β auxiliary subunits purified in almost equimolar ratios to the α1A,B proteins. However, α2δ subunits were present in only 10% molar abundance and Nrxn1α even <3% (Müller et al., 2010). Another report arrived at the conclusion that the Caenorhabditis elegans α2δ orthologue UNC-36 interacts physically and tightly with the CaV2.2 complex and with Nrxn1 (Tong et al., 2017). Using the same assay of immunoprecipitating epitope-tagged α2δ subunits and Nrxn1α, we could in fact detect binding (Fig. 9A). Unfortunately, the binding turned out to be unspecific because validation with several unrelated membrane proteins failed as all could be bound by overexpressed α2δ subunits (Fig. 9A). Moreover, for previous studies we purified soluble extracellular Nlgn1/Nrxn1α-, Nxph1/Nrxn1α- or dystroglycan/Nrxn1α-complexes using Nrxn1α::Fc, which allowed us to reliably map their interfaces (Reissner et al., 2008, 2014). The same approach failed for α2δ/Nrxn1α::Fc because we observed that α2δ binds to the Fc-tag but not to Nrxn1α (Fig. 9B–D). These data indicate that unlike Nlgn, Nxph, dystroglycan, or LRRTM, α2δ subunits do not form stable complexes with Νrxn1α. Instead, we used single-molecule tracking in neurons to explore their association because surface diffusion is a sensitive measure for Nrxn-dependent molecular interactions (Biermann et al., 2014; Neupert et al., 2015). We observed earlier that the surface mobility of α2δ-1 is much higher than of α1 pore-forming subunits and that they are only transiently confined together, supporting the idea of nonstatic, weak associations (Schneider et al., 2015; Voigt et al., 2016). We now extend these results by demonstrating that α2δ-3 shows higher surface mobility than α2δ-1 and that both subunits depend in their surface mobility on αNrxn in opposite ways (Fig. 10). In absence of αNrxn, α2δ-1 becomes less confined, suggesting that their facilitating effect on CaV2.1 function (Figs. 6, 7) requires more confined VGCC complexes (Fig. 10). In support, mobile α2δ subunits are not peculiar to mammalian neurons since labeling of C. elegans UNC-36 by split-GFP tags confirms surface dynamics of these calcium channel subunits even in vivo (Zhan et al., 2014). Thus, the processes underlying presynaptic Ca2+ influx appear to be highly dynamic in nature but also very sensitive to transient associations.
Our study does not cover all putative effects of any Nrxn variant on CaV2.1 function. For example, we cannot exclude that βNrxn, still present in TKO (Missler et al., 2003), may partially compensate by changing relative contributions of VGCC subtypes. In fact, conditional deletion of all three βNrxns in cultured cortical neurons reduced presynaptic Ca2+ transients, albeit it does not include decreased abundance of CaV2.1 (Anderson et al., 2015). Similarly, it remains to be determined which Nrxn is responsible for diminished Ca2+ transients in somatostatin interneurons following ablation of all αNrxn and βNrxn variants (L. Y. Chen et al., 2017). Because no experiments with pharmacological VGCC inhibitors and rescue constructs have been performed for Ca2+ influx in these studies, they could not arrive at conclusions whether CaV2.1 contributions are affected. Also, it is necessary to remain open for changes in the association of distinct Nrxn and α2δ variants during development because many synapses undergo changes in expression and clustering of different VGCCs (Wu et al., 1999; Iwasaki et al., 2000; Nakamura et al., 2015). Finally, any presynaptic Ca2+ signal appears as the result of a modular arrangement that depends on the identity, abundance, and flexibility of distinct partner molecules within the microenvironment of the presynaptic membrane. It remains to be explored whether the modulation of CaV2.1 by Nrxn1α and α2δ-1 is part of the larger architecture of nanocolumns (Perez de Arce et al., 2015; Tang et al., 2016) that contain alignments of active zone molecules, synaptic cell adhesion complexes and postsynaptic receptors within subregions of synapses.
In summary, our results propose Nrxn1α as a supportive modulator of presynaptic Ca2+ influx in interaction with α2δ-1. Together they may regulate the presence of activatable Ca2+ channels in the bouton, adding another way for neurons to adjust synaptic strength and efficiency.
Footnotes
This work was supported by Grants from the Deutsche Forschungsgemeinschaft (SFB1348-TPA03 and CiM FF-2015–05 to M.M.), the Land Sachsen-Anhalt (LSA research group “Molecular Physiology” to M.H.) as well as Schram Foundation (M.H.). D.R. was supported by a postdoctoral fellowship from the Fritz Thyssen Stiftung. We thank J. Aoto and M. Ahmad for comments on an earlier version of the manuscript, J. Klingauf and P. Coulon for discussions, C. Neupert and R. Freund for cloning experiments, and I. Wolff and K. Kerkhoff for technical assistance with cell culture and mouse genotyping.
The authors declare no competing financial interests.
References
- Anderson GR, Aoto J, Tabuchi K, Földy C, Covy J, Yee AX, Wu D, Lee SJ, Chen L, Malenka RC, Südhof TC (2015) Beta-neurexins control neural circuits by regulating synaptic endocannabinoid signaling. Cell 162:593–606. 10.1016/j.cell.2015.06.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Martinelli DC, Malenka RC, Tabuchi K, Südhof TC (2013) Presynaptic neurexin-3 alternative splicing trans-synaptically controls postsynaptic AMPA receptor trafficking. Cell 154:75–88. 10.1016/j.cell.2013.05.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biermann B, Sokoll S, Klueva J, Missler M, Wiegert JS, Sibarita JB, Heine M (2014) Imaging of molecular surface dynamics in brain slices using single-particle tracking. Nat Commun 5:3024. 10.1038/ncomms4024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born G, Grayton HM, Langhorst H, Dudanova I, Rohlmann A, Woodward BW, Collier DA, Fernandes C, Missler M (2015) Genetic targeting of NRXN2 in mice unveils role in excitatory cortical synapse function and social behaviors. Front Synaptic Neurosci 7:3. 10.3389/fnsyn.2015.00003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucurenciu I, Bischofberger J, Jonas P (2010) A small number of open Ca2+ channels trigger transmitter release at a central GABAergic synapse. Nat Neurosci 13:19–21. 10.1038/nn.2461 [DOI] [PubMed] [Google Scholar]
- Calderon-Rivera A, Andrade A, Hernández-Hernández O, González-Ramírez R, Sandoval A, Rivera M, Gomora JC, Felix R (2012) Identification of a disulfide bridge essential for structure and function of the voltage-gated Ca2+ channel α2δ-1 auxiliary subunit. Cell Calcium 51:22–30. 10.1016/j.ceca.2011.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantí C, Nieto-Rostro M, Foucault I, Heblich F, Wratten J, Richards MW, Hendrich J, Douglas L, Page KM, Davies A, Dolphin AC (2005) The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc Natl Acad Sci U S A 102:11230–11235. 10.1073/pnas.0504183102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao YQ, Piedras-Rentería ES, Smith GB, Chen G, Harata NC, Tsien RW (2004) Presynaptic Ca2+ channels compete for channel type-preferring slots in altered neurotransmission arising from Ca2+ channelopathy. Neuron 43:387–400. 10.1016/j.neuron.2004.07.014 [DOI] [PubMed] [Google Scholar]
- Cassidy JS, Ferron L, Kadurin I, Pratt WS, Dolphin AC (2014) Functional exofacially tagged N-type calcium channels elucidate the interaction with auxiliary α2δ-1 subunits. Proc Natl Acad Sci U S A 111:8979–8984. 10.1073/pnas.1403731111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. (2000) Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol 16:521–555. 10.1146/annurev.cellbio.16.1.521 [DOI] [PubMed] [Google Scholar]
- Chen LY, Jiang M, Zhang B, Gokce O, Südhof TC (2017) Conditional deletion of all neurexins defines diversity of essential synaptic organizer functions for neurexins. Neuron 94:611–625.e4. 10.1016/j.neuron.2017.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Chen TW, Wardill TJ, Sun Y, Pulver SR, Renninger SL, Baohan A, Schreiter ER, Kerr RA, Orger MB, Jayaraman V, Looger LL, Svoboda K, Kim DS (2013) Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature 499:295–300. 10.1038/nature12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeira JW, Felsted JA, Teillon S, Daftary S, Panessiti M, Wirth J, Sena-Esteves M, Rios M (2014) Hypothalamic dysfunction of the thrombospondin receptor α2δ-1 underlies the overeating and obesity triggered by brain-derived neurotrophic factor deficiency. J Neurosci 34:554–565. 10.1523/JNEUROSCI.1572-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean C, Scholl FG, Choih J, DeMaria S, Berger J, Isacoff E, Scheiffele P (2003) Neurexin mediates the assembly of presynaptic terminals. Nat Neurosci 6:708–716. 10.1038/nn1074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Juan-Sanz J, Holt GT, Schreiter ER, de Juan F, Kim DS, Ryan TA (2017) Axonal endoplasmic reticulum Ca2+ content controls release probability in CNS nerve terminals. Neuron 93:867–881.e6. 10.1016/j.neuron.2017.01.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. (2012) Calcium channel auxiliary α2δ and beta subunits: trafficking and one step beyond. Nat Rev Neurosci 13:542–555. 10.1038/nrn3311 [DOI] [PubMed] [Google Scholar]
- Dudanova I, Sedej S, Ahmad M, Masius H, Sargsyan V, Zhang W, Riedel D, Angenstein F, Schild D, Rupnik M, Missler M (2006) Important contribution of alpha-neurexins to Ca2+-triggered exocytosis of secretory granules. J Neurosci 26:10599–10613. 10.1523/JNEUROSCI.1913-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis SB, Williams ME, Ways NR, Brenner R, Sharp AH, Leung AT, Campbell KP, McKenna E, Koch WJ, Hui A (1988) Sequence and expression of mRNAs encoding the alpha 1 and alpha 2 subunits of a DHP-sensitive calcium channel. Science 241:1661–1664. 10.1126/science.2458626 [DOI] [PubMed] [Google Scholar]
- Ermolyuk YS, Alder FG, Surges R, Pavlov IY, Timofeeva Y, Kullmann DM, Volynski KE (2013) Differential triggering of spontaneous glutamate release by P/Q-, N- and R-type Ca2+ channels. Nat Neurosci 16:1754–1763. 10.1038/nn.3563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA (2009) Gabapentin receptor α2δ-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell 139:380–392. 10.1016/j.cell.2009.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etherton MR, Blaiss CA, Powell CM, Südhof TC (2009) Mouse neurexin-1α deletion causes correlated electrophysiological and behavioral changes consistent with cognitive impairments. Proc Natl Acad Sci U S A 106:17998–18003. 10.1073/pnas.0910297106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felix R, Gurnett CA, De Waard M, Campbell KP (1997) Dissection of functional domains of the voltage-dependent Ca2+ channel alpha2δ subunit. J Neurosci 17:6884–6891. 10.1523/JNEUROSCI.17-18-06884.1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink K, Dooley DJ, Meder WP, Suman-Chauhan N, Duffy S, Clusmann H, Göthert M (2002) Inhibition of neuronal Ca2+ influx by gabapentin and pregabalin in the human neocortex. Neuropharmacology 42:229–236. 10.1016/S0028-3908(01)00172-1 [DOI] [PubMed] [Google Scholar]
- Fuccillo MV, Földy C, Gökce Ö, Rothwell PE, Sun GL, Malenka RC, Südhof TC (2015) Single-cell mRNA profiling reveals cell-type-specific expression of neurexin isoforms. Neuron 87:326–340. 10.1016/j.neuron.2015.06.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Khvotchev M, Krasnoperov V, Goda Y, Missler M, Hammer RE, Ichtchenko K, Petrenko AG, Südhof TC (1998) Neurexin I α is a major α-latrotoxin receptor that cooperates in α-latrotoxin action. J Biol Chem 273:1705–1710. 10.1074/jbc.273.3.1705 [DOI] [PubMed] [Google Scholar]
- Glebov OO, Jackson RE, Winterflood CM, Owen DM, Barker EA, Doherty P, Ewers H, Burrone J (2017) Nanoscale structural plasticity of the active zone matrix modulates presynaptic function. Cell Rep 18:2715–2728. 10.1016/j.celrep.2017.02.064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graf ER, Zhang X, Jin SX, Linhoff MW, Craig AM (2004) Neurexins induce differentiation of GABA and glutamate postsynaptic specializations via neuroligins. Cell 119:1013–1026. 10.1016/j.cell.2004.11.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grauel MK, Maglione M, Reddy-Alla S, Willmes CG, Brockmann MM, Trimbuch T, Rosenmund T, Pangalos M, Vardar G, Stumpf A, Walter AM, Rost BR, Eickholt BJ, Haucke V, Schmitz D, Sigrist SJ, Rosenmund C (2016) RIM-binding protein 2 regulates release probability by fine-tuning calcium channel localization at murine hippocampal synapses. Proc Natl Acad Sci U S A 113:11615–11620. 10.1073/pnas.1605256113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groc L, Lafourcade M, Heine M, Renner M, Racine V, Sibarita JB, Lounis B, Choquet D, Cognet L (2007) Surface trafficking of neurotransmitter receptor: comparison between single-molecule/quantum dot strategies. J Neurosci 27:12433–12437. 10.1523/JNEUROSCI.3349-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannan S, Gerrow K, Triller A, Smart TG (2016) Phospho-dependent accumulation of GABABRs at presynaptic terminals after NMDAR activation. Cell reports 16:1962–1973. 10.1016/j.celrep.2016.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J, Wratten J, Davies A, Dolphin AC (2008) Pharmacological disruption of calcium channel trafficking by the α2δ ligand gabapentin. Proc Natl Acad Sci U S A 105:3628–3633. 10.1073/pnas.0708930105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holderith N, Lorincz A, Katona G, Rózsa B, Kulik A, Watanabe M, Nusser Z (2012) Release probability of hippocampal glutamatergic terminals scales with the size of the active zone. Nat Neurosci 15:988–997. 10.1038/nn.3137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppa MB, Lana B, Margas W, Dolphin AC, Ryan TA (2012) α2δ expression sets presynaptic calcium channel abundance and release probability. Nature 486:122–125. 10.1038/nature11033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichtchenko K, Hata Y, Nguyen T, Ullrich B, Missler M, Moomaw C, Südhof TC (1995) Neuroligin 1: a splice site-specific ligand for beta-neurexins. Cell 81:435–443. 10.1016/0092-8674(95)90396-8 [DOI] [PubMed] [Google Scholar]
- Iwasaki S, Momiyama A, Uchitel OD, Takahashi T (2000) Developmental changes in calcium channel types mediating central synaptic transmission. J Neurosci 20:59–65. 10.1523/JNEUROSCI.20-01-00059.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Lacinová L, Marais E, Hobom M, Hofmann F (1999) Molecular diversity of the calcium channel alpha2δ subunit. J Neurosci 19:684–691. 10.1523/JNEUROSCI.19-02-00684.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koester HJ, Sakmann B (2000) Calcium dynamics associated with action potentials in single nerve terminals of pyramidal cells in layer 2/3 of the young rat neocortex. J Physiol 529:625–646. 10.1111/j.1469-7793.2000.00625.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurshan PT, Oztan A, Schwarz TL (2009) Presynaptic α2δ-3 is required for synaptic morphogenesis independent of its Ca2+-channel functions. Nat Neurosci 12:1415–1423. 10.1038/nn.2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Bischofberger J, Jonas P (2007) Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J Neurosci 27:13420–13429. 10.1523/JNEUROSCI.1709-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin MZ, Schnitzer MJ (2016) Genetically encoded indicators of neuronal activity. Nat Neurosci 19:1142–1153. 10.1038/nn.4359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallmann RT, Elgueta C, Sleman F, Castonguay J, Wilmes T, van den Maagdenberg A, Klugbauer N (2013) Ablation of Ca(V)2.1 voltage-gated Ca2+ channels in mouse forebrain generates multiple cognitive impairments. PloS One 8:e78598. 10.1371/journal.pone.0078598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missler M, Zhang W, Rohlmann A, Kattenstroth G, Hammer RE, Gottmann K, Südhof TC (2003) α-Neurexins couple Ca2+ channels to synaptic vesicle exocytosis. Nature 423:939–948. 10.1038/nature01755 [DOI] [PubMed] [Google Scholar]
- Müller CS, Haupt A, Bildl W, Schindler J, Knaus HG, Meissner M, Rammner B, Striessnig J, Flockerzi V, Fakler B, Schulte U (2010) Quantitative proteomics of the Cav2 channel nano-environments in the mammalian brain. Proc Natl Acad Sci U S A 107:14950–14957. 10.1073/pnas.1005940107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Harada H, Kamasawa N, Matsui K, Rothman JS, Shigemoto R, Silver RA, DiGregorio DA, Takahashi T (2015) Nanoscale distribution of presynaptic Ca2+ channels and its impact on vesicular release during development. Neuron 85:145–158. 10.1016/j.neuron.2014.11.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neupert C, Schneider R, Klatt O, Reissner C, Repetto D, Biermann B, Niesmann K, Missler M, Heine M (2015) Regulated dynamic trafficking of neurexins inside and outside of synaptic terminals. J Neurosci 35:13629–13647. 10.1523/JNEUROSCI.4041-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermair GJ, Szabo Z, Bourinet E, Flucher BE (2004) Differential targeting of the L-type Ca2+ channel alpha 1C (CaV1.2) to synaptic and extrasynaptic compartments in hippocampal neurons. Eur J Neurosci 19:2109–2122. 10.1111/j.0953-816X.2004.03272.x [DOI] [PubMed] [Google Scholar]
- Perez de Arce K, Schrod N, Metzbower SW, Allgeyer E, Kong GK, Tang AH, Krupp AJ, Stein V, Liu X, Bewersdorf J, Blanpied TA, Lucić V, Biederer T (2015) Topographic mapping of the synaptic cleft into adhesive nanodomains. Neuron 88:1165–1172. 10.1016/j.neuron.2015.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese AL, Kavalali ET (2016) Single synapse evaluation of the postsynaptic NMDA receptors targeted by evoked and spontaneous neurotransmission. eLife 5:e21170. 10.7554/eLife.21170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner C, Klose M, Fairless R, Missler M (2008) Mutational analysis of the neurexin/neuroligin complex reveals essential and regulatory components. Proc Natl Acad Sci U S A 105:15124–15129. 10.1073/pnas.0801639105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner C, Runkel F, Missler M (2013) Neurexins. Genome Biol 14:213. 10.1186/gb-2013-14-9-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reissner C, Stahn J, Breuer D, Klose M, Pohlentz G, Mormann M, Missler M (2014) Dystroglycan binding to alpha-neurexin competes with neurexophilin-1 and neuroligin in the brain. J Biol Chem 289:27585–27603. 10.1074/jbc.M114.595413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Burnashev N, Sakmann B, Neher E (2001) Transmitter release modulation by intracellular Ca2+ buffers in facilitating and depressing nerve terminals of pyramidal cells in layer 2/3 of the rat neocortex indicates a target cell-specific difference in presynaptic calcium dynamics. J Physiol 531:807–826. 10.1111/j.1469-7793.2001.0807h.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlick B, Flucher BE, Obermair GJ (2010) Voltage-activated calcium channel expression profiles in mouse brain and cultured hippocampal neurons. Neuroscience 167:786–798. 10.1016/j.neuroscience.2010.02.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E (2000) Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature 406:889–893. 10.1038/35022702 [DOI] [PubMed] [Google Scholar]
- Schneider R, Hosy E, Kohl J, Klueva J, Choquet D, Thomas U, Voigt A, Heine M (2015) Mobility of calcium channels in the presynaptic membrane. Neuron 86:672–679. 10.1016/j.neuron.2015.03.050 [DOI] [PubMed] [Google Scholar]
- Sheng J, He L, Zheng H, Xue L, Luo F, Shin W, Sun T, Kuner T, Yue DT, Wu LG (2012) Calcium-channel number critically influences synaptic strength and plasticity at the active zone. Nat Neurosci 15:998–1006. 10.1038/nn.3129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sons MS, Busche N, Strenzke N, Moser T, Ernsberger U, Mooren FC, Zhang W, Ahmad M, Steffens H, Schomburg ED, Plomp JJ, Missler M (2006) α-Neurexins are required for efficient transmitter release and synaptic homeostasis at the mouse neuromuscular junction. Neuroscience 138:433–446. 10.1016/j.neuroscience.2005.11.040 [DOI] [PubMed] [Google Scholar]
- Südhof TC. (2017) Synaptic neurexin complexes: a molecular code for the logic of neural circuits. Cell 171:745–769. 10.1016/j.cell.2017.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AH, Chen H, Li TP, Metzbower SR, MacGillavry HD, Blanpied TA (2016) A trans-synaptic nanocolumn aligns neurotransmitter release to receptors. Nature 536:210–214. 10.1038/nature19058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong XJ, López-Soto EJ, Li L, Liu H, Nedelcu D, Lipscombe D, Hu Z, Kaplan JM (2017) Retrograde synaptic inhibition is mediated by α-neurexin binding to the α2δ subunits of N-type calcium channels. Neuron 95:326–340.e5. 10.1016/j.neuron.2017.06.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran-Van-Minh A, Dolphin AC (2010) The α2δ ligand gabapentin inhibits the Rab11-dependent recycling of the calcium channel subunit α2δ-2. J Neurosci 30:12856–12867. 10.1523/JNEUROSCI.2700-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voigt A, Freund R, Heck J, Missler M, Obermair GJ, Thomas U, Heine M (2016) Dynamic association of calcium channel subunits at the cellular membrane. Neurophotonics 3:041809. 10.1117/1.NPh.3.4.041809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SS, Held RG, Wong MY, Liu C, Karakhanyan A, Kaeser PS (2016) Fusion competent synaptic vesicles persist upon active zone disruption and loss of vesicle docking. Neuron 91:777–791. 10.1016/j.neuron.2016.07.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Westenbroek RE, Borst JG, Catterall WA, Sakmann B (1999) Calcium channel types with distinct presynaptic localization couple differentially to transmitter release in single calyx-type synapses. J Neurosci 19:726–736. 10.1523/JNEUROSCI.19-02-00726.1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Liu N, He Y, Liu Y, Ge L, Zou L, Song S, Xiong W, Liu X (2018) Improved calcium sensor GCaMP-X overcomes the calcium channel perturbations induced by the calmodulin in GCaMP. Nat Commun 9:1504. 10.1038/s41467-018-03719-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarotskyy V, Gao G, Peterson BZ, Elmslie KS (2009) The timothy syndrome mutation of cardiac CaV1.2 (L-type) channels: multiple altered gating mechanisms and pharmacological restoration of inactivation. J Physiol 587:551–565. 10.1113/jphysiol.2008.161737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarotskyy V, Gao G, Peterson BZ, Elmslie KS (2012) Domain III regulates N-type (CaV2.2) calcium channel closing kinetics. J Neurophysiol 107:1942–1951. 10.1152/jn.00993.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A, Dolphin AC (2015) The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol Rev 67:821–870. 10.1124/pr.114.009654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan H, Stanciauskas R, Stigloher C, Dizon KK, Jospin M, Bessereau JL, Pinaud F (2014) In vivo single-molecule imaging identifies altered dynamics of calcium channels in dystrophin-mutant C. elegans. Nat Commun 5:4974. 10.1038/ncomms5974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Rohlmann A, Sargsyan V, Aramuni G, Hammer RE, Südhof TC, Missler M (2005) Extracellular domains of alpha-neurexins participate in regulating synaptic transmission by selectively affecting N- and P/Q-type Ca2+ channels. J Neurosci 25:4330–4342. 10.1523/JNEUROSCI.0497-05.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]